 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arresting consecutive steps of a photochromic reaction: studies of β-thioxoketones combining laser photolysis with NMR detection†
Mariusz
Pietrzak
*a,
Jacek
Dobkowski
a,
Alexandr
Gorski
a,
Sylwester
Gawinkowski
a,
Michał
Kijak
a,
Roman
Luboradzki
a,
Poul Erik
Hansen
b and
Jacek
Waluk
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: mpietrzak@ichf.edu.pl; jwaluk@ichf.edu.pl; Fax: +48 223433333; Tel: +48 223433332
bDepartment of Science, Systems and Models, Roskilde University, DK-4000 Roskilde, Denmark. E-mail: poulerik@ruc.dk; Fax: +45 46742011; Tel: +45 46742432
First published on 28th March 2014
Abstract
Photochromism of monothiodibenzoylmethane has been studied in a number of environments at different temperatures. Direct laser irradiation of a sample located in the NMR magnet allowed in situ monitoring of the phototransformation products, determining their structure, and measuring the kinetics of the back reaction. These observations, along with the data obtained using electronic and vibrational spectroscopies for rare gas matrix-isolated samples, glasses, polymers, and solutions, as well as the results of quantum-chemical calculations, provide insight into the stepwise mechanism of the photochromism in β-thioxoketones. At low temperature in rigid matrices the electronic excitation leads to the formation of the –SH exorotamer of the (Z)-enethiol tautomer. In solutions, further steps are possible, producing a mixture of two other non-chelated enethiol forms. Photoconversion efficiency strongly depends on the excitation wavelength. Analysis of the mechanisms of the photochromic processes indicates a state-specific precursor: chelated thione–enol form in the excited S2(ππ*) electronic state. The results show the potential of using laser photolysis coupled with NMR detection for the identification of phototransformation products.
Introduction
Huge interest in photochromism is stimulated by numerous possible applications of this phenomenon.1–4 Photochromic transformations quite often involve proton transfer and/or conformational changes. A suitable, widely investigated system for studying the interplay between tautomeric and rotameric interconversion processes is provided by β-thioxoketones.5–28 In principle, these compounds can assume many different forms, as depicted in Fig. 1, which makes a reliable structural assignment of the forms involved in photochromism a very challenging task. After some controversies,6,9,12,13 it has now been established that β-thioxoketones exist in solution primarily as the thione–enol form (e-CCC) in equilibrium with the t-CCC form of Scheme 1, although the equilibrium constant does depend on the flanking groups (aliphatic or aromatic), temperature, and solvent.18,23 Irradiating at 410 nm, the wavelength corresponding to the S2(ππ*) transition of the e-CCC form of monothiodibenzoylmethane (1), carried out for rare gas, glass, polymers, and KBr matrices, leads to proton transfer and trapping of the non-hydrogen bonded t-TCC species, a form that can be kept almost indefinitely below 90 K.24 Increasing the temperature from 90 to 115 K results in over a thousand-fold decrease in the lifetime of the photochromic product. For instance, for 1 dissolved in 3-methylpentane, the lifetime drops from 1260 to 1 min.27 It can be expected that, in a less rigid environment, other photochromic channels can be activated, e.g., those involving the rotation about the C–C bond. No such structures have been reported so far. Therefore, the main goal of this work was to check for the presence of the currently unknown photochromic channels and to determine the structure of possible photochromic products.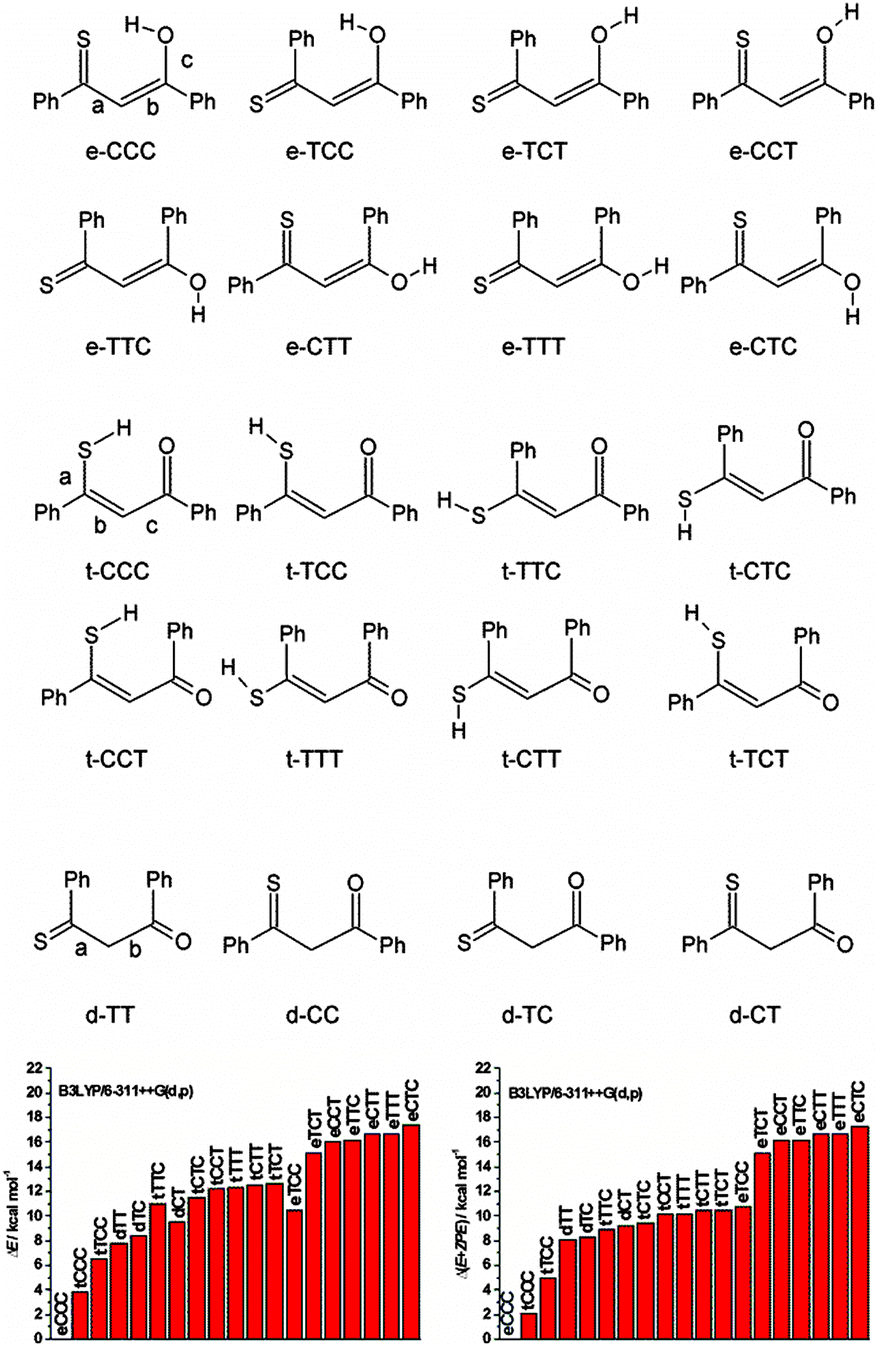 | ||
| Fig. 1 Top, possible structures of monothiodibenzoylmethane. Bottom, calculated relative ground state energies without (left) and with (right) zero-point-energy correction. | ||
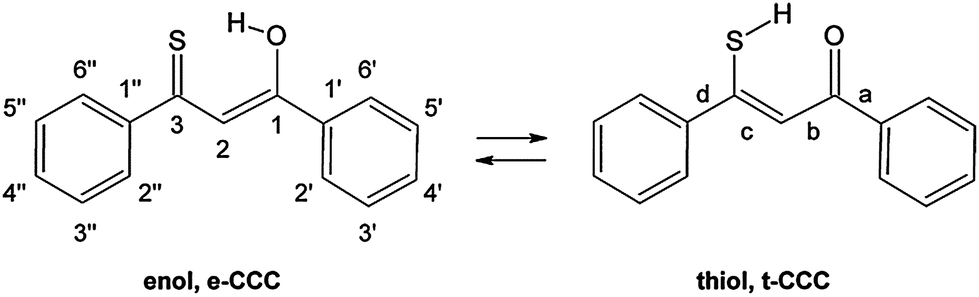 | ||
| Scheme 1 Two main tautomeric forms of 1. The relevant dihedral angles are labeled a–d. | ||
In the above-mentioned cases the species were identified by means of IR and UV measurements.24–28 Here, we present another, novel approach, based on laser irradiation in the magnet combined with in situ NMR detection. This methodology provides an interesting possibility for studies of photoinduced reactions, although a number of technical difficulties have to be overcome. So far, the technique has been used to demonstrate the occurrence of syn–anti photoisomerization in the electron donor–acceptor molecule, 2-(N-methyl-N-isopropylamino)-5-cyanopyridine.29 Other applications included studies of photosensitized oxidation of imidazole and guanosine derivatives,30–32 conformational changes in proteins,33,34 and RNA refolding.35,36 In the present investigation, monothiodibenzoylmethane 1 (Scheme 1) is investigated, as this compound is stable and many derivatives exist. The results obtained by combination of laser photolysis, NMR, electronic absorption, IR and Raman techniques, supported by density functional theory (DFT) and time-dependent DFT (TDDFT) calculations, provide information on geometric and vibrational structure, relative and electronic transition energies, as well as on NMR chemical shifts. We find that photochromic transformation of 1 involves several steps. In contrast to the situation encountered in a rigid environment, where the photoconversion leads to the t-TCC form, irradiation of solutions produces a mixture of t-CTC and t-TTC. These forms revert back to the most stable e-CCC and t-CCC structures on the time scale of days at 183 K and minutes at 293 K.
Experimental and computational details
Synthesis and preparation of samples
1 was synthesized as described in ref. 19. The concentration of the NMR sample was usually 2 millimolar. The NMR solvents used, i.e. CD2Cl2, CDCl3, CD3CN, CD3OD, THF-d8, toluene-d8, acetone-d6, were obtained from Cambridge Isotopes Laboratories, Inc. or from Armar Chemicals. Each sample was prepared from fresh solvent from an ampoule without further purification. Solutions were bubbled with dry nitrogen for at least 10 min inside NMR tubes directly before measurements.NMR-laser setup
The setup with the laser is depicted in Fig. 4. The laser beam is transmitted via three aluminium mirrors and focused with a quartz lens on the end of the quartz rod (length 1 m and diameter 3.8 mm) inserted into the NMR magnet. The other end of the quartz rod is immersed in solution in a NMR tube hanging on the rod (for details see ESI†). It is placed 1.5 mm above the upper end of the 1H receiving coil as a compromise between the best irradiation of a measured volume and good magnetic field homogeneity.NMR experiments
NMR measurements were performed on a Bruker AVANCE II 300 spectrometer operating at 300.1 MHz for 1H and 125.0 MHz for 13C. For practical reasons (the solvents were thoroughly deoxygenated) TMS was not added and the 1H and 13C solvent signals were used as a chemical shift reference over the whole temperature range. This, of course, could lead to a small variation in the chemical shifts (<0.1 ppm for 1H). In the case of mixed solvents (Fig. 8) the chemical shift was referenced directly to the internal TMS signal.The temperature experiments were controlled by the BVT-3200 unit. Temperature was set with use of the methanol thermometer as a probe before each experiment. The temperature during irradiation was increased by at most 2 °C.
Irradiation time was controlled using a NMR spectrometer by means of a shutter. After a period of laser irradiation a 1H spectrum was recorded (16 scans, 2 min) and the procedure was repeated until a steady-state was achieved. The reverse reaction was monitored by measuring 1H spectra every 10–30 minutes. The product was examined using the following NMR techniques: 1H–1H NOESY – (noesygpph, mixing time 1 s), 1H–1H DQF COSY (cosygpmfph), 1H–13C correlations, i.e. HSQC – (hsqcgpph, optimized for 170 Hz), HMBC – (hmbcgplpndqf, zero and double quantum coherence, optimized for 8 Hz with low-pass J-filter for 170 Hz).
UV-Vis experiments
The UV/Vis spectra were recorded on a Shimadzu UV2700 spectrophotometer, equipped with a home-made temperature control system. To monitor changes in the electronic spectra upon irradiation, the sample was cooled down to 205 K using a mixture of solid carbon dioxide and ethanol. DHOM-M-445-1000 and DHOM-M-532-300 diode lasers with 1 W and 300 mW of maximum output power, respectively, were used as irradiation sources.IR/Raman experiments
The IR spectra were recorded for room temperature dichloromethane solutions with 1 cm−1 resolution on a Nicolet Magna 560 FTIR spectrometer, equipped with an MCT/B liquid-nitrogen-cooled detector. While recording the spectra of the photochromic products, the IR cell, placed in the sample chamber of the spectrometer, was irradiated using the DHOM-M-445-1000 laser.For the measurements of Raman spectra, a Renishaw InVia microscopic system was used, with two excitation sources: an HPNIR785 diode laser emitting at 785 nm and Stellar Pro Modu-Laser (LLC) emitting the Ar+ 514.5 nm line. The laser light was focused on a sample with a 30 mm focusing distance objective. The laser power at the sample was about 100 mW. The Raman-scattered light was collected by the same objective through a cutoff filter to eliminate Rayleigh scattering. Gratings of 1800 and 1200 grooves mm−1 were used for 514.5 and 785 nm laser lines, respectively. The resolution was 5 cm−1 and the wavenumber accuracy was 2 cm−1, both calibrated with the Rayleigh line and the 520.6 cm−1 line of silicon. The Raman spectra were recorded using a 1024 × 256 pixel Peltier-cooled RenCam CCD detector. A Linkam THMS600 climatic chamber coupled to the system translation stage was used for measurements at low temperatures, down to 183 K.
X-ray studies
Single crystal X-ray diffraction measurements were carried out on an Agilent Supernova diffractometer, at 100 K with graphite monochromated Cu Ka radiation (1.54184 Å). The data reduction was made by using CrysAlisPRO37 software. The structures were solved by direct methods and refined on F2 by full-matrix least-squares by using SHELXS97 and SHELXL97.38 All non-hydrogen atoms were refined as anisotropic while hydrogen atoms were placed in calculated positions, and refined in riding mode. 1: MONOCLINIC, P21/c, a = 12.4853(3), b = 7.2462(2), c = 13.1368(3) Å, V = 1176.59(5) Å3, Z = 4, Dcalc = 1.357 g cm−1, μ = 2.254 cm−1, R1 = 0.0330 for 2099[ Fo > 4s(Fo)] and 0.0354 for all data, wR2 = 0.0939, S = 1.076.CCDC 974809 contains the supplementary crystallographic data for this paper. ESI.†
Calculations
All the theoretical calculations were carried out using the Gaussian 09 code.39 Molecular geometries were fully optimized using the B3LYP variant of the density functional theory (DFT)40,41 with the 6-311++(d,p) basis set. Time-dependent DFT (TD-DFT) method has been used for geometry optimization of the excited singlet states. For the triplet state geometry optimizations, both TD-DFT/B3LYP/6-311++(d,p) and DFT/UB3LYP/6-311++(d,p) methods were used, yielding similar structures and relative energies. The T1 structures presented in Fig. S6–S12 (ESI†) correspond to the UB3LYP calculations. The NMR nuclear shieldings were calculated with B3LYP/6-311++(d,p) using the GIAO method.42,43 The electronic transition energies were calculated for both ground and excited singlet and triplet state optimized geometries using TD-DFT with the same functional and basis set (B3LYP/6-311++(d,p)).Results
Irradiation experiments
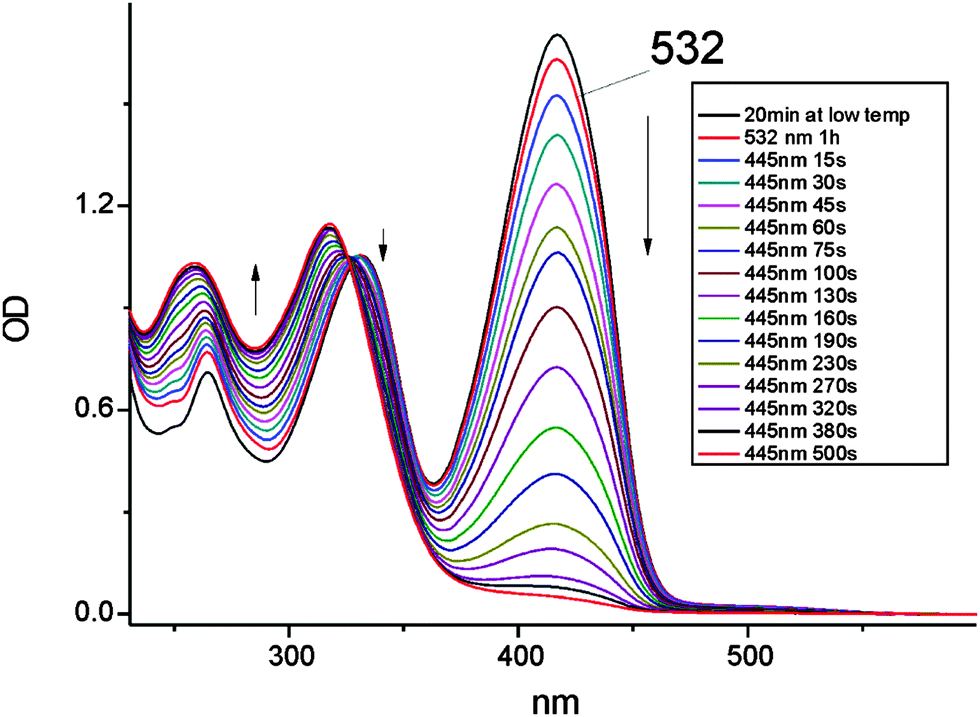 | ||
| Fig. 2 Evolution of electronic absorption of 1 in dichloromethane at 205 K. The sample was irradiated at 445 nm with a 50 mW laser line at intervals ranging between 15 and 120 s. For comparison, the spectrum labeled “532” corresponds to 1 hour of irradiation at 532 nm with a laser power of 200 mW. | ||
Contrary to the behavior observed for 445 nm excitation, when using the 532 laser line, the photochromic process is extremely slow. Correcting for the amount of photons absorbed at 445 and 532 nm, the yield of photoconversion was found to be at least 100 times lower when irradiating into the lower energy band. The lower and higher energy regions correspond to S1(nπ*) and S2(ππ*) transitions of the enol e-CCC form, respectively. The observed difference in photoconversion efficiencies raises an important question about the precursor state of the photochromic product: it can be either the enol tautomer in its S2(ππ*) electronic state or the t-CCC thiol form, for which, as discussed below, the electronic absorption is predicted to be blue-shifted with respect to the enol.
The electronic spectrum of the photoproduct after irradiation at 445 nm (Fig. 2), with the highest intensity peak at 318 nm, resembles that obtained for the product of irradiation of 1 in high-viscosity media, such as rare gas matrices, polymers and glasses. However, the spectra are not identical, suggesting that the photochromic species produced under the two regimes are different. This is proven by the decay kinetics of the photoproduct. It disappears in less than a minute in a rigid environment at 120 K, whereas in dichloromethane at 205 K the photochromic form persists for several hours; even at room temperature the photoproduct can be observed on the time scale of minutes. These results unequivocally show that the barrier for the depopulation of the photochromic species generated in solution is much higher than that required for the back reaction starting from the photoproduct obtained in the low temperature/rigid matrix regime.
On the other hand, the IR spectrum of the photoproduct could be registered even at room temperature. This spectrum was obtained while irradiating the solution of 1 in CH2Cl2 contained in the sample chamber of the FTIR spectrometer with 445 nm laser light. This led to the establishment of a photostationary regime, with about 20% depletion of the enol e-CCC form. The difference IR spectrum, presented in Fig. 3, was compared with the patterns theoretically predicted for various forms. Nice agreement between experiment and simulation is obtained for three thiol forms: t-TTC, t-CTC, and t-TCC. We note, however, that the IR spectrum of the latter, obtained in rare gas matrices, is definitely different from the spectrum obtained for room temperature solution (Fig. 3). This suggests that the photoproduct generated at higher temperatures/lower viscosities may correspond to either t-TTC or t-CTC, or the mixture of both. Based on the IR spectra alone, this problem cannot be solved unambiguously.
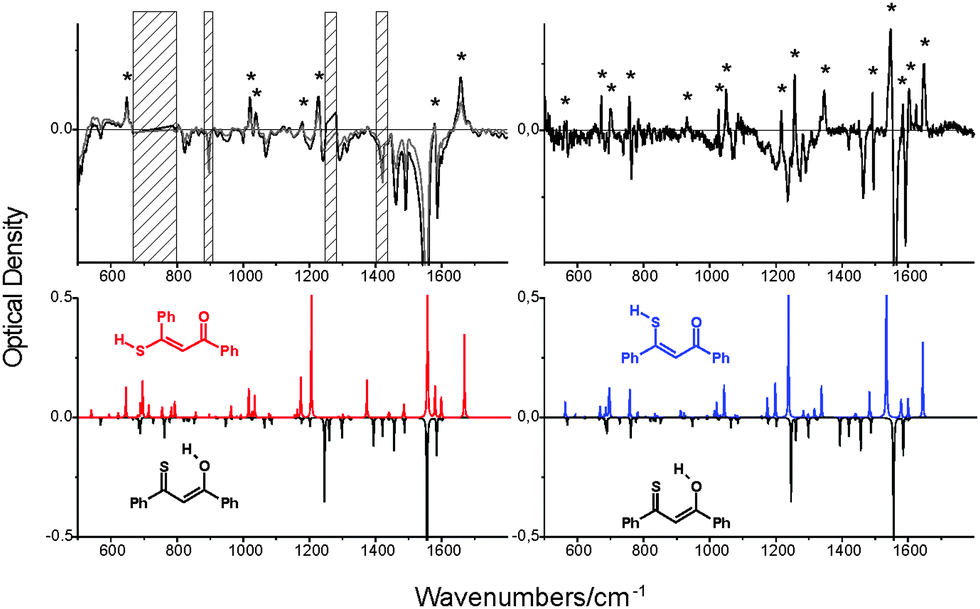 | ||
| Fig. 3 Top left, a difference IR spectrum at 293 K, obtained after subtracting the spectrum of non-irradiated CH2Cl2 solution from the spectrum recorded while irradiating the sample with a 455 nm laser line (800 mW). Top right, a difference spectrum obtained for an Ar matrix at 15 K. The asterisks denote the bands corresponding to the photoproduct. For the Ar matrix, it has been identified as t-TCC.24 The hatched regions correspond to solvent absorption. Bottom, the IR spectra simulated for e-CCC (black, inverted), t-TTC (red, left) and t-TCC (blue, right). The spectrum simulated for t-CTC is practically identical to that of t-TTC. | ||
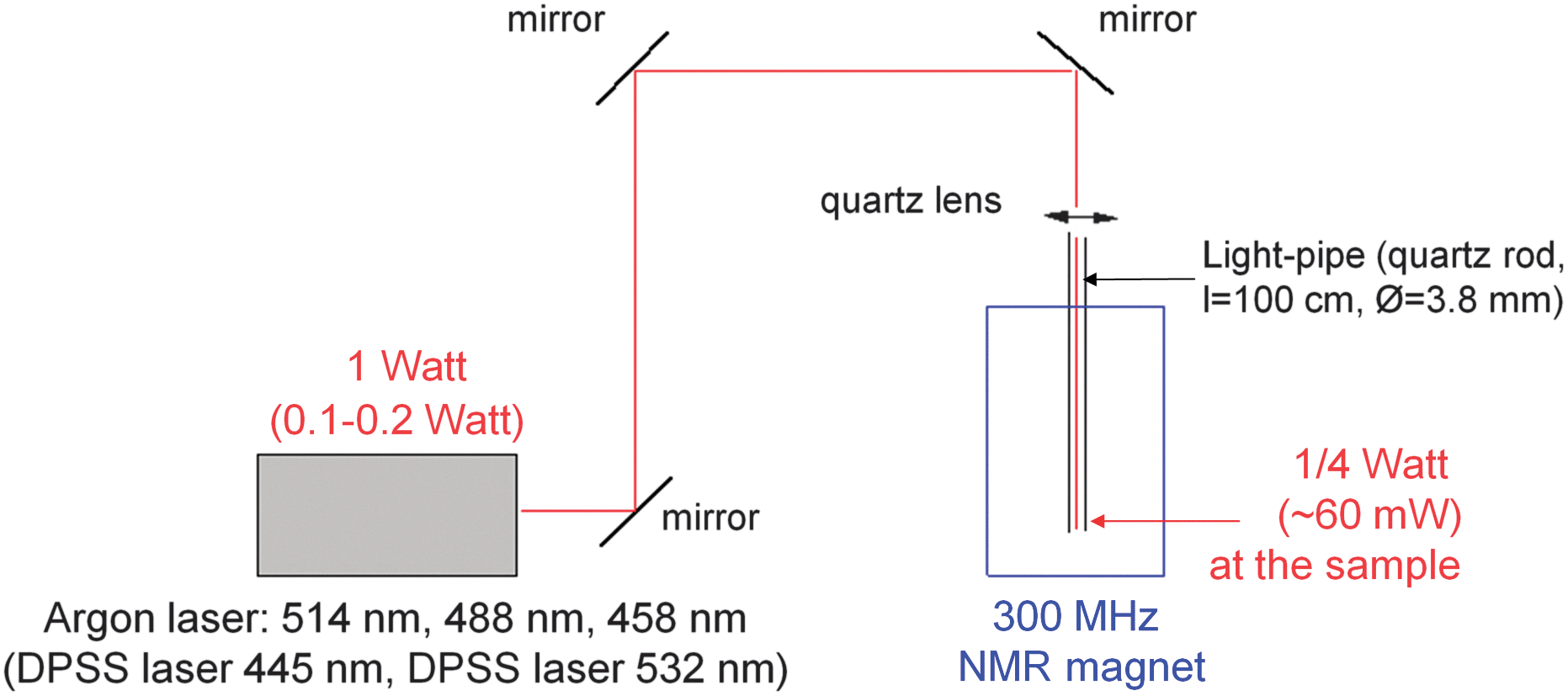 | ||
| Fig. 4 Block diagram of the hyphenated NMR setup. For a full description see Experimental. | ||
Using CD3OD, CD3CN or acetone-d6 as solvent no product was observed. Using CD2Cl2 as solvent one well defined species (1id) was produced (see Fig. 5). Typical irradiation time for maximal conversion of the starting material using the argon laser (0.3 W at the sample) was 2–3 hours in CD2Cl2. Using a laser at 445 nm (0.1 W at the sample) the time for full conversion was reduced to less than 20 min. Using a laser at 532 nm no product could be observed. Discontinuing irradiation led to conversion to the original compound, 1, as seen from the spectrum in Fig. 5.
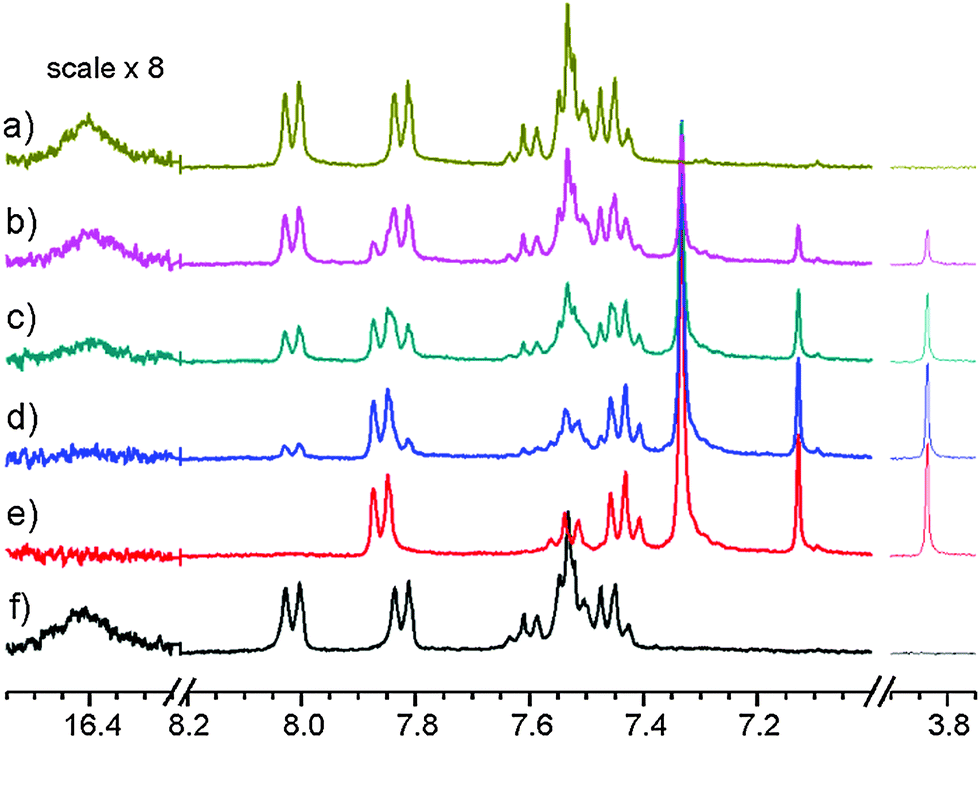 | ||
| Fig. 5 1H NMR spectrum of 1 in CD2Cl2 at 208 K. Irradiation at 445 nm (100 mW at the sample): (a) starting material; (b) irr. for 5 min; (c) 10 min; (d) 15 min; (e) 20 min; (f) after 30 min warm up to the room temp, recorded at 208 K. | ||
A very similar situation was found in CDCl3. In this case the formation of the product (1ic) upon irradiation was observed with similar speed to that for CD2Cl2. Also the reverse reaction in the darkness took place.
Irradiation of 1 in a thoroughly degassed toluene-d8 solution at 183 K for hours resulted in a product 1it as seen in Fig. 6. Discontinuation of the irradiation led to 1: at 183 K only slowly (lifetime of a day), but quicker at higher temperatures (lifetime of an hour at 273 K and of minutes at 298 K).
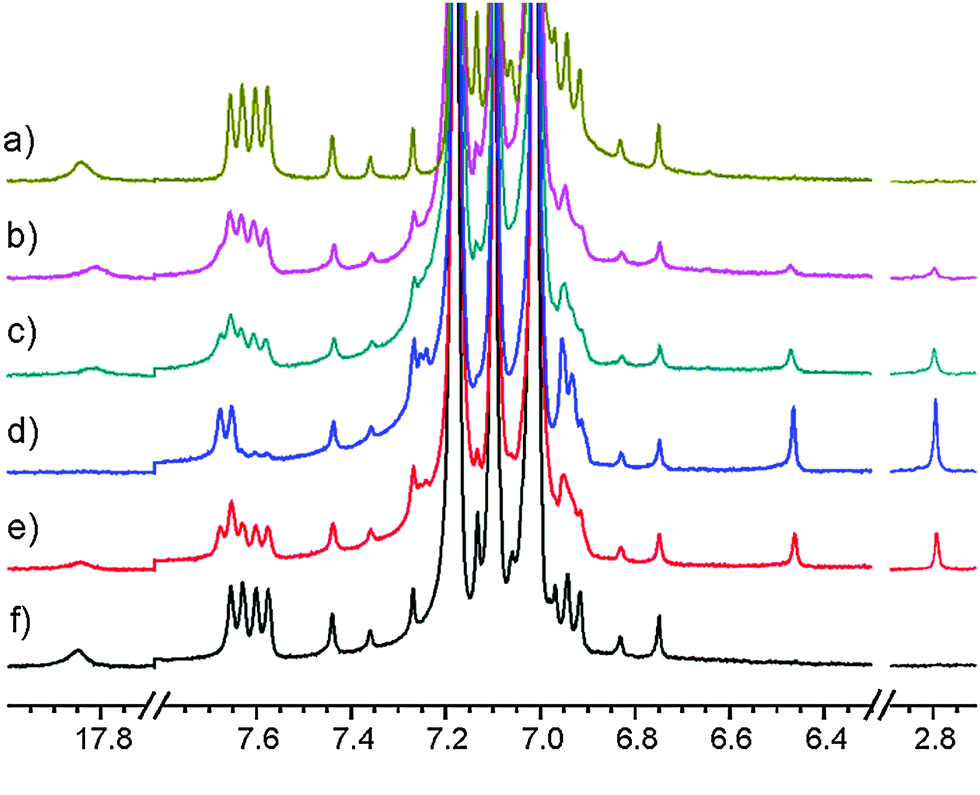 | ||
| Fig. 6 1H NMR spectrum of 1 in toluene-d8 at 183 K. Irradiation by an argon laser (ca. 0.3 W at the sample): (a) starting material; (b) irr. for 20 min; (c) 1 hour; (d) 24 hours; (e) after 24 hours at 183 K in the dark; (f) after 10 min warm up to 273 K, recorded at 183 K. | ||
Back reaction rates 1/k [s] are as follows: CD2Cl2 at 183 K: 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; CDCl3 at 213 K 40000 and toluene-d8 at 183 K: 30000. For CD2Cl2 at 183 K a much slower back reaction (1/k = 200000) is found if the solvent is purified by passing through a column of basic aluminum oxide. For CD3CN, methanol-d4 and acetone-d6 no products could be observed, so the back reaction rates could not be estimated.
000; CDCl3 at 213 K 40000 and toluene-d8 at 183 K: 30000. For CD2Cl2 at 183 K a much slower back reaction (1/k = 200000) is found if the solvent is purified by passing through a column of basic aluminum oxide. For CD3CN, methanol-d4 and acetone-d6 no products could be observed, so the back reaction rates could not be estimated.
NMR assignments
Based on the set of the following NMR spectra: 1H, 1H NOESY, 1H–1H correlation (COSY), 1H–13C correlations (HSQC, HMBC) we were able to assign most of the 1H and 13C signals. A complete analysis was done for the spectra recorded in CD2Cl2, a partial one, for CDCl3 and toluene-d8. A proper starting point for the analysis of the NOESY spectrum (Fig. 7) is the singlet at 7.13 ppm belonging to H-2. A cross peak between this and the doublet at 7.86 ppm assigns the latter as H-2′. This again is linked to the triplet at 7.43 ppm being therefore H-3′. H-4′ is by default the less intense triplet at 7.52 ppm. All resonances belonging to hydrogens of the double prime ring are found in the pseudo singlet at 7.32 ppm (confirmed in the HSQC spectrum). Cross peaks are also seen between the SH proton at 3.86 ppm and those of H-2 and the protons of the double prime ring.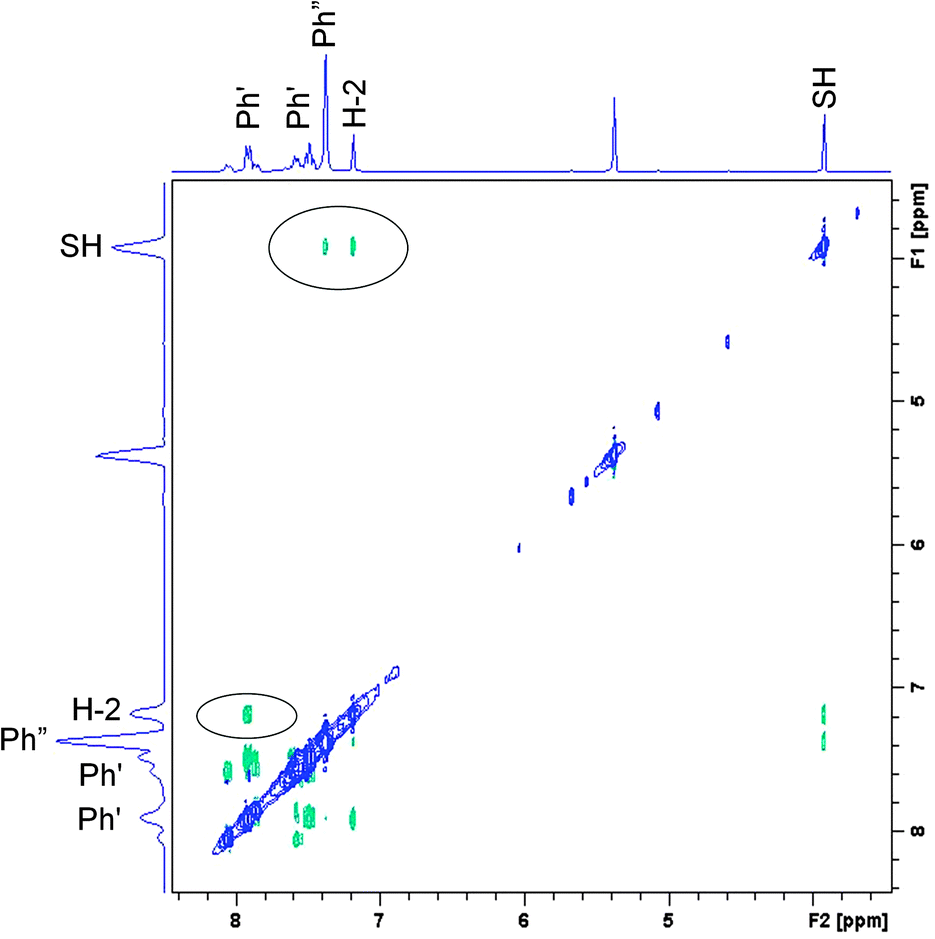 | ||
| Fig. 7 NOESY spectrum of 1id in CD2Cl2 at 183 K. | ||
The 1H NMR spectrum of the compound formed in CD2Cl2 (1id) at 183 K can be summarized as (value in ppm, multiplicity, integral): 7.86 (H-2′, d, 2H), 7.52 (H-4′, t, 1H), 7.43 (H-3′, t, 2H), 7.32 (H-2′′, H-3′′, and H-4′′, s, 5H), 7.13 (H-2, s, 1H), 3.86 (SH, s, 1H). The 1H NMR spectrum of the non-irradiated compound in CD2Cl2 (1d) at 183 K can be summarized as: 8.00 (H-2′, d, 2H), 7.80 (H-2′′, d, 2H), 7.60 (H-4′, t, 1H), 7.52 (H-2, s, 1H), 7.52 (H-4′′, t, 1H), 7.51 (H-3′, t, 2H), 7.44 (H-3′′, t, 2H), 16.86 (OH, br, 1H). The 1H NMR spectrum of the compound formed in CDCl3 (1ic) at 213 K can be summarized as: 7.89 (H-2′, d, 2H), 7.51 (H-4′, t, 1H), 7.43 (H-3′, t, 2H), 7.36 (H-2′′, H-3′′, and H-4′′, s, 5H), 7.13 (H-2, s, 1H), 3.8 (SH, s, 1H).
A NOESY spectrum of the 1it product (irradiation in toluene-d8) is depicted in Fig. S3 (ESI†). At 183 K, it only shows a few cross peaks and these are of the positive phase due to the high viscosity of toluene at low temperature. Therefore we have repeated this experiment at 213 K to get negative cross peaks. Furthermore, the spectrum is dominated by very strong signals from toluene. A cross peak is seen from the doublet at 7.67 ppm to a triplet at 6.92 ppm and one from a doublet at 7.26 ppm to a signal at 6.95 ppm. In addition, a cross peak is seen from the SH signal at 2.8 ppm to the singlet at 6.5 ppm. The latter is the signal from the H-2 proton. Based on these and supported by the theoretical calculations the following assignment can be made: 7.67 (H-2′, d), 7.26 (H-2′′, d), 6.92 (H-3′, t), 6.95 (H-3′′), 6.5 (H-2, s) and 2.8 (SH, s).
13C NMR. Based on HSQC and HMBC spectra the 13C spectrum of 1id at 183 K can be assigned as follows [ppm]: C-1 187.2, C-2 118.9, C-3 154.0, C-1′′ 139.2, C-3′′ 127.9, C-2′′ and C-4′′ at 127.4 and 128.9; C-1′ 137.3, C-2′ 128.0, C-3′ 128.3, C-4′ 132.6. The compound measured under the same conditions before irradiation (in CD2Cl2 at 183 K 1d) exhibited the following signals: C-1 177.3, C-2 109.7, C-3 211.5, C-1′′ at 145.3, C-2′′ 126.6, C-3′′ 128.7, C-4′′ 131.4, C-1′ 133.8, C-2′ 126.8, C-3′ 128.2, C-4′ 133.8.
DFT calculations of the NMR parameters
As described above, many different enol and thioenol structures can be constructed together with several diketo forms. Ground state structures, energies, and 1H and 13C chemical shifts have been calculated. The NMR parameters were not computed for the diketo forms, as the NMR spectra clearly show their absence. It is interesting to notice that all but two lowest thiol forms have very similar energies, especially when zero point energies are taken into account (Fig. 1). Moreover, except for the lowest energy e-CCC structure, all other enol forms have energies higher than the thiol species.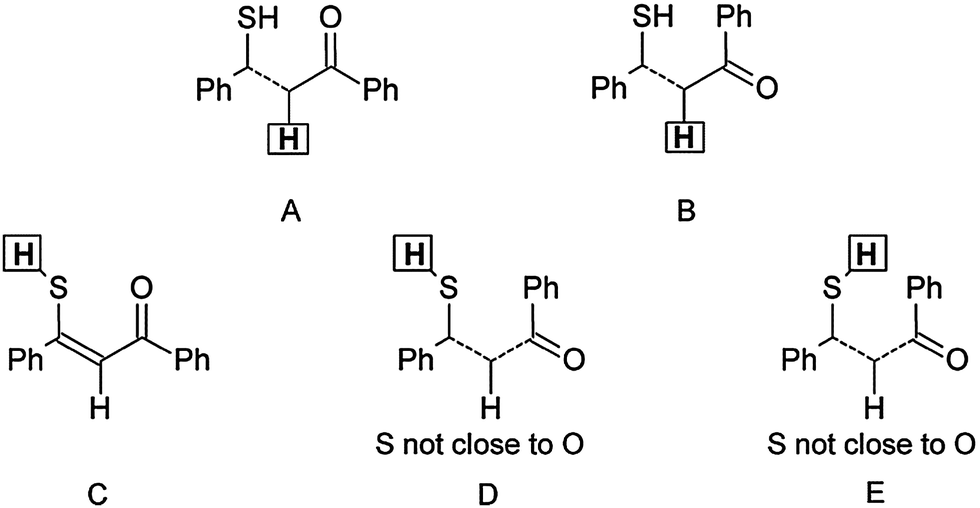 | ||
| Scheme 2 Possible tautomeric/rotameric structures (A–E), classified according to the calculated 1H chemical shielding of H-2 and SH (indicated by squares). Dashed line corresponds to both cis and trans forms. SH means orientation of no importance. Structures A, B are analysed for δ(H-2): A = t-CCC, t-TCC, t-TTC, t-CTC; B = t-CCT, t-TTT, t-CTT, t-TCT. Structures C, D, E are analysed for δ(SH): C = t-TCC; D = t-TCT, t-TTC, t-TTT; E = t-CCT, t-CTT, t-CTC. | ||
Plots of the 1H observed vs. calculated chemical shifts for 1d and 1id are given in Fig. S4 and S5 (ESI†), respectively.
Discussion
Structure of 1id
It is obvious from the 1H NMR data that 1id is not a diketo form, as no resonances are found in the 5–6 ppm range. In order to distinguish whether the enol or thiol tautomer is formed, we have to analyse the 13C chemical shifts of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X. The finding of the 13C resonance of CX at 187.2 ppm is a clear proof that this chemical shift corresponds to CO. The signals of CS are typically shifted more downfield44 and the 1d form, dominated by e-CCC, exhibits a signal at 211.5 ppm, corresponding to CS. The calculated chemical shifts are around 230–250 ppm for CS (enol forms) and around 190–200 ppm for CO (thiol forms). Naturally, hydrogen bonding to the solvent can also take place influencing the chemical shifts to some extent.23
X. The finding of the 13C resonance of CX at 187.2 ppm is a clear proof that this chemical shift corresponds to CO. The signals of CS are typically shifted more downfield44 and the 1d form, dominated by e-CCC, exhibits a signal at 211.5 ppm, corresponding to CS. The calculated chemical shifts are around 230–250 ppm for CS (enol forms) and around 190–200 ppm for CO (thiol forms). Naturally, hydrogen bonding to the solvent can also take place influencing the chemical shifts to some extent.23
The finding that the SH form is dominant is also supported by the 1H chemical shifts of the XH proton at 3.86 ppm, because such a value is characteristic rather for SH, whereas OH signals are usually shifted more downfield, as shown from the NMR calculations (Table S1, ESI†). The hydrogen-bonded thiol form t-CCC is likewise excluded. The t-TCC form found in the experiment irradiating at 415 nm at 90 K is likewise not found, in good agreement with the low barrier to return found for this intermediate. The structural assignment of 1id is based on a combination of chemical shifts and NOE data. The finding of a cross peak between the SH proton and H-2 clearly shows that those two protons are on the same side of the double bond. The chemical shifts of the H-2 proton (7.13 ppm) assign this proton and the oxygen to be on opposite sides of the double bond (structure A in Scheme 2). These two criteria only leave two possible structures for 1id, t-CTC and t-TTC (see Fig. 1). The finding of a NOE cross peak between the SH proton and the protons of the double prime ring (Scheme 1) points towards the t-TTC form. On the other hand, the SH chemical shift (3.86 ppm) points towards E of Scheme 2, corresponding to t-CTC and, most probably, both species exist in equilibrium. The relevant proton–proton distances for all calculated enol and thiol tautomers are presented in Table S2 (ESI†).
The product obtained in CDCl3, 1ic, is identical to 1id, as judged from the 1H NMR spectrum and from the value of the reverse reaction rate.
Structure of 1it
As seen in the Results section, the 1H chemical shifts of 1it and 1id are rather different, which could suggest that both structures are not the same. On the other hand, the solvent effect on the chemical shifts in toluene is specific. Ring currents of toluene lead to the low-frequency shifts of 1H signals, which makes the comparison to the spectra in the other solvents complicated. However, the 2D-NOESY experiment exhibits cross-peaks between the same protons as in the case of 1id (see Fig. 7 and Fig. S3, ESI†). The identical structures of 1id and 1it are also supported by the experiment in mixed solvents (CD2Cl2: toluene-d8 5:1 and 1:1), as seen in Fig. 8. The changes in the SH and CH chemical shifts at growing concentration of toluene have a smooth character and are similar to the behaviour of solvent peaks. Moreover, the reverse reaction in the dark is as slow as for 1id. Therefore, we can conclude that 1it has the same structure as 1id.
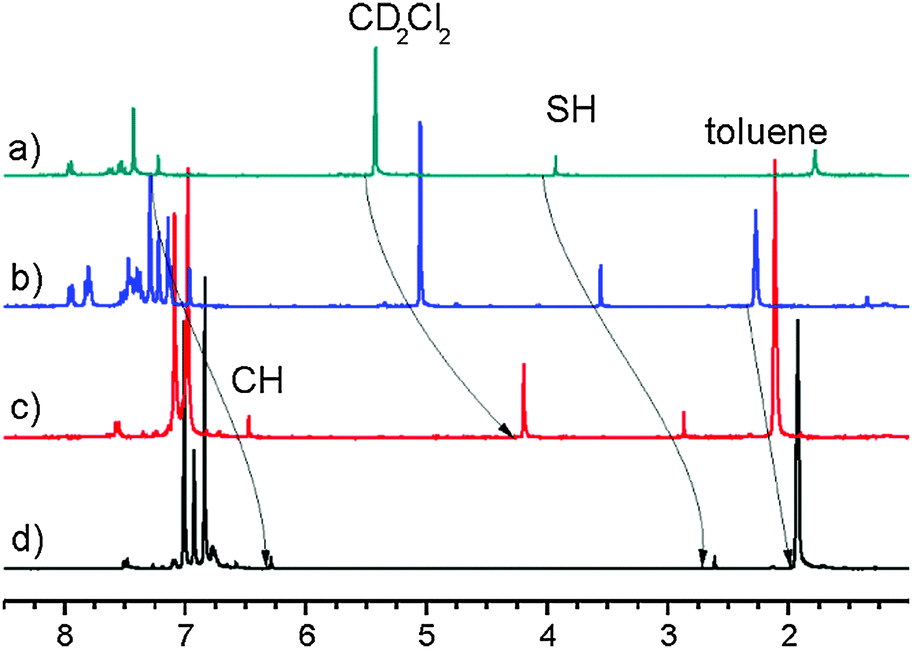 | ||
| Fig. 8
1H NMR spectra of 1 after irradiation at 183 K. Scale referenced to internal TMS; (a) CD2Cl2; (b) CD2Cl2 and toluene-d8 (5:1); (c) CD2Cl2 and toluene-d8 (1:1); (d) toluene-d8. | ||
Conversion back to 1
Is the conversion of 1id and 1it back to 1 feasible? In the context it should be considered that both 1id and 1it are so-called twisted push–pull ethylenes. The calculated twist (the S,CC,C dihedral angle) is 10° and 8° for t-TTC and t-CTC, respectively. Sandström and co-worker have found that barriers to rotation around the double bonds in such systems are not very high.45
Modeling of the photoreaction path
In order to understand the mechanisms of the multistep photochromic transformation in 1, we considered all five experimentally observed species as well as the relevant transition states. Ground state structure optimizations were followed by calculations of transition energies originating at S0 geometries. In the next step, geometries of the lowest excited singlet and triplet states were optimized. The calculated ground state geometry of the e-CCC form is in very good agreement with the X-ray structure (Fig. S6, ESI†). The optimized S0, S1, T1, and transition state geometries of all five forms are presented in Fig. S7 (ESI†). We have also repeated the calculations for thiomalonaldehyde (Fig. S8–S12, ESI†), a model system, for which all five forms are predicted to be planar in the ground state. The patterns of the relative energies obtained for both molecules turned out to be very similar, which indicates that the origin of the behavior of 1 is due to the electronic structure of the thiomalonaldehyde core, not to the symmetry-lowering perturbation of the substitution by phenyl rings. In this context, we also note that the X-ray study of 1-(1-methylcyclopropyl)-3-thioxobutan-l-one, a β-thioxoketone with aliphatic substituents,46 revealed a very similar core geometry to that of 1 (the differences in bond lengths of about 0.01 Å, the angles between bonds agreeing within 1 degree).The relative energies calculated for different tautomers/rotamers of 1 are shown in Scheme 3. The first important finding concerns the relative energies of the two most stable forms, e-CCC and t-CCC. In agreement with experiment, their energies are predicted to be similar, with the chelated thiol form higher by 4 kcal mol−1 (2 kcal mol−1 after ZPE correction). In contrast, for the S1 and T1 excited states the calculations predict a large destabilization, of the order of 10 kcal mol−1 or more, of the thiol with respect to the enol tautomer. This prediction is crucial for the understanding of photochromism, as the first photoproduct, “open” thiol t-TCC is obviously generated from t-CCC. Since the excited state e-CCC → t-CCC reaction is predicted to be strongly uphill in S1 and T1, the effective way to produce t-TCC is either by direct excitation of t-CCC or via the S2(ππ*) state of e-CCC. This conclusion is in excellent agreement with the observed huge dependence of the photoconversion efficiency on the excitation wavelength. It is well established for many thiocarbonyl compounds that the ability of sulfur to form hydrogen bonds decreases upon excitation to S1(nπ*), but increases in S2(ππ*).47–49 Moreover, the lifetime of S2(ππ*) can be as long as hundreds of picoseconds,50,51 but it can be effectively quenched by specific interactions with such solvents as acetonitrile52,53 or water.54 These findings not only provide a strong indication that the main precursor of the photochromism is the S2(ππ*) state of e-CCC: they also explain why there is no photoreaction in methanol or acetonitrile. Naturally, the reaction can also be initiated by direct excitation of t-CCC, but the ground state population is dominated by the enol, which also has a stronger absorption than the thiol at the wavelengths used. Therefore the contribution by direct excitation of this species should be minor, especially at lower temperatures. We finally recall that the open thiol can be generated photochemically at very low temperatures, when only the enol form is initially present in the ground state.24
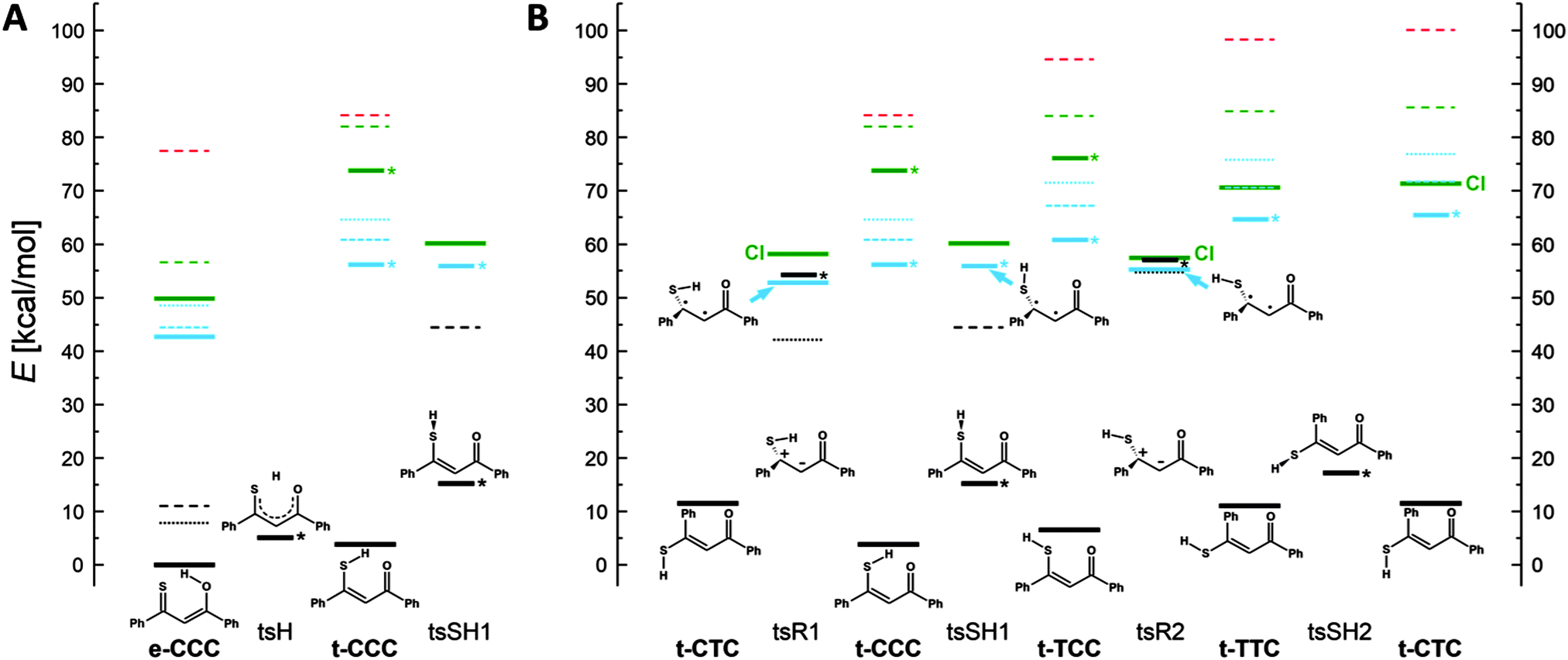 | ||
| Scheme 3 Calculated relative ground (black) and T1 (blue), S1 (green) and S2 (red) excited state energies of the experimentally observed forms of 1 and of the predicted transition states. Solid lines indicate optimized geometries. The vertical energies computed for the optimized geometries are indicated by dashed (S1 and S2 at S0, S0 at S1) or dotted (T1 at S0 and S0 at T1) lines. The vertical T1 energies were computed by both TD-DFT (short dashed lines) and UB3LYP (dots). Transition states are indicated by asterisks. CI indicates non-fully optimized structure, due to the proximity of states. | ||
The calculations predict that the e-CCC to t-CCC conversion is strongly endoenergetic in both S1 and T1 (Scheme 3A). Attempts to optimize S2 were not successful. For model molecules with hydrogen atoms or methyl groups instead of phenyls, S2 optimization assuming Cs symmetry of t-CCC ends up in e-CCC. These results indicate the e-CCC(S2) → t-CCC(S1) as the most probable path.
Once the excited t-CCC has been produced, it can be transformed into t-TCC. The S1 optimized t-CCC and t-TCC structures converge to identical geometries, with the SH bond almost perpendicular to the OC1C2C3S plane. This structure (tsSH1 in Scheme 3A and B) corresponds in S0 to the transition state between t-CCC and t-TCC. Thus, the S1–S0 relaxation (favourable because of a small energy gap) can lead to both forms.
In order to understand the next step of photochromism, the transformation into t-TTC and/or t-CTC, we note that the optimized S1/T1 (and probably also S2) states have in these structures higher energies than tsSH1. However, the structures in which the molecule is twisted about the C2C3 bond (tsR1 and tsR2 in Scheme 3) have in S1 a lower energy than tsSH1. These structures correspond in the ground state to high energy (40–50 kcal mol−1) transition states for rotation about a double bond. Full geometry optimization of the twisted forms was not possible, because of close-lying S0 and S1 states, characteristic of this type of structure and possibly leading to a conical intersection (CI). The S1 energies presented in Scheme 3 correspond therefore to upper limits. One can conclude that once the molecule overcomes the barrier from tsSH1 to tsR1 or tsR2, it can immediately convert (via conical intersection) into t-CTC and t-CCC (from tsR1) or into t-TTC and t-CTC (from tsR2). The t-TTC and t-CTC are practically isoenergetic in S0 (0.5 kcal mol−1 difference) and separated by a small barrier for the SH group rotation (6 kcal mol−1). Most probably the equilibrium is established between the two forms.
The behaviour predicted for the triplet states using UB3LYP is somewhat similar to that described above for S1. Except e-CCC, no structures with planar skeleton are obtained: they all relax to the twisted tsR1 and tsR2 forms (Scheme 3B). Contrary to the situation in S1, also the tsSH1 form is nonplanar; it now corresponds to the transition state between tsR1 and tsR2. Thus, the fate of excited molecules which underwent rapid proton transfer is similar to those in S1. Now, however, the twisting leading to one of the tsR forms is barrierless. Even though no CI is predicted for these forms, the T1–S0 energy gap is very small.
On the basis of calculations we conclude that photochromism involving large amplitude motion is possible both in singlet and triplet manifolds. Because of expected large efficiency of intersystem crossing to the triplet and the barrierless character of tsSH1 → tsR forms, the triplet path seems to be more probable.
The crucial parameter for the t-TCC → t-TTC, t-CTC conversion is the C2C3 bond length (Scheme 1), as, formally, the process involves a rotation about a double bond. The values calculated for ground and excited states are very instructive in this regard. For S0, the calculations predict 136, 135, and 135 pm for t-TCC, t-TTC, and t-CTC, respectively. For the relevant S1 structure, tsSH1, a value of 139 pm is obtained, whereas the calculations for the triplet tsR1 and tsR2 forms yield 146 pm. These results clearly demonstrate the weakening of the double bond character in the triplet state. Together with the relative energies calculated for the three forms, they provide strong arguments that the large amplitude transformation leading to the photochromic products observed in solution occurs via the triplet state.
As was already mentioned above, the rotation about the double bond in the ground state involves a large energy barrier. In principle, an alternative mechanism is possible, with the participation of the diketo form. First, the SH hydrogen atom is transferred from t-CTC to C2 carbon with partial twisting, and then back to either sulphur or oxygen, with further twisting. However, the calculated barriers to these processes are only slightly lower (still above 40 kcal mol−1). The experimental finding that this reaction takes minutes at room temperature indicates that the real mechanism is different. The large change of the reaction rate in CD2Cl2 after removal of acid traces from the solvent supports the reaction pathway involving protonation of the CO group of t-CTC and/or t-TTC. Initial calculations support this hypothesis, yielding for the protonated form a barrier of 20 kcal mol−1.
The barrier to rotation about the C1C2 single bond is very small (ca. 2 kcal mol−1). The t-TTT form is calculated to lie higher in energy than t-TTC. The experiment suggests that if it is formed, its concentration is below that required for NMR detection.
Conclusions
The use of “hyphenated techniques” such as laser irradiation combined with NMR detection at low temperature enables the observation and identification of new unstable species. By varying the temperature and viscosity, it was possible to observe the consecutive steps of photochromic transformation in monothiodibenzoylmethane. Irradiation of 1 in a rigid environment at temperatures of about 100 K or lower leads to the formation of t-TCC, a structure characterized by opening of the intramolecular hydrogen bond. Such a rotamer is stable below 100 K, but returns to the main e-CCC form upon heating up. The situation in a non-rigid environment, i.e. liquid solution, is different. Now, the end products of the irradiation are t-CTC and t-TTC. The reaction path we propose includes photoinduced transformation from e-CCC in the S2(ππ*) state to t-CCC, followed by the hydrogen bond opening which leads to t-TCC. This step is possible even at very low temperatures and in rigid media, contrary to the second stage of the photochromic transformation, rotation about the C2C3 bond of the thiol t-TCC form. This reaction can only proceed in less viscous environments. The opening of the hydrogen bond may occur in both the excited singlet and the triplet state of t-CCC, but the next stage most probably goes only through T1. The dark reaction, i.e., the recovery of the most stable form of 1 is much longer than the photoinduced reaction. It is because the rotation along the double CC bond in the ground state has a relatively high barrier. The proposed scheme of the photochromism of 1 is shown in Scheme 4.
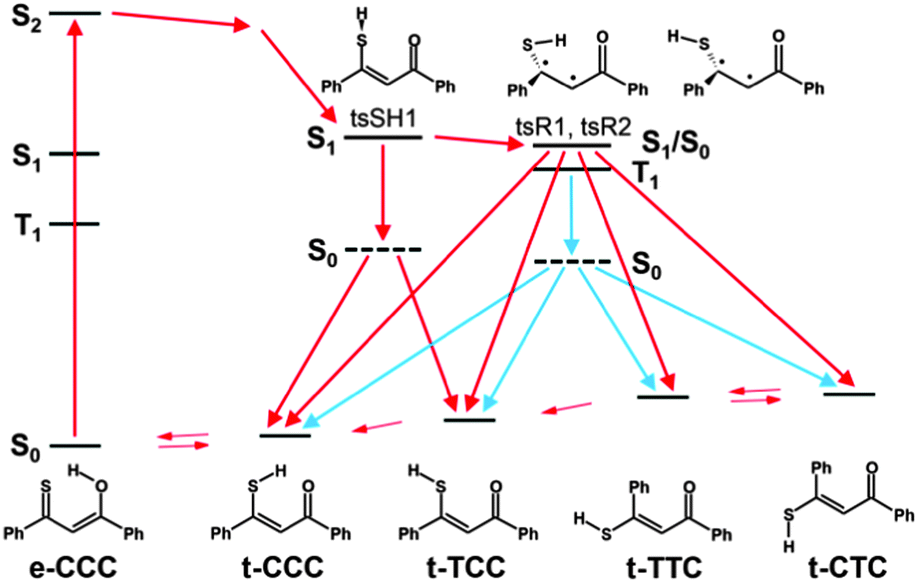 | ||
| Scheme 4 The proposed photochromic pathways. Red and blue arrows denote singlet and triplet routes, respectively. Dashed lines indicate hot ground state molecules with geometry corresponding to the S1 or the T1 excited state. | ||
An observation relevant for the understanding of the mechanism of photochromism is the extremely small yield for excitation into the low energy region where only e-CCC absorbs into its S1(nπ*) state. Combined with the theoretical prediction of a large shift of the enol–thiol equilibrium towards the enol form, this finding strongly suggests lack of proton transfer from e-CCC to t-CCC in the S1 and T1 excited states. In consequence, the precursor of the photochromism is state-specific: e-CCC enol in S2(ππ*). Another argument for this postulate is the lack of photochromism in methanol and acetonitrile, as for these solvents the S2 lifetime can be drastically reduced. The directly excited t-CCC can only contribute at high temperatures, and even then the absorption will be dominated by e-CCC.
The structural assignments of t-CTC and t-TTC provided by NMR for CD2Cl2 and CDCl3 solutions seem to be very reliable. The product in toluene-d8 is most likely the same. Quantitative description of the kinetics of photochromism in β-thioxoketones requires time-resolved experiments spanning many orders of magnitude, from femtoseconds to minutes or even days. Such studies are now underway. We note the application potential of compounds for which the photochromic properties depend on solvent, temperature, viscosity, and excitation wavelength. Finally, one should stress that using the NMR detection for direct, in situ monitoring of unstable products of photochemical transformations opens up many new avenues in all areas requiring unequivocal structural assignments.
We hope that the complicated scheme of processes responsible for the photochromism in β-thioxoketones can provide a challenge for the rapidly growing area of theoretical description of photoinduced proton/hydrogen transfer phenomena.55–65
Acknowledgements
The authors wish to thank Professor Fritz Duus, Roskilde for advice. This work has been supported by the Polish Ministry of Science and Higher Education grant no. N N204 239 134, by the Polish National Science Centre grants no. DEC-2011/01/B/ST4/01130 and DEC-2011/03/B/ST4/02621, and the PL-Grid Infrastructure grant.Notes and references
- Photochromism. Molecules and Systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier, 2003 Search PubMed.
- Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed.
- M. Irie, Photochem. Photobiol. Sci., 2010, 9, 1535–1542 CAS.
- W. Schnabel, Polymers and Light: Fundamentals and Technical Applications, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- J. Fabian, Tetrahedron, 1973, 29, 2449–2456 CrossRef CAS.
- O. Siiman, J. Fresco and H. B. Gray, J. Am. Chem. Soc., 1974, 96, 2347–2353 CrossRef CAS.
- W. F. Rowe, R. W. Duerst and E. B. Wilson, J. Am. Chem. Soc., 1976, 98, 4021–4023 CrossRef CAS.
- L. Carlsen and F. Duus, J. Am. Chem. Soc., 1978, 100, 281–282 CrossRef CAS.
- L. Carlsen and F. Duus, J. Chem. Soc., Perkin Trans. 2, 1979, 1532–1534 RSC.
- L. Carlsen and F. Duus, J. Chem. Soc., Perkin Trans. 2, 1980, 1768–1773 RSC.
- F. S. Jørgensen, L. Carlsen and F. Duus, J. Am. Chem. Soc., 1981, 103, 1350–1353 CrossRef.
- J. Gebicki and A. Krantz, J. Chem. Soc., Chem. Commun., 1981, 486–488 RSC.
- J. Gebicki and A. Krantz, J. Am. Chem. Soc., 1981, 103, 4521–4526 CrossRef CAS.
- P. E. Hansen, F. Duus and P. Schmitt, Org. Magn. Reson., 1982, 18, 58–61 CrossRef CAS.
- F. S. Jørgensen, R. S. Brown, L. Carlsen and F. Duus, J. Am. Chem. Soc., 1982, 104, 5922–5926 CrossRef.
- U. Berg, J. Sandström, L. Carlsen and F. Duus, J. Chem. Soc., Perkin Trans. 2, 1983, 1321–1325 RSC.
- L. Nørskov-Lauritsen, L. Carlsen and F. Duus, J. Chem. Soc., Chem. Commun., 1983, 496–498 RSC.
- F. Duus, J. Am. Chem. Soc., 1986, 108, 630–638 CrossRef CAS.
- P. E. Hansen, U. Skibsted and F. Duus, J. Phys. Org. Chem., 1991, 4, 225–232 CrossRef CAS.
- L. González, O. Mó and M. Yáñez, J. Phys. Chem. A, 1997, 101, 9710–9719 CrossRef.
- L. González, O. Mó and M. Yáñez, J. Org. Chem., 1999, 64, 2314–2321 CrossRef.
- N. Došlić, K. Sundermann, L. González, O. Mó, J. Giraud-Girard and O. Kühn, Phys. Chem. Chem. Phys., 1999, 1, 1249–1257 RSC.
- B. Andresen, F. Duus, S. Bolvig and P. E. Hansen, J. Mol. Struct., 2000, 552, 45–62 CrossRef CAS.
- Y. Posokhov, A. Gorski, J. Spanget-Larsen, F. Duus, P. E. Hansen and J. Waluk, Chem. Phys. Lett., 2001, 350, 502–508 CrossRef CAS.
- Y. Posokhov, A. Gorski, J. Spanget-Larsen, F. Duus, P. E. Hansen and J. Waluk, ChemPhysChem, 2004, 5, 495–502 CrossRef CAS PubMed.
- A. Gorski, Y. Posokhov, B. K. V. Hansen, J. Spanget-Larsen, J. Jasny, F. Duus, P. E. Hansen and J. Waluk, Chem. Phys., 2006, 328, 205–215 CrossRef CAS.
- A. Gorski, Y. Posokhov, B. K. V. Hansen, J. Spanget-Larsen, J. Jasny, F. Duus, P. E. Hansen and J. Waluk, Chem. Phys., 2007, 338, 11–22 CrossRef CAS.
- B. K. V. Hansen, A. Gorski, Y. Posokhov, F. Duus, P. E. Hansen, J. Waluk and J. Spanget-Larsen, Vib. Spectrosc., 2007, 43, 53–63 CrossRef CAS.
- J. Dobkowski, J. Wójcik, W. Koźmiński, R. Kołos, J. Waluk and J. Michl, J. Am. Chem. Soc., 2002, 124, 2406–2407 CrossRef CAS PubMed.
- C. Sheu, P. Kang, S. Khan and C. S. Foote, J. Am. Chem. Soc., 2002, 124, 3905–3913 CrossRef CAS PubMed.
- P. Kang and C. S. Foote, J. Am. Chem. Soc., 2002, 124, 4865–4873 CrossRef CAS PubMed.
- P. Kang and C. S. Foote, J. Am. Chem. Soc., 2002, 124, 9629–9638 CrossRef CAS PubMed.
- T. l. Kühn and H. Schwalbe, J. Am. Chem. Soc., 2000, 122, 6169–6174 CrossRef.
- S. M. Harper, L. C. Neil, I. J. Day, P. J. Hore and K. H. Gardner, J. Am. Chem. Soc., 2004, 126, 3390–3391 CrossRef CAS PubMed.
- P. Wenter, B. Furtig, A. Hainard, H. Schwalbe and S. Pitsch, Angew. Chem., Int. Ed., 2005, 44, 2600–2603 CrossRef CAS PubMed.
- J. Buck, B. Furtig, J. Noeske, J. Wohnert and H. Schwalbe, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15699–15704 CrossRef CAS PubMed.
- M. Lausmann, I. Zimmer, J. Lex, H. Lueken, K. Wieghardt and E. Vogel, Angew. Chem., Int. Ed. Engl., 1994, 33, 736–739 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. Frisch, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- A. R. Katritzky, S. Sobiak and C. M. Marson, Magn. Reson. Chem., 1988, 26, 665–670 CrossRef CAS.
- J. Sandström, U. Sjöstrand and I. Wennerbeck, J. Am. Chem. Soc., 1977, 99, 4526–4527 CrossRef.
- L. Nørskov-Lauritsen, L. Carlsen and F. Duus, J. Chem. Soc., Chem. Commun., 1983, 496–498 RSC.
- E. Krystkowiak, J. Koput and A. Maciejewski, Phys. Chem. Chem. Phys., 2012, 14, 8842–8851 RSC.
- G. J. Zhao and K. L. Han, ChemPhysChem, 2008, 9, 1842–1846 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- A. Maciejewski, D. R. Demmer, D. R. James, A. Safarzadeh-Amiri, R. E. Verrall and R. P. Steer, J. Am. Chem. Soc., 1985, 107, 2831–2837 CrossRef CAS.
- A. Maciejewski and R. P. Steer, Chem. Rev., 1993, 93, 67–98 CrossRef CAS.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat and C. Lefumeux, Chem. Phys. Lett., 2004, 384, 332–338 CrossRef CAS.
- M. Lorenc, A. Maciejewski, M. Ziolek, R. Naskrecki, J. Karolczak, J. Kubicki and B. Ciesielska, Chem. Phys. Lett., 2001, 346, 224–232 CrossRef CAS.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat, P. Toele, H. Zhang and M. Glasbeek, Chem. Phys. Lett., 2004, 393, 102–106 CrossRef CAS.
- Y. Syetov, J. Fluoresc., 2013, 23, 689–696 CrossRef CAS PubMed.
- M. Savarese, P. A. Netti, C. Adamo, N. Rega and I. Ciofini, J. Phys. Chem. B, 2013, 117, 16165–16173 CrossRef CAS PubMed.
- Y. Houari, A. Charaf-Eddin, A. D. Laurent, J. Massue, R. Ziessel, G. Ulrich and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 1319–1321 RSC.
- H.-H. G. Tsai, H.-L. S. Sun and C.-J. Tan, J. Phys. Chem. A, 2010, 114, 4065–4079 CrossRef CAS PubMed.
- Y. Shigemitsu, T. Mutai, H. Houjou and K. Araki, J. Phys. Chem. A, 2012, 116, 12041–12048 CrossRef CAS PubMed.
- M. Barbatti, A. J. A. Aquino, H. Lischka, C. Schriever, S. Lochbrunner and E. Riedle, Phys. Chem. Chem. Phys., 2009, 11, 1406–1415 RSC.
- N. Kungwan, R. Daengngern, T. Piansawan, S. Hannongbua and M. Barbatti, Theor. Chem. Acc., 2013, 132, 1–10 CrossRef CAS.
- N. Kungwan, F. Plasser, A. J. A. Aquino, M. Barbatti, P. Wolschann and H. Lischka, Phys. Chem. Chem. Phys., 2012, 14, 9016–9025 RSC.
- R. De Vivie-Riedle, V. De Waele, L. Kurtz and E. Riedle, J. Phys. Chem. A, 2003, 107, 10591–10599 CrossRef CAS.
- J. Jankowska, M. F. Rode, J. Sadlej and A. L. Sobolewski, ChemPhysChem, 2012, 13, 4287–4294 CrossRef CAS PubMed.
- A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 1999, 1, 3065–3072 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated structures and NMR parameters. CCDC 974809. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cp00530a |
| This journal is © the Owner Societies 2014 |