 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photophysical and structural characterisation of in situ formed quantum dots
A. K.
Bansal
a,
F.
Antolini
b,
M. T.
Sajjad
a,
L.
Stroea
b,
R.
Mazzaro
cd,
S. G.
Ramkumar
e,
K.-J.
Kass
e,
S.
Allard
e,
U.
Scherf
e and
I. D. W.
Samuel
*a
aOrganic Semiconductor Centre, School of Physics and Astronomy, University of St Andrews, North Haugh, St Andrews Fife, KY16 9SS, UK. E-mail: idws@st-andrews.ac.uk
bENEA UTTMATF, Via Ravegnana 186, 48018 Faenza (RA), Italy
cChemistry Department “G. Ciamician”, Bologna University, via Selmi 2, 40126 Bologna, Italy
dCNR-IMM Section of Bologna, via Gobetti 101, 40129 Bologna, Italy
eInstitut für Polymertechnologie Bergische Universität Wuppertal, Gauss-Strasse 20, 42097 Wuppertal, Germany
First published on 11th April 2014
Abstract
Conjugated polymer–semiconductor quantum dot (QD) composites are attracting increasing attention due to the complementary properties of the two classes of materials. We report a convenient method for in situ formation of QDs, and explore the conditions required for light emission of nanocomposite blends. In particular we explore the properties of nanocomposites of the blue emitting polymer poly[9,9-bis(3,5-di-tert-butylphenyl)-9H-fluorene] together with cadmium sulphide (CdS) and cadmium selenide (CdSe) precursors. We show the formation of emissive quantum dots of CdSe from thermally decomposed precursor. The dots are formed inside the polymer matrix and have a photoluminescence quantum yield of 7.5%. Our results show the importance of appropriate energy level alignment, and are relevant to the application of organic–inorganic systems in optoelectronic devices.
1. Introduction
The development of low-cost solution-processable hybrid materials is of increasing interest for the manufacturing of optoelectronic devices.1,2 The easy processability of conjugated polymers together with the tunable properties of quantum dots (QDs) makes them attractive candidates for hybrid devices. Their complementary properties have led to their use in many applications including: light emitting displays,3,4 solar cells,2,5 printable transistors,6 biological labeling7 and sensing.8 To date significant attention has been paid to the use of QDs made from metal sulphides, selenides and tellurides in optoelectronic devices fabricated using conjugated polymers due to their high charge mobility and broad spectral coverage from the visible to the near infrared.9 The conventional method for preparing such a conjugated polymer–QD nanocomposite is by mixing a polymer with previously prepared nanocrystals. The QDs are capped with ligands, and the combination of the use of ligands and surfactants confers solubility and prevents excessive aggregation. However, the ligand and surfactant may also impede charge transport.10 This approach furthermore requires the use of common solvents for nanocrystal and conjugated polymer, which can adversely affect the nanocrystal solubility and the orientation of the polymer chains. This approach also can be detrimental for the homogeneity of the sample surface quality resulting in poor device performance.11 In this paper we explore an alternative approach in which QDs can be directly grown inside the conductive polymer matrix itself.12,13 The advantage of this approach is that there is no need to perform a surface ligand exchange in the growth and nucleation of the QDs. The conjugated polymer itself controls the growth of nanoparticles and thereby removes the need for additional capping agents. Varying the decomposition time and temperature of the molecular precursor can control the size of the QDs formed. Although this method does not allow exact control of the QD size and shape, it does provide intimate contact between the conjugated polymer and the QDs, enhancing the electronic coupling between them. As demonstrated in recent reports, luminescent CdS nanoparticles have been successfully grown in situ within a polystyrene matrix14,15 by thermal decomposition of precursor or within a polymethylmethacrylate (PMMA) matrix by laser processing.16 The in situ synthetic method was also employed to synthesize CdS nanoparticles in the conducting polymer P3HT for hybrid solar cells applications.13In situ formation of light emitting dots by thermolysis of precursor inside the conjugated polymer matrix is not reported yet.The present work is motivated by the objective to obtain efficient light emission from in situ formed QDs in the conductive polymer matrix for hybrid OLED as this could lead to light-emitting devices. To do this we explore the photophysics of precursor–conducting polymer nanocomposites by studying the role of the alignment of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of the organic and inorganic components. The mechanism behind emissive QD formation has been investigated by studying the dominance of charge separation or energy transfer between polymer and nanocrystals in the nanocomposites.
In this paper, the blue emitting polymer poly[9,9-bis(3,5-di-tert-butylphenyl)-9H-fluorene], PBPFO, (see Fig. 1(a)) was selected because of its high thermal stability. It was studied with precursors cadmium diethylxanthate (CdDEX) and cadmium-2-(N,N-dimethylamino)ethylselenolate CdDAMSe (Fig. 1(b) and (c)) that can be thermally converted to CdS and CdSe respectively. Hence nanocomposite films could be made by blending one of the precursors with the polymer, followed by heating. The formation of the CdS/CdSe QDs in the polymer matrix was studied by absorption and photoluminescence measurements and additional understanding of the QD growth and quality was achieved by high-resolution transmission electron microscopy (TEM) techniques.
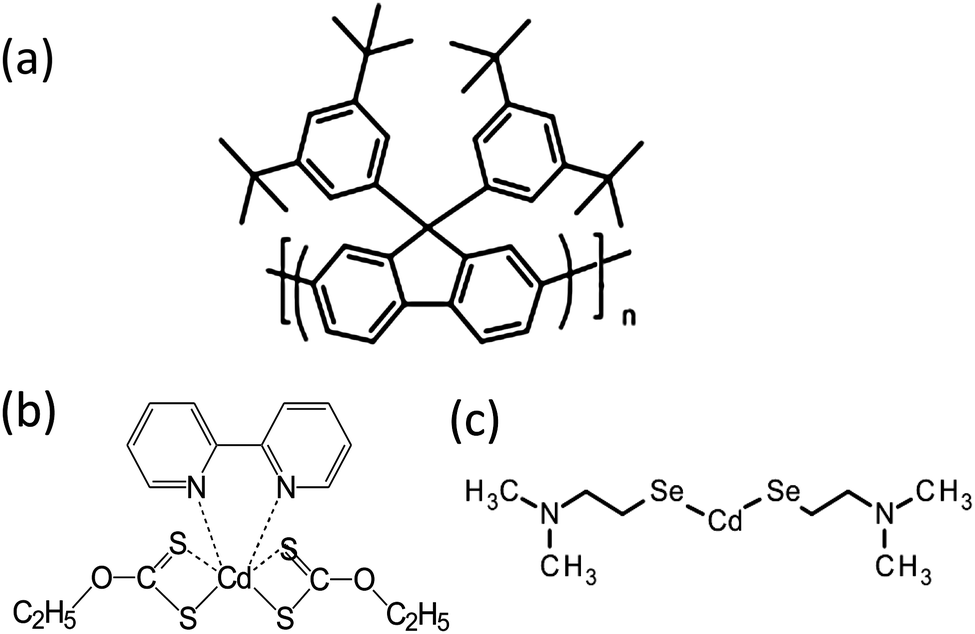 | ||
| Fig. 1 Molecular structure of (a) poly[9,9-bis(3,5-di-tert-butylphenyl)-9H-fluorene] (PBPFO) polymer and (b) CdDEX and (c) CdDMASe precursor. | ||
The paper is organized as follows. First, we discuss the photophysical properties of the polymer itself. Second, thermolysis properties of precursors are discussed using TEM and absorption and emission results. Finally, the detailed transformation of the precursor molecules into CdS/CdSe nanoparticles in the polymer matrix during the thermolysis process is explained. In the discussion, we rationalize the molecular mechanism of the emissive nanoparticle formation based on energy level alignment of donor and acceptor.
2. Materials and methods
2.1 Materials synthesis and film preparation
PBPFO homo-polymer was synthesized as reported in the literature17 with minor modifications. Specifically it was synthesized in a Yamamoto-type cross coupling reaction under microwave conditions, using the monomer 2,7-dibromo-9,9-bis(3,5-di-tert-butylphenyl)-9H-fluorene. The polymer has an average molecular weight of Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 and Mw = 18500. The monomer itself was synthesized using 2,7-dibromofluorenone in four steps with an overall yield of 45%. Thermogravimetric analysis (TGA) of the polymer has showed it was stable up to 400 °C. The CdS precursor cadmium diethylxanthate (CdDEX) was synthesized as reported in the literature18 with minor changes. Specifically the synthesis is performed in the presence of 2,2′-bipyridyl (bipyridyl) acting as additional chelating ligand. This precursor belongs to the metal xanthate family, which are known to decompose cleanly at low temperatures into metal sulphide, here by generating only volatile side products.19 The bipyridyl ligand in this precursor enables sufficient solubility of the xanthate in common organic solvents which otherwise would be insoluble. The protocol for the synthesis of the CdSe precursor cadmium-2-(N,N-dimethylamino)ethylselenolate (CdDMASe) was adapted from the work20 of Kedarnath et al. It involved first making of an aliphatic diselenide compound and then reacting it with a Cd[II] salt to give the metalorganic precursor. For CdSe this single source precursor binds two Me2NCH2CH2Se fragments, leading to good solubility, clean thermal decomposition under generation of cadmium selenide (CdSe) QDs. Both precursors are soluble in toluene, stable at room temperature conditions and can be stored for weeks.
400 and Mw = 18500. The monomer itself was synthesized using 2,7-dibromofluorenone in four steps with an overall yield of 45%. Thermogravimetric analysis (TGA) of the polymer has showed it was stable up to 400 °C. The CdS precursor cadmium diethylxanthate (CdDEX) was synthesized as reported in the literature18 with minor changes. Specifically the synthesis is performed in the presence of 2,2′-bipyridyl (bipyridyl) acting as additional chelating ligand. This precursor belongs to the metal xanthate family, which are known to decompose cleanly at low temperatures into metal sulphide, here by generating only volatile side products.19 The bipyridyl ligand in this precursor enables sufficient solubility of the xanthate in common organic solvents which otherwise would be insoluble. The protocol for the synthesis of the CdSe precursor cadmium-2-(N,N-dimethylamino)ethylselenolate (CdDMASe) was adapted from the work20 of Kedarnath et al. It involved first making of an aliphatic diselenide compound and then reacting it with a Cd[II] salt to give the metalorganic precursor. For CdSe this single source precursor binds two Me2NCH2CH2Se fragments, leading to good solubility, clean thermal decomposition under generation of cadmium selenide (CdSe) QDs. Both precursors are soluble in toluene, stable at room temperature conditions and can be stored for weeks.
Neat films of the precursor alone and of nanocomposite films of polymer–precursor blend were prepared by spin coating from toluene solution at 1500 rpm onto fused silica substrates. The film used for spin-coating consisted of either precursor powder alone or precursor/polymer in ratio of 4:1 in toluene solvent at 50 mg ml−1 concentration. As prepared uniform films were baked at different temperatures inside a low vacuum 8 × 10−2 mbar for thermolysis.
2.2 Photophysical and structural measurements
The solution photoluminescence quantum yield (PLQY) of the polymer was measured by a relative method using quinine sulfate in 0.5 M sulfuric acid, which has a PLQY of 0.546,21 as the standard. Photoluminescence spectra were recorded in a JY Horiba Fluoromax 2 fluorimeter, with the solutions excited at 360 nm. The optical densities of the standard and sample were similar and small (∼0.1). The accuracy of these PLQY measurements is estimated to be ±10% of the stated value. Solid-state PLQY measurements of thin films were measured in an integrating sphere under a nitrogen purge22 in a Hamamatsu C9920-02 luminescence measurement system.For time-resolved fluorescence measurements samples were excited with 100 fs light pulses at 385 nm from the second harmonic of a Ti:sapphire oscillator (Spectra-Physics Mai-Tai) output at 80 MHz. The emission from the samples was passed through a long-pass filter to remove the remaining excitation light and focused onto the slit of a spectrograph (Chromex 250i). The output from the spectrograph was time-resolved with a Hamamatsu streak camera operating in synchroscan mode. The fluorescence decays were captured on a number of time ranges of the streak camera to ensure that any initial decay was resolved to a resolution of 2 ps. The excitation density used was 5 nJ cm−2 or less to make sure all measurements were within the linear regime of the response.
The transmission electron microscopy (TEM) measurements were performed with a Philips Tecnai F20 Schottky Field emission gun (FEG) instrument operating at 200 kV. The images were processed with Digital micrograph and ImageJ software. The QD size was determined manually on the basis of the obtained high-resolution transmission electron microscope (HRTEM) images. The chemical composition was verified by means of energy dispersive spectrometry (EDS) with an EDAX Phoenix spectrometer equipped with an ultra-thin window detector and TEM image and analysis software. The samples for TEM characterization were deposited over a TEM grid with spin-coating using the same conditions. Copper TEM grids covered by Quantifoil® Holey Carbon film were used to maximize the area of the freestanding sample.
3. Results and discussion
3.1 Photophysical properties of polymer
Fig. 2a shows the absorption and fluorescence spectra of the polymer PBPFO in the solutions and as a thin film. The absorption has broad peaks at 380 nm in both solutions and thin film. The fluorescence peak is at 416 nm in solution and for films is red shifted by 9 nm to 425 nm. The photoluminescence quantum yield (PLQY) of the polymer in toluene solution is 65 ± 5% and in the film spun with toluene is 32 ± 3%. The time-resolved fluorescence results are shown in Fig. 2b and show that the film decay is faster than the solution. Both decays can be fitted with single exponential functions with fluorescence lifetime of 220 ps in thin film and 425 ps in solution. The observed fast decay in films in comparison to solutions is due to aggregation of the molecules within the thin film which increases non-radiative decay. The single exponential decays for both solution and film suggest that there is only one emitting chromophore in the polymer. As there is only a single emitting chromophore the natural radiative lifetime can be calculated using the PLQY and the measured fluorescence lifetime. Using solution or film measurements gives the same natural radiative lifetime of 650 ± 70 ps, indicating the identical nature of the emitting species in both solutions and films. This very short radiative lifetime is similar to the other fluorene-based polymers23 reported in the literature with advantage that this homopolymer has better thermal stability and better color purity. The thermal stability of this polymer has been checked by annealing the neat film up to 300 °C inside a low vacuum oven at 8 × 10−2 mbar and there is no green emission corresponding to fluorenone24 formation whereas we find that in other fluorene based polymers like poly(9,9-dioctylfluorene) (PFO) which produce huge green emission at such high temperatures. The PLQY of the polymer only slightly reduced to 28 ± 3% when thin film of the polymer was baked at 200 °C for 10 minutes in comparison to 32 ± 3% at room temperature. Color purity is another advantage of PBPFO polymer because in contrast to film of PFO it does not produce any red shifted emission associated with beta phase.25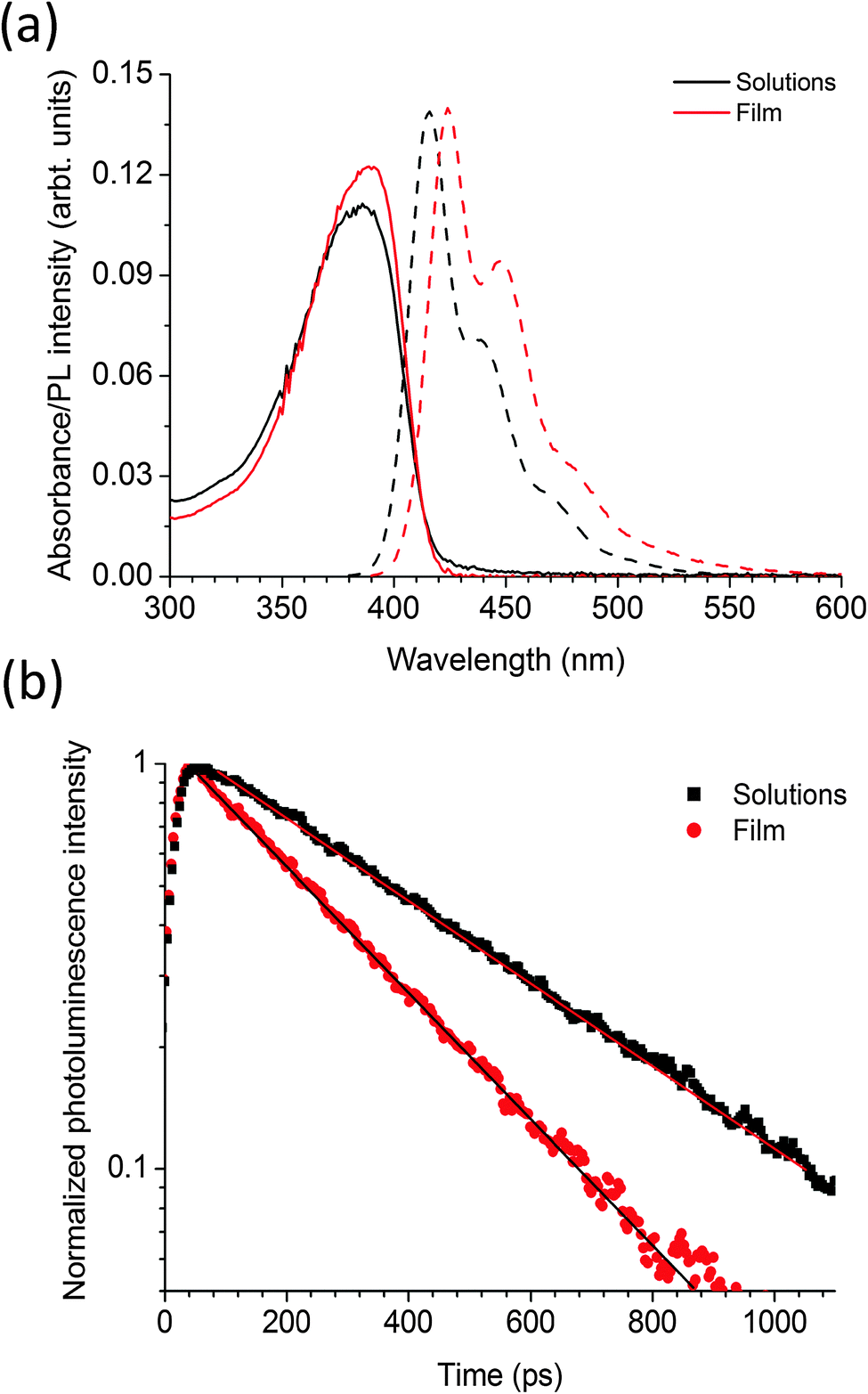 | ||
| Fig. 2 (a) Absorption (solid lines) and fluorescence spectra (dotted) of PBPFO in toluene solution and as a thin film. Fluorescence was excited at 375 nm. (b) Fluorescence lifetime of the polymer is solution and in film with samples excited at 385 nm and emission collected in the range 410 to 520 nm. | ||
3.2 Optical properties of neat films of precursors CdDEX and CdDMASe
The thermal decomposition properties of the single source precursors CdDEX and CdDMASe at various temperatures were studied in neat thin films using UV-Vis spectroscopy. Fig. 3a shows the absorption spectra of CdDEX precursor neat film, which absorbed only below 350 nm before annealing and has a peak at 305 nm due to the bipyridine group in the precursor. Heating causes longer wavelength absorption to appear. This is due to conversion of the precursor into CdS QDs. From the graph it is clear that at lower temperatures precursor does not decompose completely and there is still a shoulder at 305 nm, but as films are heated stronger, the shoulder at 305 nm start disappearing with occurrence of strong absorbance at longer wavelength in the region 440–480 nm with indication of QD formation. This longer wavelength evolution can be assigned to the optically allowed transition between the electron state in the conduction band and hole state in the valence band. The QD formation was also indicated by a color change of the films after baking. The precursor film itself was completely transparent before baking but after heating the color of the film turns to yellowish. The band edge of the formed QDs after precursor decomposition varies from 435 nm to 455 nm according to baking conditions as described in figure caption, which is corresponding to the increase in the size of the CdS nanoparticles. A similar study was performed for the CdDMASe precursor, and the resulting absorption spectra are shown in Fig. 3b. This precursor is also transparent above 400 nm before baking but after heating absorption develops at longer wavelength. This is due to the formation of CdSe QDs, and the onset wavelength of the absorption is in the range 460−520 nm, depending on the baking conditions. The neat films of both precursors before and after baking were also studied with fluorescence spectroscopy, where samples were excited at 320 nm for CdDEX precursor and 380 nm for CdDMASe precursor and also at absorption band edge of each baked film, but no fluorescence emission obtained.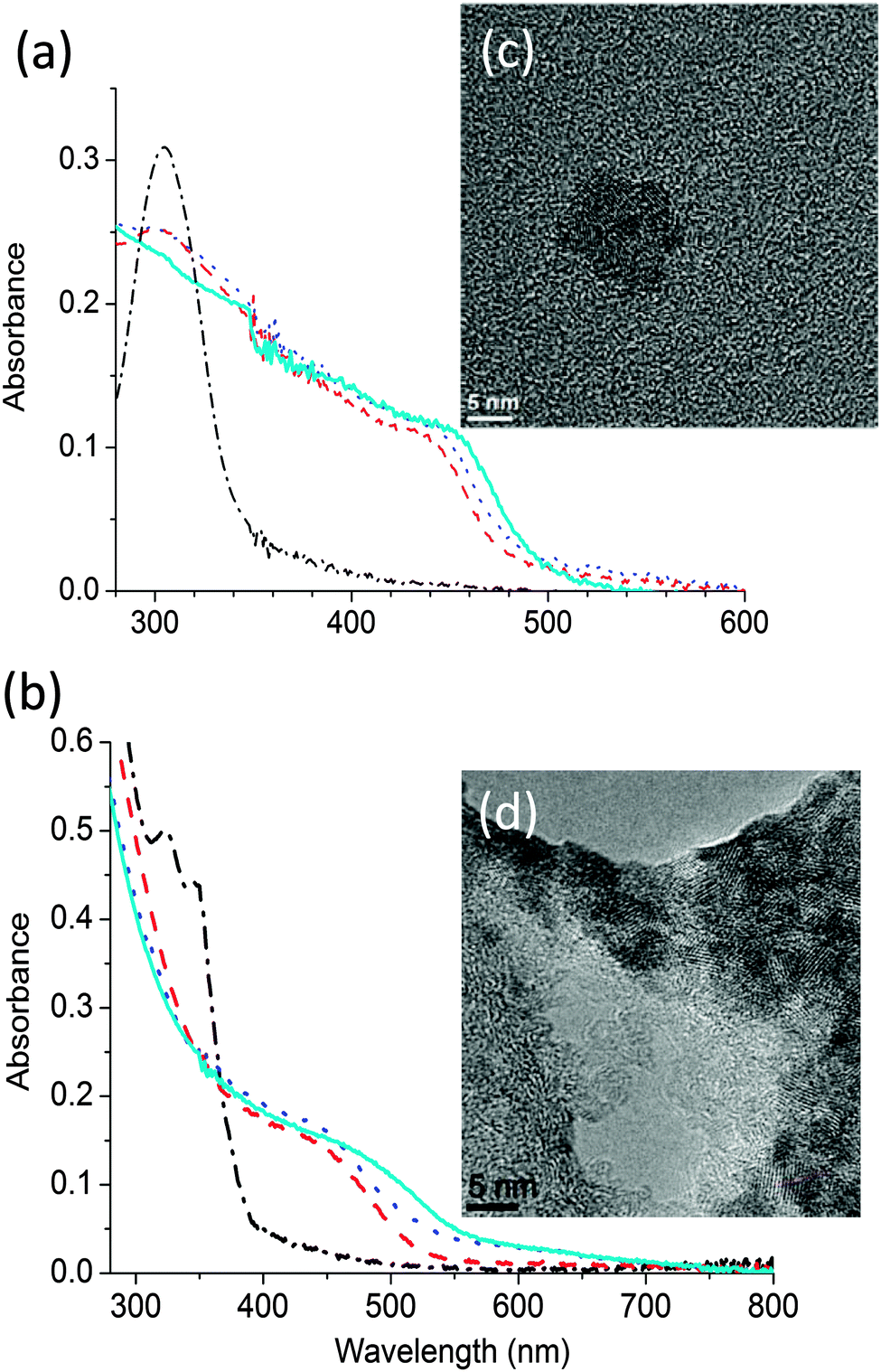 | ||
| Fig. 3 Absorption spectra of (a) CdDEX and (b) CdDMASe precursor before (dash dotted line) and after baking at one of 3 different temperatures 150 °C (dashed line), 160 °C (dotted line) and 180 °C (solid line) for 10 minutes. High resolution transmission electron microscope (HRTEM) images of the (c) CdDEX and (d) CdDMAse precursor thin films after baking at 150 °C for 10 minutes. | ||
3.3 Structural characterisation of neat films of precursors CdDEX and CdDMASe
To obtain further evidence of nanoparticle growth and nucleation, transmission electron microscopy (TEM) measurements were performed. These analyses were carried out on films deposited and annealed directly on a TEM grid. The TEM images of the films before baking did not show any of the crystalline structures ascribable to the CdS or CdSe QDs. However, the formation of CdS QDs (Fig. 3c) and CdSe QDs (Fig. 3d) was visible in the films which had been baked. Fig. 3c shows an example of the TEM results in which a neat film of precursor CdDEX was heated at 150 °C for 10 minutes under low vacuum conditions. The high-resolution image shows the QD distribution in the film that forms clusters of 6–7 QDs with the presence of both the cubic and the hexagonal crystal phase in the QD population. The average size of the CdS QDs is 4.9 nm with a standard deviation of 0.9 nm and is in the range of reported literature values26 of CdS QD sizes. Fig. 3d shows the high resolution TEM results for CdDMASe precursor film baked at 160 °C for 10 minutes, which evidenced mono-dispersed crystalline QD in clusters of 5–10 CdSe QDs. The analysis of the average size determined on 70 single QDs gives an average diameter of 4.5 nm of nanocrystals and which is below the threshold of the Bohr exciton radius for CdSe.10The above results indicate that there is nucleation and growth of CdS and CdSe QDs but fluorescence spectroscopy shows they are non-emissive. A likely reason for the lack of emission from the QDs formed might be due to lack of passivation27 of the surface of the QDs. As there is no capping agent, surface traps dominate due to S or Se ions, and this leads to non-radiative decay of the excited states. To investigate this further, we blended the precursors into the polymer matrix, as discussed in the next section.
3.4 Optical properties of nanocomposite film of polymer and CdDEX precursor
In the nanocomposites the polymer and precursor were blended in a weight/weight ratio of 1:4 respectively. Fig. 4 shows the absorption and fluorescence spectra of the polymer–precursor CdDEX nanocomposites films before and after annealing at different temperatures from 140 °C to 180 °C. It is evident from Fig. 4a that before baking the nanocomposite films the absorption spectra has contributions from both the polymer and precursor. The shoulder at 380 nm is due to the π–π* transition of the polymer, and the peak at 305 nm (not shown) is due to the bipyridine on the precursor (as discussed earlier in Fig. 3a). After baking the blend films at selected temperatures, there is an evolution of the shoulder at 450 nm. The shift of the shoulder towards longer wavelength at higher baking temperature indicates an increase in nanocrystal size and concentration. The absence of well-resolved peak like structures can be attributed to the polydispersity28 of QD size. These results show that the precursor decomposed and QDs formed similar to neat precursor film as explained in earlier section. Fig. 4b shows the fluorescence spectra of the nanocomposite films where blend films were excited at 375 nm. The results indicate that in the blend before baking the fluorescence spectrum is similar to that of neat polymer film as shown in Fig. 2a with all emission coming from polymer itself. After baking the blend films at different temperatures the spectra become narrower with sharp peaks at longer wavelengths in annealed films. This may happen due to polymer chain reorganisation at higher annealing temperatures. There is no emission corresponding to the QD formation even though there is overlap of the PL spectrum of the polymer with the absorption spectrum of the CdS QDs, so it should favour long-range energy transfer from polymer to QDs. The PLQY was also measured in the blend before and after annealing when the sample has been excited at 375 nm. Before baking the PLQY of the polymer is only 2.7%, compared with 32% for a neat film of the polymer. After baking it remains approximately 2.5% like before annealing the nanocomposites film, even though at higher temperatures the precursor mostly decomposed in the blended system. These results indicate that precursor works as a quencher in the nanocomposite films even though it does not absorb much at the selected excitation wavelength. To understand the quenching after baking, TEM studies were performed on the nanocomposite.
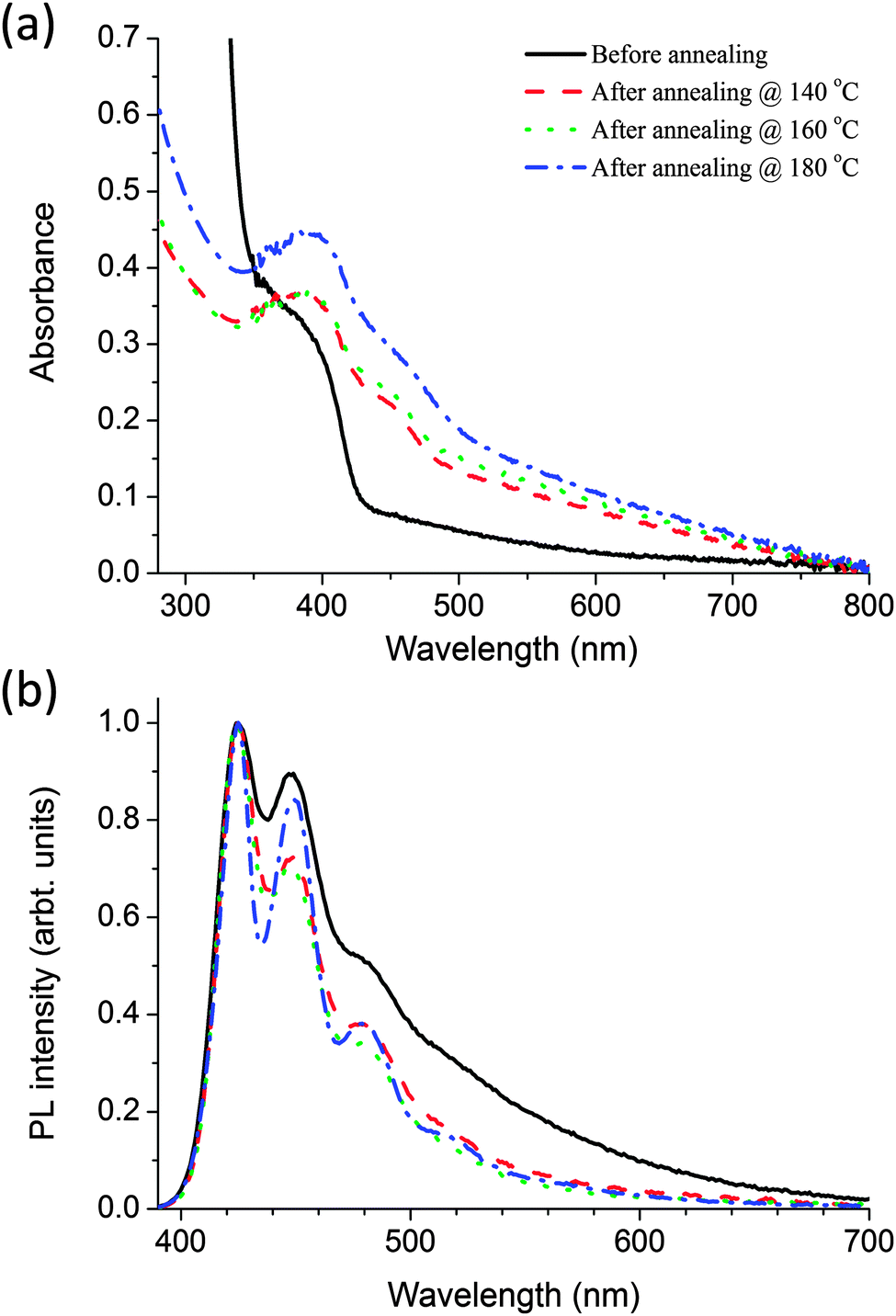 | ||
| Fig. 4 (a) Absorption (b) fluorescence spectra of the CdDEX precursor–polymer blend film before baking (solid line) and after annealing at 3 different temperatures 140 °C (dashed line), 160 °C (dotted line) and 180 °C (dash dotted line) each for 10 minutes. Samples were excited at 375 nm. | ||
3.5 Structural characterization of nanocomposite film of polymer and CdDEX precursor
The particle size and crystalline structure of the formed QDs in the nanocomposites films after baking were investigated by TEM studies and are shown in Fig. 5. Both low and high magnification micrographs (Fig. 5a and b respectively) before heating the nanocomposite show that the organic–inorganic matrix is uniformly distributed over the carbon film and no aggregates of QDs are observable under these conditions. The SAED (Selected Area Electron Diffraction) in Fig. 5c also shows that in the sample crystalline particles are absent and the observed radial distribution is typical of the polymer only. Fig. 5d shows a low magnification image of the nanocomposite film after heating the film at 160 °C for 10 minutes. The results show that there is the formation of nanocrystals in the polymer matrix and these particles within the polymer tend to aggregate in clusters with a size between 20–60 nm. The HREM image in Fig. 5e show the particle size is between 3 and 6 nm. Applying the FFT algorithm to the HREM images it was possible to recognize the typical periodicity of CdS hexagonal phase. For further confirmation, the SAED can be indexed as hexagonal CdS as shown in Fig. 5f. It is not possible to exclude the cubic phase because the reflections of the two phases overlap. | ||
| Fig. 5 (a) Low magnification and (b) high magnification images of polymer–CdDEX precursor blend before baking (c) SAED image before baking. (d) Low and (e) high magnification images of QDs generated by thermal annealing of the polymer–precursor film at 160 °C for 10 minutes; the SAED (f) confirms the presence of crystalline species. The diffraction rings indicated by a red and an orange arrow are compatible with both CdS cubic or hexagonal phase, while the one highlighted in green is typical of the hexagonal phase only. | ||
The absorption and TEM results show that nanoparticles form and yet there is no fluorescence from the nanoparticles in the blend. Possible explanations could be that the nanoparticles are either non-emissive or energy transfer does not occur to them from the polymer. Non-emissive QDs can be formed if they are not passivated well or if they form clusters in which aggregation leads to quenching of the QD emission. As the nucleation and growth of the CdS particle was inside the polymer matrix13 so we can rule out this scenario of non-passivation of QDs. TEM results show the formation of clusters so it might be possible for emission quenching due to aggregation of QDs. No energy transfer from polymer to the CdS nanocrystal is the likely phenomena. Fig. 6 shows the energy levels of the polymer from cyclic voltammetry and CdS QDs from literature. The HOMO of the polymer is at 5.83 eV and the LUMO is at 3.06 eV. The energy level band for CdS nanoparticles is shown based on literature values.29,30 This scenario suggests the type 2 energy level alignment between the polymer and thermally decomposed CdS QDs. In this case as reported by Lutich et al. there is a possibility of competition between charge separation and energy transfer and their dominance strongly depends on the nanoscale geometry of the system.31 In our system thermolysis of precursor occurs inside the polymer matrix so there is no hindrance of minimum separation due to surface ligands between the nanocrystal core and the polymer molecules. As there is no minimum separation so it favours the occurrence of the charge transfer and limits the efficiency of energy transfer, and that define the non-emissive behaviour of the QDs in this particular system.
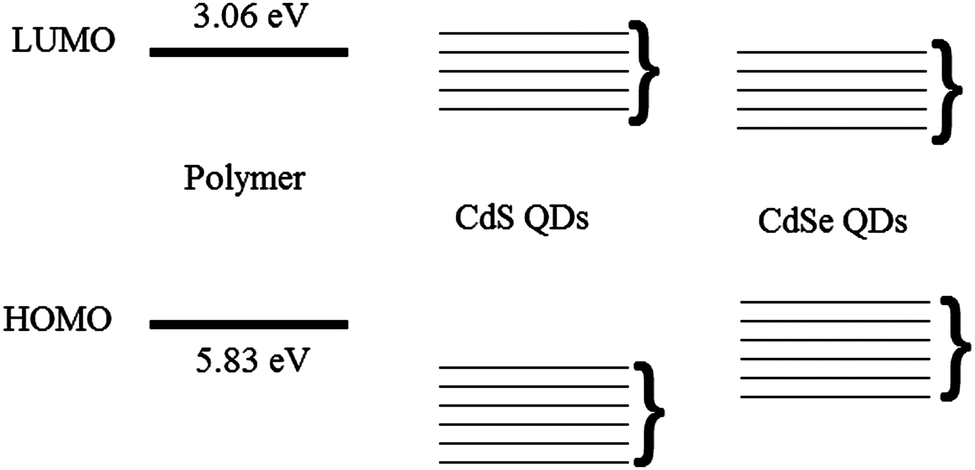 | ||
| Fig. 6 A schematic of the energy level diagrams of the polymer and Cadmium Sulphide (CdS) and Cadmium Selenide (CdSe) QDs. Here the brackets represent the variation in energy levels of QDs. | ||
3.6 Optical properties of nanocomposite film of polymer and CdDMASe precursor
Fig. 7 shows the absorption and fluorescence spectra of the nanocomposite films prepared using polymer with CdDMASe precursor. Before annealing the absorption spectrum is dominated by the polymer, with a small contribution at shorter wavelengths from the precursor. After baking a shoulder appears in the region of 450 nm, which grows, and shifts towards longer wavelength at higher baking temperature. This behaviour is due to decomposition of the precursor to form QDs as discussed in the previous section. Due to surface modifications of the nanoparticles by the polymer the absorption edge are shallow in comparison to Fig. 3b. The red shift of the absorption edge compared to the uncapped CdSe nanocrystals reveals that the growth of particle is influenced by the presence of polymer capping agent. Fig. 7b shows the fluorescence spectra of the nanocomposite films following excitation at 375 nm. The results indicate that before thermolysis fluorescence spectrum of blend is similar to that of the neat polymer film as shown in Fig. 2a, which indicates that all emission is coming from polymer itself with no contribution from the precursor. After baking the films there are some interesting features in the emission spectra. For instance the nanocomposite film annealed at 150 °C for 10 minutes shows emission of the polymer and an extra emission at longer wavelengths. As the polymer is thermally stable at this temperature we assign the extra emission at longer wavelength to QD formation only.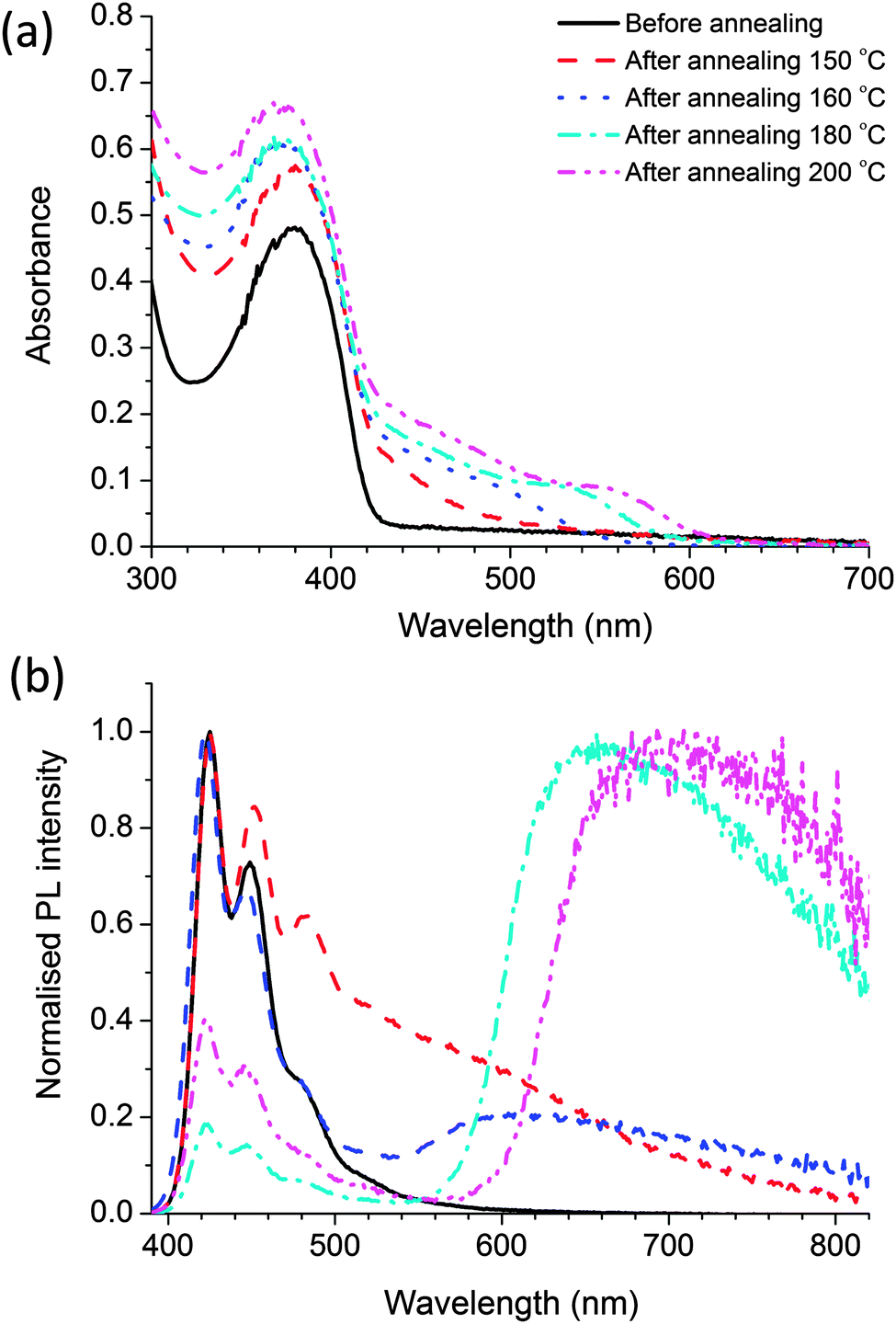 | ||
| Fig. 7 (a) Absorption (b) fluorescence spectra of the precursor CdDMAse–polymer blend film before baking (solid line) and after annealing at 4 different temperatures 150 °C (dashed line), 160 °C (dotted line), 180 °C (dash dotted line) and 200 °C (dash double dotted line) each for 10 minutes. Samples were excited at 375 nm. | ||
To understand clearly the behaviour of the emissive QDs, the emission spectra of the QDs were extracted from emission spectrum of the nanocomposite film by subtracting the polymer contribution to the spectrum. Fig. 8 shows the resulting normalised photoluminescence signal at different temperatures. The results clearly show broad emission from the QDs. The peak of the QDs emission varies from 510 nm obtained after baking at 150 °C to 690 nm after heating to 200 °C.
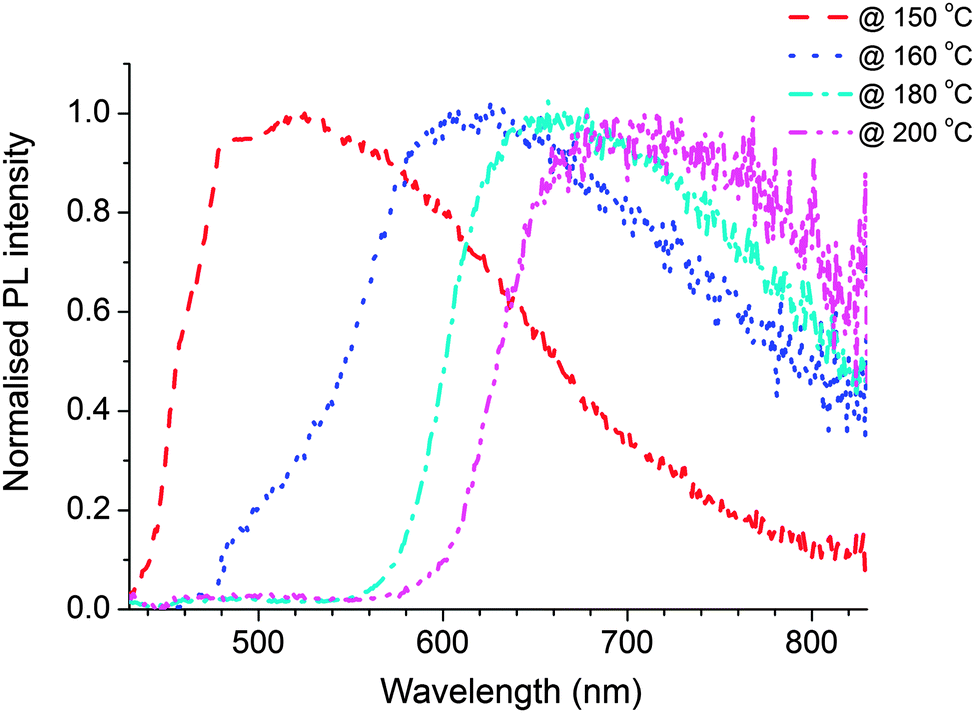 | ||
| Fig. 8 Emission spectrum of QDs obtained after subtracting the polymer contribution from the polymer–precursor nanocomposite annealed at temperatures 150 °C (dashed line), 160 °C (dotted line), 180 °C (dash dotted line) and 200 °C (dash double dotted line) each for 10 minutes. | ||
PLQY measurements give further insight into the properties of these nanocomposite films and were performed with excitation at 375 nm. The blend film has 29% PLQY before heating and which is very similar to that value of neat polymer of 32% when the sample has been excited at 375 nm. These results are different from the results of the nanocomposite films of the CdS based precursor where the PLQY of the polymer was quenched by the precursor. This is an intriguing result as it suggests the precursor CdDMASe interacts differently with the blue emitting polymer. After thermolysis of blend, PLQY of the QDs is extracted from the total PLQY of the nanocomposites films by calculating the emission contribution from 500 to 900 nm. The maximum PLQY obtained for the QDs is 7.5% after heating to 160 °C. The PLQY decreases to 1% for heating at 200 °C. As discussed earlier, the decrease of the PLQY can be attributed to trapping of electrons in the surface-defect states with further increase of the density of trap states at higher temperature. In some cases, trap emission has been related to low crystallinity or to selenide excess or other defects at the interface of the nanocrystals. However, band edge and trap emissions are often observed together in CdSe nanocrystals. Increasing the amount of surface defects of the nanoparticles can enhance enormously the trap state emission with respect to the band-edge recombination mechanism that can be completely suppressed. The other scenario of reduction in PLQY might be due to aggregate of the QDs at the higher annealing temperature, which has been investigated by TEM studies.
3.7 Structural characterization of nanocomposite film of polymer and CdDMASe precursor
TEM images of nanocomposite films at medium and high magnification are shown in Fig. 9. Image 9a shows that heating to 160 °C for 10 minutes leads to the formation of particles in the polymer matrix. From the HREM micrograph displayed in Fig. 9b it is clear that most of the particles are well dispersed and have a diameter about 4 to 8 nm, but there is also a population of aggregated clusters, containing from 5 to 10 units. The particle size is smaller or in the similar range of the uncapped QDs shown earlier in Section 3.2. Fig. 9c and d shows the TEM results of the film baked at 180 °C for 10 minutes and it is clear from the figure that the particle starts to aggregate under these annealing conditions by making clusters of 20–30 units. SAED analysis (not reported) shows for both the annealed samples the presence of crystalline CdSe with a mixture of hexagonal and cubic phase. Aggregate formation at higher baking temperature explains the reduction in the PLQY of the emissive QDs at higher temperatures. | ||
| Fig. 9 (a) Medium and (b) high magnification images of QDs generated by 10 minutes of thermal annealing at 160 °C of the polymer–CdAMSe precursor film. (c) Medium and (d) high magnification images of QDs generated by 10 minutes of thermal annealing at 180 °C of the polymer–precursor film. | ||
The behaviour of these nanocomposite films is completely different from those made from the other precursor. This can be understood in terms of the energy levels. Efficient transfer has been observed from conjugated polymers to colloidal semiconductor QD in several reports with having comparison of short and long chain ligands,32 dopant concentration in core–shell nanoparticles33 and the dominance of long-range energy transfer has been reported in them.34 In our results on the basis of energy level alignment of polymer and QDs we can identify the dominant mechanism. Fig. 6 shows the position of the energy levels of CdSe QDs taken from the literature11,35,36 with respect to polymer energy levels. This scenario suggests type 1 energy level alignment between polymer and CdSe nanocrystals. As discussed earlier in our system thermolysis of precursor occurs inside the polymer matrix so there is no minimum separation due to surface ligands between the nanocrystal core and the polymer molecules so it favours the occurrence of the charge transfer/Dexter type energy transfer. We know that to allow the simultaneous injection of electron and hole to the donor, Dexter type energy transfer requires type-I alignment of HOMO and LUMO levels of the donor–acceptor. This scenario suggests that we have Dexter type energy transfer from polymer to the QDs. But there is good spectral overlap of polymer emission with absorbance of the QDs so there is a possibility of long-range energy transfers as well.
4. Conclusions
In summary, we have demonstrated the in situ formation of QDs inside the conjugated polymer matrix. We show that the polymer PBPFO having solid state PLQY up to 32% and natural radiative lifetime of 650 ps is suitable for thermolysis of QDs due to its excellent thermal stability. Our studies show that the thermal decomposition of the precursor CdDEX and CdDAMSe inside a polymer matrix produces different characteristics of the QDs due to differences in energy level alignment. We also describe here the use of time and temperature to achieve size (and spectroscopic) tuning of the final particles. Our results show that in situ emissive QDs can be made inside the conjugated polymer matrix and in our studied system, a PLQY of CdSe QDs of 7.5% is achieved. Our results open up the possibility for laser writing of emissive QDs in optoelectronic devices.Acknowledgements
We acknowledge financial support from FP7 project “Laser Induced Synthesis of Polymeric Nanocomposite Materials and Development of Micro-patterned Hybrid Light Emitting Diodes (LED) and Transistors (LET)” – LAMP project (G.A. 247928). AKB and IDWS also acknowledge financial support from EPSRC Programme grant “Challenging the limits of photonics: Structured light” EP/J01771X/1.References
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425 CrossRef CAS PubMed.
- A. L. Briseno, T. W. Holcombe, A. I. Boukai, E. C. Garnett, S. W. Shelton, J. J. M. Frechet and P. Yang, Nano Lett., 2010, 10, 334 CrossRef CAS PubMed.
- S. Coe, W. K. Woo, M. Bawendi and V. Bulovis, Nature, 2002, 420, 800 CrossRef CAS PubMed.
- N. Tessler, V. Medvedev, M. Kazes, S. H. Kan and U. Banin, Science, 2002, 295, 1506 CrossRef PubMed.
- S. Dayal, N. Kopidakis, D. C. Olson, D. S. Ginley and G. Rumbles, Nano Lett., 2010, 10, 239–242 CrossRef CAS PubMed.
- F. Hetsch, N. Zhao, S. V. Kershaw and A. L. Rogach, Mater. Today, 2013, 16, 312–325 CrossRef CAS PubMed.
- S. J. Rosenthal, J. C. Chang, O. Kovtun, J. R. McBride and I. D. Tomlison, Chem. Biol., 2011, 18, 10–24 CrossRef CAS PubMed.
- H. Dhyani, C. Dhand, B. D. Malhotra and P. Sen, J. Biosens. Bioelectron., 2011, 3, 112 Search PubMed.
- P. O. Anikeeva, J. E. Halpert, M. G. Bawendi and V. Bulovic, Nano Lett., 2009, 9, 2532–2536 CrossRef CAS PubMed.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS PubMed.
- C. Querner, P. Reiss, S. Sadki, M. Zagorskab and A. Pron, Phys. Chem. Chem. Phys., 2005, 7, 3204–3209 RSC.
- H. C. Leventis, S. P. King, A. Sudlow, M. S. Hill, K. C. Molloy and S. A. Haque, Nano Lett., 2010, 10, 1253–1258 CrossRef CAS PubMed.
- N. Bansal, L. X. Reynolds, A. MacLachlan, T. Lutz, R. S. Ashraf, W. Zhang, C. B. Nielsen, I. McCulloch, D. G. Rebois, T. Kirchartz, M. S. Hill, K. C. Molloy, J. Nelson and S. A. Haque, Sci. Rep., 2013, 3, 1531 CAS.
- F. Antolini, T. Di Luccio, A. M. Laera, L. Mirenghi, E. Piscopiello, M. Re and L. Tapfer, Phys. Status Solidi B, 2007, 244, 2768–2781 CrossRef CAS.
- F. Antolini, E. Burresi, L. Stroea, V. Morandi, L. Ortolani, G. Accorsi and M. Blosi, J. Nanomater., 2012, 2012, 1–11 CrossRef PubMed.
- V. Resta, A. M. Laera, A. Camposeo, E. Piscopiello, L. Persano, D. Pisignano and L. Tapfer, J. Phys. Chem. C, 2012, 116, 25119–25125 CAS.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Mullen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946–953 CrossRef CAS PubMed.
- D. Barreca, A. Gasparotto, C. Maragno, R. Seraglia, E. Tondello, A. Venzo, V. Krishnan and H. Bertagnolli, Appl. Organomet. Chem., 2005, 19, 59–67 CrossRef CAS.
- N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS.
- G. Kedarnath, S. Dey, V. K. Jain, G. K. Dey and B. Varghese, Polyhedron, 2006, 25, 2383–2391 CrossRef CAS PubMed.
- A. N. Fletcher, Photochem. Photobiol., 1969, 9, 439–444 CrossRef CAS.
- N. C. Greenham, I. D. W. Samuel, G. R. Hyes, R. T. Philips, Y. A. R. Kessener, S. C. Moratti, A. B. Holmes and R. H. Friend, Chem. Phys. Lett., 1995, 241, 89–96 CrossRef CAS.
- F. Laquai, A. K. Mishra, M. R. Ribas, A. Petrozza, J. Jacob, L. Akcelrud, K. Müllen, R. H. Friend and G. Wegner, Adv. Funct. Mater., 2007, 17, 3231–3240 CrossRef CAS.
- K. Becker, J. M. Lupton, J. Feldmann, B. S. Nehls, F. Galbrecht, D. Gao and U. Scherf, Adv. Funct. Mater., 2006, 16, 364–370 CrossRef CAS.
- A. K. Bansal, A. Ruseckas, P. E. Shaw and I. D. W. Samuel, J. Phys. Chem. C, 2010, 114, 17864–17867 CAS.
- T. Rath and G. Trimmel, Hybrid Mater., 2013, 3, 15–36 Search PubMed.
- S. Kango, S. Kaila, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS PubMed.
- D. Zhitomirsky, I. J. Kramer, A. J. Labelle, A. Fischer, R. Debnath, J. Pan, O. M. Bakr and E. H. Sargent, Nano Lett., 2012, 12, 1007–1012 CrossRef CAS PubMed.
- S. K. Haram, B. M. Quinn and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 8860–8861 CrossRef CAS.
- C. Yang, A. Tang and F. Teng, J. Electrochem. Soc., 2013, 160, H121–H125 CrossRef CAS PubMed.
- A. A. Lutich, G. Jiang, A. S. Susha, A. L. Rogach, F. D. Stefani and J. Feldmann, Nano Lett., 2009, 9, 2636–2640 CrossRef CAS PubMed.
- T.-W. F. Chang, S. Musikhin, L. Bakueva, L. Levina, M. A. Hines, P. W. Cyr and E. H. Sargent, Appl. Phys. Lett., 2004, 84, 4295 CrossRef CAS PubMed.
- M. Anni, L. Manna, R. Cingolani, D. Valerini, A. Creti and M. Lomascolo, Appl. Phys. Lett., 2004, 85, 4169–4171 CrossRef CAS PubMed.
- T. Stoferle, U. Scherf and R. F. Mahrt, Nano Lett., 2009, 9, 453–456 CrossRef CAS PubMed.
- M. Amelia, C. Lincheneau, S. Silvi and A. Credi, Chem. Soc. Rev., 2012, 41, 5728–5743 RSC.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
| This journal is © the Owner Societies 2014 |