 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile and generic method to improve cathode materials for lithium-ion batteries via utilizing nanoscale surface amorphous films of self-regulating thickness†
Jiajia
Huang
and
Jian
Luo
*
Department of NanoEngineering, Program of Materials Science and Engineering, University of California, San Diego, La Jolla, CA 92093-0448, USA. E-mail: jluo@alum.mit.edu
First published on 6th March 2014
Abstract
As a facile and generic surface modification method, a unique class of surface amorphous films (SAFs) is utilized to significantly improve the rate performance and cycling stability of cathode materials for lithium-ion batteries. These nanoscale SAFs form spontaneously and uniformly upon mixing and annealing at a thermodynamic equilibrium, and they exhibit self-regulating or “equilibrium” thickness due to a balance of attractive and repulsive interfacial interactions acting on the films. Especially, spontaneous formation of nanoscale Li3PO4-based SAFs has been demonstrated in two proof-of-concept systems, LiCoO2 and LiMn1.5Ni0.5O4, which have an equilibrium thickness of ∼2.9 nm and ∼2.5 nm, respectively. At a high discharge rate of 25 C, these Li3PO4-based SAFs improve the discharge capacity by ∼130% for LiCoO2 and by ∼40% for LiMn1.5Ni0.5O4, respectively. Furthermore, these SAFs improve the cycling stability and reduce capacity fading of both LiCoO2 and LiMn1.5Ni0.5O4. At an elevated temperature of 55 °C, Li3PO4-based SAFs can help to maintain ∼90 mA h g−1 discharge capacity of the high-voltage material LiMn1.5Ni0.5O4 after 350 cycles at a relatively high charge–discharge rate of 1 C. Further mechanistic studies suggest that these SAFs reduce the interfacial charge transfer resistance and suppress the growth of the solid–electrolyte interphase. This facile method can be utilized to improve a broad range of cathode and anode materials. A thermodynamic framework is proposed, which can be used to guide future experiments of other material systems.
1. Introduction
Surface coatings and modifications are widely used to improve the performance of electrode materials for lithium-ion batteries. Notably, a series of recent studies have demonstrated that nanoscale surface oxide coatings made by atomic layer deposition (ALD) can improve the cycling stability and other performance properties of cathode materials.1–8 Although ALD can be used to make uniform nanoscale oxide coatings with high levels of control, this technique requires special equipment. Alternatively, numerous prior studies attempted to “coat” battery materials by mixing the active materials with coating materials by wet chemistry methods or simply dry mixing, where the specimens were typically subjected to subsequent annealing.9–27 It is hoped that uniform nanoscale surface coatings might form, which were not always guaranteed. Consequently, substantial trials and errors were required and the success of such an approach largely depended on luck.This study aims to establish an innovative coating strategy through a facile “mixing and annealing” route via utilizing a unique class of equilibrium-thickness surface amorphous films (SAFs). Compared with conventional approaches, these nanoscale SAFs form spontaneously with self-regulating and uniform thickness. Using LiCoO2 and LiMn1.5Ni0.5O4 as two proof-of-concept systems, we have demonstrated that nanoscale Li3PO4-based SAFs can form these spontaneously and uniformly at thermodynamic equilibria, which have subsequently improved the rate performance and cycling stability of the two cathode materials via reducing the interfacial charge transfer resistance and suppressing the growth of the solid–electrolyte interphase (SEI).
This study was primarily motivated by the discovery of the thermodynamic stabilization of nanoscale SAFs in a variety of oxide systems.28–36 These SAFs are free-surface counterparts to a class of equilibrium-thickness intergranular films (IGFs) that have been widely observed at ceramic grain boundaries and metal–oxide interfaces.35–40 Thermodynamically, these equilibrium-thickness SAFs (or IGFs) can be considered as two-dimensional surface (or interfacial) “phases,” which have been named as “complexions” by Tang, Carter and Cannon based on arguments that they are not “phases” according to the rigorous Gibbs definition and they cannot exist without abutting bulk phases.35,41–46 These SAFs (and analogous IGFs) have several distinct characteristics. First, they form spontaneously by mixing and annealing at a thermodynamic equilibrium. Second, they adopt a self-regulating or “equilibrium” thicknesses on the order of 1 nm. Third, they are neither fully crystalline nor completely amorphous (despite being called “amorphous” films). Fourth, they can possess structures and compositions that are neither found nor stable as bulk phases (e.g., the average film composition can lie in a bulk miscibility gap). Fifth, they can form at a thermodynamic equilibrium when the corresponding bulk liquid or glass phase is no longer stable. Thus, they can be utilized to achieve superior properties unattainable by conventional bulk phases or nanomaterials.28
In 2005, Li and Garofalini47 first suggested that such nanoscale “amorphous” interfacial films can act as rapid Li ion transport pathways via molecular dynamics simulations of V2O5. In 2008, De Jonghe and co-workers showed that the formation of 1–4 nm thick, impurity-based IGFs in lanthanum phosphate solid-state electrolytes increased the proton conductivity by more than an order of magnitude.48 Later, nanoscale, phosphate-based IGFs, along with SAFs of similar character, have also been observed in partially-sintered LiFePO4 and LiMn1.5Ni0.5O4 electrodes.29,49,50 In 2009, Tang, Chiang and Carter suggested that nanoscale SAFs can form in LiMPO4 (M = Fe, Mn, Co, Ni) olivines and critically affect the phase transformation during electrochemical cycling via a diffuse-interface (phase-field) model.51 In the same month, Kang and Ceder reported that the formation of a glassy Li4P2O7-like “fast ion-conducting surface phase” (<5 nm) in “off-stoichiometric” LiFePO4 can help to achieve ultrafast discharging.52 Although this report52 led to great excitement, it too resulted in a debate.53,54 A technical comment53 suggested: “There is no reason to believe that Li4P2O7 impurity will coat the particles. Instead, impurities usually form nanoparticles that stick on the surfaces.” The follow-up study by Kayyar et al.29 showed that such coatings can form and they are likely equilibrium-thickness SAFs. In 2012, Chong et al. carefully re-examined Kang and Ceder's material and benchmarked it with carbon-coated LiFePO4; their study confirmed the effects of the Li4P2O7-based “fast ion-conducting surface phase” in improving the rate performance despite that the electronic conductivity was not increased.49
It is also worth noting that a series of prior studies coated amorphous oxides (such as Al2O3, ZnO, Bi2O3, AlPO4, MgO, CoPO4, CeO2, ZrO2, and SiO2) on a variety of cathode particles to improve cycling and rate performances.11–20 These coatings were made by a special solution-based sol–gel coating method. Some of these coatings remained uniform and amorphous after subsequent annealing at 400–600 °C, so that they must be at least metastable. However, it is unknown whether they are true equilibrium SAFs; in fact, many of these systems are unlikely equilibrium SAFs because of the high surface energies of the film-forming oxides (as discussed in the next section). Thus, these surface coatings were likely kinetically stabilized (a.k.a. they were not the equilibrium SAFs that would form spontaneously with self-regulating thickness upon annealing). Nonetheless, this series of studies11–20 demonstrated the great potential of using nanoscale amorphous coatings to improve the performance of cathode materials.
Three more recent studies attempted to coat lithium phosphates on the surfaces of various cathode materials with some success. In 2011, Sun and Dillon showed that Li3PO4-based surficial films could form on LiCoO2 in specimens annealed and quenched from 850 °C, which improved the rate performance; however, the authors noted that the surficial films formed under that specific condition were “not necessarily continuous or constant thickness” and were thicker (∼10 nm) where they were present.22 In 2012, Li et al. coated “nano-Li3PO4” on LiMn2O4 to enhance the cycling stability at an evaluated temperature;25 in their work, the specimens were calcined at a lower temperature of 450 °C, which resulted in a ∼10 nm thick crystalline nano-Li3PO4 phase (presumably not uniform films) on LiMn2O4. In 2013, Chong et al. reported “surface stabilized LiMn1.5Ni0.5O4” by Li4P2O7-based coatings with improved rate capability and cycling stability at room temperature;50 in this study, the surface coatings appear to be uniform and thicker (8–10 nm), which the authors referred to as “a coating layer of Li4P2O7 crystallite coexisting with a little Li3PO4”. It appeared that these phosphate-based coatings obtained under the specific processing conditions used in the three above-mentioned studies were not fully equilibrium SAFs that would from continuously with nearly constant thickness in a typical range of 0.5–5 nm.28
The assembly of the above-discussed recent studies motivated us to use Li3PO4-based SAFs on LiCoO2 and LiMn1.5Ni0.5O4 as two proof-of-concept systems to conduct a systematic and definite study, where we successfully found the processing conditions to form SAFs with a self-regulating (equilibrium) thickness of ∼2.9 nm and ∼2.5 nm, respectively. It is imperative to conduct careful statistical measurements to prove the formation of uniform SAFs with nearly constant (equilibrium) thickness and then demonstrate that such SAFs can be utilized to improve the rate performance and cycling stability; such a critical and definite study has never been conducted before, which warrants the current work.
A further objective of this study is to investigate the underlying mechanisms of how such surface films/phases (regardless they are equilibrium or not) improve the performance of cathode materials. Kang and Ceder proposed that the Li4P2O7-like “fast ion-conducting surface phase” can serve as a “beltway” to effectively improve ion transport because of the one-dimensional lithium ion conduction in LiFePO4.52 Sun and Dillon showed that Li3PO4-based surficial films, although they were not continuous with a constant thickness, significantly improved the rate performance of a more isotropic material, LiCoO2, with two-dimensional ion conduction; thus, they suggested that these films might enhance the rate capability by reducing concentration polarization at the particle surfaces.22 In this study, we found the processing conditions to form significantly more uniform Li3PO4-based SAFs on LiCoO2 with an equilibrium thickness of ∼2.9 nm. Furthermore, we adopted a special technique developed by Creager and co-workers55 to show that the enhanced rate capability is not due to reduction in the concentration polarization; further impedance measurements suggested that these nanoscale SAFs may enhance the rate performance by reducing the interfacial charge transfer resistance. We further demonstrated that Li3PO4-based SAFs can enhance the rate performance of an even more isotropic material, spinel LiMn1.5Ni0.5O4, as well as significantly improve its cycling stability at an elevated temperature by protecting the electrode surfaces and suppressing the SEI growth.
2. Thermodynamic model and theoretical analysis
Here, we first present a thermodynamic model28 to assess the stabilization of Li3PO4-based SAFs in the two proof-of-concept systems and to explain the origin of the equilibrium thickness. This model can also be used to analyze and forecast the formation and stability of equilibrium-thickness SAFs in other battery materials in future studies.A film will spontaneously “coat” on the surface of crystalline electrode particles if replacing a “clean” crystal-vapor surface of the electrode (γ(0)cv) with a film-vapor surface (γfv) and a crystal–film interface (γcf) lowers the free energy:
| γcf + γfv < γ(0)cv. | (1) |
Eqn (1) suggests that we should select a coating material with a lower surface energy than that of the electrode material (γfv < γ(0)cv) in order to form uniform coatings spontaneously. Moreover, the film–electrode interfacial energy (γcf) should be small; consequently, it is easier to make structurally-disordered coatings than crystalline coatings because the incoherent crystal–crystal interfacial energy is typically great. In the following text, we only consider structurally-disordered surface films; thermodynamically, we treat them as an undercooled quasi-liquid; thus, we replace all “f” with “l” in the subscripts (i.e., we rename γlv = γfv and γcl = γcf) in the following text.
We should recognize three important nanoscale wetting phenomena, as follows.28,39 First, as schematically illustrated in Fig. 1(a), a nanometer-thick undercooled liquid film of thickness h can be thermodynamically stabilized on a surface below the bulk solidus line if
| −Δγ ≡ γ(0)cv − (γcl + γlv) > ΔG(vol)amorph·h, | (2) |
 | ||
| Fig. 1 (a) Schematic illustration of the thermodynamic principle for stabilizing a nanometer-thick SAF (being treated as an undercooled quasi-liquid film) below the bulk solidus line. The two micrographs used in this schematic illustration are actual HRTEM images of the LiMn1.5Ni0.5O4 specimens with and without a Li3PO4-based SAF. (b) Schematic illustration of an excess free energy vs. film thickness curve. The equilibrium thickness (heq) corresponds to the minimum in Gx(h), which is determined by a trade-off between the reduction of surface energy as the thermodynamic driving force (Δγ·f(h)) and the free-energy penalty for forming the undercooled liquid (ΔG(vol)amorph·h). | ||
Second, when the quasi-liquid film is nanometer-thick, the abutting crystal will inevitably impose significant partial structural order into the film.28,62 Thus, these SAFs are not fully liquid/amorphous, despite that they were named as surface “amorphous” films.28 Interestingly, recent experimental and theoretical studies suggest that such partial structural order near the crystal–glass interfaces can promote ion transport to achieve higher ionic conductivity than both bulk crystal and glass phases.63,64
Third, when the film thickness is in the nanometer range, short-range, van der Waals (vdW) London dispersion, electrostatic, and other interfacial interactions will arise. Thus, the excess surface free energy can be written as:
| Gx(h) = (γcl + γlv) + ΔG(vol)amorph·h + σshort-range(h) + σvdW(h) + σelec(h) + …, | (3) |
| γ(eq)cv = γSAF = min{Gx(h)} = Gx(heq) ≤ γ(0)cv ≡ Gx(0). | (4) |
The sum of the total interfacial pressure 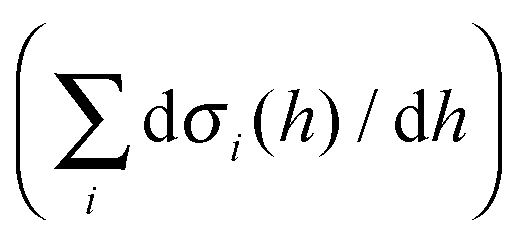 is the well-known Deryaguin disjoining pressure. Quantifying all interfacial interactions in eqn (3) for an oxide or phosphate system is infeasible. Alternatively, we can define a dimensionless interfacial coefficient (f(h)) based on the following equation:
is the well-known Deryaguin disjoining pressure. Quantifying all interfacial interactions in eqn (3) for an oxide or phosphate system is infeasible. Alternatively, we can define a dimensionless interfacial coefficient (f(h)) based on the following equation:
 | (5) |
Then, eqn (3) can be simplified to:
| Gx(h) − γ(0)cv = Δγ·f(h) + ΔG(vol)amorph·h, | (6) |
| −Δγ·f(heq) > ΔG(vol)amorph·heq. | (7) |
By definitions, the dimensionless interfacial coefficient changes from zero to one as the film thickness is varied from zero to infinity, i.e., f(0) = 0 and f(+∞) = 1.
Then, we can use the above framework to assess the possible stabilization of Li3PO4-based, equilibrium-thickness SAFs on LiCoO2 and LiMn1.5Ni0.5O4, the two proof-of-concept systems adopted for this study. On one hand, the reduction in the interfacial energy (represented by Δγ·f(h), which should be negative for SAF formation,) is the thermodynamic driving force to form an SAF. In the current case, first-principle calculations estimated the γ(0)cv to be ∼1–3 J m−2 for LiCoO2 (ref. 68) and ∼1.7–3.1 J m−2 for LiMn1.5Ni0.5O4 (ref. 69), respectively. Thus, we adopt a median value of γ(0)cv ≈ 2 J m−2 for our estimation. In comparison, the crystalline Li3PO4 surface energy was calculated to be ∼0.6–1.2 J m−2 by first-principle calculations,70 and the liquid/amorphous γlv for Li3PO4 should be less because some broken bonds can be satisfied by more relaxation; thus, we adopt the lower end value of the computed crystalline Li3PO4 surface energy for the liquid/amorphous surface energy: γlv ≈ 0.6 J m−2. Since we know that 0 < −Δγ < (γ(0)cv − γlv) ≈ 1.4 J m−2 for SAF formation, we adopt a median value of Δγ ≈ −0.7 J m−2 as a rough estimation for the driving force for stabilizing an SAF in our systems.
On the other hand, there are two major attractive interactions that act to thin (diminish) the SAF. First, an attractive vdW London dispersion force is believed to restrain SAFs and IGFs from thickening above the bulk solidus line.28,30,37,39,40 In the current cases, the specific refractive indices and dielectric constants are not available for estimating the sign and strength of the dispersion force; however, the refractive index of the LiPO3 glass (n ≈ 1.5) is less than those of transition metal oxides (n > 2 for MnO and NiO),71 so the dispersion force is likely to be repulsive and insignificant for the current case.72,73 More importantly, we aimed to form SAFs well below the corresponding bulk solidus lines and prior studies suggested that the dispersion force is typically overwhelmed by the other (second) most common attractive interaction resulted from the ΔG(vol)amorph·h term.28,66 Under such conditions, we can safely neglect the dispersion interaction. Then, we can introduce a thermodynamic parameter (λ) to represent the thermodynamic tendency to stabilize a nanoscale SAF well below the bulk solidus line, as follows:74–76
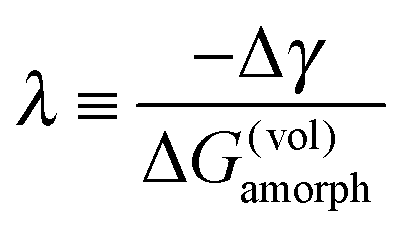 | (8) |
The computed λ scales the actual film thickness. In the current case, where the ΔG(vol)amorph·h term is the dominant term that controls the film thickness well below the bulk solidus line, λ can be used as a first-order estimate of the film thickness. More details about the derivation and justifications of this model and estimation method can be found in earlier studies of analogous subsolidus IGFs in metallic alloys,74–78 where the basic interfacial thermodynamic model and analysis methods are applicable to the current case.
To quantify ΔG(vol)amorph and accurately estimate the film thickness, we need full thermodynamic functions (typically from CALPHAD data) for the multicomponent systems involved,74–78 which are not available for the current case. However, we can roughly estimate this term by using a one-component equation, ΔG(vol)amorph ≈ ΔS(vol)fusion·ΔT, where ΔS(vol)fusion is the volumetric fusion entropy and ΔT is the effective undercooling. There is no reported fusion entropy for Li3PO4. The fusion entropy is ∼23 J mol−1 K−1 for K3PO4, ∼33 J mol−1 K−1 for H3PO4, and ∼17 J mol−1 K−1 for NaPO3 (ref. 79); thus, we estimate the fusion entropy for Li3PO4 to be ∼15–35 J mol−1 K−1 (ref. 79), resulting in ΔS(vol)fusion ≈ ∼3–7 × 105 J m−3 K−1. The melting temperature for Li3PO4 is 1205 °C (ref. 80 and 81). We select annealing temperatures of 600–800 °C to form SAFs so that the effective ΔT ≈ 400–600 K since there is no known intermediate compound or eutectic reaction in either binary system. Subsequently, ΔG(vol)amorph is estimated to be ∼1–4 × 108 J m−3, which is equivalent to an attractive pressure of 100–400 MPa that acts to thin the SAF; this estimate also further justifies that dispersion forces, which are typically on the order of 1–10 MPa for similar cases,28,66 can be safely neglected for the current cases. The actual effective ΔT and ΔG(vol)amorphcan be reduced somewhat if there are some solubilities of other oxide components in the Li3PO4-based liquid or SAFs.
Eqn (2), (7) and Fig. 1(b) show that ΔG(vol)amorph·h is the free-energy penalty to form an SAF, which adds 0.1–0.4 J m−2 per nanometer (of the SAF thickness) to the total excess surface energy in eqn (3) or (6). The analysis above estimates the median value of Δγ to −0.7 J m−2, which provides the thermodynamic driving force that is significant enough for stabilizing an SAF (Fig. 1(b)). Combining the estimation of the driving force and penalty, eqn (8) produces an estimated λ value of ∼2–7 nm (which may be somewhat greater if the actual effective ΔT is less). Furthermore, comparing eqn (7) and (8) produces: heq < λ·f(heq); thus, it is reasonable to estimate the actual equilibrium thickness to be in the low end of the estimated range of 2–7 nm. In the experiments that will be presented subsequently, we have observed the formation of SAFs with equilibrium thicknesses of ∼2.9 nm and ∼2.5 nm, respectively, for the two proof-of-concept systems, which are well consistent with the model prediction.
3. Experimental
3.1 Materials synthesis
To prepare Li3PO4-coated LiCoO2, 2 or 5 vol% of Li3PO4 powder was added to 5 g of as-received LiCoO2 (Alfa Aesar, 99.5%) and dispersed in 10 ml of acetone; the mixture was placed in a silicon nitride grinding vial with two silicon nitride balls. Before sealing the jar, the corprene gasket was taped by Teflon to prevent it from acetone corrosion and precursor contamination. High energy ball milling was carried out using a SPEX 8000D mill for a duration of 10 min followed by a 15 min resting interval, and this milling process was repeated for 3 times. The mixture was dried in a vacuum oven. The dried powder was placed in a covered alumina crucible, isothermally annealed at 600 °C for 4 h in a box furnace with a heating rate of 5 °C min−1, and air quenched. As a reference, controlled specimens of uncoated LiCoO2 were prepared using exactly the same ball milling and annealing procedures described above without the addition of Li3PO4.LiMn1.5Ni0.5O4-based specimens were prepared using a similar procedure. To prepare the reference uncoated specimens, as-received LiMn1.5Ni0.5O4 (Sigma-Aldrich, >99%) was ball milled for 60 min, isothermally annealed at 800 °C for 8 h (with 5 °C min−1 heating rate), and quickly cooled down in the furnace with power shut down. Li3PO4-coated LiMn1.5Ni0.5O4 specimens were prepared by mixing 2 vol% Li3PO4 with the pristine LiMn1.5Ni0.5O4, which were subsequently subjected to the exact same ball milling and heat treatment procedures. Since the as-received LiMn1.5Ni0.5O4 is non-uniform in particle size distribution and agglomerated, an additional reference LiMn1.5Ni0.5O4 specimen was prepared by annealing as-received LiMn1.5Ni0.5O4 at 800 °C for 8 h without prior ball milling.
3.2 Material characterization
X-ray diffraction (XRD) was carried out on a Scintag 2000 diffractometer using Cu Kα radiation (λ = 1.5418 Å) operating at 40 kV and 35 mA with a step size of 0.02° and a step time of 1 s. Particle sizes and morphologies were characterized using a Hitachi 4800 scanning electron microscope (SEM). Particle surfaces were characterized by high-resolution transmission electron microscopy (HRTEM) using a Hitachi 9500 microscope. HRTEM specimens were prepared by dispersing powders ultrasonically in acetone and dropping a small amount of the suspension onto carbon coated copper grids; the specimens were then dried overnight in a desiccator. Minimum exposure was used during HRTEM to reduce electron beam damage. A large number of images of randomly-selected particle surfaces were recorded for each specimen for a fair statistical analysis.3.3 Electrochemical measurements
To prepare cathodes, 80 wt% active materials, 15 wt% carbon black (MTI), 5 wt% PVDF (MTI), and an appropriate amount of NMP (Alfa Aesar, anhydrous, 99.5%) were mixed in a glass vial by a vibrating mixer, followed by further ultrasonic dispersion. The mixture was coated on an aluminum foil, which was subsequently dried in a vacuum oven at 120 °C for 12 h. Cathode electrodes with a diameter of 10 mm were punched out, pressed at ∼187 MPa, and transferred into an Ar-filled glovebox for battery construction. Half cells were made using a cathode electrode, a metal Li chip (MTI, 99.9%) as the anode, 1 M LiPF6 electrolyte, C480 separator (Celgard), and 2032 coin cell cases (SS304, MTI). The detailed procedure of assembling coin cells can be found in ref. 82. To make the electrolyte, EC/DEC/DMC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 vol, BASF) solvent was used for LiCoO2 batteries that were charged up to 4.3 V, and EC/DMC (1:1 vol, BASF) solvent was used for LiCoO2 batteries that were charged up to 4.5 V and LiMn1.5Ni0.5O4 batteries that were charged to 5.0 V. To probe whether the interfacial polarization of the electrolyte near the particle surfaces is the rate control step in LiCoO2-based cathodes, we also prepared and tested coin cells using the 0.1 M LiPF6 electrolyte following a procedure developed by Creager and co-workers.55
:1:1 vol, BASF) solvent was used for LiCoO2 batteries that were charged up to 4.3 V, and EC/DMC (1:1 vol, BASF) solvent was used for LiCoO2 batteries that were charged up to 4.5 V and LiMn1.5Ni0.5O4 batteries that were charged to 5.0 V. To probe whether the interfacial polarization of the electrolyte near the particle surfaces is the rate control step in LiCoO2-based cathodes, we also prepared and tested coin cells using the 0.1 M LiPF6 electrolyte following a procedure developed by Creager and co-workers.55
Electrochemical cycling tests were carried out on an Arbin 2143 tester. The rate capabilities of LiCoO2 were tested at charge and discharge rates of C/5 for 4 cycles, followed by discharging at 1 C, 2 C, 5 C, 10 C, and 25 C sequentially (2 cycles at each discharge rate) while keeping the charge rate at C/5. An external pressure of ∼40 MPa was added on coin cells during rate performance tests to reduce internal contact resistance. The rate performance of LiMn1.5Ni0.5O4 specimens was tested at the discharge rates of C/5, 1 C, 5 C, 25 C, 45 C, 65 C, and 85 C sequentially (1 cycle at each discharge rate) with a constant charge rate of C/5. Before the rate performance test, all fresh LiCoO2 cells were charged and discharged at C/10 for 1 cycle and C/5 for 10 cycles to allow cells reaching a steady state. The cycling stability test of LiCoO2 was performed between 3.0 V and 4.5 V at a rate of 1 C and room temperature after charging and discharging from 3 V to 4.2 V at a rate of C/10 for 1 cycle. The cycling stability of LiMn1.5Ni0.5O4 specimens was measured at a charge and discharge rate of 1 C between 3.2 V and 5.0 V at an elevated temperature of 55 ± 3 °C in an isothermal dry oven after first idling at 55 ± 3 °C for 2 h.
Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed using a Solartron 1287A/1255B analyzer. CV of LiCoO2 was performed between 3.3 V and 4.5 V at a scan rate of 0.1 mV s−1. Electrochemical impedance was measured from 1 MHz to 0.05 Hz at 10 mV. Fresh cells of LiCoO2 with Li metal as the counter electrode were cycled for 4 cycles at a rate of C/5 and finally charged to 4.2 V before the impedance measurements. Impedance measurements were also conducted for LiMn1.5Ni0.5O4 cells that were cycled at room temperature for 10 cycles (before the first measurement) and then cycled at 55 ± 3 °C for 50 additional cycles (before the second measurement). All cells for impedance measurements were kept for more than 10 h after the charging to reach steady states.
4. Results and discussion
4.1 Li3PO4-based equilibrium-thickness SAFs on LiCoO2
XRD revealed only LiCoO2 peaks in both uncoated and 2 vol% Li3PO4 coated LiCoO2 specimens (Fig. S1, ESI†); some crystalline Li3PO4 phase might exist in the coated specimens as the secondary phase, but the amount and crystallinity were below the detection limit of XRD. SEM characterization showed that uncoated and 2 vol% Li3PO4 coated LiCoO2 specimens have similar particle sizes on the order of ∼1 μm (Fig. S2, ESI†).Thin layers were found on the surfaces of uncoated LiCoO2 particles, which were presumably Li2CO3 formed during annealing (that was commonly seen in prior studies29,83). As shown in Fig. 2, thicker and more uniform amorphous films were observed to form on particle surfaces of Li3PO4 added LiCoO2 specimens, in comparison with the reference uncoated LiCoO2 that had been subjected to the exactly the same milling and annealing conditions. The observed surface films in Li3PO4-coated LiCoO2 specimens appeared to be largely “amorphous” in HRTEM imaging (Fig. 2(b)), despite the equilibrium temperature (600 °C) was likely well below the solidus temperature (Tmelt = 1205 °C for Li3PO4 according to ref. 80 and 81 and there is no known deep eutectic reaction between Li3PO4 and LiCoO2). As discussed in Section 2, the stabilization of the “amorphous” or quasi-liquid surface films with large structural disorder was likely driven by the −Δγ term as the formation of crystalline surface films was frustrated by the high crystal–crystal interfacial energy that would occur.
 | ||
| Fig. 2 Representative HRTEM images of the particle surfaces in (a) uncoated and (b) 2 vol% Li3PO4 coated LiCoO2 specimens equilibrated at 600 °C. (c) Measured film thickness vs. volume percentage of Li3PO4 added in LiCoO2 for specimens equilibrated at 600 °C. Each horizontal bar represents the average thickness of an SAF formed on the surface of one particle (measured at multiple points around the particles and averaged). Each solid dot represents the measured mean thickness for the SAFs on all particle surfaces in a specimen and the connecting vertical bar represents ± one standard deviation. Open circles, upper triangles and lower triangles, respectively, represent the medians, lower quartiles and upper quartiles, respectively. These SAFs exhibit a self-limiting thickness that is independent of the excess volume fraction of Li3PO4, after an equilibration is achieved, which unequivocally proved the existence of a thermodynamically-determined equilibrium thickness. | ||
To determine whether the observed SAFs have an equilibrium thickness, a large number of HRTEM images were recorded for randomly selected particle surfaces in three specimens (uncoated, 2 vol% and 5 vol% Li3PO4-coated LiCoO2) that were equilibrated at and quenched from 600 °C, and careful statistical analysis was performed subsequently. The key results are summarized in Fig. 2(c). In the uncoated LiCoO2 specimen, surface (carbonate) films (>∼0.3 nm thick to be clearly discerned by HRTEM) were found on ∼78% particle surfaces among 18 surfaces characterized. The mean film thickness was measured to be 0.88 nm with a large relative standard deviation of 0.75 nm; this specimen was referred to as “uncoated LiCoO2” or “LiCoO2 without SAFs” interchangeably despite the presence of thin carbonate layers. Discernible SAFs were observed on 62 out of 64 (∼97%) independent particle surfaces in the Li3PO4-coated LiCoO2 specimens. In the 2 vol% Li3PO4 added LiCoO2 specimen, the mean film thickness was measured to be 2.90 nm with a standard deviation of 2.17 nm from a population of 35 independent particle surfaces characterized. When the addition of Li3PO4 was increased to 5 vol%, the mean measured film thickness remained at 2.97 nm with a standard deviation of 2.00 nm (measured from 29 independent particle surfaces). The fact that the mean and distribution of the measured film thickness were independent of the extra amount of added Li3PO4 (2 vs. 5 vol%) after reaching equilibration (Fig. 2(c)) unequivocally proved that these Li3PO4-based SAFs exhibited a self-limiting (equilibrium) thickness of ∼2.9 nm, which was thermodynamically-determined (as discussed in Section 2).
As shown in Fig. 3(a), Li3PO4-based SAFs appreciably improved the rate performance of LiCoO2 for specimens tested in the normal 1 M electrolyte. At 25 C, the average discharge capacity was measured to be 25.0 mA h g−1 for the LiCoO2 specimen without SAFs, which was increased by ∼130% to 56.1 mA h g−1 in the 2 vol% Li3PO4-coated LiCoO2 specimen with ∼2.9 nm thick SAFs. Since the SEM measurements showed that both specimens have comparable particles sizes (Fig. S2, ESI†), the comparison of rate performances was fair. Interestingly, the average discharge capacity was measured to be 37.0 mA h g−1 for the 5 vol% Li3PO4 added LiCoO2 specimen, which represented only a ∼50% increase from the uncoated specimen. This result suggested that adding extra amounts of Li3PO4 beyond that was needed for forming equilibrium-thickness SAFs provided no additional benefits; in fact, the extra Li3PO4 existed as a secondary phase, which might cause detrimental sintering and agglomeration effects during the 600 °C annealing. The substantial improvement of the rate performance was consistent with Sun and Dillon's prior results obtained in specimens quenched from 800 °C with thicker and non-uniform surficial films.22
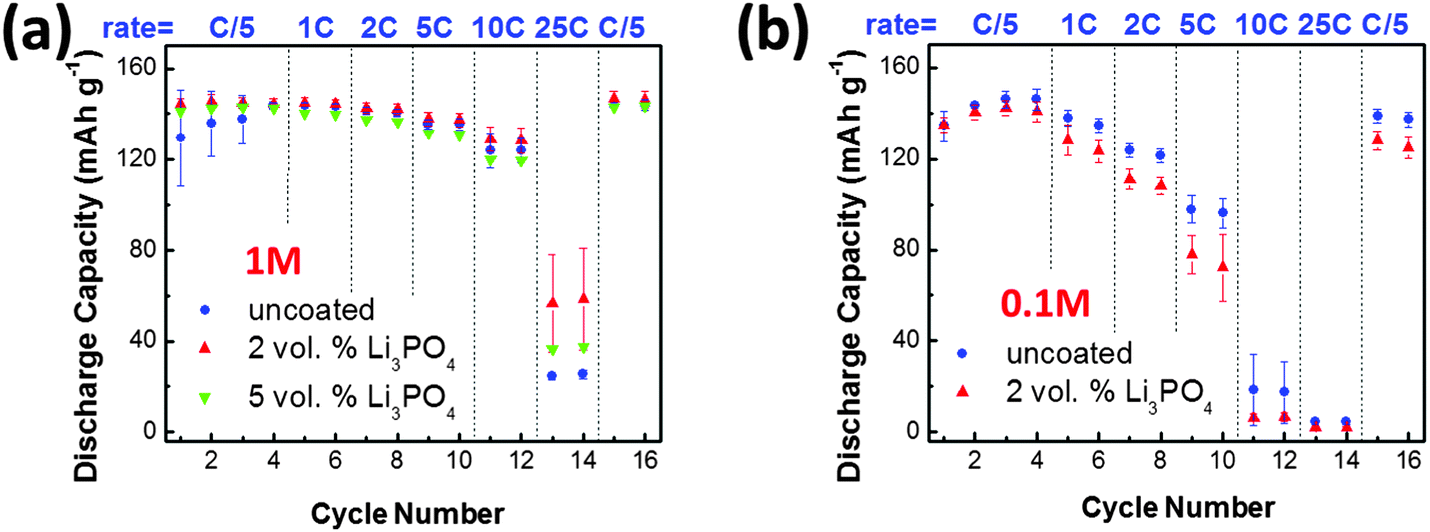 | ||
| Fig. 3 Measured rate performance of uncoated and coated LiCoO2 at the electrolyte concentration of (a) 1 M and (b) 0.1 M, respectively. Three coin cells were made and tested for each condition; the means are presented in the graphs and error bars represent ± one standard deviations. All cells were charged at C/5 and discharged at various rates (labeled in the graphs) at room temperature. | ||
Kang and Ceder proposed that the Li4P2O7-based surface phase improved the rate performance of one-dimensional ionic conducting LiFePO4 by enhancing fast surface ion conduction around the particle.52 Since LiCoO2 is a more isotropic two-dimensional ion conductor, Sun and Dillon suspected that the enhanced rate performance was achieved by reducing concentration polarization of the electrolyte at the participle surfaces.22 In a recent work, Creager and co-workers55 suggested that if the interfacial concentration polarization was the rate limiting step, the benefit would be increased after intentionally lowering the concentration of the electrolyte. Following this idea, we tested our specimens with and without SAFs in the 0.1 M LiPF6 electrolyte for a mechanistic study. As shown in Fig. 3(b), the SAFs became detrimental and lowered the discharge capacity at all rates. This result suggested that the concentration polarization of the electrolyte at the interface was not the rate limiting step for the current case according to Creager and co-workers' theory,55 though we should recognize that Sun and Dillon's specimens had thicker (but non-uniform) surficial films, which might exhibit a greater ability to reduce the concentration polarization of the electrolyte by creating a through-thickness lithium gradient in the film (that was ∼10 nm or ∼3× thicker, according to their HRTEM images) as they suspected.22
EIS measurements were carried out to further understand how SAFs improve the rate performance. The measured spectra are shown in Fig. 4(a) and fitted to an equivalent circuit shown in Fig. 4(b) that was proposed and tested by Liu and Manthiram.14 In this model (Fig 4(b)), RO refers to the Ohmic resistance arising from the electrolyte, internal contact of different cell parts, separator, and cell cases; Rf and Cf refer to film resistance and capacitance of SEI and SAFs, which produce the high-frequency semicircle in the spectra; Rct and Cdl represent the charge transfer resistance and capacitance of the double layer, which produce the middle-frequency semicircle. The fitted Rf and Rct values are summarized in Table 1. It was found that the sum resistance of Rf and Rct inversely correlated with the measured discharge capacities at 25 C (by comparing the last two columns in Table 1), with the following order (decreasing rate performance or increasing Rf + Rct): the coated cathode in the 1 M electrolyte > the uncoated cathode in the 1 M electrolyte > the uncoated cathode in the 0.1 M electrolyte > the coated sample cathode in the 1 M electrolyte, which suggested that (Rf + Rct) might control the rate performance. If this was true, the EIS measurements suggested that the formation of SAFs (at ∼97% of surfaces with an average thickness of 2.9 nm) improved rate performance (at the normal electrolyte concentration of 1 M) largely by reducing interfacial charge transfer resistance (Rct), as shown in Table 1.
 | ||
| Fig. 4 (a) Measured electrochemical impedance spectra of the LiCoO2 specimens with and without Li3PO4-based SAFs, respectively, tested in 1 M and 0.1 M electrolytes, respectively. Dots represent experimental data and solid lines represent fitting curves using (b) an equivalent circuit model that was proposed by Liu and Manthiram.14 | ||
| Specimen | Electrolyte concentration (M) | R f (Ω) | R ct (Ω) | R f + Rct (Ω) | Discharge capacity at 25 C (mA h g−1) |
|---|---|---|---|---|---|
| LiCoO2 with SAFs | 1 | 14.0 ± 0.5 | 26.0 ± 0.2 | 39.9 | 57.6 |
| LiCoO2 without SAFs | 1 | 11.0 ± 1.2 | 37.0 ± 0.5 | 48.1 | 25.0 |
| LiCoO2 without SAFs | 0.1 | 12.1 ± 0.9 | 42.2 ± 0.5 | 54.3 | 4.3 |
| LiCoO2 with SAFs | 0.1 | 24.4 ± 0.7 | 38.8 ± 0.7 | 63.2 | 2.0 |
It is well known that LiCoO2 exhibits good cycling stability at room temperature when it charged up to 4.2 V, but the capacity fades when it is charged to a higher voltage (with the occurrence of a hexagonal–monoclinic–hexagonal phase transition accompanying anisotropic lattice distortion84 along with possibly more significant Co dissolution and HF corrosion). To investigate the effects of SAFs on capacity fading, we measured cycling performances of the LiCoO2 specimens with and without Li3PO4-based SAFs by charging and discharging between 3.0 and 4.5 V for 200 cycles. The measured results suggested that SAFs did appreciably reduce the capacity decaying (Fig. S3, ESI†). It was presumed that SAFs protected the electrode by reducing HF attack and Co dissolution. In the current case, phase transitions and associated strains and fractures likely make significant contributions to the capacity fading,85 which are presumably less effected by SAFs; this is partially confirmed by CV studies of cycling stability mechanism described below.
As shown in Fig. 5(a), CV curves of LiCoO2 specimens with and without Li3PO4-based SAFs after the first cycle indicate that both materials underwent similar Li ion extraction/insertion and phase transition. Specifically, the peaks at 4.20/4.16 V corresponded to the phase transition with large lattice expansion,84 and Fig. 5(a) illustrated that Li3PO4-based SAFs had no effect on suppressing this phase transition. Fig. 5(b)–(d) displays the CV results of these two materials after 10, 50 and 100 cycles, respectively. It was evident that the peak current of LiCoO2 with Li3PO4-based SAFs was higher than that of the reference LiCoO2 specimen without Li3PO4-based SAFs at all cycles, suggesting a protection effect from the SAFs; these CV results were consistent with cycling stability performance shown in Fig. S3 (ESI†). Furthermore, the peak currents of both coated and uncoated LiCoO2 decreased with increasing number of cycles, indicating the performance decaying in both specimens; this was presumably due to the strain accumulation and possible micro-fractures for which SAFs would have little protection effect; these results were also consistent with the gradual decaying of discharge capacities for both materials (Fig. S3, ESI†). Consequently, we conducted a more thorough study to examine the effects of Li3PO4-based SAFs on protecting LiMn1.5Ni0.5O4 at an elevated temperature, where HF attack and Mn dissolution are the major concerns (so SAF formation is more beneficial), which is described in the next section.
 | ||
| Fig. 5 Comparison of cyclic voltammograms of the LiCoO2 specimens with and without Li3PO4-based SAFs after (a) 1 cycle, (b) 10 cycles, (c) 50 cycles, and (d) 100 cycles, which were measured at a scan rate of 0.1 mV s−1 from 3.3 V to 4.5 V. The first charge–discharge cycle was conducted from 3 V to 4.2 V at C/10, and the subsequent cycles were all conducted from 3 V to 4.5 V at 1 C. | ||
4.2 Li3PO4-based equilibrium-thickness SAFs on LiMn1.5Ni0.5O4
XRD characterization of LiMn1.5Ni0.5O4 specimens revealed the presence of minor impurities of LixNi1−xO in the milled and annealed specimens (Fig. S4, ESI†), the formation of which was reported in literature for specimens annealed under similar conditions.86 LiMn1.5Ni0.5O4 was the majority phase (while the amount of LixNi1−xO was fairly minor) and no other impurity phase was detected by XRD (Fig. S4, ESI†). Again, a minor secondary crystalline phase of Li3PO4 might be present in the Li3PO4 added specimens, but below the detection limit of XRD. SEM characterization showed that the particles were well dispersed with comparable and normal distributions of particle sizes (both about 1 μm) for uncoated and coated specimens after milling and annealing, even if the starting powder has a non-uniform particle size distribution and was agglomerated (Fig. S5, ESI†).HRTEM showed the formation of nanoscale Li3PO4-based SAFs in the 2 vol% Li3PO4 added specimen, which appeared to be largely “amorphous” in HRTEM imaging (Fig. 6(b)); this was again consistent with the thermodynamic model presented in Section 2. The statistical results of the film thickness measurements are displayed in Fig. 6(c). On one hand, ultrathin surface layers were identified on 17 out of 19 particle surfaces in the reference uncoated specimen; the mean thickness was measured to be 0.44 nm with a standard deviation of 0.26 nm, which was significantly thinner than the case of uncoated LiCoO2. Again, this specimen is referred to as “uncoated LiMn1.5Ni0.5O4” or “LiMn1.5Ni0.5O4 without SAFs” in the following text despite the presence of thin layers of presumably carbonate. On the other hand, discernable SAFs were observed on 28 out of 29 (∼97%) independent particle surfaces in the 2 vol% Li3PO4 added LiMn1.5Ni0.5O4 specimen that was milled and annealed under identical conditions. The mean film thickness was measured to be 2.53 nm with a standard deviation of 1.31 nm. The measured film thicknesses for the Li3PO4-coated LiMn1.5Ni0.5O4 specimen have a relative narrow distribution (given the possible broadening of the thickness distribution due to anisotropy, which is discussed below), indicating that equilibrium (constant) thicknesses were achieved for these Li3PO4-based SAFs.
 | ||
| Fig. 6 Representative HRTEM images of the particle surfaces in the (a) LiMn1.5Ni0.5O4 and (b) LiMn1.5Ni0.5O4 + 2 vol% Li3PO4 specimens that were ball milled and subsequently annealed at 800 °C for 8 h. (c) The corresponding distributions of measured thicknesses of the surface carbonate layers or Li3PO4-based SAFs for these two specimens, where each bar represents an average thickness measured from multiple points of one particle surface (and each error bar represents + one standard deviation of those multiple measurements, which signifies the uniformity of the surface film). | ||
It is important to note that all reported standard deviations in this study include variations in the equilibrium thicknesses due to anisotropic effects, i.e., the equilibrium thickness is a function of surface orientation as a result of anisotropy in surface/interface energies. Specifically, the surface energy of LiMn1.5Ni0.5O4 was computed to be 1.7 J m−2 for the (111) facet and 3.1 J m−2 for the (110) facet,69 suggesting significant anisotropy. The measured average thickness of ∼2.5 nm and standard deviation of ∼1.3 nm represent the overall mean and distribution of thicknesses for SAFs that form on all different surface orientations. The anisotropic formation and thicknesses of SAFs were reported for Bi2O3 on ZnO and V2O5 on TiO2 systems in prior studies.31,66
The rate performances of LiMn1.5Ni0.5O4 specimens with and without Li3PO4-based SAFs, which were subjected to identical milling and annealing conditions and have comparable particle sizes of ∼1 μm, are summarized in Table 2. Representative discharging curves are displayed in Fig. 7. Similar to the case of LiCoO2, Li3PO4-based SAFs appreciably improved the rate performance of LiMn1.5Ni0.5O4 at all rates. The increases were ∼5–6% at 0.2–5 C, ∼40% at 25 C, and ∼360% at 45 C, respectively, which were presumably due to the formation of ∼2.5 nm thick Li3PO4-based SAFs. Small (but non-zero) discharge capacities of ∼15.4 mA h g−1 at 65 C and ∼7.3 mA h g−1 at 85 C were measured for the specimen with SAFs, while the capacity almost vanished for the specimen without SAFs at the same rates. At the same nominal rate of 25 C, the achieved discharge capacity with SAFs was ∼70.7 mA h g−1 for the LiMn1.5Ni0.5O4 specimen with ∼2.5 nm thick Li3PO4-based SAFs, which was greater than the discharge capacity of 56.1 mA h g−1 for the LiCoO2 specimen with ∼2.9 nm thick Li3PO4-based SAFs, although the percentage increase was less (∼40% for LiMn1.5Ni0.5O4vs. ∼ 130% for LiCoO2 with the SAF formation). LiMn1.5Ni0.5O4 has a cubic spinel structure, which is even more isotropic than the layered LiCoO2. The SAFs still enhanced the rate performance, suggesting that anisotropic ion conduction was not a necessary condition for SAFs to improve the rate performance.22,52
| Rate | Discharge capacity (mA h g−1) | Increased percentage with SAFs (%) | ||
|---|---|---|---|---|
| Annealed only | Without SAFs | With SAFs | ||
| C/5 | 116.9 ± 0.6 | 110.5 ± 0.1 | 116.3 ± 3.1 | 5.2 |
| 1 C | 115.5 ± 1.0 | 109.2 | 114.5 ± 3.3 | 4.8 |
| 5 C | 99.6 ± 8.4 | 96.9 ± 2.4 | 102.6 ± 6.1 | 5.9 |
| 25 C | 58.3 ± 6.0 | 50.1 ± 6.1 | 70.7 ± 7.1 | 41 |
| 45 C | 8.7 ± 5.7 | 7.9 ± 2.7 | 36.3 ± 7.9 | ∼360 |
| 65 C | 3.0 ± 2.9 | 2.2 ± 1.0 | 15.4 ± 3.8 | ∼600 |
| 85 C | 1.3 ± 1.3 | ∼0.2 | 7.3 ± 2.0 | — |
| C/5 | 118.6 ± 1.1 | 112.4 | 118.1 ± 2.3 | 5.1 |
 | ||
| Fig. 7 Comparisons of discharge curves of LiMn1.5Ni0.5O4 specimens without (solid lines) and with (dash lines) Li3PO4-based SAFs. All cells were first charged and discharged at C/10 for 1 cycle and C/5 for 10 cycles to reach steady states before testing at the various discharge rates labeled in the graph. | ||
It is well-known that the high-voltage material LiMn1.5Ni0.5O4 is prone to capacity fading due to Mn dissolution and unstable SEI, particularly at elevated temperatures.87–89 We have measured the cycling stability at an elevated temperature of 55 °C (with a relatively high charge–discharge rate of 1 C) for five cells of LiMn1.5Ni0.5O4 specimens without Li3PO4-based SAFs and five cells of LiMn1.5Ni0.5O4 specimens without Li3PO4-based SAFs, respectively; the results clearly showed that the formation of Li3PO4-based SAFs reduced the capacity fading at 55 °C and improved cycling stability and consistence substantially. As shown in Fig. 8, all five cells of LiMn1.5Ni0.5O4 with Li3PO4-based SAFs produced very consistent and stable cycling behaviors. In contrast, the cycling behaviors of the LiMn1.5Ni0.5O4 specimens without Li3PO4-based SAFs (that was subjected to identical milling and annealing processes) showed large variations in their capacities and capacity fading rates. Two cells of the uncoated LiMn1.5Ni0.5O4 died after ∼150 and ∼250 cycles, respectively; other cells also exhibited lower capacities and greater capacity fading rates. Fig. 8 clearly shows that all five LiMn1.5Ni0.5O4 specimens with Li3PO4-based SAFs consistently exhibited more superior cycling stability than the five reference specimens without Li3PO4-based SAFs. Notably, the LiMn1.5Ni0.5O4 specimen with ∼2.5 nm thick SAFs retained ∼90 mA h g−1 or ∼80% of the initial capacity after 350 cycles at an elevated temperature of 55 °C and a relatively high charge–discharge rate of 1 C, which was an exceptional performance. Fig. S6 (ESI†) further showed that LiMn1.5Ni0.5O4 with ∼2.5 nm thick SAFs also performed better than the as-received LiMn1.5Ni0.5O4 and an additional reference specimen that was annealed at 800 °C without prior ball milling.
 | ||
| Fig. 8 Comparison of the cycling stabilities of five LiMn1.5Ni0.5O4 specimens without Li3PO4-based SAFs and five LiMn1.5Ni0.5O4 specimens with Li3PO4-based SAFs; all ten specimens were ball milled and subsequently annealed at 800 °C for 8 h. All fresh cells were charged and discharged at 1 C at 55 °C. “X” indicates that the battery died at that point. | ||
We further conducted EIS measurements to investigate how SAFs improved the cycling stability. Four electrochemical impedance spectra for the specimens with and without SAFs, respectively, after 10 cycles at room temperature (as the start point before raising the temperature) and after 50 additional cycles at an elevated temperature of 55 °C, respectively, were collected and are shown in Fig. 9. The semicircles in the spectra represented the responses from the film resistance, which were mainly contributed from the SEI formed during the cycling. While the film resistance should always increase with cycling due to the SEI formation, it was clearly evident from Fig. 9 that Li3PO4-based SAFs suppressed the growth of SEI, which was likely related to the improvement of the cycling stability at 55 °C; presumably, this was because the Li3PO4-based SAFs protected the electrode surfaces from HF attacks and reduced Mn dissolution, which resulted in more stable SEI that grew slower with the high-temperature cycling. It is worth noting that Kobayashi et al. already showed that the formation of ∼100 nm thick Li3PO4 films on the surfaces of LiMn1.5Ni0.5O4 protected the electrode from reacting with the solid polymer electrolyte.99 In the current study, Li3PO4-based SAFs (surface coatings) formed spontaneously via a facile mixing and annealing process, with self-regulating (ultra-thin) thicknesses of 2–3 nm, which are significantly thinner than ∼100 nm thick Li3PO4 films made by electrostatic spray deposition in Kobayashi et al.'s study. Nonetheless, these ultra-thin SAFs protected the electrode surfaces while simultaneously improving the rate performance.
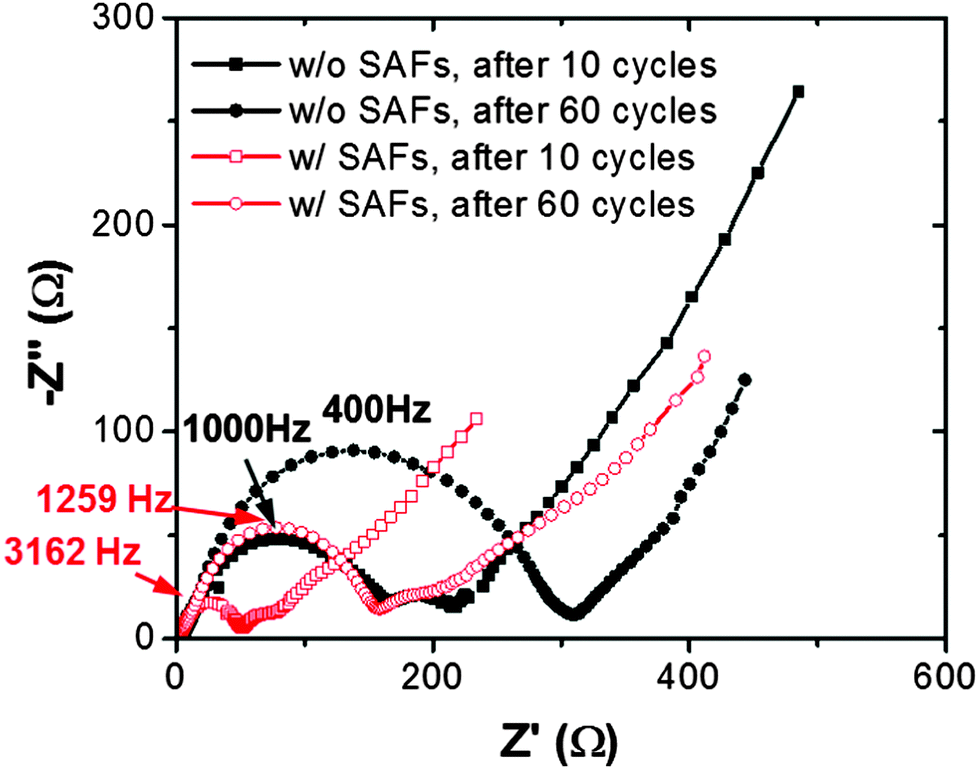 | ||
| Fig. 9 Electrochemical impedance spectra of LiMn1.5Ni0.5O4 specimens with and without Li3PO4-based SAFs. Specimens were firstly charged and discharged at 1 C for 10 cycles at room temperature followed by charging and discharging at 1 C for 50 cycles at 55 °C. | ||
5. Concluding remarks
This study unequivocally demonstrated that equilibrium-thickness SAFs can form spontaneously via a facile mixing and annealing method in selected systems and processing conditions that satisfy certain thermodynamic criteria. A thermodynamic framework is presented, which can be used to forecast the formation and stability of such nanoscale equilibrium-thickness SAFs in a broad range of battery electrode-coating systems if the relevant thermodynamic data are available or can be estimated. Using Li3PO4-based SAFs on LiCoO2 and LiMn1.5Ni0.5O4 as two proof-of-concept systems, we demonstrated that in each system/equilibrium condition, the spontaneously-formed SAFs formed uniformly (on ∼97% of surfaces) with rather a narrow distribution of film thicknesses (around the corresponding thermodynamically-determined “equilibrium” thickness). Adding extra film-forming materials beyond that was needed for forming equilibrium-thickness SAFs did not change the mean and distribution of the measured film thicknesses, which definitely proved that these SAFs have self-limiting (equilibrium) thickness.The formation of SAFs significantly improved the rate performance and cycling stability of both LiCoO2 and LiMn1.5Ni0.5O4. At a high rate of 25 C, SAF formation improved the discharge capacity by ∼130% for LiCoO2 and by ∼40% for LiMn1.5Ni0.5O4, respectively. A specially-designed mechanistic study suggested that interfacial polarization of the electrolyte was not the rate-limiting step for LiCoO2 specimens, and EIS measurements further suggested that SAFs improved the rate performance of LiCoO2 by facilitating interfacial charge transfer. Li3PO4-based SAFs also significantly improved the cycling stability of both cathodes. With the formation of ∼2.5 nm thick equilibrium-thickness SAFs on the high-voltage material LiMn1.5Ni0.5O4, a discharge capacity of ∼90 mA h g−1 has been successfully retained after 350 cycles at an elevated temperature 55 °C and a relatively high charge–discharge rate of 1 C. EIS measurements suggested that Li3PO4-based SAFs reduced the capacity fading and improved the cycling stability and consistence by suppressing the SEI growth.
This study established a facile and generic surface modification method via an innovative use of equilibrium-thickness SAFs (a.k.a. a type of equilibrium surface “phases”) to improve the performance of battery electrodes, and this method can be readily applied to many other cathode as well as anode materials. The key idea is to let thermodynamics make the uniform nanoscale coating for us. On the top of this, a potentially-transformative concept is to utilize surface “phases” or complexions (equilibrium-thickness SAFs as only one example) to achieve superior structures and properties that are unattainable by using conventional bulk phases or normal materials fabrication methods. It is interesting to note that there was spot evidence in recent literature showing that facile heat treatments in controlled chemical environments can sometimes substantially improve the performance of both cathode90–92 and anode93–96 materials for lithium-ion batteries via the formation of surface defects or disordered structures including segregation or adsorption of impurity or doping species91–92,96 (and similar surface “phases” can also be utilized to improved other properties, e.g., catalytic and photocatalytic activities31–32,97,98), though it was unknown whether equilibrium surface “phases” or complexions truly formed (and if so, what were their particular types and characters) in those cases. The success of the current study called for systematic and in-depth studies to explore the innovative concept of utilizing surface “phases” or complexions to achieve distinct surface structures with superior properties that may be unattainable otherwise.
Acknowledgements
This work is financially supported by an NSF grant no. DMR-1006515/DMR-1320615 in the Ceramics program, managed by Dr Lynnette D. Madsen.Notes and references
- X. Meng, X.-Q. Yang and X. Sun, Adv. Mater., 2012, 24, 3589–3615 CrossRef CAS PubMed.
- L. A. Riley, S. Van Ana, A. S. Cavanagh, Y. Yan, S. M. George, P. Liu, A. C. Dillon and S.-H. Lee, J. Power Sources, 2011, 196, 3317–3324 CrossRef CAS PubMed.
- Y. S. Jung, A. S. Cavanagh, A. C. Dillon, M. D. Groner, S. M. George and S.-H. Lee, J. Electrochem. Soc., 2010, 157, A75–A81 CrossRef CAS PubMed.
- I. D. Scott, Y. S. Jung, A. S. Cavanagh, Y. An, A. C. Dillon, S. M. George and S.-H. Lee, Nano Lett., 2011, 11, 414–418 CrossRef CAS PubMed.
- D. Guan, J. A. Jeevarajan and Y. Wang, Nanoscale, 2011, 3, 1465–1469 RSC.
- J. Zhao and Y. Wang, J. Phys. Chem. C, 2012, 116, 11867–11876 CAS.
- J. Zhao and Y. Wang, J. Solid State Electrochem., 2013, 17, 1049–1058 CrossRef CAS PubMed.
- J. Zhao and Y. Wang, Nano Energy, 2013, 2, 882–889 CrossRef CAS PubMed.
- C. Li, H. P. Zhang, L. J. Fu, H. Liu, Y. P. Wu, E. Ram, R. Holze and H. Q. Wu, Electrochim. Acta, 2006, 51, 3872–3883 CrossRef CAS PubMed.
- L. J. Fu, H. Liu, C. Li, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Solid State Sci., 2006, 8, 113–128 CrossRef CAS PubMed.
- J. Liu and A. Manthiram, J. Electrochem. Soc., 2009, 156, A833–A838 CrossRef CAS PubMed.
- J. Liu and A. Manthiram, J. Phys. Chem. C, 2009, 113, 15073–15079 CAS.
- Q. Y. Wang, J. Liu, A. V. Murugan and A. Manthiram, J. Mater. Chem., 2009, 19, 4965–4972 RSC.
- J. Liu and A. Manthiram, Chem. Mater., 2009, 21, 1695–1707 CrossRef CAS.
- Y. Wu and A. Manthiram, Solid State Ionics, 2009, 180, 50–56 CrossRef CAS PubMed.
- J. Liu and A. Manthiram, J. Electrochem. Soc., 2009, 156, A66–A72 CrossRef CAS PubMed.
- Y. Wu, A. V. Murugan and A. Manthiram, J. Electrochem. Soc., 2008, 155, A635–A641 CrossRef CAS PubMed.
- Y. Wu and A. Manthiram, Electrochem. Solid-State Lett., 2006, 9, A221–A224 CrossRef CAS PubMed.
- A. M. Kannan, L. Rabenberg and A. Manthiram, Electrochem. Solid-State Lett., 2003, 6, A16–A18 CrossRef CAS PubMed.
- A. M. Kannan and A. Manthiram, Electrochem. Solid-State Lett., 2002, 5, A167–A169 CrossRef CAS PubMed.
- J. Cho, Y. J. Kim and B. Park, Chem. Mater., 2000, 12, 3788–3791 CrossRef CAS.
- K. Sun and S. J. Dillon, Electrochem. Commun., 2011, 13, 200–202 CrossRef CAS PubMed.
- J. Chong, S. D. Xun, X. Y. Song, G. Liu and V. S. Battaglia, Nano Energy, 2013, 2, 283–293 CrossRef CAS PubMed.
- T. F. Yi, X. Y. Li, H. P. Liu, J. Shu, Y. R. Zhu and R. S. Zhu, Ionics, 2012, 18, 529–539 CrossRef CAS.
- X. W. Li, R. Yang, B. Cheng, Q. Hao, H. Y. Xu, J. Yang and Y. T. Qian, Mater. Lett., 2012, 66, 168–171 CrossRef CAS PubMed.
- K. T. Lee, S. Jeong and J. Cho, Acc. Chem. Res., 2013, 46, 1161–1170 CrossRef CAS PubMed.
- T. F. Yi, Y. R. Zhu, X. D. Zhu, J. Shu, C. B. Yue and A. N. Zhou, Ionics, 2009, 15, 779–784 CrossRef CAS.
- J. Luo and Y.-M. Chiang, Annu. Rev. Mater. Res., 2008, 38, 227–249 CrossRef CAS.
- A. Kayyar, H. J. Qian and J. Luo, Appl. Phys. Lett., 2009, 95, 221905 CrossRef PubMed.
- H. J. Qian, J. Luo and Y. M. Chiang, Acta Mater., 2008, 56, 862–873 CrossRef CAS PubMed.
- H. Qian and J. Luo, Acta Mater., 2008, 56, 4702–4714 CrossRef CAS PubMed.
- H. J. Qian and J. Luo, Appl. Phys. Lett., 2007, 91, 061909 CrossRef PubMed.
- J. Luo, H. Wang and Y.-M. Chiang, J. Am. Ceram. Soc., 1999, 82, 916 CrossRef CAS.
- J. Luo and Y.-M. Chiang, J. Eur. Ceram. Soc., 1999, 19, 697–701 CrossRef CAS.
- M. Baram, D. Chatain and W. D. Kaplan, Science, 2011, 332, 206–209 CrossRef CAS PubMed.
- M. Baram and W. D. Kaplan, J. Mater. Sci., 2006, 41, 7775–7784 CrossRef CAS.
- D. R. Clarke, J. Am. Ceram. Soc., 1987, 70, 15–22 CrossRef CAS.
- R. M. Cannon and L. Esposito, Z. Metallkd., 1999, 90, 1002–1015 CAS.
- J. Luo, Crit. Rev. Solid State Mater. Sci., 2007, 32, 67–109 CrossRef CAS.
- D. R. Clarke, T. M. Shaw, A. P. Philipse and R. G. Horn, J. Am. Ceram. Soc., 1993, 76, 1201–1204 CrossRef.
- M. P. Harmer, Science, 2011, 332, 182–183 CrossRef CAS PubMed.
- M. Tang, W. C. Carter and R. M. Cannon, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 024102 CrossRef.
- W. D. Kaplan, D. Chatain, P. Wynblatt and W. C. Carter, J. Mater. Sci., 2013, 48, 5681–5717 CrossRef CAS.
- P. R. Cantwell, M. Tang, S. J. Dillon, J. Luo, G. S. Rohrer and M. P. Harmer, Acta Mater., 2014, 62, 1–48 CrossRef CAS PubMed.
- J. Luo, H. Cheng, K. M. Asl, C. J. Kiely and M. P. Harmer, Science, 2011, 333, 1730–1733 CrossRef CAS PubMed.
- S. J. Dillon, M. Tang, W. C. Carter and M. P. Harmer, Acta Mater., 2007, 55, 6208–6218 CrossRef CAS PubMed.
- W. Q. Li and S. H. Garofalini, J. Electrochem. Soc., 2005, 152, A364–A369 CrossRef CAS PubMed.
- G. J. Zhang, R. Yu, S. Vyas, J. Stettler, J. A. Reimer, G. Harley and L. C. De Jonghe, Solid State Ionics, 2008, 178, 1811–1816 CrossRef CAS PubMed.
- J. Chong, S. D. Xun, X. Y. Song, P. Ridgway, G. Liu and V. S. Battaglia, J. Power Sources, 2012, 200, 67–76 CrossRef CAS PubMed.
- J. Chong, S. Xun, X. Song, G. Liu and V. S. Battaglia, Nano Energy, 2013, 2, 283–293 CrossRef CAS PubMed.
- M. Tang, H. Y. Huang, N. Meethong, Y. H. Kao, W. C. Carter and Y. M. Chiang, Chem. Mater., 2009, 21, 1557–1571 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- K. Zaghib, J. B. Goodenough, A. Mauger and C. Julien, J. Power Sources, 2009, 194, 1021–1023 CrossRef CAS PubMed.
- G. Ceder and B. Kang, J. Power Sources, 2009, 194, 1024–1028 CrossRef CAS PubMed.
- J. M. Oh, O. Geiculescu, D. DesMarteau and S. Creager, J. Electrochem. Soc., 2011, 158, A207–A213 CrossRef CAS PubMed.
- J. G. Dash and J. S. Wettlaufer, Phys. Rev. Lett., 2005, 94, 235301 CrossRef CAS.
- J. G. Dash, Contemp. Phys., 1989, 30, 89–100 CrossRef CAS.
- J. F. van der Veen, B. Pluis and A. W. Denier, in Chemistry and Physics of Solid Surfaces, ed. R. Vanselow and R. F. Howe, Springer, Berlin, 1988, vol. 7, pp. 455–467 Search PubMed.
- J. W. Cahn, J. Chem. Phys., 1977, 66, 3667–3672 CrossRef CAS PubMed.
- M. Tang, W. C. Carter and R. M. Cannon, Phys. Rev. Lett., 2006, 97, 075502 CrossRef.
- J. Luo, M. Tang, R. M. Cannon, W. C. Carter and Y.-M. Chiang, Mater. Sci. Eng., A, 2006, 422, 19–28 CrossRef PubMed.
- W. D. Kaplan and Y. Kauffmann, Annu. Rev. Mater. Res., 2006, 36, 1–48 CrossRef CAS.
- C. Li, L. Gu and J. Maier, Adv. Funct. Mater., 2012, 22, 1145–1149 CrossRef CAS.
- A. Schirmeisen, A. Taskiran, H. Fuchs, H. Bracht, S. Murugavel and B. Roling, Phys. Rev. Lett., 2007, 98, 225901 CrossRef.
- M. Bobeth, D. R. Clarke and W. Pompe, J. Am. Ceram. Soc., 1999, 82, 1537–1546 CrossRef CAS.
- J. Luo and Y.-M. Chiang, Acta Mater., 2000, 48, 4501–4515 CrossRef CAS.
- R. M. Cannon, M. Rühle, M. J. Hoffmann, R. H. French, H. Gu, A. P. Tomsia and E. Saiz, Ceram. Trans., 2000, 118, 427–444 CAS.
- D. Kramer and G. Ceder, Chem. Mater., 2009, 21, 3799–3809 CrossRef CAS.
- E. Lee and K. A. Persson, Nanotechnology, 2013, 24, 424007 CrossRef PubMed.
- N. D. Lepley, N. A. W. Holzwarth and Y. J. A. Du, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 104103 CrossRef.
- G. V. Samsonov, The Oxide Handbook, Plenum Data Company, New York, 1982 Search PubMed.
- R. H. French, V. A. Parsegian, R. Podgornik, R. F. Rajter, A. Jagota, J. Luo, D. Asthagiri, M. K. Chaudhury, Y. M. Chiang, S. Granick, S. Kalinin, M. Kardar, R. Kjellander, D. C. Langreth, J. Lewis, S. Lustig, D. Wesolowski, J. S. Wettlaufer, W. Y. Ching, M. Finnis, F. Houlihan, O. A. von Lilienfeld, C. J. van Oss and T. Zemb, Rev. Mod. Phys., 2010, 82, 1887–1944 CrossRef.
- R. H. French, J. Am. Ceram. Soc., 2000, 83, 2117–2146 CrossRef CAS.
- J. Luo, J. Am. Ceram. Soc., 2012, 95, 2358–2371 CrossRef CAS.
- X. Shi and J. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 014105 CrossRef.
- J. Luo and X. M. Shi, Appl. Phys. Lett., 2008, 92, 101901 CrossRef PubMed.
- X. Shi and J. Luo, Phys. Rev. Lett., 2010, 105, 236102 CrossRef.
- J. Luo, Curr. Opin. Solid State Mater. Sci., 2008, 12, 81–88 CrossRef CAS PubMed.
- B. E. Poling, G. H. Thomson, D. G. Friend, R. L. Rowley and W. V. Wilding, Perry's Chemical Engineers' Handbook, The McGraw-Hill Companies, 2008 Search PubMed.
- L. N. Ji, J. B. Li, Y. Q. Chen, J. Luo, J. K. Liang and G. H. Rao, J. Alloys Compd., 2009, 486, 352–356 CrossRef CAS PubMed.
- J. Nakano, T. Yamada and S. Miyazawa, J. Am. Ceram. Soc., 1979, 62, 465–467 CrossRef CAS.
- A. Kayyar, J. Huang, M. Samiee and J. Luo, J. Visualized Exp., 2012, e4104 Search PubMed.
- D. Aurbach, B. Markovsky, M. D. Levi, E. Levi, A. Schechter, M. Moshkovich and Y. Cohen, J. Power Sources, 1999, 81, 95–111 CrossRef.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef CAS PubMed.
- H. F. Wang, Y. I. Jang, B. Y. Huang, D. R. Sadoway and Y. M. Chiang, J. Electrochem. Soc., 1999, 146, 473–480 CrossRef CAS PubMed.
- F. G. B. Ooms, E. M. Kelder, J. Schoonman, M. Wagemaker and F. M. Mulder, Solid State Ionics, 2002, 152, 143–153 CrossRef.
- W. Choi and A. Manthiram, J. Electrochem. Soc., 2006, 153, A1760–A1764 CrossRef CAS PubMed.
- T. F. Yi, Y. Xie, M. F. Ye, L. J. Jiang, R. S. Zhu and Y. R. Zhu, Ionics, 2011, 17, 383–389 CrossRef CAS.
- D. Liu, W. Zhu, J. Trottier, C. Gagnon, F. Barray, A. Guerfi, A. Mauger, H. Groult, C. M. Julien, J. B. Goodenough and K. Zaghi, RSC Adv., 2014, 4, 154–167 RSC.
- D. W. Liu, Y. Y. Liu, A. Q. Pan, K. P. Nagle, G. T. Seidler, Y. H. Jeong and G. Z. Cao, J. Phys. Chem. C, 2011, 115, 4959–4965 CAS.
- D. W. Shin, C. A. Bridges, A. Huq, M. P. Paranthaman and A. Manthiram, Chem. Mater., 2012, 24, 3720–3731 CrossRef CAS.
- D. W. Shin and A. Manthiram, Electrochem. Commun., 2011, 13, 1213–1216 CrossRef CAS PubMed.
- K.-S. Park, A. Benayad, D.-J. Kang and S.-G. Doo, J. Am. Chem. Soc., 2008, 130, 14930–14931 CrossRef CAS PubMed.
- K. S. Han, J. W. Lee, Y. M. Kang, J. Y. Lee and J. K. Kang, Small, 2008, 4, 1682–1686 CrossRef CAS PubMed.
- H. Han, T. Song, J.-Y. Bae, L. F. Nazar, H. Kim and U. Paik, Energy Environ. Sci., 2011, 4, 4532–4536 CAS.
- M. Samiee and J. Luo, J. Power Sources, 2014, 245, 594–598 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- M. Samiee and J. Luo, Mater. Lett., 2013, 98, 205–208 CrossRef CAS PubMed.
- Y. Kobayashi, H. Miyashiro, K. Takei, H. Shigemura, M. Tabuchi, H. Kageyama and T. Iwahori, J. Electrochem. Soc., 2003, 150, A1577–A1582 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp00869c |
| This journal is © the Owner Societies 2014 |