 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The first tyrosyl radical intermediate formed in the S2–S3 transition of photosystem II†
Marius
Retegan
,
Nicholas
Cox
,
Wolfgang
Lubitz
,
Frank
Neese
and
Dimitrios A.
Pantazis
*
Max Planck Institute for Chemical Energy Conversion, Stiftstr. 34-38, 45470 Mülheim an der Ruhr, Germany. E-mail: dimitrios.pantazis@cec.mpg.de
First published on 3rd April 2014
Abstract
The EPR “split signals” represent key intermediates of the S-state cycle where the redox active D1-Tyr161 (YZ) has been oxidized by the reaction center of the photosystem II enzyme to its tyrosyl radical form, but the successive oxidation of the Mn4CaO5 cluster has not yet occurred (SiYZ˙). Here we focus on the S2YZ˙ state, which is formed en route to the final metastable state of the catalyst, the S3 state, the state which immediately precedes O–O bond formation. Quantum chemical calculations demonstrate that both isomeric forms of the S2 state, the open and closed cubane isomers, can form states with an oxidized YZ˙ residue without prior deprotonation of the Mn4CaO5 cluster. The two forms are expected to lie close in energy and retain the electronic structure and magnetic topology of the corresponding S2 state of the inorganic core. As expected, tyrosine oxidation results in a proton shift towards His190. Analysis of the electronic rearrangements that occur upon formation of the tyrosyl radical suggests that a likely next step in the catalytic cycle is the deprotonation of a terminal water ligand (W1) of the Mn4CaO5 cluster. Diamagnetic metal ion substitution is used in our calculations to obtain the molecular g-tensor of YZ˙. It is known that the gx value is a sensitive probe not only of the extent of the proton shift between the tyrosine–histidine pair, but also of the polarization environment of the tyrosine, especially about the phenolic oxygen. It is shown for PSII that this environment is determined by the Ca2+ ion, which locates two water molecules about the phenoxyl oxygen, indirectly modulating the oxidation potential of YZ.
1. Introduction
All oxygenic life on Earth is sustained by biological water oxidation performed in higher plants, algae and cyanobacteria.1–5 Understanding and mimicking this process in artificial systems is a central challenge for energy research.6–11 Photosystem II, the enzyme responsible for water splitting, functions by coupling one-electron photochemical charge separation with the four-electron process of water oxidation by storing oxidizing equivalents (“electron holes”) on the inorganic Mn4CaO5 cluster known as the oxygen evolving complex (OEC), the active site of catalysis. The OEC cycles through five oxidation states Si, where i = 0–4 indicates the number of stored oxidizing equivalents. S4 is not observable and spontaneously decays to S0 with evolution of triplet dioxygen. As shown in Fig. 1, absorption of sunlight by PSII results in charge separation at the reaction center of the enzyme, with the electron being transported through several cofactors to the second exchangeable acceptor, the plastoquinone QB. The highly oxidizing radical cation (P680˙+) is re-reduced by D1-Tyr161 (YZ), which subsequently extracts one electron from the Mn4Ca cluster, advancing it to the next Si state of the catalytic cycle.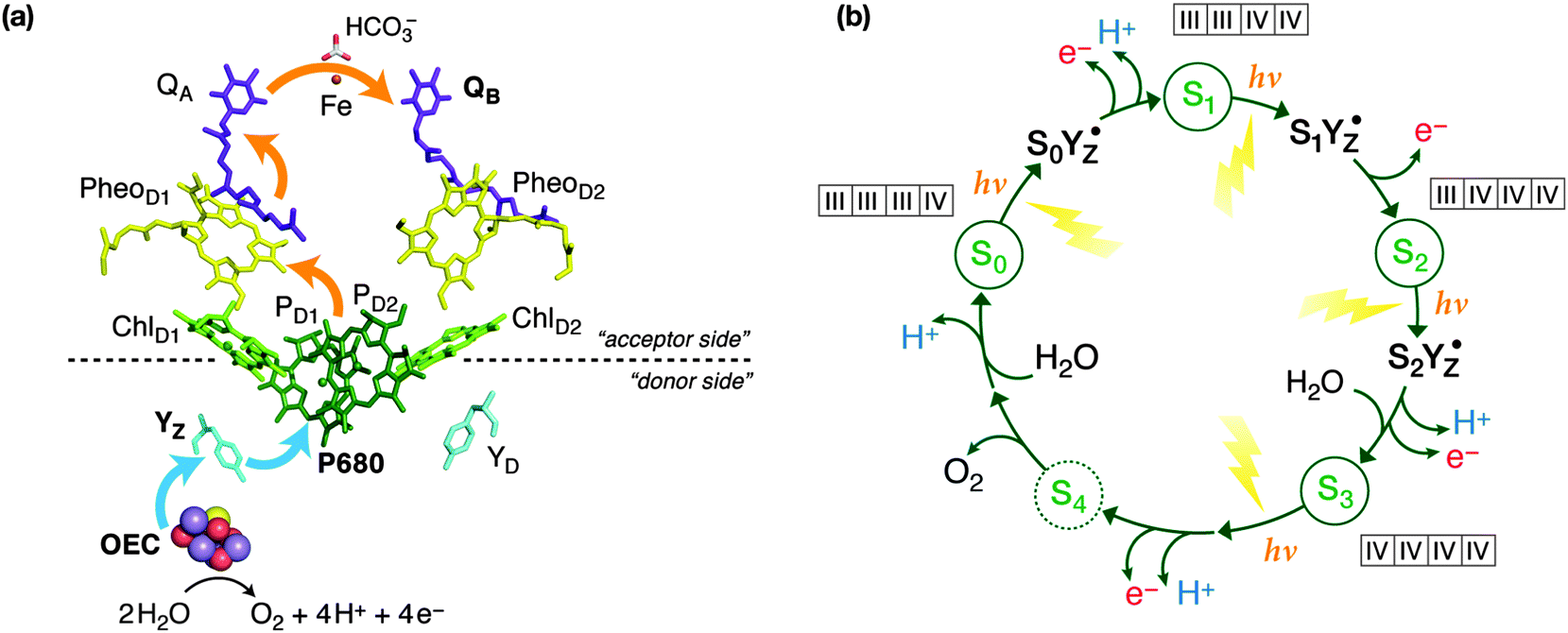 | ||
| Fig. 1 (a) Schematic depiction of electron flow among the major cofactors of PSII: orange arrows indicate the flow of electrons from P680 after photo-excitation towards the acceptor plastoquinone (QB), blue arrows indicate the flow of electrons to P680 from the Mn4Ca cluster via D1-Tyr161 (YZ). Water is the ultimate source of electrons, through its oxidation at the OEC. (b) The S-state cycle of the OEC, indicating formation of YZ radical intermediates between the first three transitions; the most probable Mn oxidation states are shown for the S0–S3 states. | ||
YZ oxidation by P680˙+ occurs on a 1–10 ns timescale. The formed transient radical state (YZ˙) then decays on the μs to ms timescale, with the concomitant oxidation of the Mn4CaO5 cluster under physiological conditions. Cryogenic temperatures (<20 K) alter this single electron progression. At these temperatures, electron donation to P680˙+ by YZ can still occur in a minority of centers (<50%), but the subsequent electron transfer from the Mn4CaO5 cluster to YZ˙ is blocked, arresting catalytic progression from SiYZ˙ to Si+1YZ.12–29 Similar trapped intermediate states can be generated in chemically modified PSII, with examples seen in Ca2+ depleted PSII,30–32 acetate treated PSII,13,33 and in PSII poised at high pH.34,35 It is also noted that in the higher S-states (S2, S3) infrared light excitation of the Mn4CaO5 cluster also allows the “backwards” cycling, i.e. inducing the one electron oxidation of YZ by the Mn cluster.15,20,36–38
From the perspective of electron paramagnetic resonance (EPR) spectroscopy, the tyrosine radical is not isolated from the Mn4CaO5 cluster.39,40 This means that the tyrosyl radical senses the structure of the Mn4CaO5 cluster, and hence each metastable SiYZ˙ state has a characteristic spectral lineshape. Importantly, these “metallo-radical” states have the potential to resolve the concerted structural changes that occur in-between S-state transitions, which are coupled to cluster deprotonation and substrate binding.41–45 This is particularly important for the most complicated S-state transition, from S2 to S3. While experimental and theoretical efforts have produced a quite robust understanding of the S2 state itself,2 the S3 state is arguably the least well understood state of the cycle, with even the nature of the oxidation event, Mn-centered vs. ligand-centered oxidation, still being debated.46–48 As shown in Fig. 1, S2–S3 is a complex transition that must involve several intermediates because it likely combines water binding43 with deprotonation of the OEC.41,42 It is the most crucial step of the cycle, because it activates the catalyst allowing the O–O bond formation step to occur. The precise sequence of events within the S2–S3 transition is currently unknown and several strands of observations must be rationalized simultaneously for it to be understood.49
A quantum chemical description of the electronic structure of the split signal states has been lacking because of (a) the large size of molecular models which must include both the OEC with its first and second coordination spheres and the YZ residue with its environment and (b) the sufficiently high levels of theory required to obtain an adequate description of the electronic structure of the coupled system and to predict its spectroscopic properties. In the present work we employ large quantum cluster models to describe the S2YZ˙ state. We report its electronic structure and spectroscopic properties, including the g-tensor of the tyrosine radical. The present results suggest deprotonation of a terminal water ligand as the most likely next step in the S2–S3 transition. We also resolve the influence of hydrogen bonding on the phenolic oxygen of the YZ˙, suggesting a role for the Ca2+ ion in modulating the oxidation potential of the tyrosine by structuring its local hydration environment.
2. Methodology
Optimized models of the S2 state were used as starting points for geometry optimizations of the one-electron oxidized species.50 The structures contain all first-sphere residues of the Mn4CaO5 core, second-sphere residues that hydrogen-bond to the inorganic core or its first-sphere residues, several vicinal water molecules and the D1-Tyr161–His190 pair, along with complete backbone loops where necessary. The BP86 functional51,52 was employed for optimizations, while the TPSSh functional53,54 was used for single-point calculations of different spin configurations in the context of broken-symmetry density functional theory55–61 and for the calculation of all spectroscopic properties reported in this work. D3 dispersion corrections62 were used throughout, and the influence of the environment was simulated using the conductor-like screening model (COSMO)63 assuming a dielectric constant of ε = 8, as in previous studies. The scalar relativistic effects were included with the zeroth order regular approximation (ZORA)64,65 with one-center terms and using ZORA-recontracted polarized triple-ζ basis sets.66,67 Fully decontracted auxiliary basis sets were used for the resolution of the identity (RI) and chain-of-spheres (COSX) approximations68 for the Coulomb and exact exchange, as implemented in ORCA.69 Fine general integration grids with additionally increased radial accuracy and tight convergence criteria were used for all calculations. The extraction of pairwise exchange coupling constants from the individual broken-symmetry solutions and calculation of the complete energy ladder of spin eigenstates through diagonalization of the Heisenberg Hamiltonian followed established procedures.50,70–77The same level of theory was used for the calculation of the magnetic and spectroscopic parameters. The g-matrix describes the coupling between the molecular magnetic moment and the external field. In the framework of DFT and Hartree–Fock theory the g-matrix can be evaluated using a coupled-perturbed self-consistent field approach.78 For the evaluation of the spin–orbit coupling operator we used an efficient implementation of the spin–orbit mean-field approximation to the Breit–Pauli operator.79 In determining the g-matrix components of the YZ radical we followed the pragmatic approach of substituting the open-shell Mn ions of the cluster with diamagnetic ions of the same formal charge (Ga3+ and Ge4+), as the interaction between YZ and the Mn cluster is small (less than 500 MHz). We note that more elaborate approaches have been proposed for the calculation of g-values in magnetically strongly coupled dimers.80 In the present case, namely the very weak interaction between the hypothetical spin pair, the YZ and the Mn cluster, only the electrostatic influence of the inorganic cluster on the tyrosine needs to be considered and can be fully captured using diamagnetic ion substitution.
3. Results and discussion
3.1 The S2 state prior to the oxidation of YZ
The models used in the present work are based on previously optimized models for the S2 state of the OEC.50 Recent efforts towards refining the 1.9 Å resolution crystallographic model81 of PSII for the S2 state led to the realization that the S2 state of the cluster can exist in two interconvertible and approximately isoenergetic forms. These two forms of S2, which can be described as “open cubane” (S2A) and “closed cubane” (S2B) owing to their core connectivity (see Fig. 2), are valence isomers that differ in the position of the unique Mn(III) ion. This difference in the oxidation state distribution results in distinct magnetic topologies and hence distinct ground spin states for the two structures (S = 1/2 for the open cubane and S = 5/2 for the closed cubane). These two states correspond to the well-known S2 state EPR signatures, the g ≈ 2.0 multiline signal for the open cubane and the g ≈ 4.1 signal for the closed cubane, with the computed hyperfine coupling constants of the metal ions and the first coordination sphere ligands matching experiment. These two structures also rationalize a series of recent experiments aimed at determining the protonation states of Mn-bound water-derived ligands and identifying one of the substrate oxygen atoms.2,73,74,76,82–84 The protonation state assignment for the cluster in the S2 state is as follows: none of the five oxo bridges is protonated (O1–O5) and all four terminal water-derived ligands (W1 and W2 on Mn4, W3 and W4 on Ca2+) represent H2O ligands, with the exception of W2, which is bound as OH−.72 | ||
| Fig. 2 (a) Model of the OEC including the YZ–His190 pair (most of the hydrogen atoms are omitted for clarity). (b) Structure of the S2 state of the Mn4CaO5 core in its two interconvertible forms. | ||
These experimentally validated models represent a well-defined starting point for the search of the corresponding split signal state. This was initiated by oxidation of the open and closed cubane forms, thus mimicking the first step upon illumination of PSII poised in the S2 state, i.e. the oxidation of YZ by P680˙+. This forms S2YZ˙, where the tyrosine radical is weakly coupled to the S2-state Mn cluster (i.e. with the same Mn oxidation states as in S2). As mentioned in the Introduction, this triggers a series of events that results in the formation of the S3 state, which involves in an unspecified sequence: (i) the reduction of the YZ radical by either the last remaining Mn(III) ion of the cluster to yield an all-Mn(IV) configuration or the oxidation of a first coordination sphere ligand; (ii) the binding of the second H2O substrate molecule; and (iii) the loss of a proton either from the incoming H2O molecule or from a pre-existing titratable group.
As described in the Introduction, under physiological advancement YZ oxidation must occur prior to OEC deprotonation as YZ oxidation by P680˙+ is 2–3 orders of magnitude faster than OEC deprotonation. However, it has been suggested25 that deprotonation of the Mn cluster may be required to trap/accumulate the S2YZ˙ intermediate at low temperatures. In this scenario, deprotonation of the cluster presumably serves to retard the decay, via charge recombination, of the arrested S2YZ˙ intermediate. The enhanced lifetime of the YZ˙ state may come about because the lower positive charge of the OEC cluster would stabilize the electron hole on YZ. Evidence for this stabilization mechanism is the observation that a pre-illumination of functional PSII samples at elevated temperatures (200 K) increases the subsequent yield of S2YZ˙ formed at cryogenic (<20 K) temperatures. At this higher temperature the S2YZ˙ state forms but decays very quickly. Importantly though, YZ˙ formation at this temperature is thought to trigger cluster deprotonation, as the protein is flexible enough for proton transfer but not enough to allow S3 formation, which requires a protein conformational change.24,30,31,85 While we cannot exclude this possibility, we consider it unlikely. The S2 state multiline EPR signal is the same with or without this pre-illumination. As demonstrated by Ames et al.,72 deprotonation of the S2-state cluster should clearly manifest itself in terms of an altered55 Mn-hyperfine structure, but no such drastic change is observed experimentally. Therefore, we assume that no modification of the OEC needs to occur to trap the first S2YZ˙ intermediate state, but instead occurs at a later stage (see Section 3.4).
3.2 Geometric structure of the S2YZ˙ states
The optimized open and closed cubane forms of the S2-state (S2A and S2B) described above50 were used as the starting point for optimization of the one-electron oxidized forms. For both structural forms, calculations on the corresponding oxidized species converge to states that contain the YZ radical, S2AYZ˙ and S2BYZ˙. The Mn ions of each structure retain the same electronic configuration and local oxidation states as in the S2-state models, i.e. Mn1 is still a Mn(III) ion in S2AYZ˙ and Mn4 is a Mn(III) ion in S2BYZ˙. Oxidation of a Mn ion instead of the YZ residue was found to be energetically inaccessible. The S2A and S2B forms were confirmed using multiple computational approaches50,86 to be very close in energy, less than 2 kcal mol−1; the same is observed here for the S2AYZ˙ and S2BYZ˙ forms, the latter being 1.3 kcal mol−1 higher in energy than the former for the BP86 optimized structures.As the manganese ions do not change their electronic state, the geometric changes observed upon YZ oxidation are insignificant in the region of the inorganic core. However, this is not the case for YZ. In the S2AYZ˙ model, the tyrosine loses the proton (the O–H distance is 1.639 Å compared to 1.096 Å in the S2A state), which shifts spontaneously to the τ-N (Nε) atom of the imidazole ring of His190 (N–H = 1.058 Å vs. 1.439 Å in S2A). Similar changes are observed in the S2BYZ˙ form, where the O–H and N–H distances are 1.592 and 1.067 Å, respectively, compared with 1.109 and 1.412 in the S2B state. This proton shift suggests that the tyrosyl in S2YZ˙ is best described as a neutral radical coupled to an imidazolium cation. It is noted that such a proton transfer was proposed for the D2-Tyr160 (YD) radical (to D2-His189),87,88 but a recent work of Saito et al. instead suggested that the proton acceptor partner for the homologous YD residue is a nearby water molecule, explaining why its oxidation is not reversible.89 This same proton shift has been demonstrated in biomimetic model systems.90,125
3.3 Electronic structure of the S2YZ˙ states
Fig. 3(a) shows the frontier spin-α and spin-β orbitals of the S2 state of the OEC in the multiline g ≈ 2 (S2A) conformation. For both spin manifolds, the HOMO − 1 is a π-type orbital localized mostly on the imidazole ring of His190 and the HOMO is localized on Tyr161. This orbital takes the form of an antibonding combination of the second-lowest occupied π orbital of the benzyl ring with the pπ orbital of the oxygen atom. All dominantly manganese-based orbitals lie lower in energy. The LUMOs are different for the two manifolds: the α-LUMO is a metal–ligand σ-antibonding orbital localized on Mn4 whereas the β-LUMO is exclusively of metal character, being almost equally distributed over Mn2, Mn3 and Mn4. A similar configuration is obtained for the closed cubane form S2B (see ESI†). In the context of this one-particle picture, the tyrosine-based HOMO should be the orbital involved in the one-electron oxidation of the system. This is precisely what the calculations on the S2YZ˙ states demonstrate: Fig. 3(b) depicts the frontier orbitals of the oxidized S2AYZ˙ state (for the corresponding orbitals of S2BYZ˙, see the ESI†), while Fig. 4 shows the spin density distributions for both forms of the cluster with a clear localization of the π-type unpaired spin density on the phenoxyl ring. The spin populations are also consistent with the localization of one unpaired electron on YZ; as in the S2 state, the spin populations on the Mn ions are close to the values expected for the formal oxidation states of III and IV, ca. 3.8–3.9 and 2.9–3.0 unpaired electrons, respectively. | ||
| Fig. 3 Frontier orbitals of the S2A model of the OEC (a) and the oxidized S2AYZ˙ model (b); hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 4 Spin density maps and Mulliken spin populations on the tyrosyl fragment for models of the open cubane S2AYZ˙ (left) and closed cubane S2BYZ˙ (right) states of the OEC in their respective lowest-energy broken-symmetry spin configurations. | ||
It is stressed that no valence isomers could be found where the Mn(III) ion is oxidized instead of YZ. This suggests that the S2 state cannot proceed to the S3 state but instead is arrested in the S2YZ˙ state. It is also important to note that the S2 state structures were developed using backbone constraints from X-ray crystallographic data and are fully consistent with the EPR results which constrain the protonation state. Thus our results require a conformational change during the S2 to S3 transition to allow the oxidation of the cluster, in line with earlier experimental results. It is noted that if the Tyr161–His190 pair is absent from the S2 state model, the HOMO is fundamentally different by construction from the one described above. Oxidation of such an S2-state model without further modifications (water binding and/or deprotonation) leads, by definition, to an “unphysical” oxidation event that cannot have any correspondence to the natural system.
3.4 YZ oxidation reorients the dipole moment of the OEC
An important question regarding the mechanistic details of the S2–S3 transition is what happens after the oxidation of YZ by P680˙+ and the formation of S2YZ˙. Although the next steps are not explicitly studied in the present work, an important observation is that the dipole moment of the model is reoriented after YZ oxidation. Fig. 5 compares the dipole moment vector before and after oxidation of YZ. It can be seen that in the “split signal” state the dipole moment is directed in such a way that it points approximately from the cationic imidazolium of His190 to Asp61. The same result is seen for both S2AYZ˙ and S2BYZ˙, suggesting that this region of the OEC is now the locus of the negative charge regardless of the formal oxidation state of Mn4.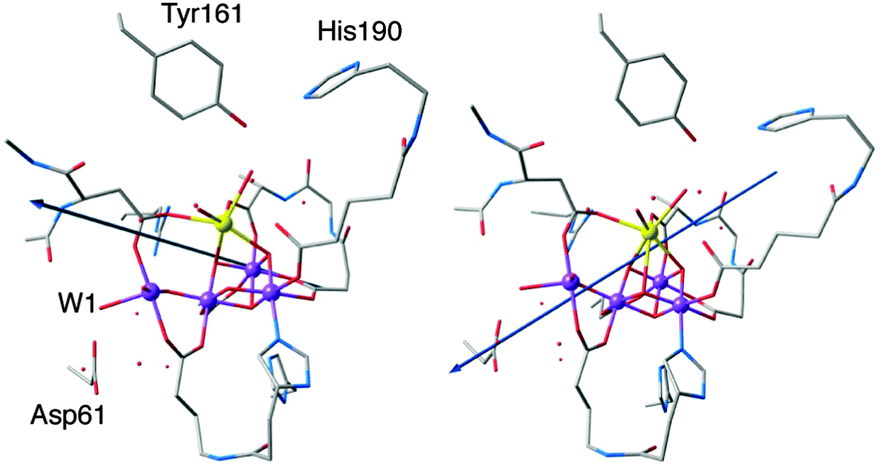 | ||
| Fig. 5 Dipole moment in the S2B state (left) and in the oxidized S2BYZ˙ state (right). | ||
In the context of the putative requirements for progression to the S3 state (see Fig. 1), this dipole reorientation can be interpreted as indicating the likely direction of proton removal from the system. Given that the dipole is almost coincident with the Mn4–W1 bond and Asp61, it is likely that YZ oxidation triggers the loss of a proton from W1 to the acceptor Asp61. This was suggested also in previous computational studies91–93 and would be consistent with the proposal that: (i) proton release occurs prior to formation of the S3 state42 and (ii) Asp61 participates in a proton-transfer pathway involving the proximal chloride ion.94–97 Note that a continuous network of hydrogen bonds formed by a chain of water molecules between YZ and Mn4 establishes a communication pathway across the cluster.89,98–101
3.5 Magnetism of the S2YZ˙ states
The magnetism of each S2YZ˙ model is defined by Mn–Mn ion exchange interactions within the inorganic cluster, and the tyrosine–OEC interaction which is orders of magnitude smaller. For the two configurations discussed above (S2AYZ˙ and S2BYZ˙) an overdetermined system of equations, which is derived from the set of broken-symmetry solutions, can be solved by singular value decomposition to yield pairwise exchange coupling constants, Jij, shown in Fig. 6. Based on these values, diagonalization of the Heisenberg Hamiltonian then yields the complete spectrum of energy levels produced by the coupling of the four local spins of each model. In line with the small structural perturbations of the inorganic core upon YZ oxidation, the exchange coupling constants for the S2AYZ˙ and S2BYZ˙ models are similar to those obtained previously for the corresponding S2 state models: in the open cubane form there are two antiferromagnetic couplings, between Mn1–Mn2 and Mn3–Mn4, whereas in the closed cubane form all interactions within the cuboidal part are ferromagnetic.50,75 | ||
| Fig. 6 Exchange coupling constants (cm−1) and ground spin states of the Mn4CaO5 subunit for the open-cubane (left) and closed-cubane (right) forms of the S2YZ˙ state. | ||
These results require that the two clusters retain the same ground spin state seen in the corresponding S2 state structure, i.e. S = 1/2 for the open cubane and S = 5/2 for the closed cubane form. The first excited states and their separation are also similar: S = 3/2 at 23.2 cm−1 and S = 7/2 at 8.6 cm−1, respectively. In view of these results, it can be surmised that the split signals arising from the S2AYZ˙ and S2BYZ˙ forms reflect the interaction of the S = 1/2 tyrosyl radical with the OEC exhibiting a net spin state of different magnitudes in each case. As the two oxidized forms remain as close in energy as in S2, it might be expected that illumination of PSII poised in the S2 state with both S2A and S2B forms being populated, would lead to formation of both S2AYZ˙ and S2BYZ˙, and therefore of two split signals. However, only a signal attributed to an interaction of YZ˙ with a spin 1/2 state of the manganese cluster has been experimentally observed. The above results do not allow us to address the question why the putative S2BYZ˙ split signal has not yet been observed, since the present calculations reveal nothing about the lifetimes of the states or their possible decay pathways. Nevertheless, the results allow us to positively identify the experimentally observed split signal of the S2 state with the open cubane conformation of the manganese cluster, i.e. with the S2AYZ˙ model. It is noted though that the closed cubane conformation may be associated with the NIR-induced split signals (see Introduction).
Turning now to the coupling between the inorganic cluster and the oxidized tyrosine, according to simulations of EPR spectra, this interaction is of the order of −400 MHz.13 Since this value is very small, it becomes important to eliminate all numerical noise from the calculations and for this reason a higher threshold for convergence was used for the computed broken-symmetry energies. Nevertheless, when confronted with such energy differences, agreement of the calculation with experiment even in terms of the order of magnitude should be considered satisfactory. In the present case the calculated values for the coupling of YZ˙ with the manganese cluster are −819 MHz for S2AYZ˙ and −626 MHz for S2BYZ˙, when the lowest-energy spin configuration is used in the broken-symmetry calculations for each of the manganese clusters (∣αββα>, MS = 1/2 for the open cubane and ∣αααβ>, MS = 5/2 for the closed cubane form). If instead the high-spin (MS = 13/2) configuration of the clusters is used, then the corresponding values become −149 MHz for S2AYZ˙ and −324 MHz for S2BYZ˙. In either case the models used in this work correctly capture the essential physics of the interaction.
3.6 The g-matrix of the YZ˙ radical
The calculation of the g-matrix in tyrosine radicals has been attempted previously using various approaches. The majority of these initial studies used simplified models such as the phenoxyl–water or phenoxyl–imidazolium pair to obtain structural and spectroscopic properties of the tyrosine radical.102–108 Recently, more elaborate models of PSII that include the surrounding residues were used to study the histidine–tyrosine pair for both YZ and YD.88,89,109,110 In addition to considering the effects of the protein on the calculated properties, these larger models have allowed the environment of the two redox active tyrosine residues of PSII (YD and YZ) to be differentiated.Our g-matrix calculations match earlier model system studies regarding the g orientation with respect to the phenoxyl ring: the gx axis is oriented almost perfectly along the C–O bond (θ = 3.8°) and gz is perpendicular to the plane of the ring (Fig. 7). The values of the individual g-matrix components are identical for the two forms of the cluster, indicating the absence of any effect derived from the different magnetic topologies of the inorganic core.
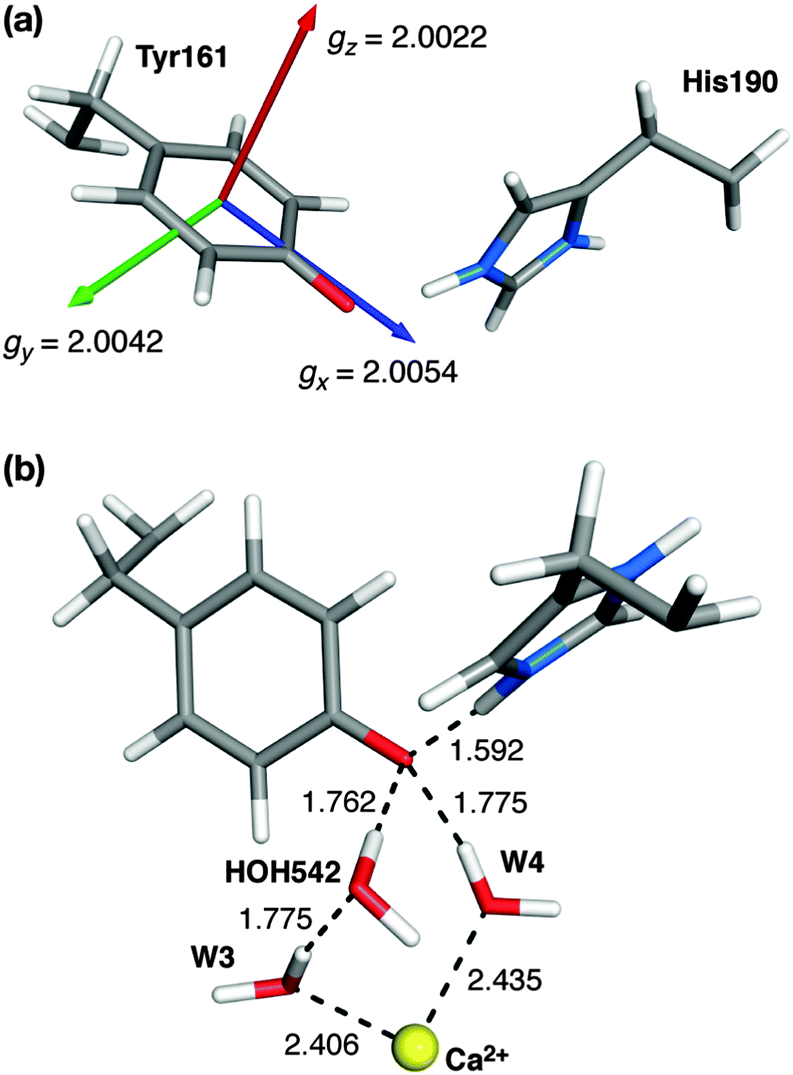 | ||
| Fig. 7 (a) Orientation and components of the g-matrix for the S2BYZ˙ state. (b) Water molecules involved in hydrogen bonding to YZ˙ (distances, in Å, shown for the S2BYZ˙ model). | ||
The dependence of the g-matrix elements on the position of the proton between the YZ–His190 pair is depicted in Fig. 8. The gx component shows the strongest dependence: the largest calculated value corresponds to the largest O⋯H distance (gx = 2.0057), while the smallest value is obtained for the structure in which the hydrogen is closest to the oxygen atom of the YZ˙ radical (gx = 2.0041). For the gy and gz components no such dependence is observed, in agreement with the previous tyrosyl radical g-tensor studies.
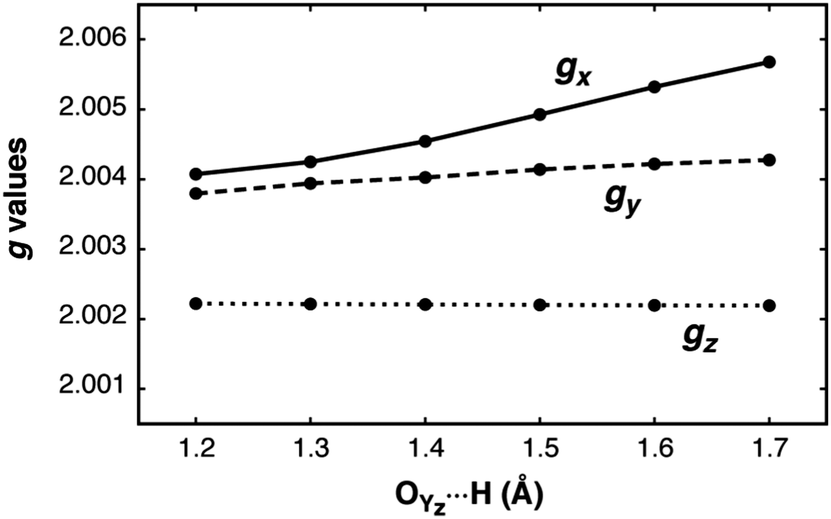 | ||
| Fig. 8 Scan of the g values for the S2BYZ˙ model with respect to the YZ˙O⋯H distance. | ||
EPR studies of YZ˙ have been performed by replacing YD by phenylalanine and removing the Mn cluster (see ref. 39 and references therein for a review of the relevant literature). In contrast to YD˙, the g-matrix anisotropy of YZ (in the absence of the OEC cluster) has only recently been measured.108 The values of gx = 2.00714 in frozen solution and 2.00705 in single crystals have been reported,108 while the experimental values of gy and gz match the computed ones of Fig. 7a almost exactly. These higher values of gx are similar to the ones reported for the much more extensively studied Tyr160 (YD) of the D2 protein of PSII; a compilation of YD˙ values for different organisms is given in Hofbauer et al. (gx ≈ 2.0074–2.0078).111 Compared with these values and those of other systems, the computed value of 2.0054 for gx appears to be somewhat small. However, gx is known to strongly depend on hydrogen bonding and the local electrostatic interactions. To give a few representative examples, the gx value of the tyrosyl radical Y122˙ in E. coli ribonucleotide reductase (RNR) shifts from 2.0087 to 2.0091 in the absence of any hydrogen bonding to ca. 2.0076 in a more polar environment,104,112,113 similar to the value for the YD˙ of PSII. At the other extreme, a value of gx = 2.0052 for the NH2Y730˙ radical in E. coli RNR has been observed,114 resulting from the combined influence of three hydrogen bonds.115 Similarly, in phenoxyl radical compounds that contain a strong intramolecular hydrogen bond the gx values fall within 2.0063–2.0067.116 In the present case the low gx value of 2.0054 for S2YZ˙ can be interpreted as being due to the existence of three hydrogen-bonding interactions, two from adjacent water molecules (see Fig. 7b) and one from the proton that shifts to the imidazole of His190, which remains at a distance of approximately 1.6 Å from the O atom of YZ˙.
To test this hypothesis, three additional calculations were carried out on fragments of the full model. First, the inorganic part of the model and all its associated ligands were removed, leaving a model containing only the YZ–His190 pair and the two water molecules that form direct hydrogen bonds to the phenoxyl oxygen, the calcium-bound W4 (HOH540 in the 3ARC PDB structure) and the HOH542 that forms a hydrogen-bonding bridge between the other calcium-bound water (W3 or HOH541) and the tyrosine. The structure was not allowed to relax, essentially isolating the effect of the inorganic cluster while retaining the immediate environment of the tyrosine. As shown in Table 1, the effect of the Mn cluster on the low gx value is insignificant. Removing also the HOH542 molecule results in an increase of gx value to 2.0063. However, the largest effect was observed when both H-bonding water molecules were removed; the model that contains only the two amino acid residues displays a marked increase in the gx value to 2.0072, showing how the local electropositive environment plays a crucial role in suppressing the expected effect on gx of the proton shift to His190.
| Model | g x | g y | g z | ρ O |
|---|---|---|---|---|
| Full model | 2.0054 | 2.0042 | 2.0022 | 0.254 |
| YZ˙–His with W4, HOH542 | 2.0055 | 2.0043 | 2.0023 | 0.267 |
| YZ˙–His with W4 | 2.0063 | 2.0044 | 2.0022 | 0.321 |
| YZ˙–His | 2.0072 | 2.0046 | 2.0022 | 0.360 |
As demonstrated previously for a series of synthetic phenoxyl radical models, the value of gx tracks closely the change in the unpaired spin population on the oxygen atom (Table 1 and Fig. S3, ESI†), the atom with the largest spin–orbit coupling constant.116 An almost linear correlation is observed between the O spin population and gx, with R2 = 0.993. The change in spin population can be interpreted as the result of increased hydrogen bonding “pulling” the electron density towards the oxygen and hence “pushing” the unpaired density over the phenyl ring, an effect also seen for the in-plane proton alone (Fig. 8). A detailed discussion of this effect is provided by Sinnecker et al. for hydrogen bonding in semiquinones117 and for the case of the primary quinone (QA/QA˙−) in bacterial reaction centers.118
It is noted that the larger value of gx for the model where the hydrogen-bonded water molecules are removed agrees with the experimental value reported for YZ˙ in Mn-depleted PSII and with that reported for the YD radical. The above observation regarding gx implies that the removal of the OEC cluster in the experiments also perturbs the native hydration environment of YZ. As a corollary, and given the highly ordered hydrogen bonding network shown in Fig. 7, it can be suggested that in addition to other roles,3,119–121 the Ca2+ ion serves to structure the water molecules in that region and adjust their acidities in order to (a) modulate the electronic structure and hence the redox potential of the tyrosine residue and (b) optimize the hydrogen-bond-mediated communication between YZ and the inorganic cluster.
Given that the hydrogen bonding network into which YZ is embedded must be highly optimized for a proper function, even small perturbations on the structure and pKA value of the constituent water molecules may have a significant impact on the function of the catalyst. The role of the structured water environment in modulating the redox potential of the tyrosyl radical can be inferred by estimating the electron affinities of the simplified models of Table 1. DFT calculations suggest that the absence or presence of the water molecules at the optimized positions shifts the electron affinity by ca. 0.46/0.28 eV (vertical/adiabatic values; Fig. S4, ESI†). The above suggestion for the role of the Ca2+ ion is consistent with the measurements of frequency shifts of the proximal peptide carbonyl bands as markers for hydrogen-bonding changes between the S1–S2 transition in the presence of different cations.122 It is also in line with the recent proposal of Boussac and coworkers regarding the Ca2+ ion acting as an entropic regulator for the S3–S0 transition based on its involvement in structuring the environment of YZ in the S3 state.123
4. Conclusions
In this work we developed structural models for the S2YZ˙ “split signal” state of the OEC, based on the open-cubane (S2A) and closed-cubane (S2B) forms of the S2 state. It was observed that no proton removal was required to form the oxidized states S2AYZ˙ and S2BYZ˙. In addition, no valence isomers could be found where the Mn(III) ion is oxidized instead of YZ, implying that a chemical modification of the OEC is necessary for the subsequent S2 to S3 transition to occur. The reorientation of the dipole moment suggests that this most probably involves the deprotonation of W1.The calculations of the tyrosyl g-matrix provide estimates (gx = 2.0054, gy = 2.0042, and gz = 2.0022) that form a good predictive basis regarding the environment of the tyrosine. The response of the g-matrix components was examined with respect to the extent of proton transfer to the τ-N of His190 and the contribution of different subsets of the model. The predicted gx value is due to the presence of three hydrogen bonding interactions in which calcium-bound water molecules are involved. Given that the existing experimental g-anisotropy values were obtained from measurements on Ca-depleted PSII samples, it is suggested that the measured values do not necessarily reflect the natural environment of YZ because they presumably miss the structuring effect of the Ca2+ ion which operates in the native system. Thus, the Ca2+ ion, besides its possible role in adjusting the redox potential of the inorganic cluster itself, as also indicated in studies of synthetic complexes,124 has an additional role; it organizes the water environment and optimizes the hydrogen-bonding network around YZ. Since, as demonstrated by the present results, hydrogen bonding to W4 and HOH542 strongly affects the electronic structure of the tyrosyl radical through modulation of its spin and charge density distribution, this ordering effect of the Ca2+ ion indirectly fine-tunes the function of the tyrosine residue by regulating its redox potential and electron transfer properties.
References
- J. Messinger and G. Renger, in Primary Processes of Photosynthesis, Part 2: Principles and Apparatus, The Royal Society of Chemistry, Cambridge, 2008, vol. 9, pp. 291–349 Search PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell, Oxford, 2001 Search PubMed.
- N. Cox and W. Lubitz, in Chemical Energy Storage, ed. R. Schlögl, De Gruyter, Berlin, 2013, pp. 185–224 Search PubMed.
- S. DeBeer, M. van Gastel, E. Bill, S. Ye, T. Petrenko, D. A. Pantazis and F. Neese, in Chemical Energy Storage, ed. R. Schlögl, De Gruyter, Berlin, 2013, pp. 353–377 Search PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 CAS.
- M. M. Najafpour, A. N. Moghaddam, S. I. Allakhverdiev and Govindjee, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1110–1121 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- V. Petrouleas, D. Koulougliotis and N. Ioannidis, Biochemistry, 2005, 44, 6723–6728 CrossRef CAS PubMed.
- J. M. Peloquin, K. A. Campbell and R. D. Britt, J. Am. Chem. Soc., 1998, 120, 6840–6841 CrossRef CAS.
- J. H. A. Nugent, I. P. Muhiuddin and M. C. W. Evans, Biochemistry, 2002, 41, 4117–4126 CrossRef CAS PubMed.
- N. Ioannidis and V. Petrouleas, Biochemistry, 2000, 39, 5246–5254 CrossRef CAS PubMed.
- C. Zhang and S. Styring, Biochemistry, 2003, 42, 8066–8076 CrossRef CAS PubMed.
- C. Zhang, A. Boussac and A. W. Rutherford, Biochemistry, 2004, 43, 13787–13795 CrossRef CAS PubMed.
- K. G. V. Havelius, J.-H. Su, Y. Feyziyev, F. Mamedov and S. Styring, Biochemistry, 2006, 45, 9279–9290 CrossRef CAS PubMed.
- N. Ioannidis, G. Zahariou and V. Petrouleas, Biochemistry, 2006, 45, 6252–6259 CrossRef CAS PubMed.
- J.-H. Su, K. G. V. Havelius, F. M. Ho, G. Han, F. Mamedov and S. Styring, Biochemistry, 2007, 46, 10703–10712 CrossRef CAS PubMed.
- A. Boussac, M. Sugiura, T.-L. Lai and A. W. Rutherford, Philos. Trans. R. Soc., B, 2008, 363, 1203–1210 CrossRef CAS PubMed.
- G. Sioros, D. Koulougliotis, G. Karapanagos and V. Petrouleas, Biochemistry, 2006, 46, 210–217 CrossRef PubMed.
- M. Chrysina, G. Zahariou, N. Ioannidis and V. Petrouleas, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 487–493 CrossRef CAS PubMed.
- M. Chrysina, G. Zahariou, Y. Sanakis, N. Ioannidis and V. Petrouleas, J. Photochem. Photobiol., B, 2011, 104, 72–79 CrossRef CAS PubMed.
- J. Sjöholm, S. Styring, K. G. V. Havelius and F. M. Ho, Biochemistry, 2012, 51, 2054–2064 CrossRef PubMed.
- J.-H. Su, K. G. V. Havelius, F. Mamedov, F. M. Ho and S. Styring, Biochemistry, 2006, 45, 7617–7627 CrossRef CAS PubMed.
- N. Cox, F. M. Ho, N. Pewnim, R. Steffen, P. J. Smith, K. G. V. Havelius, J. L. Hughes, L. Debono, S. Styring, E. Krausz and R. J. Pace, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 882–889 CrossRef CAS PubMed.
- K. G. V. Havelius, J.-H. Su, G. Han, F. Mamedov, F. M. Ho and S. Styring, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 11–21 CrossRef CAS PubMed.
- K. G. V. Havelius, J. Sjöholm, F. Ho, F. Mamedov and S. Styring, Appl. Magn. Reson., 2010, 37, 151–176 CrossRef.
- A. Boussac and A. W. Rutherford, Biochemistry, 1988, 27, 3476–3483 CrossRef CAS.
- A. Boussac, J. L. Zimmermann and A. W. Rutherford, Biochemistry, 1989, 28, 8984–8989 CrossRef CAS.
- M. Sivaraja, J. Tso and G. C. Dismukes, Biochemistry, 1989, 28, 9459–9464 CrossRef CAS.
- D. A. Force, D. W. Randall and R. D. Britt, Biochemistry, 1997, 36, 12062–12070 CrossRef CAS PubMed.
- P. Geijer, F. Morvaridi and S. Styring, Biochemistry, 2001, 40, 10881–10891 CrossRef CAS PubMed.
- J. Sjöholm, K. G. V. Havelius, F. Mamedov and S. Styring, Biochemistry, 2010, 49, 9800–9808 CrossRef PubMed.
- N. Ioannidis, J. H. A. Nugent and V. Petrouleas, Biochemistry, 2002, 41, 9589–9600 CrossRef CAS PubMed.
- N. Ioannidis and V. Petrouleas, Biochemistry, 2002, 41, 9580–9588 CrossRef CAS PubMed.
- D. Koulougliotis, J.-R. Shen, N. Ioannidis and V. Petrouleas, Biochemistry, 2003, 42, 3045–3053 CrossRef CAS PubMed.
- W. Lubitz, in Electron Paramagnetic Resonance, ed. B. C. Gilbert, M. J. Davies and D. M. Murphy, The Royal Society of Chemistry, 2004, vol. 19, pp. 174–242 Search PubMed.
- A. Haddy, Photosynth. Res., 2007, 92, 357–368 CrossRef CAS PubMed.
- H. Dau and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 472–483 CrossRef CAS PubMed.
- A. Klauss, M. Haumann and H. Dau, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16035–16040 CrossRef CAS PubMed.
- T. Noguchi, Philos. Trans. R. Soc., B, 2008, 363, 1189–1195 CrossRef CAS PubMed.
- J. Messinger, T. Noguchi and J. Yano, in Molecular Solar Fuels, ed. T. J. Wydrzynski and W. Hillier, The Royal Society of Chemistry, Cambridge, 2012, pp. 163–207 Search PubMed.
- W. Hillier and T. Wydrzynski, Coord. Chem. Rev., 2008, 252, 306–317 CrossRef CAS PubMed.
- J. Messinger, J. H. Robblee, U. Bergmann, C. Fernandez, P. Glatzel, H. Visser, R. M. Cinco, K. L. McFarlane, E. Bellacchio, S. A. Pizarro, S. P. Cramer, K. Sauer, M. P. Klein and V. K. Yachandra, J. Am. Chem. Soc., 2001, 123, 7804–7820 CrossRef CAS PubMed.
- M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke and H. Dau, Biochemistry, 2005, 44, 1894–1908 CrossRef CAS PubMed.
- P. Glatzel, H. Schroeder, Y. Pushkar, T. Boron, S. Mukherjee, G. Christou, V. L. Pecoraro, J. Messinger, V. K. Yachandra, U. Bergmann and J. Yano, Inorg. Chem., 2013, 52, 5642–5644 CrossRef CAS PubMed.
- N. Cox and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1020–1030 CrossRef CAS PubMed.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS PubMed.
- K. Yamaguchi, Y. Takahara and T. Fueno, in Applied Quantum Chemistry, ed. V. H. Smith Jr., H. F. Schaefer III, K. Morokuma and D. Reidel, Boston, 1986, p. 155 Search PubMed.
- S. Yamanaka, T. Kawakami, H. Nagao and K. Yamaguchi, Chem. Phys. Lett., 1994, 231, 25–33 CrossRef CAS.
- A. Bencini, F. Totti, C. A. Daul, K. Doclo, P. Fantucci and V. Barone, Inorg. Chem., 1997, 36, 5022–5030 CrossRef CAS.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS PubMed.
- M. Orio, D. A. Pantazis and F. Neese, Photosynth. Res., 2009, 102, 443–453 CrossRef CAS PubMed.
- F. Neese, W. Ames, G. Christian, M. Kampa, D. G. Liakos, D. A. Pantazis, M. Roemelt, P. Surawatanawong and S. F. Ye, Adv. Inorg. Chem., 2010, 62, 301–349 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- A. Klamt and D. Schüürman, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS PubMed.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS PubMed.
- D. A. Pantazis, X. Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- D. A. Pantazis, M. Orio, T. Petrenko, S. Zein, E. Bill, W. Lubitz, J. Messinger and F. Neese, Chem. – Eur. J., 2009, 15, 5108–5123 CrossRef CAS PubMed.
- D. A. Pantazis, M. Orio, T. Petrenko, S. Zein, W. Lubitz, J. Messinger and F. Neese, Phys. Chem. Chem. Phys., 2009, 11, 6788–6798 RSC.
- W. Ames, D. A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz and F. Neese, J. Am. Chem. Soc., 2011, 133, 19743–19757 CrossRef CAS PubMed.
- N. Cox, L. Rapatskiy, J.-H. Su, D. A. Pantazis, M. Sugiura, L. Kulik, P. Dorlet, A. W. Rutherford, F. Neese, A. Boussac, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2011, 133, 3635–3648 CrossRef CAS PubMed.
- J.-H. Su, N. Cox, W. Ames, D. A. Pantazis, L. Rapatskiy, T. Lohmiller, L. V. Kulik, P. Dorlet, A. W. Rutherford, F. Neese, A. Boussac, W. Lubitz and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 829–840 CrossRef CAS PubMed.
- V. Krewald, F. Neese and D. A. Pantazis, J. Am. Chem. Soc., 2013, 135, 5726–5739 CrossRef CAS PubMed.
- M. Pérez Navarro, W. M. Ames, H. Nilsson, T. Lohmiller, D. A. Pantazis, L. Rapatskiy, M. M. Nowaczyk, F. Neese, A. Boussac, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15561–15566 CrossRef PubMed.
- M. Retegan, F. Neese and D. A. Pantazis, J. Chem. Theory Comput., 2013, 9, 3832–3842 CrossRef CAS.
- F. Neese, J. Chem. Phys., 2001, 115, 11080–11096 CrossRef CAS PubMed.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- L. D. Slep, A. Mijovilovich, W. Meyer-Klaucke, T. Weyhermüller, E. Bill, E. Bothe, F. Neese and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 15554–15570 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- T. Lohmiller, N. Cox, J.-H. Su, J. Messinger and W. Lubitz, J. Biol. Chem., 2012, 287, 24721–24733 CrossRef CAS PubMed.
- L. Rapatskiy, N. Cox, A. Savitsky, W. M. Ames, J. Sander, M. M. Nowaczyk, M. Rögner, A. Boussac, F. Neese, J. Messinger and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 16619–16634 CrossRef CAS PubMed.
- T. Lohmiller, V. Krewald, M. Pérez Navarro, M. Retegan, L. Rapatskiy, M. M. Nowaczyk, A. Boussac, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Phys. Chem. Chem. Phys., 2014 10.1039/C3CP55017F.
- Y. Pushkar, J. Yano, K. Sauer, A. Boussac and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1879–1884 CrossRef CAS PubMed.
- D. Bovi, D. Narzi and L. Guidoni, Angew. Chem., Int. Ed., 2013, 52, 11744–11749 CrossRef CAS PubMed.
- S. Styring, J. Sjöholm and F. Mamedov, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 76–87 CrossRef CAS PubMed.
- R. Hart and P. J. O'Malley, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 250–254 CrossRef CAS PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7690–7695 CrossRef CAS PubMed.
- G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466–10467 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2012, 14, 4849–4856 RSC.
- P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135, 9442–9449 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- F. M. Ho, in Molecular Solar Fuels, ed. T. J. Wydrzynski and W. Hillier, The Royal Society of Chemistry, Cambridge, 2012, pp. 208–248 Search PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- H. Ishikita, W. Saenger, B. Loll, J. Biesiadka and E.-W. Knapp, Biochemistry, 2006, 45, 2063–2071 CrossRef CAS PubMed.
- I. Rivalta, M. Amin, S. Luber, S. Vassiliev, R. Pokhrel, Y. Umena, K. Kawakami, J. R. Shen, N. Kamiya, D. Bruce, G. W. Brudvig, M. R. Gunner and V. S. Batista, Biochemistry, 2011, 50, 6312–6315 CrossRef CAS PubMed.
- K. Linke and F. M. Ho, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 14–32 CrossRef CAS PubMed.
- R. J. Service, W. Hillier and R. J. Debus, Biochemistry, 2010, 49, 6655–6669 CrossRef CAS PubMed.
- R. J. Service, W. Hillier and R. J. Debus, Biochemistry, 2014, 53, 1001–1017 CrossRef CAS PubMed.
- M. Shoji, H. Isobe, S. Yamanaka, Y. Umena, K. Kawakami, N. Kamiya, J. R. Shen and K. Yamaguchi, Catal. Sci. Technol., 2013, 3, 1831–1848 CAS.
- P. J. O'Malley, J. Am. Chem. Soc., 1998, 120, 11732–11737 CrossRef CAS.
- Y.-N. Wang and L. A. Eriksson, Int. J. Quantum Chem., 2001, 83, 220–229 CrossRef CAS.
- M. Engström, F. Himo, A. Gräslund, B. Minaev, O. Vahtras and H. Agren, J. Phys. Chem. A, 2000, 104, 5149–5153 CrossRef.
- M. Brynda and R. David Britt, Res. Chem. Intermed., 2007, 33, 863–883 CrossRef CAS PubMed.
- M. Engström, F. Himo and H. Ågren, Chem. Phys. Lett., 2000, 319, 191–196 CrossRef.
- M. Kaupp, T. Gress, R. Reviakine, O. L. Malkina and V. G. Malkin, J. Phys. Chem. B, 2002, 107, 331–337 CrossRef.
- H. Matsuoka, J.-R. Shen, A. Kawamori, K. Nishiyama, Y. Ohba and S. Yamauchi, J. Am. Chem. Soc., 2011, 133, 4655–4660 CrossRef CAS PubMed.
- K. Saito, J.-R. Shen, T. Ishida and H. Ishikita, Biochemistry, 2011, 50, 9836–9844 CrossRef CAS PubMed.
- K. Saito and H. Ishikita, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 159–166 CrossRef CAS PubMed.
- W. Hofbauer, A. Zouni, R. Bittl, J. Kern, P. Orth, F. Lendzian, P. Fromme, H. T. Witt and W. Lubitz, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6623–6628 CrossRef CAS PubMed.
- M. Högbom, M. Galander, M. Andersson, M. Kolberg, W. Hofbauer, G. Lassmann, P. Nordlund and F. Lendzian, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3209–3214 CrossRef PubMed.
- G. Bleifuss, M. Kolberg, S. Pötsch, W. Hofbauer, R. Bittl, W. Lubitz, A. Gräslund, G. Lassmann and F. Lendzian, Biochemistry, 2001, 40, 15362–15368 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, T. Argirević, E. C. Minnihan, J. Stubbe and M. Bennati, J. Am. Chem. Soc., 2009, 131, 15729–15738 CrossRef CAS PubMed.
- T. Argirević, C. Riplinger, J. Stubbe, F. Neese and M. Bennati, J. Am. Chem. Soc., 2012, 134, 17661–17670 CrossRef PubMed.
- L. Benisvy, R. Bittl, E. Bothe, C. D. Garner, J. McMaster, S. Ross, C. Teutloff and F. Neese, Angew. Chem., Int. Ed., 2005, 44, 5314–5317 CrossRef CAS PubMed.
- S. Sinnecker, E. Reijerse, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2004, 126, 3280–3290 CrossRef CAS PubMed.
- S. Sinnecker, M. Flores and W. Lubitz, Phys. Chem. Chem. Phys., 2006, 8, 5659–5670 RSC.
- H. J. van Gorkom and C. F. Yocum, in Photosystem II. The Light-Driven Water: Plastoquinone Oxidoreductase, ed. T. Wydrzynski and K. Satoh, Springer, Dordrecht, 2005, vol. 22, pp. 307–328 Search PubMed.
- C. F. Yocum, Coord. Chem. Rev., 2008, 252, 296–305 CrossRef CAS PubMed.
- M. Miqyass, H. J. Gorkom and C. F. Yocum, Photosynth. Res., 2007, 92, 275–287 CrossRef CAS PubMed.
- B. C. Polander and B. A. Barry, J. Phys. Chem. Lett., 2013, 786–791 CrossRef CAS.
- F. Rappaport, N. Ishida, M. Sugiura and A. Boussac, Energy Environ. Sci., 2011, 4, 2520–2524 CAS.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef PubMed.
- T. Irebo, S. Y. Reece, M. Sjödin, D. G. Nocera and L. Hammarström, J. Am. Chem. Soc., 2007, 129, 15462–15464 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp00696h |
| This journal is © the Owner Societies 2014 |