 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reductive activation and structural rearrangement in superoxide reductase: a combined infrared spectroscopic and computational study†
M.
Horch
*a,
A. F.
Pinto‡
b,
T.
Utesch
a,
M. A.
Mroginski
a,
C. V.
Romão
b,
M.
Teixeira
b,
P.
Hildebrandt
a and
I.
Zebger
*a
aTechnische Universität Berlin, Institut für Chemie, Sekr. PC14, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: marius.horch@gmx.de; ingo.zebger@tu-berlin.de
bInstituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Av. da República (EAN), P-2780-157 Oeiras, Portugal
First published on 29th May 2014
Abstract
Superoxide reductases (SOR) are a family of non-heme iron enzymes that limit oxidative stress by catalysing the reduction of superoxide to hydrogen peroxide and, thus, represent model systems for the detoxification of reactive oxygen species. In several enzymes of this type, reductive activation of the active site involves the reversible dissociation of a glutamate from the proposed substrate binding site at the iron. In this study we have employed IR spectroscopic and theoretical methods to gain insights into redox-linked structural changes of 1Fe-type superoxide reductases, focusing on the enzyme from the archaeon Ignicoccus hospitalis. Guided by crystal structure data and complemented by spectra calculation for an active site model, the main IR difference signals could be assigned. These signals reflect redox-induced structural changes in the first coordination sphere of the iron centre, adjacent loop and helical regions, and more remote β-sheets. By comparison with the spectra obtained for the E23A mutant of Ignicoccus hospitalis SOR, it is shown that glutamate E23 dissociates reversibly from the ferrous iron during reductive activation of the wild type enzyme. Moreover, this process is found to trigger a global conformational transition of the protein that is strictly dependent on the presence of E23. Similar concerted structural changes can be inferred from the IR spectra of related SORs such as that from Archaeoglobus fulgidus, indicating a widespread mechanism. A possible functional role of this process in terms of synergistic effects during reductive activation of the homotetrameric enzyme is proposed.
Introduction
In living systems, the superoxide anion radical, O2˙−, and other reactive oxygen species (ROS) are formed from molecular oxygen by reactions with transition metals or radical species, most notably via incomplete reduction of O2 in the aerobic membrane-bound electron transfer chain. These species may have a beneficial role, e.g. in cell signalling, immune response, and redox homeostasis. On the other hand, increased levels of ROS severely harm cellular systems including DNA damage, lipid peroxidation, oxidation of amino acids, and degradation of protein metal centres. As a consequence, ROS have also been associated with ageing and a wide range of diseases in humans1 and, thus, biochemical reactions and detoxification of these compounds are of general relevance.To counteract oxidative stress, all known organisms harbour molecular systems to detoxify ROS. In particular, the disproportionation of superoxide, yielding O2 and H2O2, is catalysed by superoxide dismutases (SODs),2 which are almost ubiquitously present. Superoxide reductase (SOR) provides an alternative detoxification system in anaerobic and microaerophilic bacteria or archaea (for recent reviews, see ref. 3 and 4) as well as unicellular eukaryotes, such as the human pathogenic protozoan Giardia intestinalis.5 In contrast to SOD, superoxide reductase catalyses only the reduction of superoxide:3–10
| SORred + O2˙− + 2H+ → SORox + H2O2 |
A detailed catalytic cycle,3,9,10 based on an inner sphere mechanism, has been proposed for this process mainly on the basis of pulse radiolysis assays coupled to visible spectroscopy,10–12 corroborated by crystallographic9,13 and spectroscopic14–17 data. Oxidized SOR is re-activated by cellular reductases, such as NAD(P)H oxidoreductases, involving in several cases rubredoxins as electron shuttles,18–21 while H2O2 may be further decomposed by catalase,22 peroxiredoxin,23 or rubrerythrin.24,25 In many cases, however, the physiological electron donors are not known.
Superoxide reductases are non-heme iron metalloproteins, which can be divided into two major classes according to the number of metal centres: neelaredoxin (Nlr), denoted as 1Fe-SOR, and homodimeric desulfoferrodoxin (Dfx), or 2Fe-SOR, both of which were first isolated from anaerobic sulphate reducing bacteria.26,27 In the ferrous state, the iron ion of the catalytic site, named centre II in 2Fe-SORs, is pentacoordinated in a square-pyramidal geometry by four equatorial histidine imidazoles (one via the Nδ and three via the Nε nitrogens) and one apical cysteinyl sulphur donor (see Fig. 1(I)). For the inactive ferric state, however, coordination of the iron is not uniform. For SORs from Pyrococcus (P.) furiosus,28P. horikoshii (unpublished, H. Yamamoto and N. Kunishima, PDB 2HVB), Desulfoarculus (D.) baarsii,29 and Treponema (T.) pallidum,29 a glutamate carboxylate was identified to bind to the sixth coordination site of the ferric ion in a monodentate fashion, thereby forming an octahedral geometry (as shown by X-ray crystallography for both Pyrococcus enzymes and infrared (IR) spectroscopy for the remaining ones). Although this residue is widely conserved, it is absent in several 1Fe-SORs, such as that from Nanoarchaeum equitans,30 indicating that the sixth coordination site of the ferric iron may be vacant or occupied by a solvent molecule in some SORs. In 2Fe-SORs, there is another iron site, named centre I, where the metal ion is coordinated by four cysteines in a desulforedoxin-like, distorted tetrahedral geometry.31 The function of this centre is so far unknown.32 Methanoferrodoxin is a recently discovered type of SOR proposed to contain another C-terminal domain harbouring a [4Fe–4S]2+/1+ cluster in addition to the common mononuclear catalytic site.33
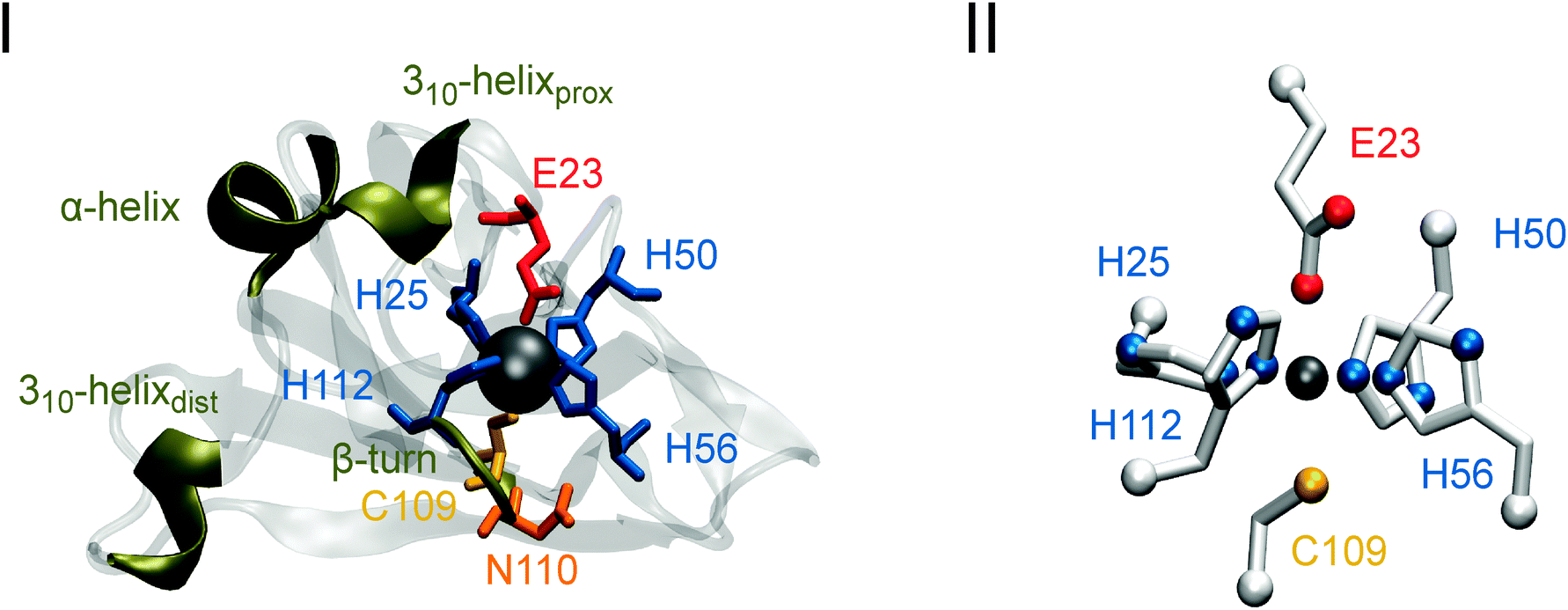 | ||
| Fig. 1 (I) Crystal structure of a single monomer of the I. hospitalis SOR. Essential amino acids (in one-letter code) and secondary structure elements involved in the redox response of the enzyme are highlighted. 310 helices proximal and distal to E23 are marked with ‘prox’ and ‘dist’, respectively. (II) Model compound of the SOR active site used for IR spectra calculations. All ligands are assigned to the respective coordinated amino acids using the nomenclature of the I. hospitalis SOR. Both structures correspond to the ferric state. | ||
IR difference spectroscopy provides information on structural changes of individual amino acid side chains and local perturbations of the peptide backbone in proteins by monitoring spectral differences between two defined states of a chemical species.34–36 This technique is sensitive towards electrostatic interactions, hydrogen bonding, and the protonation state of individual titratable groups, thereby providing detailed insights into bonding and non-bonding interactions complementary to crystallographic data.36 In contrast to the latter, these studies are performed in solution and, thus, allow probing conformational changes that might be obscured by crystal packing. In particular, electrochemically triggered IR spectroscopy monitors structural changes related to redox transitions, e.g. in metalloproteins.29,34–37
In the present study, we have focused on the 1Fe-type SOR from the hyperthermophilic, anaerobic archaeon Ignicoccus (I.) hospitalis, using IR spectroscopic techniques complemented by theoretical methods. A special interest in this enzyme comes from the fact that it lacks a quasi conserved lysine residue that has been proposed to stabilize and/or promote proton transfer to a catalytic reaction intermediate.3,4,11,38,39 Recently, a crystal structure has been solved for the ferric homotetrameric enzyme (see Fig. 1(I), PDB code 4BK8, P. Matias and T. Bandeiras, personal communication). However, in the absence of a conclusive structure for the reduced state, it remained uncertain whether the ferric iron glutamate ligand dissociates reversibly upon reduction of this enzyme (E23 in I. hospitalis SOR; unless otherwise stated, amino acid numbering for this enzyme will be used). In previous studies, electrochemically triggered IR difference spectroscopy was successfully applied to elucidate this issue for 1Fe- and 2Fe-SORs from T. pallidum and D. baarsii, respectively.29 In the present work, we use this technique to investigate reversible glutamate dissociation from the metal centre of the I. hospitalis superoxide reductase by monitoring redox-dependent perturbations of the E23 carboxylate side chain as well as structural changes of other amino acid residues and the protein backbone around the active site. As a reference, we also characterized the E23A mutant of the I. hospitalis SOR, where the active site glutamate is exchanged for alanine, thus excluding any carboxylate binding to the iron. Since the apical glutamate coordinates the iron ion of the 1Fe-type SOR from Archaeoglobus (A.) fulgidus in a reversible manner (PDB code 4BGL, P. Matias and T. Bandeiras, personal communication), this enzyme was characterized for comparison as well.
Experimental procedures
Enzyme overexpression and purification
Recombinant I. hospitalis (wild type and E23A mutant) and A. fulgidus 1Fe-SORs were overexpressed in Escherichia coli and purified as previously described.40,41Sample preparation
Samples were concentrated to 2 mM and buffered at pH 7.5/pD 7.5 in an aqueous (H2O/D2O) solution of 20 mM Tris and 150 mM NaCl using HCl/DCl for pH/pD adjustment. For measurements in D2O, the pD was estimated according to pD = pH meter reading + 0.4.42 To prevent back-exchange with atmospheric water vapour, samples in deuterated solution were kept under Argon during sample preparation. To ensure proper electron transfer in solution sample preparations were incubated with a mixture of the following redox mediators (40 μM each) for 30 min at 4 °C (potentials given vs. standard hydrogen electrode): ferrocene (Em = 422 mV), N,N-dimethyl-p-phenylenediamine (Em = 371 mV), 1,4-benzoquinone (Em = 280 mV), N,N,N′,N′-tetramethyl-p-phenylenediamine (Em = 260 mV), 2,3,4,5-tetramethyl-p-phenylenediamine (Em = 230 mV), phenazine ethosulfate (Em = 55 mV), and duroquinone (Em = 5 mV).Spectroelectrochemistry
IR spectra were recorded with a spectral resolution of 4 cm−1 on a Bruker IFS-66 v/S FTIR spectrometer equipped with a liquid N2 cooled MCT detector and a 5500 cm−1 low-pass filter. 400 scans per single channel spectrum were accumulated and five equivalent difference spectra were averaged for each sample. Samples were held in a temperature controlled (T = 10 °C), optically transparent thin-layer electrochemical (OTTLE) cell (V ∼ 10 μL) equipped with CaF2 windows, a Au mesh working electrode (d ∼ 5 μm), a Ag/AgCl reference electrode, and a Pt sheet as counter electrode.37 To prevent unspecific protein binding and denaturation, the Au mesh was coated with a mixed self assembled monolayer consisting of cysteamine and mercaptopropionic acid for 30 min under Argon atmosphere prior to measurements. Electrochemical control was accomplished using an Eco Chemie B.V. Autolab PGStat12 potentiostat. Measurements were performed under N2 atmosphere. All spectra were processed using the Bruker OPUS software version 5.5.IR spectra calculation
IR spectra were computed for a model of the ferric SOR active site (Fig. 1(II)), monoethanethiolato-monobutyrato-tri(4-ethylimidazole-N1)-mono(4-ethylimidazole-N3)-iron(III), using density functional theory (DFT). Initial geometric parameters of the complex were derived from the crystal structure of the oxidized SOR from I. hospitalis by extracting the active site and substituting the –(NH–Cα–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O))– peptide moieties with methyl groups (CαH3). After adding protons according to neutral pH, geometry optimization and spectra calculations for the model compound were performed on the BP86 level of theory43,44 in Gaussian 03, using the 6-31g* and tzvp basis sets45 for H, C, N, O, S atoms and the Fe atom, respectively. Cartesian coordinates of the Cα atoms of the coordinated amino acids were fixed in order to compensate for the missing protein backbone rigidity, thereby preserving a native-like framework for the active site geometry. Calculated IR spectra were plotted using Gaussian-shaped bands with a half-width at half height of 4 cm−1.
O))– peptide moieties with methyl groups (CαH3). After adding protons according to neutral pH, geometry optimization and spectra calculations for the model compound were performed on the BP86 level of theory43,44 in Gaussian 03, using the 6-31g* and tzvp basis sets45 for H, C, N, O, S atoms and the Fe atom, respectively. Cartesian coordinates of the Cα atoms of the coordinated amino acids were fixed in order to compensate for the missing protein backbone rigidity, thereby preserving a native-like framework for the active site geometry. Calculated IR spectra were plotted using Gaussian-shaped bands with a half-width at half height of 4 cm−1.
Aliphatic index and hydrophobicity estimation
Both quantities were determined using the ProtParam tool as implemented on the ExPASy – SIB Bioinformatics Resource Portal (http://web.expasy.org/protparam). The aliphatic index was calculated according to:46| aliphatic index = XA + a·XV + b·(XI + XL) |
Calculation of protein packing density
Overall and local protein packing densities of the ferric I. hospitalis SOR crystal structure were determined to a grid distance of 0.01 Å by an improved Voronoi cell algorithm48 as implemented in the Voronoia software package49 using standard atomic radii and volumes.50Secondary structure evaluation
Localization and quantification of hydrogen bonds was accomplished using the Define Secondary Structure of Proteins (DSSP) algorithm51 as implemented in the program of the same name.Determination of solvent accessibility
After protonating the crystal structure of the ferric I. hospitalis SOR according to neutral pH, solvent accessibility was determined by calculating the solvent accessible area (SAA) of each individual atom using Visual Molecular Dynamics (VMD) 1.8.6.52 An SAA map was generated for the whole enzyme, assuming an average molecular radius of 1.4 Å for water in the screening procedure.Calculation of collective protein motions
Atom displacements were determined for the first 100 low-frequency modes of the ferric I. hospitalis SOR using normal mode analysis in concert with an all-atom elastic network model, as implemented in the Normal Mode Analysis, Deformation and Refinement (NOMAD-Ref) server.53 Distance-dependent atomic interactions were modelled from a global elastic constant of 100 kcal mol−1 Å−2 by means of an exponential decay weighting function, using a distance-weight and cut-off parameter of 5 and 10 Å, respectively.Results and discussion
Analysis of iron ligand modes
To elucidate whether E23 binds reversibly to the ferric iron of I. hospitalis SOR, we first focused on changes in the normal modes of this amino acid. The symmetric and antisymmetric stretching modes of the carboxylate side chain, which appear at ∼1400 cm−1 [νs(COO)] and ∼1560 cm−1 [νas(COO)], respectively,54,55 are sensitive to bonding and non-bonding interactions, such that anionic, protonated, H-bonded and different metal-bound forms can be distinguished by vibrational spectroscopy.56 Monodentate metal binding is generally indicated by an increase of νas(COO) and a decrease of νs(COO) relative to the non-coordinated state, leading to an increased frequency difference Δν between these two vibrational modes.54 Trace A of Fig. 2 shows the IR difference spectrum [reduced–oxidized] of SOR from I. hospitalis, where bands with a positive sign are related to the ferrous form, while those with a negative sign are due to the ferric state. Possible candidates for νas(COO) of E23 are detected at 1554/1546 cm−1 (−/+), while bands at 1409/1386 cm−1 (+/−) may (partly) originate from the respective symmetric stretching mode. Both difference signals are absent in the spectrum of the E23A mutant protein (Fig. 2B), but observed for the wild type SOR from A. fulgidus (Fig. 2C), supporting their assignment to binding/dissociation related perturbations of the terminal COO− group of E23. These findings are in line with previous studies comparing IR spectroscopic data of D. baarsii WT SOR to the corresponding E47A mutant.29 Thus, the observed increase of Δν between νas(COO) and νs(COO) upon oxidation indicates monodentate binding of E23 to the ferric non-heme iron of SOR from I. hospitalis.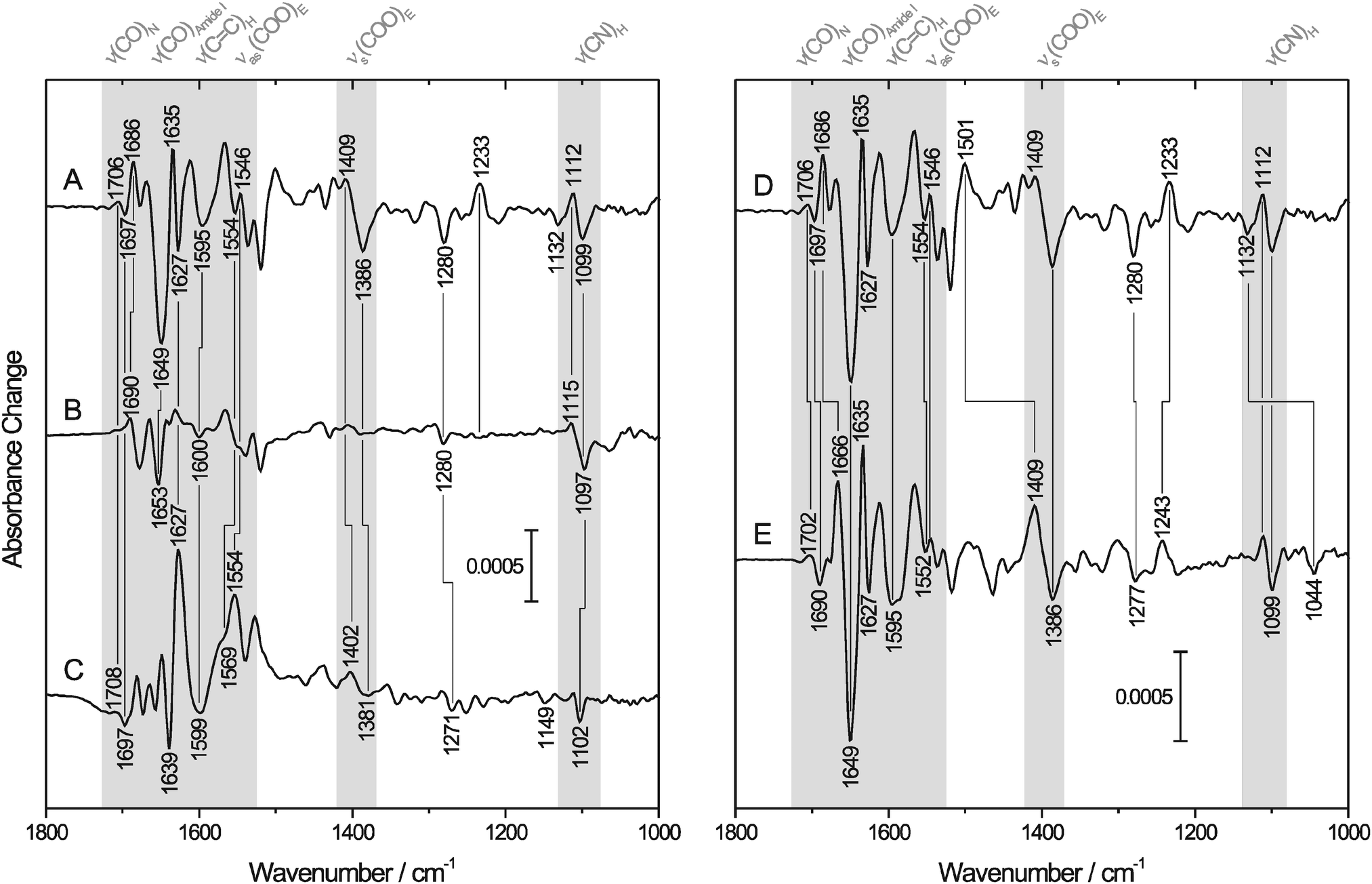 | ||
| Fig. 2 IR difference spectra [reduced–oxidized] of SOR. Positive and negative bands correspond to the ferrous and ferric state of the enzyme, respectively. Spectral regions of particular interest are highlighted in gray. Normal modes of important amino acids (in one-letter code) and the peptide backbone are indicated. For a detailed band assignment and further information, see Table 1. (Left) IR difference spectra recorded from aqueous solutions of (A) wild type SOR and (B) the E23A mutant protein from I. hospitalis as well as (C) the wild type SOR from A. fulgidus. (Right) IR difference spectra of the wild type I. hospitalis SOR recorded in (D) H2O and (E) D2O buffer. | ||
Redox-linked spectral changes of wild-type I. hospitalis SOR (Fig. 2A) are not restricted to the carboxylate side chain of E23 but also include bands attributable to other amino acid residues and the peptide backbone (Table 1). Some of these difference signals are located in crowded regions of the spectrum such that the pattern of conjugate positive and negative signals is partially obscured by overlapping bands of opposite sign and different amplitudes. These difference signals are well observed for the I. hospitalis SOR wild type enzyme (Fig. 2A), but virtually absent or less pronounced in the corresponding spectrum of the E23A mutant (Fig. 2B), revealing a significant involvement of E23 in the redox-related structural changes of the wild type enzyme. In particular, bands at 1595 (−), 1280 (−), 1233 (+), 1112 (+), and 1099 cm−1 (−) can be assigned to histidine side chain modes,56–58 suggesting perturbations of the iron histidine ligands, H25, H50, H56, and/or H112 (the only histidines of the I. hospitalis SOR, see Table 1). Comparable features, as also observed in the difference spectrum of the A. fulgidus SOR (Fig. 2C), have been monitored previously during redox-dependent glutamate binding/dissociation in SORs from T. pallidum and D. baarsii,29 indicating that these bands are general markers for changes in the coordination number and geometry of the active site, caused by the binding/dissociation of the glutamate (here E23). As a consequence, these bands are likely to monitor the binding of other ligands as well and, thus, may provide valuable insights into changes of the active site (electronic) structure resulting from substrate binding and conversion.
| Assignment | Protein | Band position (H2O)/cm−1 | Band position (D2O)/cm−1 | Ref. |
|---|---|---|---|---|
| E, νs(COO) | Ih WT | 1386(−)/1409(+) | 1386(−)/1409(+) | 54–56 |
| Af WT | 1381(−)/1402(+) | n.a. | ||
| E, νas(COO) | Ih WT | 1546(+)/1554(−) | 1546(+)/1552(−) | 54–56 |
| Af WT | 1554(+)/1569(−) | n.a. | ||
| H, ν(CN) | Ih WT | 1099(−)/1112(+) | 1099(−)/1112(+) | 56–58 |
| Ih E23A | 1097(−)/1115(+) | 1098(−)/1113(+) | ||
| Af WT | 1102(−) | 1102(−) | ||
| H, δ(CH + NH) + ν(CN) | Ih WT | 1233(+) | 1243(+) | 56 |
| H, ν(CN + CC) or γt(CH2) | Ih WT | 1280(−) | 1277(−) | 56 |
| Ih E23A | 1280(−) | 1280(−) | ||
| Af WT | 1271(−) | n.a. | ||
| H, ν(CC) |
Ih WT | 1595(−)/1580(−) | 1595(−)/1586(−) | 56–58 |
| Ih E23A | 1600(−)/1581(−) | 1597(−)/1588(−) | ||
| Af WT | 1599 | 1599 | ||
| N, ν(CO) |
Ih WT | 1697(−)/1706(+) | 1690(−)/1702(+) | 59–61 |
| Af WT | 1697(−)/1708(+) | 1687(−)/1693(+) | ||
| N, γr(NH2) | Ih WT | 1132(−) | 1044(−) | 83 |
| Af WT | 1149(−) | <1000 | ||
| Aliphatic amino acids, δs(CH3) | Ih WT | 1386(−) | 1386(−) | 56 |
| Helices, amide I | Ih WT | 1649(−) | 1649(−) | 62 |
| Ih E23A | 1653(−) | 1653(−) | ||
| Turn, amide I | Ih WT | 1686(+) | 1666(+) | 62 |
| Ih E23A | 1690(+) | n.a. | ||
| Turn, amide II | Ih WT | 1501(+) | 1409(+) | |
| β-Sheets, amide I | Ih WT | 1635(+)/1627(−) | 1635(+)/1627(−) | 62 |
| Ih WT | 1678(−)/1669(+) | 1678(−)/1669(+) | ||
| Af WT | 1639(−)/1627(+) | 1639(−)/1627(+) |
Evaluation of vibrational markers of the first coordination sphere of the active site Fe has clearly shown reversible dissociation of E23 from this metal ion. This is an important finding since this process is essential to the enzyme's catalytic function requiring a vacant coordination site for substrate binding and interconversion. Notably, glutamate displacement was not clearly observed in preliminary crystal structure data of the reduced enzyme, possibly due to crystal packing effects. Consequently, we have also turned our attention to further redox-related structural changes of the enzyme that might be restrained in the crystal state as well.
Redox-linked structural changes beyond the first coordination sphere
IR difference spectroscopy also provides evidence that reductive dissociation of E23 triggers extended structural changes of I. hospitalis WT SOR, as proven by the absence of the corresponding spectroscopic markers in the spectrum of the E23A mutant (Fig. 2A and B). In this respect, the slightly H/D sensitive difference signal at 1706/1697 cm−1 (+/−) is assigned to a mode dominated by the stretching of the carboxamide group of an asparagine side chain59–61 (Fig. 2A, D and E and Table 1), most likely N110 (see Fig. 1(I)). This amino acid is also present in the A. fulgidus SOR (N111) and, thus, a corresponding difference signal at 1708/1697 cm−1 (+/−) is observed in the [red–ox] difference spectrum of this enzyme (Fig. 2C). Since this mode exhibits a frequency close to that of the backbone CO stretching and NH bending (amide I) it may have contributions from these latter vibrations, as demonstrated for the model peptide Glu–Asn–Glu (for an animation, see ESI,† S1). Consequently, this mode may serve as a probe for changes of the N110 peptide bond and, thus, for a local perturbation of the protein backbone. In I. hospitalis SOR, N110 is located in a β-turn and adjacent to H112 and C109, both of which are ligands to the iron (see Fig. 1(I)). Hence, it is plausible to assume that the 1706/1697 cm−1 difference signal (+/−) reflects a redox-dependent rearrangement of the β-turn, mediated by structural changes in H112 and C109 as discussed above. This hypothesis is supported by a pronounced band at 1686 cm−1 (+), which is tentatively assigned to changes in the amide I mode of this β-turn62 (Fig. 2A). Interestingly, the cysteinyl thiolate of the SOR active site is generally considered to exert a strong trans influence on the opposite bond.14,63,64 Therefore, assuming a mutual influence between two trans oriented ligands,65,66 probing of Fe–S stretching vibrations is a powerful means to assess changes in the binding of E23.67 These normal modes are not directly accessible by mid-infrared spectroscopy. However, the above mentioned local perturbations in the β-turn encompassing C109 are an indirect indication for structural changes around this cysteine and, thus, a probe for alterations in the metal binding of E23 and related conformational transitions.
Further structural changes are indicated by a pronounced amide I band at 1649 cm−1 (−), which is assigned to a structural reorganization in at least one of the three consecutive helices at the N-terminus adjacent to E23 (Fig. 1(I)),62 most likely related to the binding/dissociation of this amino acid in the I. hospitalis WT enzyme. Such structural changes are neither observed for the SOR from A. fulgidus (Fig. 2C), whose N-terminus is shorter, nor for those from T. pallidum and D. baarsii,29 since this region corresponds to the linker to the desulforedoxin-like domain in these enzymes. The above findings show that dissociation of E23 involves extended structural changes in I. hospitalis WT SOR especially affecting nearby helical regions as well as the β-turn in trans position of the active site.
Another strong band, observed for the ferric I. hospitalis WT SOR at 1386 cm−1 (−) (Fig. 2A), most likely originates from the δs(CH3) modes of aliphatic amino acid side chains and possibly, to a lesser extent, from the νs(COO) mode of E23 (vide supra). Indeed, several hydrophobic amino acids are found near the active site, all of which are located close to amino acids that undergo structural reorganization upon the enzyme's redox transition. Hence, this band can be taken as a further indication for a concerted structural change around the iron site in I. hospitalis SOR due to reversible dissociation of E23. This rearrangement appears to be a wide spread feature of SORs since similar pronounced bands are also observed in the difference spectra of the enzymes from T. pallidum and D. baarsii,29 while the A. fulgidus SOR exhibits less intense yet numerous features in this spectral range (Fig. 2C). Such a concerted redox-dependent structural change in SOR is also indicated by perturbations of the peptide backbone, observed for all SORs characterized by IR spectroscopy so far (this work and ref. 29).
Hydrogen–deuterium exchange of protein backbone protons
Additional experiments were performed in D2O (Fig. 2E and Table 1) to promote band assignments. Due to limited protein stability, proper evaluation of these experiments was only possible for the I. hospitalis and A. fulgidus WT SORs.Interestingly, large parts of I. hospitalis SOR appear to be excluded from proton exchange under the present experimental conditions, as indicated by an amide II/amide II' ratio of almost one (data not shown). In general, incomplete H/D exchange may be ascribed to high protein packing densities, a large number of hydrophobic amino acids and/or a significant content of H-bonded amide protons involved in secondary structure elements. Thus, evaluation of this observation may provide valuable insights into structural aspects of the enzyme.
To estimate the contribution of hydrophobic amino acids to the incomplete proton exchange, we determined the aliphatic index and the overall hydrophobicity according to the Grand Average of Hydropathy (GRAVY). Compared to other proteins,68 the percentage of hydrophobic amino acids is only slightly increased in this enzyme by 2.6% (see also Fig. 3(I)) and no above-average values46,47 were observed for the aliphatic index (77) and the GRAVY (−0.43). The impact of secondary structure elements on H/D exchange properties can be assessed by quantifying the amount of H-bonds per residue, which was found to be slightly higher (0.69) compared to the average (0.63) of globular proteins of this type and size,69 in line with the absence of large flexible regions (Fig. 3(II)). Most H-bonds are located in the centre of the protein, consistent with a high content of β-sheets in this region (Fig. 3(II)). For the packing density, we found a low70 average value of 0.688. However, local densities strongly vary between 0.17 close to the surface and 0.92 in the core (Fig. 3(III)). Again, the latter region coincides with the central β-sheet pattern and, thus, hindered H/D exchange in I. hospitalis SOR is ascribed to limited solvent accessibility in this highly dense structural motif. This conclusion is in agreement with solvent accessibility calculations (Fig. 3(IV)) and previous observations for other proteins.71 Notably, high packing (and H-bond) densities, as observed in the β-sheet core of the I. hospitalis SOR, have been described as a marker for rigid domains and a prerequisite for non-local structural changes70,72 like those observed in this study.
 | ||
| Fig. 3 Crystal structure of the homotetrameric SOR from I. hospitalis: (I) distribution of hydrophobic amino acids (blue). (II) Location of central β-sheets (blue opaque) and other secondary structure elements (blue transparent) (III) regions exhibiting high (blue), medium (white), and low (red) packing densities. (IV) Areas with low (blue), medium (white), and high (red) solvent accessibility. | ||
For I. hospitalis SOR in D2O, redox-dependent structural changes are displayed in the [red–ox] difference spectrum in trace E of Fig. 2. Although the iron site is solvent exposed, the spectrum largely resembles that obtained in H2O, revealing a few changes of prominent absorptions assigned to the amide I and amide II modes. In particular, the dominating amide I band at 1649 cm−1 (−), ascribed to structural reorganization in at least one of the three helices close to the active site, remains unchanged in position and relative intensity in D2O. Considering the solvent exposed position of these secondary structure motifs, hindered exchange of the corresponding amide protons is most likely related to strong hydrogen bond interactions. This suggests high rigidity of these helices, which in turn may be able to transmit active site structural changes to other parts of the protein (vide supra). The negative band at 1686 cm−1 (in H2O, Fig. 2D) is considerably down-shifted by 20 cm−1 in D2O (trace E), supporting its assignment to the solvent exposed β-turn including C109, N110, and H112 (see Fig. 1(I)). Consistently, the positive band at 1501 cm−1 in H2O, shifted by −92 cm−1 in D2O, likely results from changes in the amide II mode of this β-turn. Additional, unchanged absorptions at 1635/1627 (+/−) and 1678/1669 cm−1 (−/+) are ascribed to amide I modes62 of H/D exchange-resistant β-sheets (vide supra), as also observed for A. fulgidus SOR (Fig. 2C). Redox dependent changes of these dense and rigid structural motifs, remote from the active site, support the view of a collective redox-dependent structural reorganization. Further assignments are indicated in Fig. 2 and Table 1.
Extended redox-linked structural changes
IR difference spectra clearly indicate that SOR undergoes a concerted structural change during reductive activation of the active site. Global conformational changes of this type rely upon collective thermal motions that are well captured in one or few low-frequency normal modes of the entire protein.73 Normal mode analysis based on elastic network models74 provides an efficient tool to gain insights into these large scale movements.75,76 Using this approach, we screened for normal modes that include a considerable change of the OE23–Fe distance in I. hospitalis SOR and, thus, may be involved in redox-dependent domain movements with a possible impact on enzyme activation. Accordingly, all modes fulfilling this requirement match the structural changes probed by IR difference spectroscopy. In particular, normal mode no. 62 (displayed in an animation provided in the ESI,† S2) reveals considerable in-phase movements of loops and helices close to the active site of all four subunits accompanied by a rearrangement of the β-sheet core. As a consequence, this mode may be essential for a guided structural reorganization and promoting interactions of the individual monomers during activation. Since the OE23–Fe stretching approximates the reaction coordinate of the redox-linked structural transition, the associated domain movements also provide a coarse-grained 3D illustration of the conformational changes encoded in the IR difference spectrum.Redox-linked perturbations and H/D exchange of coordinated histidine ligands
H/D shifts have been evaluated for all major bands (see Fig. 2D and E and Table 1). In contrast to expectations,57,58 the histidine side chain modes ν(CN) at 1099/1112 cm−1 (−/+) and ν(CC) at 1595 cm−1 (−) were found to remain largely unchanged in D2O (Fig. 2D and E). Similar observations were made for the A. fulgidus SOR (data not shown), the SOR from T. pallidum, and centre II of the D. baarsii SOR,29 pointing to a general feature of SOR active site histidines. Since these amino acids are highly solvent exposed, this observation cannot be related to a restricted accessibility.
To confirm band assignments and elucidate the intrinsic H/D sensitivity of these modes for the metal-coordinated histidines in SOR, we have carried out DFT calculations for monoethane-thiolato-monobutyrato-tri(4-ethylimidazole-N1)-mono(4-ethylimidazole-N3)-iron(III) (1) (Fig. 1(II)), a model compound designed to mimic essential features of the ferric active site (see Experimental procedures). Spectral regions with essential histidine side chain (imidazole) absorptions are shown in Fig. 4 while selected vibrational modes are listed in Table 2.
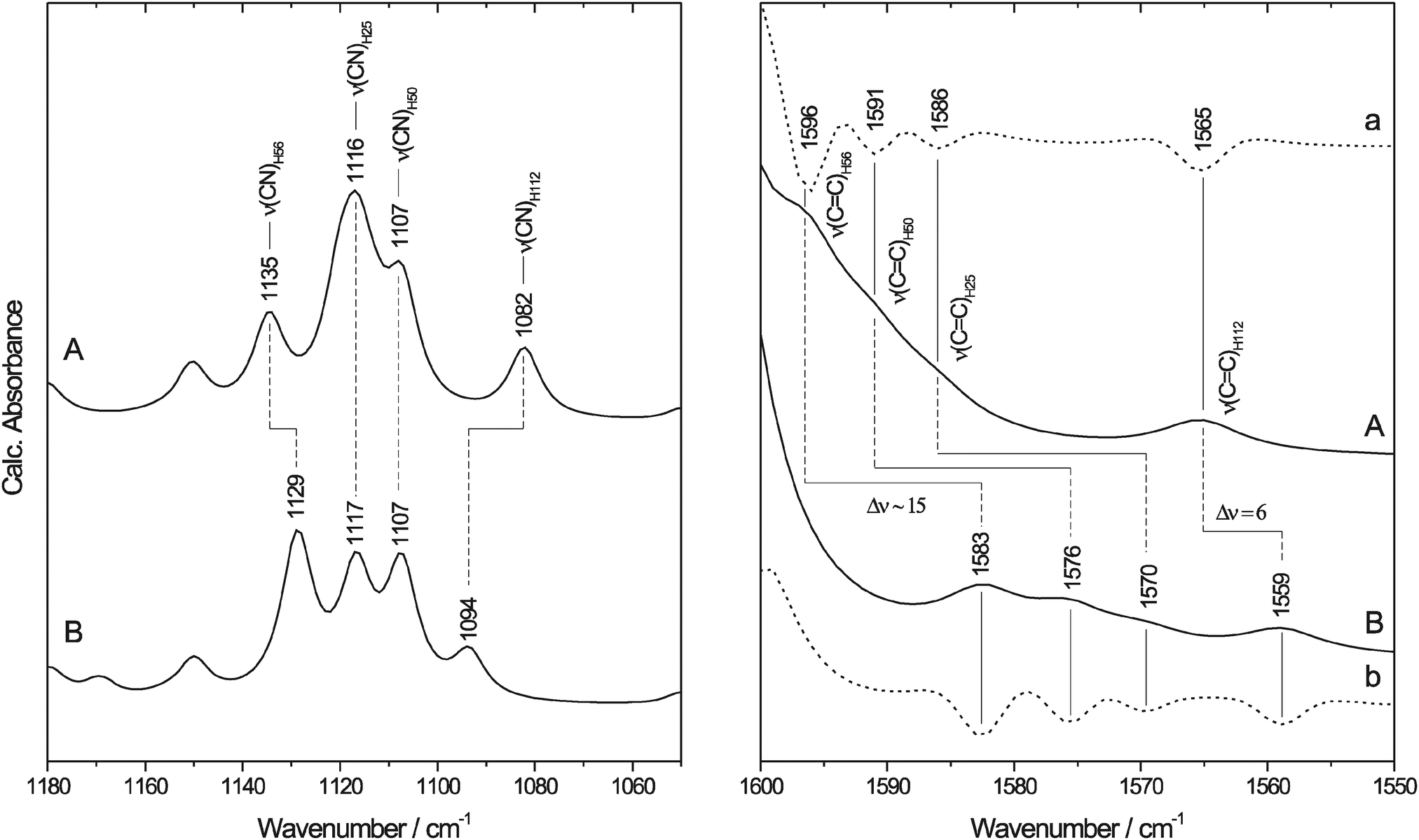 | ||
| Fig. 4 Calculated IR spectra of the SOR model (Fig. 1(II)) with neutral (A) N-protonated and (B) N-deuterated ethyl-imidazole ligands. The spectral region of imidazole ν(CN) and ν(CC) absorptions is enlarged in the left and right panel, respectively. For a better visualisation of the weak bands in the right panel, the second derivatives of the spectra are included (a and b, dotted lines). IR bands are assigned to the corresponding histidine ligands of SOR using the nomenclature of the enzyme from I. hospitalis. | ||
| Assignment | Experiment/in silico | I. hospitalis nomenclature | Band position (H2O)/cm−1 | Band position (D2O)/cm−1 | H/D shift/cm−1 |
|---|---|---|---|---|---|
| E, νs(COO) | Experiment | E23 | 1386(−)/1409(+) | 1386(−)/1409(+) | 0/0 |
| In silico | 1316/1350 | 1315/1350 | −1/0 | ||
| E, νas(COO) | Experiment | E23 | 1546(+)/1554(−) | 1546(+)/1552(−) | 0/−2 |
| In silico | 1605 | 1604 | −1 | ||
| H, ν(CN) | Experiment | ? | 1099(−)/1112(+) | 1099(−)/1112(+) | 0/0 |
| In silico | H25 | 1116 | 1117 | +1 | |
| H50 | 1107 | 1107 | 0 | ||
| H56 | 1135 | 1129 | −6 | ||
| H112 | 1082 | 1094 | +12 | ||
| H, ν(C=C) | Experiment | ? | 1595(−)/1580(−) | 1595(−)/1586(−) | 0/+6 |
| In silico | H25 | 1586 | 1570 | −16 | |
| H50 | 1591 | 1576 | −15 | ||
| H56 | 1596 | 1583 | −13 | ||
| H112 | 1565 | 1559 | −6 |
Imidazole modes with major contributions from the ν(CN) coordinates are centred around 1100 cm−1 in 1, confirming the assignment of experimental bands for the I. hospitalis SOR. Here, the corresponding modes of H56, H25, H50, and H112 are observed at 1135, 1116, 1107, and 1082 cm−1, respectively, resulting in band separations of 9–25 cm−1 in the calculated spectrum (left panel of Fig. 4). Thus, the well-defined difference signal, observed at 1099/1112 cm−1 (−/+) for I. hospitalis SOR, is most likely related to redox-linked perturbations at the level of a single histidine side chain. In agreement with the experimental data, absorption bands with major contributions from the imidazole ν(CC) modes of 1 arise at 1596, 1591, 1586, and 1565 cm−1 for H56, H50, H25, and H112, respectively (right panel of Fig. 4).
Upon replacing the hydrogen atoms at the imidazole nitrogens (Fig. 5) by deuterium (1-D), the ν(CN) modes of the Nδ-protonated imidazoles show only a small (H56) or essentially no change (H25, H50) whereas the Nε-protonated tautomer (H112) exhibits a considerable frequency upshift (left panel of Fig. 4 and Table 2). These results are in good qualitative agreement with experimental data for 4-methylimidazole (4-MeIm).57,77 Thus, solely based on these findings one might assign the H/D insensitive 1099/1112 (−/+) cm−1 signal in the experimental spectrum of I. hospitalis WT SOR to the ν(CN) containing modes of the Nδ-protonated histidines H25, H50, and/or H56. However, ν(CC) modes of both 1-D and (metal-bound) 4-MeIm57,58 exhibit considerable H/D shifts, especially in case of Nδ-protonated imidazole (right panel of Fig. 4). This observation is in contrast to experimental SOR spectra, where these modes are merely changed in D2O. Interestingly, the observed H/D insensitivity of both ν(CN) and ν(CC) modes is a typical characteristic of anionic, deprotonated imidazolate (Im−).57 In view of this finding, a deprotonated histidine (Fig. 5) at the ferric and ferrous active site may be an alternative explanation for the lack of imidazole H/D shifts in I. hospitalis WT and other SORs. Considering the solution pH of 7.5, this would indicate a considerable decrease of the pKa for imidazolate formation (pKa ∼ 14 in solution). Such an effect has been previously observed for metal-bound imidazole and ascribed to pronounced σ-donation and the resulting withdrawal of electron density from the non-coordinated imidazole nitrogen.78,79 Albeit weaker, this effect can be compared to the binding of a second proton (Fig. 5), which lowers the pKa value to approximately 6 in the imidazolium cation (ImH2+). Indeed, transition metal-induced histidine ionization was previously claimed to be functionally relevant79 and proposed to occur in a few enzymes.80–82 Consequently, the possible presence of a deprotonated histidine at the SOR active site requires further investigation, since this type of modification may have a considerable impact on electronic structure and reactivity.
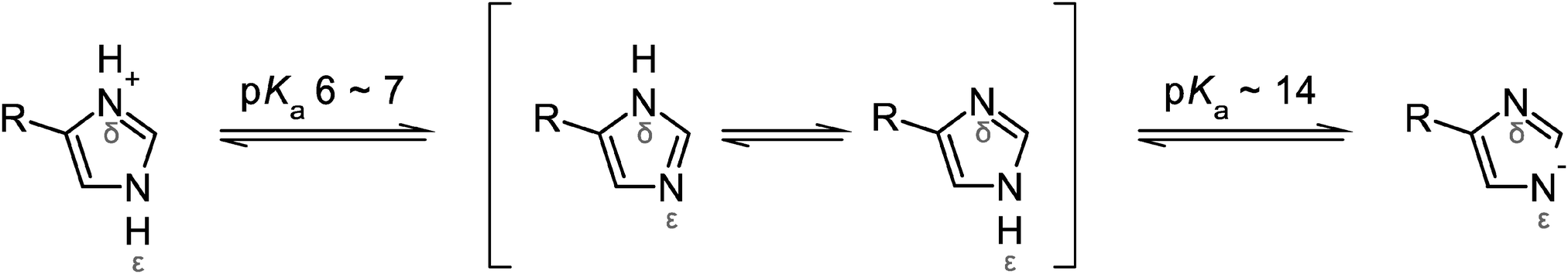 | ||
| Fig. 5 Protonation equilibria and tautomeric forms of the imidazole side chain of histidine. R refers to the NH2–CαH–CH2–COOH moiety. | ||
Conclusions
The redox-linked changes of the I. hospitalis SOR, as monitored by IR difference spectroscopy, indicate the reversible dissociation of glutamate E23 from the active site iron upon reductive activation, thereby enabling substrate binding and transformation. This change of the active site structure is linked to extended conformational perturbations of a nearby loop and especially helical regions. In view of the compact protein structure, inter alia reflected by restricted amide proton exchange, we conclude that the rearrangement of these regions couples local changes of the active site to the observed global movements of the β-sheet core. This interpretation is supported by the analysis of low-frequency modes of the entire enzyme, which are proposed to promote and guide the redox-dependent structural transition. In view of the collective character of these modes, the above mentioned structural changes might integrate the conformational states of the individual subunits, possibly constituting the basis for cooperativity in the enzymatic reductive activation process. While this hypothesis requires further experimental confirmation, the underlying structural transition appears to be a general phenomenon for SORs as indicated by similar observations in the IR difference spectra of enzymes from A. fulgidus and other organisms. In contrast, these characteristic spectroscopic signals are not observed for the E23A mutant from I. hospitalis, demonstrating that the reduction-coupled E23 dissociation from the iron centre constitutes the trigger for protein structure changes that are of potential functional relevance. This latter process may be driven by the high electron density on the ferrous iron, possibly further promoted by a deprotonated histidine ligand.Abbreviations
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| SOR | Superoxide reductase |
| (FT)IR | (Fourier transform) infrared |
| ν s | Symmetric stretching vibration |
| ν as | Antisymmetric stretching vibration |
| δ s | Symmetric bending vibration |
| γ t | Twisting vibration |
| γ r | Rocking vibration |
| DFT | Density functional theory |
| GRAVY | Grand average of hydropathy |
| DSSP | Define secondary structure of proteins |
| SAA | Solvent accessible area. |
Acknowledgements
We would like to thank Dr António Baptista, ITQB, for helpful discussions. We thank also Drs Tiago Bandeiras and Pedro Matias for supplying prior to publication the coordinates for the I. hospitalis and A. fulgidus SOR crystallographic structures. This work was supported by the DFG (Cluster of Excellence “UniCat”), the program “Acções Integradas Luso-Alemãs/DAAD”, Fundação para a Ciência e a Tecnologia (Portugal), grants PEst-OE/EQB/LA0004/2011, and PTDC/BIA-PRO/111940/2009 (to CVR).Notes and references
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44 CrossRef CAS PubMed.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049 CAS.
- A. F. Pinto, J. V. Rodrigues and M. Teixeira, Biochim. Biophys. Acta, 2010, 1804, 285 CrossRef CAS PubMed.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A. Miller, M. Teixeira and J. Selverstone Valentine, Chem. Rev., 2014, 114, 3854 CrossRef CAS PubMed.
- F. Testa, D. Mastronicola, D. E. Cabelli, E. Bordi, L. P. Pucillo, P. Sarti, L. M. Saraiva, A. Giuffre and M. Teixeira, Free Radical Biol. Med., 2011, 51, 1567 CrossRef CAS PubMed.
- S. I. Liochev and I. Fridovich, J. Biol. Chem., 1997, 272, 25573 CrossRef CAS PubMed.
- F. E. Jenney, Jr., M. F. Verhagen, X. Cui and M. W. Adams, Science, 1999, 286, 306 CrossRef.
- M. Lombard, M. Fontecave, D. Touati and V. Niviere, J. Biol. Chem., 2000, 275, 115 CrossRef CAS PubMed.
- G. Katona, P. Carpentier, V. Niviere, P. Amara, V. Adam, J. Ohana, N. Tsanov and D. Bourgeois, Science, 2007, 316, 449 CrossRef CAS PubMed.
- J. V. Rodrigues, I. A. Abreu, D. Cabelli and M. Teixeira, Biochemistry, 2006, 45, 9266 CrossRef CAS PubMed.
- J. P. Emerson, E. D. Coulter, D. E. Cabelli, R. S. Phillips and D. M. Kurtz, Jr., Biochemistry, 2002, 41, 4348 CrossRef CAS PubMed.
- V. Niviere, M. Asso, C. O. Weill, M. Lombard, B. Guigliarelli, V. Favaudon and C. Houee-Levin, Biochemistry, 2004, 43, 808 CrossRef CAS PubMed.
- V. Adam, A. Royant, V. Niviere, F. P. Molina-Heredia and D. Bourgeois, Structure, 2004, 12, 1729 CrossRef CAS PubMed.
- M. D. Clay, F. E. Jenney, Jr., P. L. Hagedoorn, G. N. George, M. W. Adams and M. K. Johnson, J. Am. Chem. Soc., 2002, 124, 788 CrossRef CAS PubMed.
- M. D. Clay, T. C. Yang, F. E. Jenney, Jr., I. Y. Kung, C. A. Cosper, R. Krishnan, D. M. Kurtz, Jr., M. W. Adams, B. M. Hoffman and M. K. Johnson, Biochemistry, 2006, 45, 427 CrossRef CAS PubMed.
- C. Mathe, V. Niviere, C. Houee-Levin and T. A. Mattioli, Biophys. Chem., 2006, 119, 38 CrossRef CAS PubMed.
- C. Mathe, T. A. Mattioli, O. Horner, M. Lombard, J. M. Latour, M. Fontecave and V. Niviere, J. Am. Chem. Soc., 2002, 124, 4966 CrossRef CAS PubMed.
- M. J. Brumlik and G. Voordouw, J. Bacteriol., 1989, 171, 4996 CAS.
- J. P. Emerson, E. D. Coulter, R. S. Phillips and D. M. Kurtz, Jr., J. Biol. Chem., 2003, 278, 39662 CrossRef CAS PubMed.
- J. V. Rodrigues, I. A. Abreu, L. M. Saraiva and M. Teixeira, Biochem. Biophys. Res. Commun., 2005, 329, 1300 CrossRef CAS PubMed.
- J. V. Rodrigues, L. M. Saraiva, I. A. Abreu, M. Teixeira and D. E. Cabelli, JBIC, J. Biol. Inorg. Chem., 2007, 12, 248 CrossRef CAS PubMed.
- W. G. Dos Santos, I. Pacheco, M. Y. Liu, M. Teixeira, A. V. Xavier and J. LeGall, J. Bacteriol., 2000, 182, 796 CrossRef CAS.
- L. B. Poole, C. M. Reynolds, Z. A. Wood, P. A. Karplus, H. R. Ellis and M. Li Calzi, Eur. J. Biochem., 2000, 267, 6126 CrossRef CAS.
- E. D. Coulter, N. V. Shenvi and D. M. Kurtz, Jr., Biochem. Biophys. Res. Commun., 1999, 255, 317 CrossRef CAS PubMed.
- H. L. Lumppio, N. V. Shenvi, A. O. Summers, G. Voordouw and D. M. Kurtz, Jr., J. Bacteriol., 2001, 183, 101 CrossRef CAS PubMed.
- L. Chen, P. Sharma, G. J. Le, A. M. Mariano, M. Teixeira and A. V. Xavier, Eur. J. Biochem., 1994, 226, 613 CrossRef CAS PubMed.
- I. Moura, M. Bruschi, G. J. Le, J. J. Moura and A. V. Xavier, Biochem. Biophys. Res. Commun., 1977, 75, 1037 CrossRef CAS.
- A. P. Yeh, Y. Hu, F. E. Jenney, Jr., M. W. Adams and D. C. Rees, Biochemistry, 2000, 39, 2499 CrossRef CAS PubMed.
- C. Berthomieu, F. Dupeyrat, M. Fontecave, A. Vermeglio and V. Niviere, Biochemistry, 2002, 41, 10360 CrossRef CAS PubMed.
- J. V. Rodrigues, B. L. Victor, H. Huber, L. M. Saraiva, C. M. Soares, D. E. Cabelli and M. Teixeira, J. Biol. Inorg. Chem., 2008, 13, 219 CrossRef CAS PubMed.
- M. Archer, R. Huber, P. Tavares, I. Moura, J. J. Moura, M. A. Carrondo, L. C. Sieker, J. LeGall and M. J. Romao, J. Mol. Biol., 1995, 251, 690 CrossRef CAS.
- J. P. Emerson, D. E. Cabelli and D. M. Kurtz, Jr., Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3802 CrossRef CAS PubMed.
- C. Kratzer, C. Welte, K. Dorner, T. Friedrich and U. Deppenmeier, FEBS J., 2011, 278, 442 CrossRef PubMed.
- W. Mäntele, Trends Biochem. Sci., 1993, 18, 197 CrossRef.
- F. Siebert, Infrared spectroscopy applied to biochemical and biological problems, in Methods in Enzymology Biochemical Spectroscopy, ed. S. Kenneth, Academic Press, 1995 Search PubMed.
- A. Barth, Biochim. Biophys. Acta, 2007, 1767, 1073 CrossRef CAS PubMed.
- F. Baymann, D. A. Moss and W. Mäntele, Anal. Biochem., 1991, 199, 269 CrossRef CAS.
- M. Lombard, C. Houee-Levin, D. Touati, M. Fontecave and V. Niviere, Biochemistry, 2001, 40, 5032 CrossRef CAS PubMed.
- A. F. Pinto, Reductive scavenging of reactive oxygen species in prokaryotes, PhD thesis, Universidade Nova de Lisboa, 2012 Search PubMed.
- I. A. Abreu, L. M. Saraiva, J. Carita, H. Huber, K. O. Stetter, D. Cabelli and M. Teixeira, Mol. Microbiol., 2000, 38, 322 CrossRef CAS.
- F. G. Pinho, C. V. Romao, A. F. Pinto, L. M. Saraiva, H. Huber, P. M. Matias, M. Teixeira and T. M. Bandeiras, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 605 CrossRef CAS PubMed.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. Ikai, J. Biochem., 1980, 88, 1895 CAS.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105 CrossRef CAS.
- A. Goede, R. Preissner and C. Frömmel, J. Comput. Chem., 1997, 18, 1113 CrossRef CAS.
- K. Rother, P. W. Hildebrand, A. Goede, B. Gruening and R. Preissner, Nucleic Acids Res., 2009, 37, D393–D395 CrossRef CAS PubMed.
- J. Tsai, R. Taylor, C. Chothia and M. Gerstein, J. Mol. Biol., 1999, 290, 253 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
- E. Lindahl, C. Azuara, P. Koehl and M. Delarue, Nucleic Acids Res., 2006, 34, W52–W56 CrossRef CAS PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- S. Yu. Venyaminov and N. N. Kalnin, Biopolymers, 1990, 30, 1243 CrossRef PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141 CrossRef CAS.
- K. Hasegawa, T. a. Ono and T. Noguchi, J. Phys. Chem. B, 2000, 104, 4253 CrossRef CAS.
- K. Hasegawa, T. a. Ono and T. Noguchi, J. Phys. Chem. A, 2001, 106, 3377 CrossRef.
- K. Gerwert, B. Hess, J. Soppa and D. Oesterhelt, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 4943 CrossRef CAS.
- A. Maeda, J. Sasaki, Y. Shichida, T. Yoshizawa, M. Chang, B. Ni, R. Needleman and J. K. Lanyi, Biochemistry, 1992, 31, 4684 CrossRef CAS.
- Y. Cao, G. Varo, A. L. Klinger, D. M. Czajkowsky, M. S. Braiman, R. Needleman and J. K. Lanyi, Biochemistry, 1993, 32, 1981 CrossRef CAS.
- D. M. Byler and H. Susi, Biopolymers, 1986, 25, 469 CrossRef CAS PubMed.
- F. Namuswe, G. D. Kasper, A. A. Sarjeant, T. Hayashi, C. M. Krest, M. T. Green, P. Moenne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2008, 130, 14189 CrossRef CAS PubMed.
- J. A. Kovacs and L. M. Brines, Acc. Chem. Res., 2007, 40, 501 CrossRef CAS PubMed.
- E. M. Shustorovich, M. A. Porai-Koshits and Y. Buslaev, Coord. Chem. Rev., 1975, 17, 1 CrossRef CAS.
- J. K. Burdett and T. A. Albright, Inorg. Chem., 1979, 18, 2112 CrossRef CAS.
- S. Todorovic, J. V. Rodrigues, A. F. Pinto, C. Thomsen, P. Hildebrandt, M. Teixeira and D. H. Murgida, Phys. Chem. Chem. Phys., 2009, 11, 1809 RSC.
- P. McCaldon and P. Argos, Proteins, 1988, 4, 99 CrossRef CAS PubMed.
- A. V. Glyakina, N. S. Bogatyreva and O. V. Galzitskaya, PLoS One, 2011, 6, e28464 CAS.
- F. M. Richards, Annu. Rev. Biophys. Bioeng., 1977, 6, 151 CrossRef CAS PubMed.
- R. Jaenicke, Eur. J. Biochem., 1991, 202, 715 CrossRef CAS PubMed.
- R. Scandurra, V. Consalvi, R. Chiaraluce, L. Politi and P. C. Engel, Biochimie, 1998, 80, 933 CrossRef CAS.
- S. E. Dobbins, V. I. Lesk and M. J. E. Sternberg, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10390 CrossRef CAS PubMed.
- M. M. Tirion, Phys. Rev. Lett., 1996, 77, 1905 CrossRef CAS.
- K. Hinsen, Proteins, 1998, 33, 417 CrossRef CAS.
- I. Bahar and A. J. Rader, Curr. Opin. Struct. Biol., 2005, 15, 586 CrossRef CAS PubMed.
- T. Noguchi, Y. Inoue and X. S. Tang, Biochemistry, 1999, 38, 10187 CrossRef CAS PubMed.
- R. J. Sundberg and R. B. Martin, Chem. Rev., 1974, 74, 471 CrossRef CAS.
- A. Blackman, Reactions of Coordinated Ligands, in Adv. Heterocycl. Chem., ed. R. K. Alan, Academic Press, 1993 Search PubMed.
- G. Smulevich, J. M. Mauro, L. A. Fishel, A. M. English, J. Kraut and T. G. Spiro, Biochemistry, 1988, 27, 5477 CrossRef CAS.
- C. Berthomieu and R. Hienerwadel, Biochemistry, 2001, 40, 4044 CrossRef CAS PubMed.
- R. Hienerwadel and C. Berthomieu, Biochemistry, 1995, 34, 16288 CrossRef CAS.
- P. Dhamelincourt and F. J. Ramirez, Appl. Spectrosc., 1993, 47, 446 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Animated Fig. S1 and S2. See DOI: 10.1039/c4cp00884g |
| ‡ Present Address: Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Scheeles Väg 2, 17177 Stockholm, Sweden. |
| This journal is © the Owner Societies 2014 |