 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The molecular configuration of a DOPA/ST monolayer at the air–water interface: a molecular dynamics study†
Chui-Peng
Kong
ab,
E. A. J. F.
Peters
b,
Qing-Chuan
Zheng
a,
G.
de With
*b and
Hong-Xing
Zhang
*a
aState Key Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, People’s Republic of China. E-mail: zhanghx@jlu.edu.cn
bLaboratory of Materials and Interface Chemistry, Eindhoven University of Technology, P. O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: G.d.With@tue.nl
First published on 26th March 2014
Abstract
In this study, surface pressure–area isotherms for N-stearoyldopamine (DOPA) and 4-stearylcatechol (ST) monolayers are obtained by means of molecular dynamics simulations and compared to experimental isotherms. The difference between DOPA and ST is an amide group, which is present in the alkyl tails of DOPA molecules. We find a large difference between the isotherms for DOPA and ST monolayers. Upon using TIP4P/2005 for water and OPLS force fields for the organic material and a relatively large system size, the simulated results are found to be consistent with experiments. With molecular dynamics simulations, the configurations of molecules in the monolayers can be directly analyzed. When the surface pressure is high, a regular molecular orientation is observed for ST molecules, whereas regular orientations are only observed in local domains for DOPA molecules. The differences between DOPA and ST monolayers are attributed to the amide groups in DOPA molecules, which are useful for both steric effects and the formation of hydrogen bonds in the DOPA monolayers. This study clearly demonstrates that hydrogen bonds, due to the presence of the amide group in DOPA, are the cause of the disorder in its Langmuir monolayers. Thus, the conclusion may be helpful in making ordered organic monolayers in the future.
Introduction
Langmuir–Blodgett films, which are obtained by transferring an air–water interface monolayer onto a solid substrate, are widely used because of their capability of building highly ordered structures on substrate surfaces.1–4 The molecular structure of the monolayer is of critical importance and is mainly controlled by compressing the monolayer at the interface of air and water.5–8 For example, the Langmuir–Blodgett films of triphenylene-based pyridinium and imidazolium salts with bromine had been studied. For Langmuir–Blodgett films transferred at different surface pressures, different configurations of molecules on hydrophilic silicon substrates are observed.9While the macroscopic results of the monolayers are usually revealed by experimental investigations, the formation of a monolayer at the air–water interface has been also extensively studied using simulation methods.10–14 Kox et al.15 carried out one of the first molecular dynamics (MD) simulations, by simulating a lipid monolayer based on a relatively simplified model. Since then a number of studies based on Lennard-Jones (LJ) and Coulomb interactions have been performed with the purpose of studying the details of the molecular structures on the surface of the water.12–14,16 Most of these simulation studies are based on well-developed force fields or coarse grained methods combined with the NVT ensemble.14,17–19
The target molecules in this study are derived from blue mussel's foot proteins.20 A number of studies21–23 have revealed that the unusually frequent appearance of 3,4-dihydroxy-L-phenylalanine in mussel's protein is the main reason for its unmatched adhesive ability.24–26 The adhesion is mainly due to the catechol and amide groups. Thus, by combining the amide and catechol groups with an aliphatic tail, a molecule with both hydrophilic and hydrophobic groups has been formed, namely N-stearoyldopamine (DOPA). The monolayer of DOPA can be potentially used as an adhesion promoter. To assess the role of the amide groups during the formation of a DOPA monolayer, a molecule with a similar structure except for the amide group, namely 4-stearylcatechol (ST), is also included for comparison. The properties of DOPA and ST on a gold surface has been studied both experimentally and theoretically in our previous studies.27,28
It has been known that the results of a molecular simulation containing water molecules can be influenced by the choice of the water model.29,30 Thus, to assess the influence of water models, DOPA/ST monolayers on a water surface are simulated with two kinds of water models (TIP4P/200531 and SPC32,33). The influence of different boundary sizes on the simulation results has also been considered. To construct surface pressure–area (π–A) isotherms, MD simulations were performed by controlling the surface tension of the whole system and the tail correction item is added.30,34 The π–A isotherms obtained using our simulation scheme are consistent with experimental results.27 The different configurations for DOPA and ST monolayers are displayed. The configurations of the hydrophilic and hydrophobic groups have been analyzed, so as to exhibit the difference quantitatively. Finally, steric effect and hydrogen bonds have been employed to explain the different configurations between DOPA and ST monolayers.
Simulation details
All simulations were carried out using GROMACS 4.5.535–39 and Visual Molecular Dynamics (VMD)40 was used for visualization. The Particle Mesh Ewald (PME) method was employed to accurately compute electrostatic interactions.41 The cut-off for the Coulomb interaction and L-J interaction was set at 10 Å. The simulation temperature was kept at 300 K with V-rescale coupling.42 We performed simulations at constant surface tension to obtain equilibrate structures and used NVT for production runs.A. Calculating surface tension
We employed the surface tension as defined in GROMACS:| γ = ½Lz[PZZ − ½(PXX + PYY)] | (1) |
The cutoff (rc) of the L-J interactions has a minor influence on the molecular arrangement, but significantly influences the computed value of the surface tension. The so-called tail corrections can be employed to improve the computed values. The dispersion correction to the surface tension for the LJ interactions is included following Chapela et al.,34
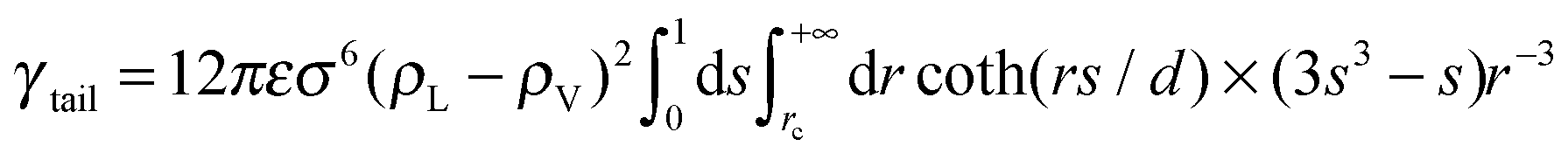 | (2) |
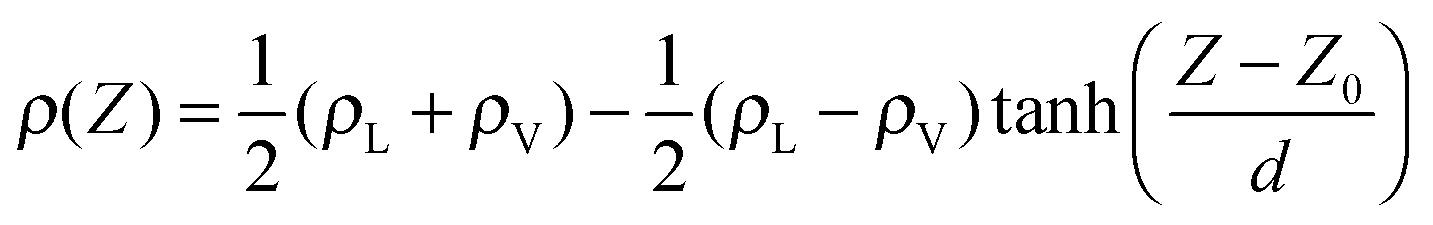 | (3) |
| Πsurf = (γwater + γtailwater) − (γset + γtailset) | (4) |
B. Simulation settings for the water interfaces
Clearly, to compute the surface pressure and have a direct comparison with experiments, we need the simulated pure water surface tension with the chosen force field, because it turns out that the surface tension of water is a quantity that is very sensitive to the details of the force field used. Moreover, the differences between the simulated surface tensions and the experimental values are significant. Thus, a simulation of a water slab containing 4320 water molecules in a periodic box (5.0 × 5.0 × 20.0 nm3) had been carried out to determine the surface tension of the applied water models (Fig. S1, ESI†). The system was equilibrated for 2 ns first. After that, another 2 ns NVT simulation run was used to calculate the surface tension. The calculation was repeated three times to obtain an average surface tension of 65.2 mN m−1 for TIP4P/2005 and 49.0 mN m−1 for SPC. Tail corrections34,45 have been applied for both TIP4P/2005 (4.3 mN m−1) and SPC (5.2 mN m−1) systems. These values are in good agreement with previous studies30,33 and are employed as the pure-water surface tension data in this study to compute the surface pressure.C. Simulation settings for the water–organic interfaces
In the molecular dynamic simulations of DOPA (ST) molecules on water, the organic molecules are simulated using the OPLS all atom force field.46–48 For comparison, both the large size system (containing 480 DOPA/STs, with TIP4P/2005 for water) and the relatively small size system (120 DOPA/STs, with TIP4P/2005 and SPC for water) have been employed. In order to simulate a monolayer at a prescribed surface tension, the system is initiated with a large enough surface area (Fig. S3, ESI†). An equal number of the organic (DOPA or ST) molecules are assigned to either of the two sides of the water slab to make two organic–water interfaces (Fig. 1A). To control the constant surface tension simulations, the Berendsen pressure coupling is used.49 For this coupling method to be effective, a value of the compressibility that is close to the real compressibility of the system,50 4.5 × 10−5 bar−1, is required. The next step is to gradually increase the surface pressure to generate the pressure–area isotherms. At each chosen surface pressure, the surface tension coupling molecular dynamics has been performed while monitoring the change of box size on x and y axes. Until the system size has reached its stable state for 2 ns, we consider the system to be in its equilibrium state (Fig. S3, ESI†, lower figure). Then, another 2 ns NVT simulation with V-rescale coupling is performed to accumulate the data for the analysis. The tail correction for these organic–water systems are considered by coupling the gas–liquid phase (see Fig. S2, ESI†). The surface tensions are then corrected according to eqn (2)–(4).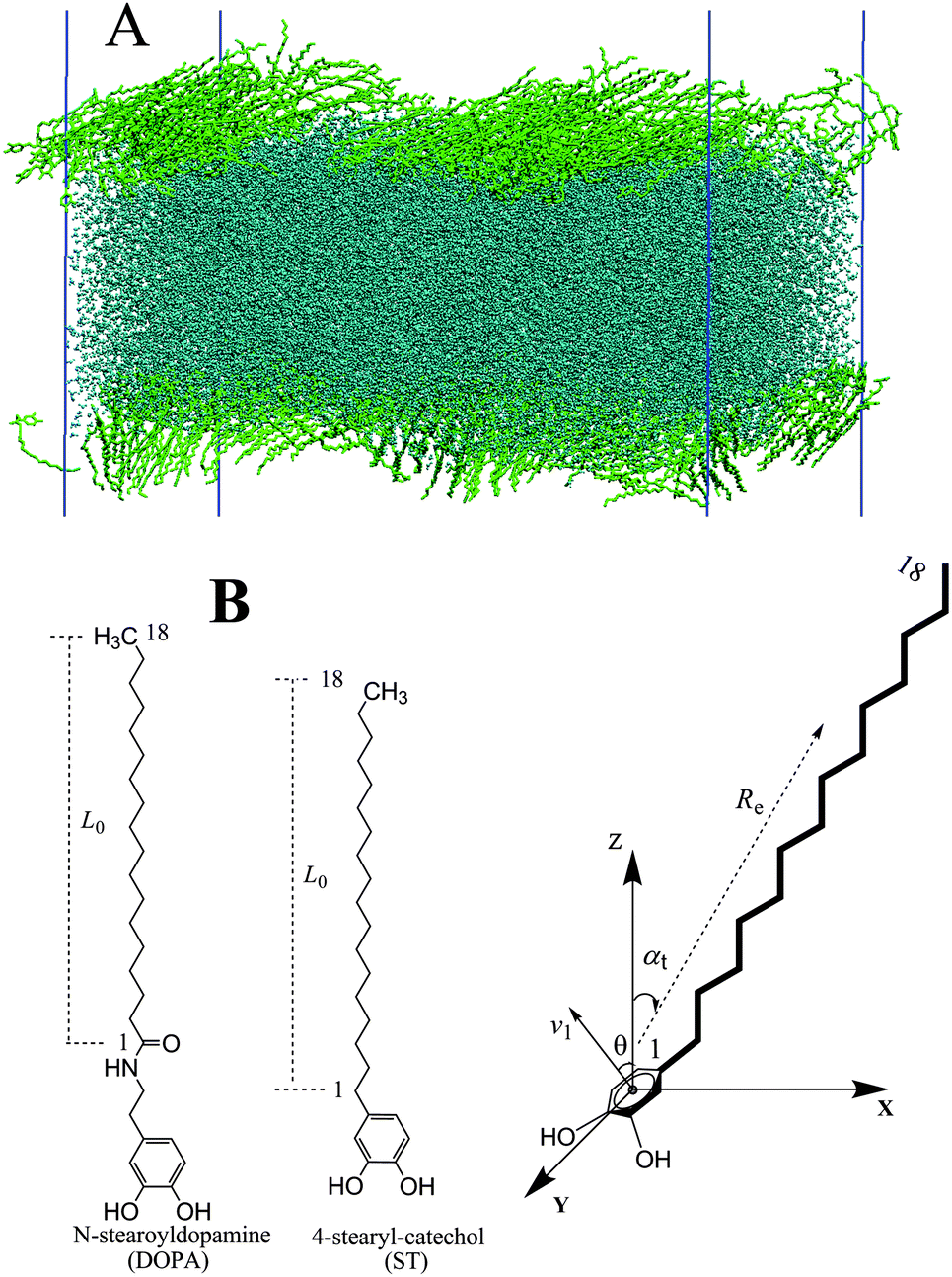 | ||
| Fig. 1 (A) Illustration of the large system set up. (B) Quantities used to characterize the molecular configuration. L0 is the contour distance of C1–C18; Re represents the C1–C18 vector; the angle θ is used to describe the orientation of aromatic ring; the angle between the Re vector and the Z vector is defined as αt. | ||
D. Structural aspects
In order to characterize the configurations of the molecules, several quantities (Fig. 1B) are evaluated.28,51 The position of the aromatic rings is described by the angle θ. To characterize the degree of orientation and stretching of the aliphatic tails, a vector Re that contains the carbon atoms labelled from 1 to 18 as shown on the right-hand side of Fig. 1B, is used.52 The projections of Re on xy plane are then employed to describe the orientations. The angle of orientation of the tails with respect to the surface normal is defined as αt. The condition αt = 0 indicates a complete perpendicular to the surface. In order to evaluate the average orientation of the alkyl tails, the order parameter has been defined as15,53Sz = ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) 〈cos2 〈cos2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θz〉 − ½ θz〉 − ½ | (5) |
Results and discussion
A. Pressure–area isotherms
The isotherms computed using both TIP4P/2005 and SPC water models as well as the isotherms with different system sizes are summarized in Fig. 2A. The surface tension of water using different water models have been discussed in a number of studies.30,32 In the present study, when comparing the two water models, only a minor shift of surface area per molecule (1 Å2 at maximum) between the two isotherms is observed (ST: SPC and ST: TIP4P/2005) for ST monolayers. For DOPA molecules, the influence of the water model on the isotherm is much more pronounced. When using the SPC model, the surface area per molecule is smaller than the area obtained using the TIP4P/2005 model (with a difference of more than 5 Å2). The TIP4P/2005 isotherms are consistent with experiments (Fig. 2B). The different results of TIP4P/2005 and SPC are mainly attributed to the non-bonded parameters and Coulomb charges applied in the water model.30–32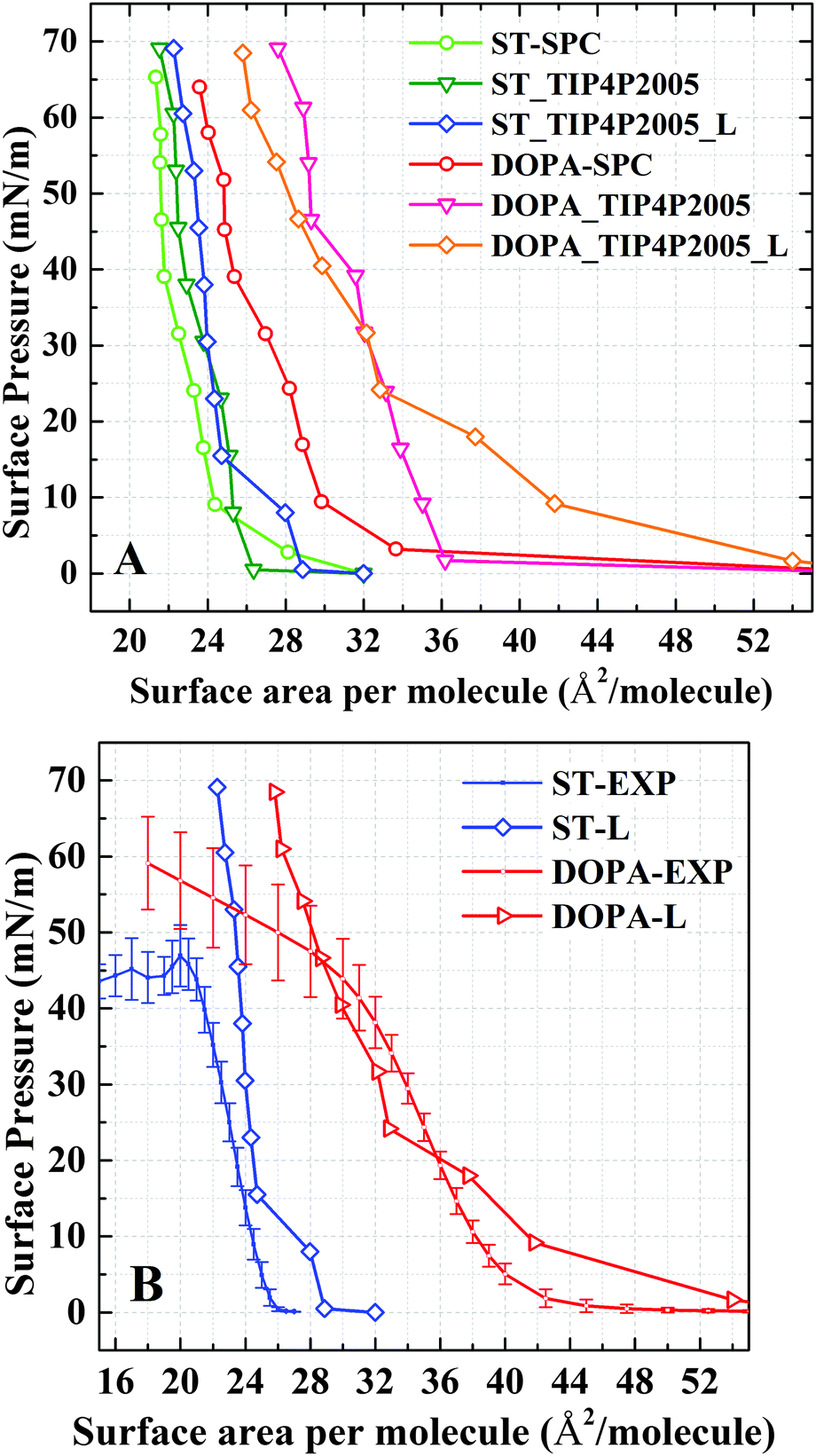 | ||
| Fig. 2 (A) Comparison of isotherms at SPC and TIP4P/2005 models, including both small and large system size (L). (B) Comparison between TIP4P/2005 large (L) isotherms with experiments. The error bars of experimental values are obtained by averaging batch results.27 | ||
For the TIP4P/2005 water model, both the small size system and the large size system have been computed. It is also worthwhile to compare the size effect in this simulation (Fig. 2A). Usually, the artificial boundary effect is decreased with a larger size system. Thus, to better understand molecular configurations for DOPA/ST monolayers, the following analysis is based on the simulation results of TIP4P/2005 large size systems. Although it seems that the system size does not change the DOPA/ST isotherms very much, the configuration of organic molecules in the monolayers are different (Fig. 3 and Fig. S4, ESI†). This issue is explicitly addressed in the later discussion.
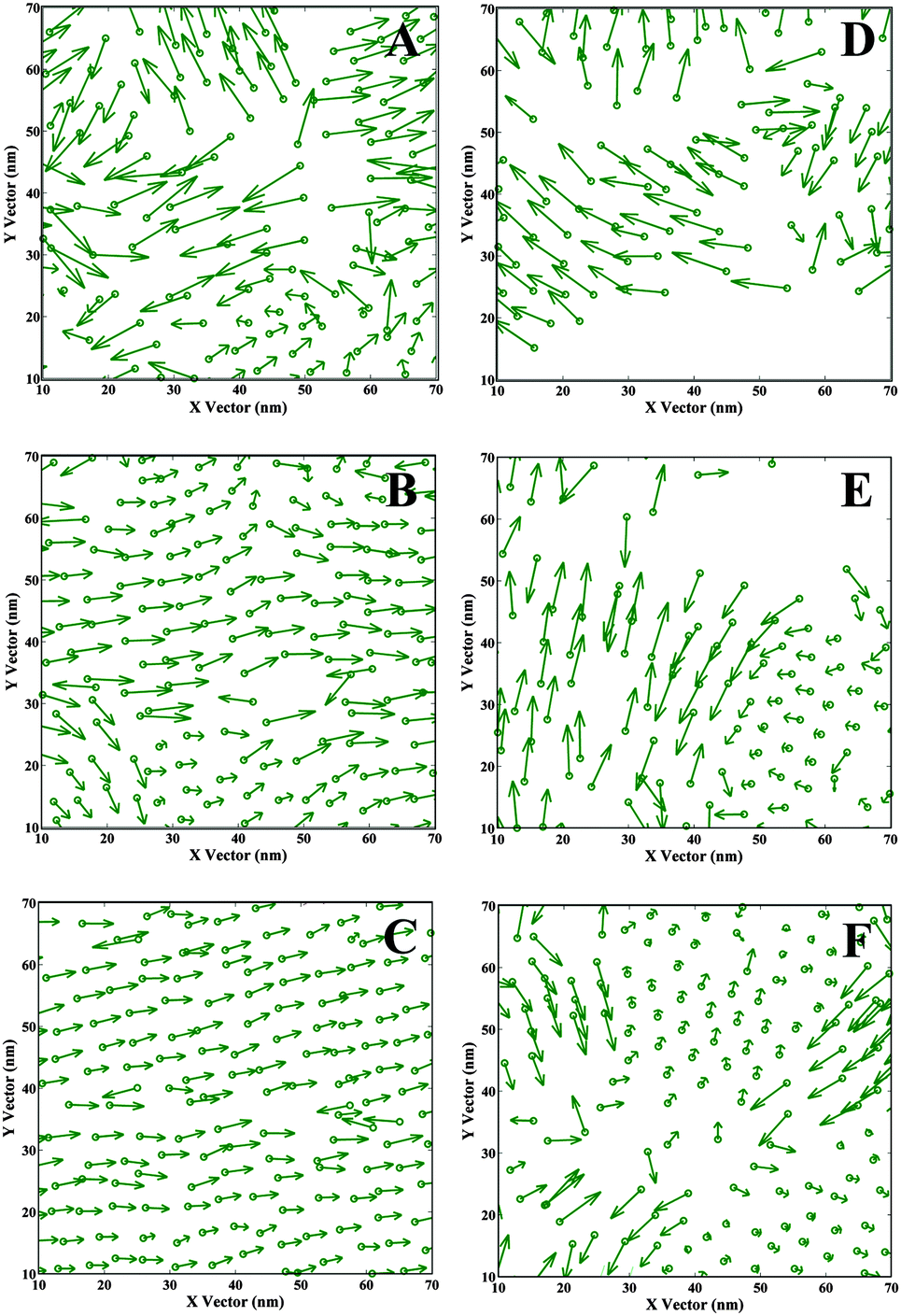 | ||
| Fig. 3 Projection of the vectors Re on x–y plane. The circles represent the catechol groups. The arrow pointing from C1 to C18 (Fig. 1) represents alkyl tails. A–C are ST results, 0.5 mN m−1, 45.5 mN m−1, and 60.5 mN m−1 correspondingly. D–F are DOPA results, 1.6 mN m−1, 46.6 mN m−1, and 61.0 mN m−1. | ||
While the π–A isotherms for both sizes (large and small) of the simulation systems are consistent with experiments, the shape of the isothermal curve of the large size DOPA system fits with experimental data, for the finite size effect is less. A comparison between simulation and experimental surface pressure–area (π–A) isotherms for DOPA and ST is shown in Fig. 2B. The experimental results have been reported before by some of the authors and their collaborators.27 In the experiments, a monolayer of DOPA/ST usually collapses at a surface pressure of around 50 mN m−1 (Fig. 2B). Most likely, the time scale that is connected to the dynamics of the monolayer is much larger than the nanosecond scale. Also the relatively small size of the system can have a stabilizing effect. Therefore it is not possible to observe the collapse in a fully atomistic simulation as ours. On the time and length scales of the simulation the compressed state at high surface pressures is metastable. Only a relatively rough water surface is observed in this simulation. The DOPA/ST monolayers at a surface pressure of around 60 mN m−1 are analyzed subsequently to show an ideal molecular configuration of the monolayer. In Fig. 2B, the TIP4P/2005 results fit the experimental results while a small difference is observed for ST in the range of 25–45 mN m−1. This difference may be due to stronger LJ interactions in TIP4P/2005 than in SPC. Thus, a more accurate water model is expected to describe water–organic interactions.
B. Orientations of DOPA/ST molecules
In order to visualize the overall orientation of molecules, a single DOPA/ST molecule has been simplified to a circle (representing the catechol group) and a vector (representing the alkyl tail). The projections of vectors on x–y plane are displayed in Fig. 3 and the water molecules are not shown. For ST monolayers, while the surface pressure is relatively low (Fig. 3A), the catechol groups (green circles) are homogeneously distributed in local patches. Because of the relatively large surface area per molecule (28.9 Å2 per molecule) some of the alkyl tails exhibit arbitrary orientation, but we also observe some locally ordered orientations. This indicates that the ST monolayer formation starts with small regions of regular structure. When the surface pressure gets higher (Fig. 3B), a more homogenous distribution of catechol groups on the water surface is observed. Also, a number of the hydrophobic groups (alkyl tails) start to orient towards the same direction. In Fig. 3C (60.5 mN m−1), nearly all the ST molecules become homogenously oriented, with the catechol groups distributed regularly and almost all the vector arrows pointing to the same direction.For DOPA monolayers, the configurations are different. For lower surface pressure (Fig. 3D, 1.6 mN m−1), some space of the water surface is not occupied with DOPA, which is probably due to the stronger intra-molecular forces incurred by the amide groups in DOPA than in ST. Thus, the applied surface tension is reached even before the water surface is fully covered with DOPA molecules. With the increase of surface pressure, the water surface is gradually covered with DOPA molecules (Fig. 3E and F). For Fig. 3F (61.0 mN m−1), the distribution of catechol groups on water surface is almost homogeneous. However, probably due to the intra-molecular forces and steric effect of amide groups, the surface area per molecule for DOPA is larger than for the corresponding ST system. Consequently, domains of regular oriented alkyl tails are only observed locally (Fig. 3F). Some of the very short vectors projected on top of the circles indicate perpendicular orientation to x–y plane, while some others seem to be pointing to certain directions. These results are different from the corresponding ST molecules (Fig. 3C), where there is a long ranged orientation order.
In previous studies,52,54–56 the necessity of using a larger cell for simulating linear molecular systems has been discussed. In a periodic system, a linear molecule is possibly interacting (non-bond interactions) with itself in the neighbouring cell, which leads to an unusual uniform structure. Thus, the chosen cell size should be large enough to avoid these interactions. In this study, it should be noticed that the vector projections also depend on the system size. The graphs for small size systems can be found in the ESI† (Fig. S4). Comparing these two cell sizes (Fig. 3 and Fig. S4, ESI†), the artificial boundary effect for the ST system is more obvious at a lower surface pressure (0.5 mN m−1). For DOPA, this effect is clearly shown when the cell sizes are small (Fig. S4c and d, ESI†). This is because the small size cell vector (about 5.0–3.0 nm) is too small. Thus, a molecule may be both interacting with itself in the neighbouring cell, and influenced by a neighbouring molecule and its images in all the nearby cells. According to our simulation, a cell size at least larger than twice the linear molecular length is preferred while simulating water–organic monolayer system composed with linear molecules.
The tilt angle distribution of alkyl tails (αt) is displayed in Fig. 4. For ST (Fig. 4A), most of the αt locate in the range of 20°–40° except for 0.5 mN m−1. This indicates that the orientation of alkyl tails becomes gradually more vertical with increasing surface pressure (According to Fig. 1, a relatively small tilt angle demonstrates a more perpendicular alkyl tails.). For DOPA (Fig. 4B), the trend is identical. The most favourite range for αt gradually moved from 50–60 to 30–40 with the surface pressure increasing from 1.6 mN m−1 to 46.6 mN m−1. However, at a surface pressure of 61.0 mN m−1, the αt are relatively equally distributed from 0°to 50° while the two most favourite ranges are 10°–20° and 30°–40° correspondingly. This difference is because of the relatively large surface area per DOPA molecule and in accordance with Fig. 3F.
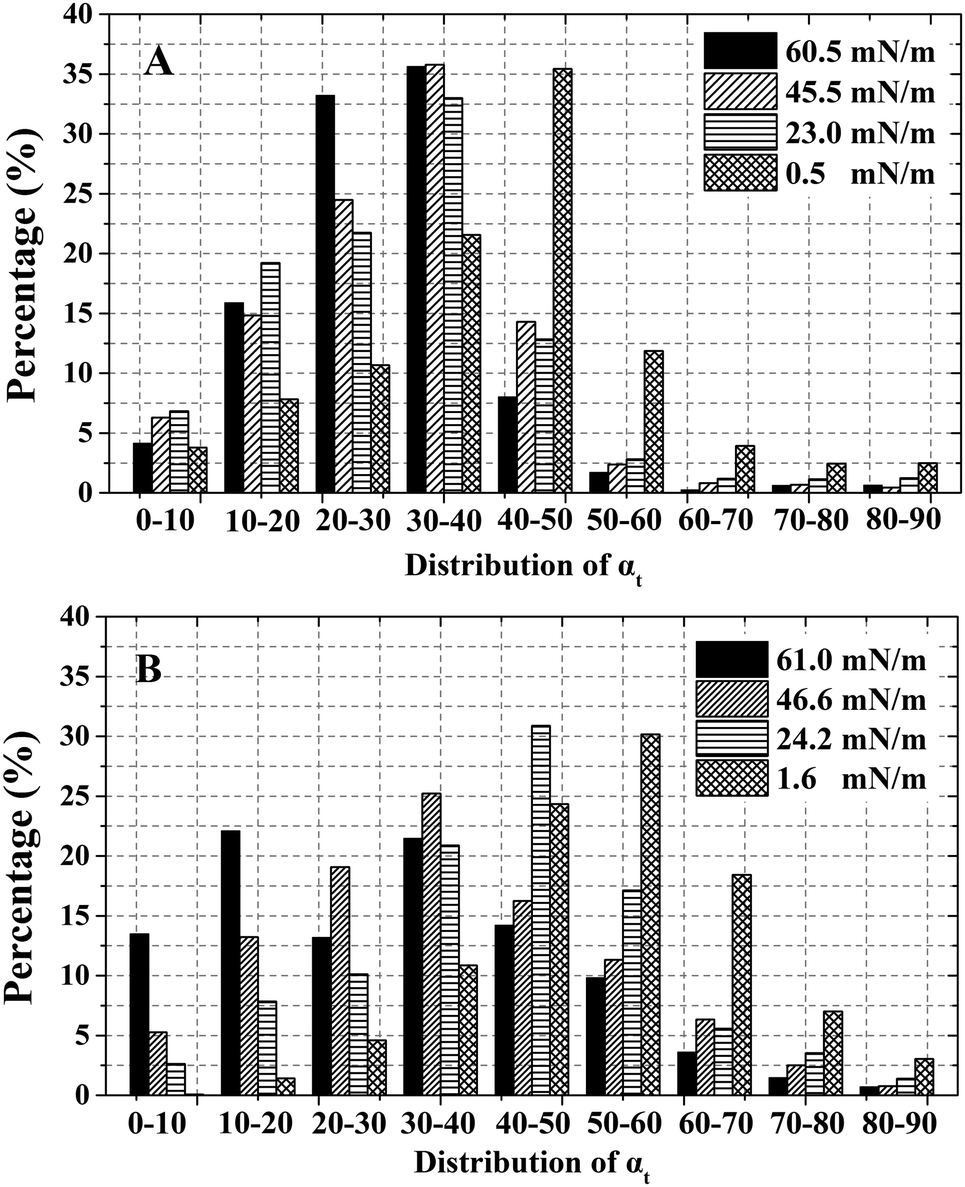 | ||
| Fig. 4 Statistics of tilt angle αt (degree) of alkyl tails with the increase of surface pressure. (A) ST; (B) DOPA. | ||
C. Configurations for DOPA/ST molecules in monolayers
The distribution and configuration of catechol groups are displayed in Fig. 5. At the beginning of the compression (Fig. 5A and C), both ST and DOPA molecules are not well distributed on the water surface. At a higher surface pressure (61.0 mN m−1 for DOPA and 60.5 mN m−1 for ST), the distributions of catechol groups become more uniform. For both surface pressures, there are always more uniformly distributed catechol groups observed for ST than for DOPA on the water surface. This is not only because of the larger surface area per molecule for DOPA than that of ST but also due to the extra amide groups in DOPA.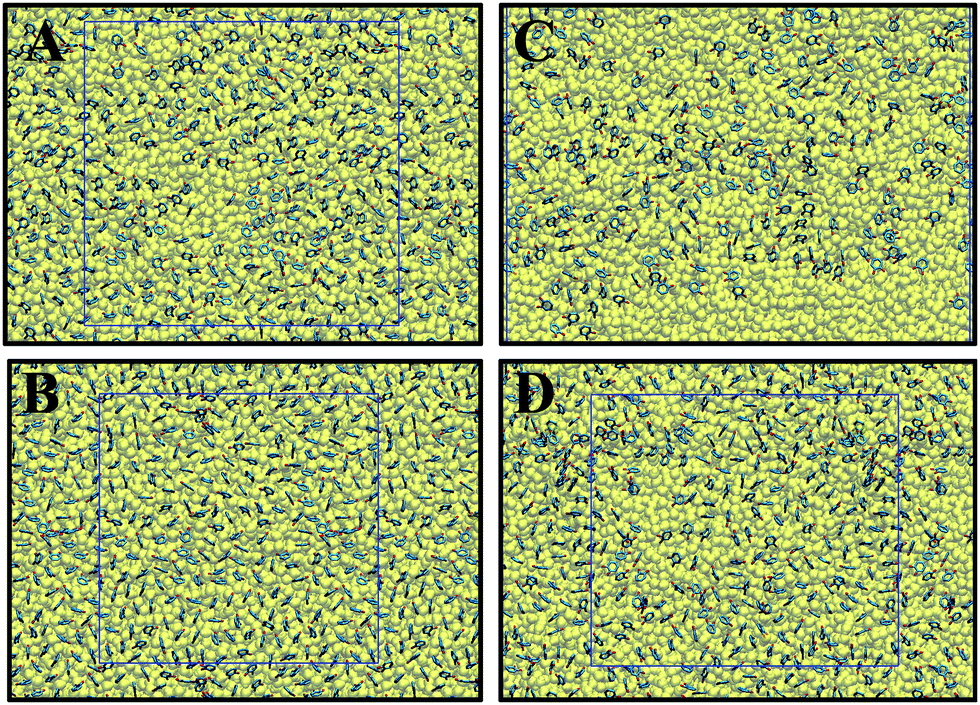 | ||
| Fig. 5 Top view of catechols distributions on top of water surfaces. The squares represent the cell boundary. (A) 0.5 mN m−1 (ST); (B) 60.5 mN m−1 (ST); (C) 1.6 mN m−1 (DOPA); (D) 61.0 mN m−1 (DOPA). The detailed tilt angle (θ) distribution for catechol groups are summarized in Fig. S5 (see ESI†). | ||
The average values of θ, αt, and |Re| (see definition in Fig. 1) corresponding to different surface pressures are listed in Table 1. For catechol groups, all θ values increase with the increase of surface pressures, and catechols groups in DOPA are more perpendicular to the z axis especially at lower surface pressure. These values are consistent with Fig. 5. For |Re|, it is seen that the values are all close to the calculated ideal counter length (21.8 Å). This indicates that the preferred configuration of the DOPA/ST molecules during compression is the stretched position. This also implies that representing alkyl tails by the vector Re (see Fig. 3) is reliable. Considering the statistics of tilt angles in DOPA (αt), first of all, the standard deviation increases with the increase of surface pressure. Secondly, the average value of αt decreases correspondingly. These results are in accordance to Fig. 3D–F and Fig. 4B. For ST molecules, the tilt angle αt decreases normally with the increase of surface pressure. This is consistent with Fig. 3A–C.
| π (mN m−1) | Area per DOPA (Å2) | θ (Degree) | |Re|b (Å) | α t (Degree) | |
|---|---|---|---|---|---|
| a The differences of surface pressures for DOPA and ST monolayers are caused by the tail correction of surface tension. b The calculated ideal counter length of Re is 21.8 Å. | |||||
| DOPA | 1.6 | 54.0 | 56.1 ± 20.5 | 20.8 | 52.6 ± 13.9 |
| 24.2 | 32.8 | 62.6 ± 18.5 | 20.6 | 41.9 ± 15.9 | |
| 30.5 | 30.5 | 63.3 ± 18.1 | 20.6 | 40.1 ± 16.2 | |
| 46.6 | 28.7 | 66.2 ± 17.5 | 20.7 | 36.0 ± 16.9 | |
| 61.0 | 26.2 | 70.5 ± 15.8 | 20.8 | 30.7 ± 18.0 | |
| π (mN m−1) | Area per ST (Å2) | θ (Degree) | |Re| (Å) | α t (Degree) | |
|---|---|---|---|---|---|
| ST | 0.5 | 28.9 | 67.4 ± 17.2 | 20.7 | 40.8 ± 16.1 |
| 23.0 | 24.3 | 72.2 ± 14.3 | 20.8 | 30.3 ± 14.6 | |
| 31.7 | 31.7 | 73.9 ± 14.0 | 21.0 | 30.2 ± 14.4 | |
| 45.5 | 23.5 | 74.8 ± 12.1 | 21.0 | 30.3 ± 12.9 | |
| 60.5 | 22.7 | 74.9 ± 11.8 | 21.1 | 29.1 ± 11.7 |
To further investigate the alkyl tails in the monolayers, order parameters with respect to z-axis are calculated for both DOPA and ST (Fig. 6). For each curve, values from C2 to C17 are located in a narrow range (less than 0.1), indicating the straightness of the alkyl tails. For both DOPA and ST, the order parameter of each carbon atom increases continuously as the surface pressure increases, indicating the more vertical orientation of alkyl tails. At each surface pressure, the order parameter of DOPA is smaller than that of ST. This is because the surface area per molecule of DOPA is larger than that of ST, which is also mainly attributed to amide groups.
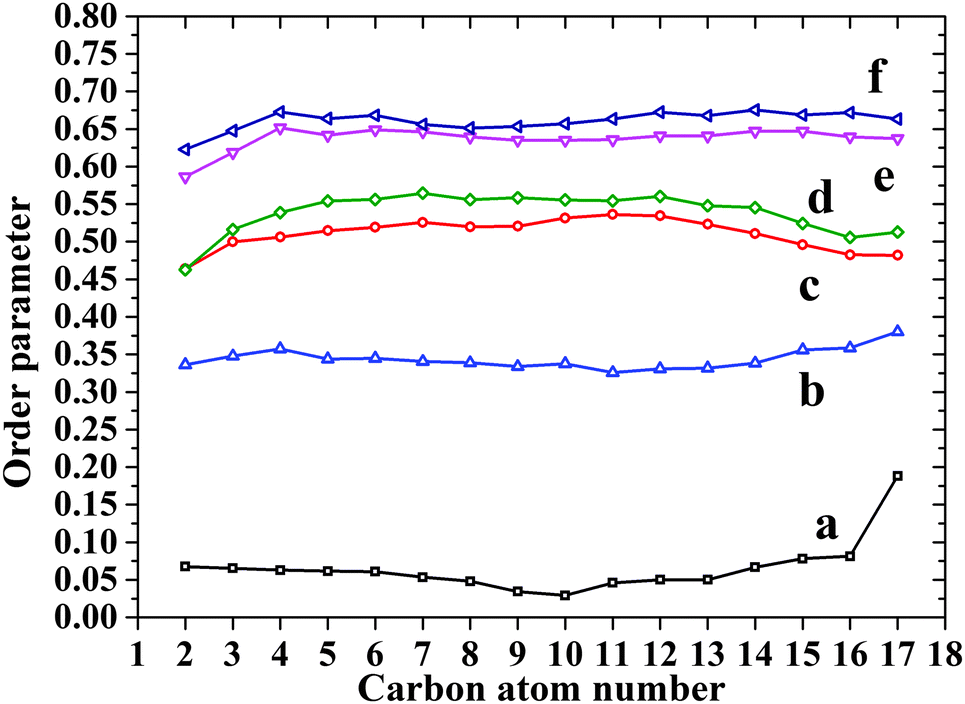 | ||
| Fig. 6 Z dimension order parameters for DOPA and ST monolayers. (a) DOPA 1.6 mN m−1; (b) ST 0.5 mN m−1; (c) DOPA 61.0 mN m−1; (d) DOPA 68.5 mN m−1; (e) ST 60.5 mN m−1; (f) ST 69.1 mN m−1. | ||
D. Hydrogen bond influences on monolayer formation
In order to understand the reason for different configurations between ST and DOPA monolayers, the role of amide groups and the formation of hydrogen bonds are analyzed. Density profiles for water and amide groups are displayed in Fig. 7. The water surface is from around 3 nm to 5 nm. The highest density for amide groups is in the range of 3.5–4.5 nm. This indicates that some of the amide groups are located on the water surface. Hence, the volume of hydrophilic group for DOPA may be enlarged by the amide groups compared to ST. Moreover, water molecules are likely to interact with amide groups because of the hydrogen bond donors and acceptors in DOPA. This may lead to stronger intra-molecular forces in the DOPA system.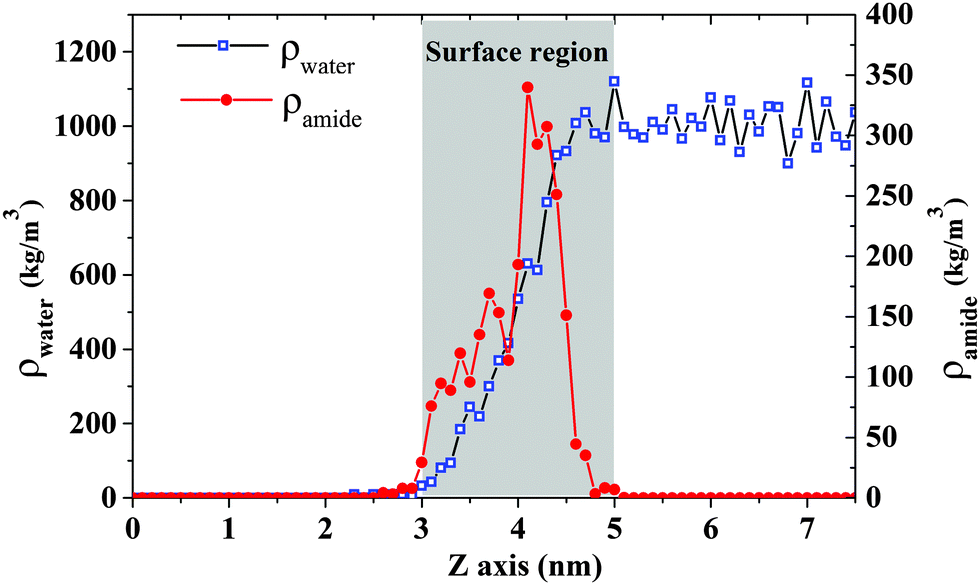 | ||
| Fig. 7 Density profile for water and amide groups in DOPA system with a surface pressure 61.0 mN m−1. The density calculation includes C O N H atoms in amide group. | ||
Hydrogen bonds are of great importance in various systems.57–59 The main function of amide groups is increasing the formation of hydrogen bonds in a monolayer. The statistics of hydrogen bonds formed in a DOPA/ST monolayer are listed in Table 2. It is clearly seen that because of the amide groups, there are many more hydrogen bonds in DOPA than in ST. According to Table 2, in a ST monolayer, the hydrogen bonds are only formed between two hydroxyl groups, with the numbers around sixty (except for the case of 0.5 mN m−1). However, in a DOPA monolayer, there are many more hydrogen bonds. Unlike ST, the total number of hydrogen bonds increases slightly (from 404.3 to 465.6) with the increase of surface pressure. Nearly each molecule in a DOPA monolayer is linked to others with hydrogen bonds. As illustrated in Fig. 8, there are three kinds of hydrogen bonds in a DOPA monolayer. The hydrogen bonds that contribute most are those between two amide groups (type 1). There are also a large number of hydrogen bonds between an amide group and a hydroxyl (type 2) group. The type 3 hydrogen bonds are the interactions between two hydroxyls. Among these three types of H-bonds, type 2 decreases slightly during compression. But type 1 and type 3 H-bonds increase in contrast to the decrease of surface area per molecule (the surface pressure increases in the meantime). The results show that during compression, the amide groups, as well as the catechol groups, get closer to each other. This indicates a relatively ordered arrangement for DOPA molecules at higher surface pressures. These observations are consistent with the previous analysis (Fig. 3 and 5).
| DOPA | ST | |||||
|---|---|---|---|---|---|---|
| π (mN m−1) | Type 1a | Type 2 | Type 3 | Totalb | π (mN m−1) | Totalb (3) |
| a The definition of hydrogen bonds is from GROMACS: 0.35 nm (donor–acceptor), 30 degree (donor–H–acceptor). Type 1: amide–amide hydrogen bonds; type 2: amide–hydroxyl hydrogen bonds; type 3: hydroxyl–hydroxyl hydrogen bond (see Fig. S7, ESI for further information). b The number of hydrogen bonds is obtained by averaging over a 2 ns NVT simulation. There are 480 DOPA (ST) molecules in total. | ||||||
| 1.6 | 231.8 | 140.8 | 31.7 | 404.3 | 0.5 | 31.7 |
| 24.2 | 245.4 | 143.1 | 38.1 | 426.6 | 23.0 | 60.2 |
| 46.6 | 269.9 | 140.4 | 39.3 | 449.6 | 45.5 | 59.3 |
| 61.0 | 288.3 | 130.3 | 47.0 | 465.6 | 60.5 | 62.6 |
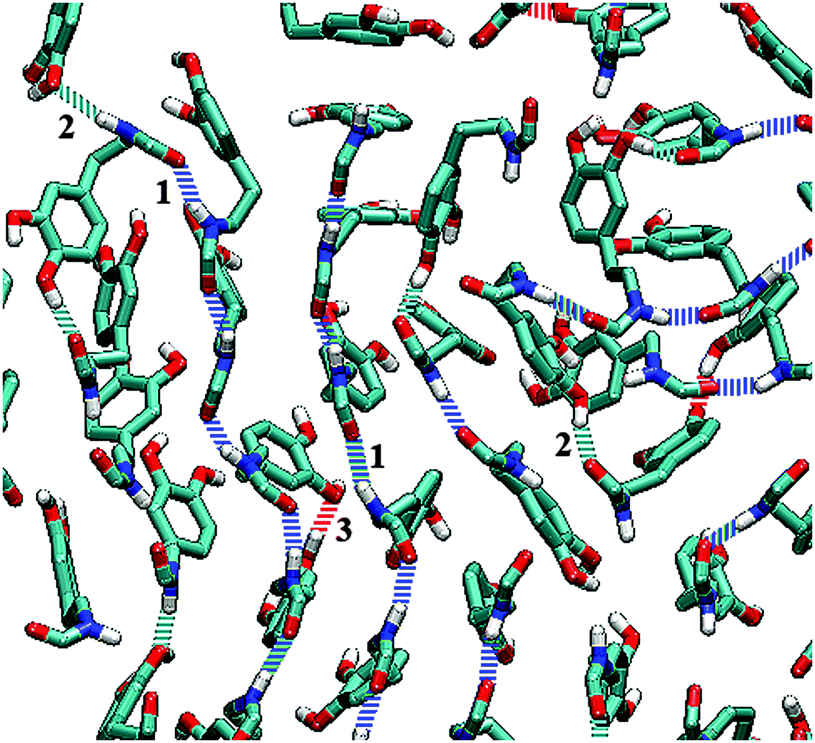 | ||
| Fig. 8 Hydrogen bonds in DOPA system. Only amide groups and catechol groups are shown. Dashed blue lines represent type 1. Dashed cyan lines represent type 2. Dashed red lines represent type 3 (see Table 2). | ||
The statistics in Table 2 indicate that during compression, the intra-molecular interactions in DOPA molecules are considerably enhanced by the hydrogen bonds introduced from amide groups compared to ST. There are also hydrogen bonds between amide groups and water molecules (Table S1, ESI†). The hydrogen bonds in DOPA are considered as the main reason for the different configuration of a DOPA monolayer as compared to ST. These hydrogen bonds may be strong enough to fix DOPA molecules to some extent and is difficult to move or rotate when the surface pressure is low. When the surface pressure gets higher, the DOPA molecules can then be moved and reoriented. But the hydrogen bonds are still effective. Finally, DOPA molecules can distribute relatively uniformly on the water surface (see Fig. 3F) at a high surface pressure.
With all the analysis given above, the influence of hydrogen bonds in DOPA can be summarized as follows. During surface compression, DOPA molecules are connected with various kinds of hydrogen bonds (Fig. 8). Sometimes, the hydrogen bonds can also connect some water molecules to amide groups (Fig. S7 and Table S1, ESI†). The hydrogen bonds are relatively strong (for non-bonded interactions) and therefore the network of hydrogen-bonds can resist being broken or compressed. Thus, one will need much higher surface pressures to overcome the non-bonded interaction in the DOPA system than in the ST system, because these kinds of interactions are not present in the ST monolayer. Consequently, the stronger interactions and steric effect in DOPA lead to a larger surface area per molecule for DOPA than for ST when the surface pressures of the two systems are similar. This is the main reason for the formation of the not well-organized monolayer of DOPA molecules (compared to the ST monolayer).
Concluding remarks
The simulations of organic monolayers on water surface have been of great interest.60–62 In this study, the experimental results of compressing DOPA/ST monolayers27 have been carefully reproduced with all-atomic force field (OPLS-AA) and the surface-tension coupling. By employing proper force field for water molecules (TIP4P/2005) and treating surface tension with tail correction, the obtained simulated surface pressure–area (π–A) isotherms for DOPA and ST are consistent with experimental results. More importantly, comparing to the coarse grain19 method and the NVT coupling,17,18 a main advantage of the simulating scheme in this study is that the detailed configurations for molecules (e.g. hydrogen bonds interactions) can be collected and the compressing process can be reproduced.According to our analysis, the distribution of DOPA molecules is less homogeneous than that of ST molecules at each point of the π–A isotherm. Because of the hydrogen bonds present in the DOPA system, DOPA molecules are restricted in their local positions when the surface pressure is not sufficient to move them. With the increase of surface pressure, only locally ordered arrangements are gradually obtained for DOPA. Thus, DOPA monolayers are not as regularly oriented as ST monolayers. Hence, the different surface pressure–area (π–A) isotherms for DOPA/ST in the experiments are well explained using our simulation. It is also worthwhile to point out that decorating hydrophobic tails (adding an amide group onto an alkyl tail in this case) may be not always helpful to the uniform formation of the monolayer on water surface. According to previous experiments63 and the present study, the added group sometimes causes the molecules to orient less regularly because of the hydrogen bonds.
To sum up, the simulating scheme used in this study has been proved effective in reproducing experimental surface pressure–area (π–A) isotherms comparing to coarse grain method or the traditional NVT coupling. The result in the present study may be useful for choosing decoration groups on alkyl tails during a monolayer synthesis. Further works based on the influences of water models or a combination of experiments and simulations are welcome to strengthen our conclusion.
Acknowledgements
We would like to thank China Scholarship Council (No. 2010617085) and National Natural Science Foundation of China (Grant No. 21303068) for the support of this work.Notes and references
- P. Sanassy and S. D. Evans, Langmuir, 1993, 9, 1024–1027 CrossRef CAS.
- K. Graf and H. Riegler, Langmuir, 2000, 16, 5187–5191 CrossRef CAS.
- K. Graf and H. Riegler, Colloids Surf., A, 1998, 131, 215–224 CrossRef CAS.
- B. Moghaddam, M. H. Ali, J. Wilkhu, D. J. Kirby, A. R. Mohammed, Q. G. Zheng and Y. Perrie, Int. J. Pharm., 2011, 417, 235–244 CrossRef CAS PubMed.
- V. M. Kaganer, H. Mohwald and P. Dutta, Rev. Mod. Phys., 1999, 71, 779–819 CrossRef CAS.
- J. Claret, R. Reigada, R. Albalat, J. Crusats, E. Abad, J. Ignes-Mullol and F. Sagues, J. Phys. Chem. B, 2004, 108, 17274–17277 CrossRef CAS.
- A. H. Su, S. S. Tan, P. Thapa, B. N. Flanders and W. T. Ford, J. Phys. Chem. C, 2007, 111, 4695–4701 CAS.
- K. Hac-Wydro and P. Dynarowicz-Latka, J. Phys. Chem. B, 2008, 112, 11324–11332 CrossRef CAS PubMed.
- A. Nayak, K. A. Suresh, S. K. Pal and S. Kumar, J. Phys. Chem. B, 2007, 111, 11157–11161 CrossRef CAS PubMed.
- J. D. McCoy and J. G. Curro, J. Chem. Phys., 2009, 130, 194708 CrossRef PubMed.
- J. J. Giner-Casares, L. Camacho, M. T. Martin-Romero and J. J. L. Cascales, Langmuir, 2008, 24, 1823–1828 CrossRef CAS PubMed.
- M. Javanainen, L. Monticelli, J. B. de la Serna and I. Vattulainen, Langmuir, 2010, 26, 15436–15444 CrossRef CAS PubMed.
- D. J. Henry, V. I. Dewan, E. L. Prime, G. G. Qiao, D. H. Solomon and I. Yarovsky, J. Phys. Chem. B, 2010, 114, 3869–3878 CrossRef CAS PubMed.
- D. Rose, J. Rendell, D. Lee, K. Nag and V. Booth, Biophys. Chem., 2008, 138, 67–77 CrossRef CAS PubMed.
- A. J. Kox, J. P. J. Michels and F. W. Wiegel, Nature, 1980, 287, 317–319 CrossRef CAS.
- S. Karaborni and S. Toxvaerd, J. Chem. Phys., 1992, 96, 5505–5515 CrossRef CAS PubMed.
- W.-X. Shi and H.-X. Guo, J. Phys. Chem. B, 2010, 114, 6365–6376 CrossRef CAS PubMed.
- G. Watanabe, J. I. Saito, N. Kato and Y. Tabe, J. Chem. Phys., 2011, 134, 054513 CrossRef PubMed.
- S. L. Duncan and R. G. Larson, Biophys. J., 2008, 94, 2965–2986 CrossRef CAS PubMed.
- M. Wiegemann, Aquat. Sci., 2005, 67, 166–176 CrossRef CAS.
- J. H. Waite, Integr. Comp. Biol., 2002, 42, 1172–1180 CrossRef CAS PubMed.
- J. L. Dalsin, B. H. Hu, B. P. Lee and P. B. Messersmith, J. Am. Chem. Soc., 2003, 125, 4253–4258 CrossRef CAS PubMed.
- M. Weinhold, S. Soubatch, R. Temirov, M. Rohlfing, B. Jastorff, F. S. Tautz and C. Doose, J. Phys. Chem. B, 2006, 110, 23756–23769 CrossRef CAS PubMed.
- K. Inoue, Y. Takeuchi, D. Miki and S. Odo, J. Biol. Chem., 1995, 270, 6698–6701 CrossRef CAS PubMed.
- H. Zhao and J. H. Waite, J. Biol. Chem., 2006, 281, 26150–26158 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- D. Rıbena, A. Alekseev, O. van Asselen, G. J. A. Mannie, M. M. R. M. Hendrix, L. G. J. van der Ven, N. A. J. M. Sommerdijk and G. de With, Langmuir, 2012, 28, 16900–16908 CrossRef PubMed.
- C.-P. Kong, E. A. J. F. Peters, G. de With and H.-X. Zhang, Phys. Chem. Chem. Phys., 2013, 15, 15426–15433 RSC.
- R. Sakamaki, A. K. Sum, T. Narumi and K. Yasuoka, J. Chem. Phys., 2011, 134, 144702 CrossRef PubMed.
- C. Vega and E. de Miguel, J. Chem. Phys., 2007, 126, 154707 CrossRef CAS PubMed.
- J. L. F. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS PubMed.
- H. Berendsen, J. Postma, W. van Gunsteren and J. Hermans, Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry held in Jerusalem, Israel, 1981.
- F. Chen and P. E. Smith, J. Chem. Phys., 2007, 126, 221101 CrossRef PubMed.
- G. A. Chapela, G. Saville, S. M. Thompson and J. S. Rowlinson, J. Chem. Soc., Faraday Trans. 2, 1977, 73, 1133–1144 RSC.
- H. Bekker, H. J. C. Berendsen, E. J. Dijkstra, S. Achterop, R. Vondrumen, D. Vanderspoel, A. Sijbers, H. Keegstra, B. Reitsma, M. K. R. Renardus, Physics Computing 92, 1993.
- H. J. C. Berendsen, D. Vanderspoel and R. Vandrunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CAS.
- D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- D. M. York, T. A. Darden and L. G. Pedersen, J. Chem. Phys., 1993, 99, 8345–8348 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- J. G. Harris, J. Phys. Chem., 1992, 96, 5077–5086 CrossRef CAS.
- L. Sun, X. Li, T. Hede, Y. Tu, C. Leck and H. Ågren, J. Phys. Chem. B, 2012, 116, 3198–3204 CrossRef CAS PubMed.
- J. Alejandre, D. J. Tildesley and G. A. Chapela, J. Chem. Phys., 1995, 102, 4574–4583 CrossRef CAS PubMed.
- M. G. Martin, Fluid Phase Equilib., 2006, 248, 50–55 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- D. van der Spoel, E. Lindahl, B. Hess, A. R. van Buuren, E. Apol, P. J. Meulenhoff, D. P. Tieleman, A. L. T. M. Sijbers, K. A. Feenstra, R. van Drunen and H. J. C. Berendsen, Gromacs User Manual version 4.5.6, 2010, www.gromacs.org Search PubMed.
- R. A. Fine and F. J. Millero, J. Chem. Phys., 1973, 59, 5529–5536 CrossRef CAS PubMed.
- J. Ignes-Mullol, J. Claret, R. Reigada and F. Sagues, Phys. Rep., 2007, 448, 163–179 CrossRef CAS PubMed.
- C.-p. Kong, H.-x. Zhang, Z.-x. Zhao and Q.-C. Zheng, Chem. Res. Chin. Univ., 2013, 29, 545–550 CrossRef CAS.
- S. K. Ghosh, Nuovo Cimento Soc. Ital. Fis., D, 1984, 4, 229–244 CrossRef.
- J. D. Honeycutt and H. C. Andersen, J. Phys. Chem., 1986, 90, 1585–1589 CrossRef CAS.
- A. H. de Vries, I. Chandrasekhar, W. F. van Gunsteren and P. H. Hünenberger, J. Phys. Chem. B, 2005, 109, 11643–11652 CrossRef CAS PubMed.
- W. Weber, P. H. Hünenberger and J. A. McCammon, J. Phys. Chem. B, 2000, 104, 3668–3675 CrossRef CAS.
- G.-J. Zhao and K.-L. Han, Acc. Chem. Res., 2011, 45, 404–413 CrossRef PubMed.
- N. Suttiwijitpukdee, H. Sato, M. Unger and Y. Ozaki, Macromolecules, 2012, 45, 2738–2748 CrossRef CAS.
- Q. Xue, J.-L. Zhang, Q.-C. Zheng, Y.-L. Cui, L. Chen, W.-T. Chu and H.-X. Zhang, Langmuir, 2013, 29, 11135–11144 CrossRef CAS PubMed.
- S. Baoukina, E. Mendez-Villuendas and D. P. Tieleman, J. Am. Chem. Soc., 2012, 134, 17543–17553 CrossRef CAS PubMed.
- T. R. Lucas, B. A. Bauer, J. E. Davis and S. Patel, J. Comput. Chem., 2012, 33, 141–152 CrossRef CAS PubMed.
- M. Tarek, D. J. Tobias and M. L. Klein, J. Phys. Chem., 1995, 99, 1393–1402 CrossRef CAS.
- U. Tasic, B. S. Day, T. Yan, J. R. Morris and W. L. Hase, J. Phys. Chem. C, 2007, 112, 476–490 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp00555d |
| This journal is © the Owner Societies 2014 |