 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kink energy and kink dipole moments on vicinal Au(001) in halide electrolytes
M.
Al-Shakran
a,
G.
Beltramo
b and
M.
Giesen
*c
aInstitut für Elektrochemie, Universität Ulm, Albert-Einstein-Allee 47, 89069 Ulm, Germany
bInstitute of Complex Systems (ICS-7), Jülich Forschungszentrum GmbH, 52425 Jülich, Germany
cPeter Grünberg Institute (PGI-6), Jülich Forschungszentrum GmbH, 52425 Jülich, Germany. E-mail: m.giesen@fz-juelich.de; Tel: +49-2461-614524
First published on 17th March 2014
Abstract
Using electrochemical scanning tunnelling microscopy, we measured the potential-dependent kink energy and the corresponding dipole moments of kinks at step edges on vicinal Au(001) surfaces in chloride and bromide containing electrolytes. Combining the results of the potential dependence of the kink energy with impedance spectroscopy data for the surface charge, we can directly deduce the dipole moment of kinks at the Au steps with co-adsorbed Cl− and Br−, respectively. We find μCl = (6.0 ± 0.7) × 10−3 eÅ and μBr = (10.1 ± 0.6) × 10−3 eÅ.
Introduction
The investigation of transport processes at electrode surfaces in an electrochemical environment has attracted scientific interest for a couple of years due to their relevance in electrochemical processes such as equilibrium and non-equilibrium dynamics,1 corrosion2,3 and catalysis4 and also in applied research such as battery-related transport phenomena.5 Recently it has been demonstrated that all migration processes have an exponential dependence on the electrode potential.6–8 In these reports, it was shown that defects at charged surfaces carry a dipole moment that contributes linearly to the energy of the defects in the electric field of the double layer and, consequently, adds the exponential dependence of the transport rates on the electrode potential to Boltzmann terms for activated transport processes or defect formation. This concept has meanwhile been confirmed in further experimental studies performed in electrolytes.9,10In order to understand the details of transport processes in electrochemical systems it is therefore of enormous importance to know the dipole moments of migrating defects. Unfortunately, experimental as well as theoretical values are still scarce: theoretical studies by Müller and Ibach considered the dipole moment of free adatoms on Au, Ag and Cu surfaces in vacuum using cluster calculations.8 The data as obtained by Müller et al. fit nicely to experimental data on the decay of islands on Au(001).6 Pötting et al. employed density functional theory (DFT) methods to calculate the dipole moments of transition states for a large number of diffusion paths on silver surfaces.11 In further studies by Cockayne and Burton, DFT calculations were used to determine the dipole moment of Pb–O vacancy pairs in PbTiO3.12 Step dipole moments on metal electrodes have been measured using impedance spectroscopy.13,14 Recently, the dipole moments of adsorbates on metal electrodes have been determined experimentally by Magnussen and coworkers.10,15
Experimental data on the dipole moment of kinks at steps that are preferred binding sites and therefore serve as sinks and sources in growth and degradation phenomena are not available to date.
The aim of this paper is to fill this gap: we present an experimental method to determine the dipole moment of kinks at steps, using a combination of potential dependent measurements of step fluctuations and experimental data on the surface charge. The method is demonstrated for the particular system of stepped Au(001) electrodes in dilute halide solutions with specific adsorption present. The method to measure the kink dipole moment as such, however, is not restricted to systems with specifically adsorbed anions, but rather can be applied to electrolyte interfaces in general.
Experimental
STM measurements
Experimental data were acquired using the electrochemical version of the Topometrix TMX 2010 Discoverer STM. Our instrument is modified to enable temperature variable STM recording as described earlier.16,17 The results reported here, however, are data obtained at 293 K exclusively. Tip and sample potentials are independently controlled using a bipotentiostat.As samples we used Au(11n) single crystal electrodes (n = 17 and 29), vicinal to Au(001) with 〈110〉-oriented steps and (001) terraces of mean width 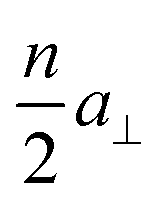 (a⊥ the nearest-neighbor distance perpendicular to 〈110〉). These surfaces have a small miscut angle (4.8° and 2.8°, respectively) to the (001) plane and hence a high density of parallel steps and are suitable electrodes to study step and kink properties with a large statistical database. For the data presented here, we typically analyzed a total step length of several μm at a distinct electrode potential. The electrodes were spark eroded from a single crystal rod, oriented by diffractometry and polished to the desired orientation to within 0.1°, which is the accuracy of high-quality single crystals. The accuracy is limited by the mosaic structure of the crystal.
(a⊥ the nearest-neighbor distance perpendicular to 〈110〉). These surfaces have a small miscut angle (4.8° and 2.8°, respectively) to the (001) plane and hence a high density of parallel steps and are suitable electrodes to study step and kink properties with a large statistical database. For the data presented here, we typically analyzed a total step length of several μm at a distinct electrode potential. The electrodes were spark eroded from a single crystal rod, oriented by diffractometry and polished to the desired orientation to within 0.1°, which is the accuracy of high-quality single crystals. The accuracy is limited by the mosaic structure of the crystal.
Prior to experiment, the Au(001) electrodes were heated in a hydrogen atmosphere and then flame annealed for 5 min to about 900 °C. The temperature was visually controlled by the color of the annealed crystal. After thoroughly rinsing the cell with Milli-Q water, the crystals were mounted on the electrochemical STM cell connected to a bipotentiostat. The crystal surface was then brought in contact with the electrolyte under potential control. This was visually checked prior to STM experiment by the presence of the well-ordered quasi-hexagonal reconstruction of the clean Au(001) surface.18
As electrolytes, we used suprapure HClO4, HCl and KBr (all Merck) and Milli-Q water from a Millipore Elix 3 and A10 Gradient system (18.2 MΩ cm−1; organic contents <3 ppb). The STM measurements were performed in 0.1 M HClO4 + 1 mM HCl and 0.1 M HClO4 + 1 mM KBr, respectively.
The tunnelling tips used in the experiments were etched from tungsten wires following standard procedures19–21 and coated with polyethylene to avoid Faraday currents at the foremost part of the tip. We used a tunnelling current of 2 nA. The typical time per image was 40 s and the STM images were recorded with a 400 × 400 pixel resolution in the constant current mode.
High-purity, flame-annealed Pt wires (Goodfellow, 99.999%) served as counter and quasi-reference electrodes. In the following, the electrode potentials of the metal sample, however, are given with respect to the saturated calomel electrode (SCE). As a caveat we emphasize that the electrode potentials denoted here are the nominal values given by the bipotentiostat. Since Pt is a quasi-reference, non-ideally polarizable electrode, we cannot exclude that the potential values as denoted here might deviate from the real potential by about 50 mV due to possible shifts in the open circuit potential of the Pt wire during measurement.
Depending on the chosen electrode potential, good experimental conditions lasted for 30 min up to 2 h. Then, generally, contamination of the electrolyte in the open STM cell became visible and the measurements were terminated.
Step profiles in STM images were analyzed using a special computer code that determines the maximum slope in the grey scale in an individual scan line of the STM image. To account for a possible drift in the STM image we analyzed exclusively step pairs,22 a procedure that proved successful in a large number of studies of step fluctuations in vacuum as well as in electrolytes (for a review see ref. 1).
Surface charge measurements
Capacitance curves were recorded using a Zahner IM5 impedance potentiostat. As electrodes we used flat Au(001) bead-type single crystals prepared according to the Clavilier method.23 Further preparation of the electrodes prior to experiments followed the same procedure as that for the STM samples described before.The crystal surface was brought in contact with the electrolyte under potential control at values well below the potential of zero charge (pzc). The contact between the electrode surface and the solution was made by means of the hanging meniscus method.24 A saturated calomel electrode served as a reference electrode and a platinum foil served as a counter electrode.
The capacity curves were recorded at a frequency of 20 Hz and at an amplitude of 5 mV. Data were obtained in the direction of negative sweeps exclusively, i.e. the capacity data are that of the unreconstructed flat Au(001) electrode.
We used electrolytes 9 mM KClO4 + 1 mM KCl and 9 mM KClO4 + 1 mM KBr made from suprapure KCl, KBr and from KClO4 twice recrystallized (all Merck) and Milli-Q water as before. The electrolytes differ from those used in STM experiments: first, the total amount of anions did not exceed 10 mM. Under this condition, the pzc can be determined via the minimum in the differential capacity25 dominated by the Gouy–Chapman contribution in this case. Second, for the analysis of the surface charge density the capacity curves must be measured at low potentials. For the given system, one would have to measure at potentials negative of the hydrogen evolution onset. The use of KClO4 rather than HClO4 shifts the onset of the hydrogen evolution to lower potentials and allows the measurement of the capacity at sufficiently low potentials. Hence, for the system considered in this work, the use of KClO4 for obtaining the capacity curve is preferred over the use of HClO4. Although the two electrolytes differ in pH, the shift in pzc is negligibly small.
In order to check for a possible influence of the different electrolytes on our results, we additionally measured cyclic voltammograms (CV) in 9 mM XClO4 + 1 mM XCl (X = H or K) and in 9 mM XClO4 (X = H or K) + 1 mM KBr with a rate of 10 mV s−1. The CVs in HClO4 and KClO4 are almost identical, safe for the shift of the onset potential of hydrogen evolution at low potentials and of oxygen evolution at high potentials (data not shown here).
For all electrolytes we used, in STM experiments, CV and capacity studies, the important content of the halides, Cl− and Br−, however, is identical.
The capacity curves were determined using various models for the interface capacity. As demonstrated in more detail for Ag(111) electrodes in dilute halide containing electrolytes, the chosen model has no significant influence on the capacity curves as long as the potential is well below the range where ordered halide adlayers are formed.26
Theory of step fluctuations
Experimental and theoretical data on step fluctuations have been widely discussed in literature throughout the last two decades. Therefore, we refer for details to review articles.1,27 As a brief summary, we recall the basic equation for the spatial correlation function G(y) used to determine the kink energy, valid under nearest-neighbor conditions: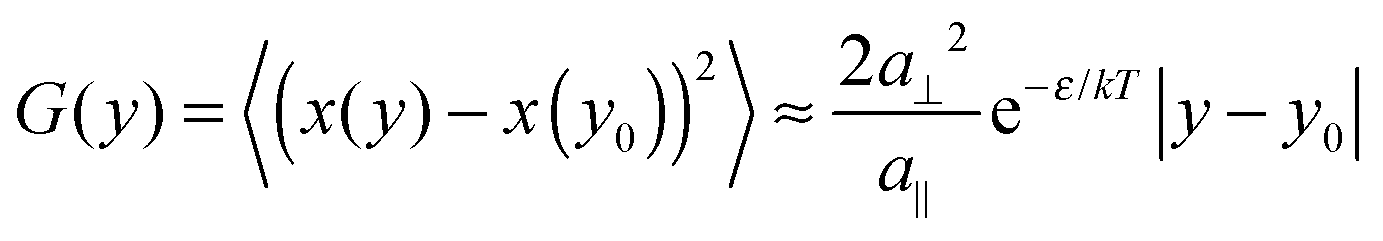 | (1) |
Recently we showed that defect migration and formation energies depend on electrode potential ϕ in electrochemical systems and are directly related to the dipole moment μ of the defects.6–8 In the case of kinks we find for the kink energy ε (with ε0 the vacuum permittivity):
 | (2) |
Results and discussion
Fig. 1 shows STM images of various vicinal Au(11n) electrodes in Cl− and Br−-containing solutions at different potentials. At high potentials, the surface is unreconstructed and the mobility of the steps may become quite large. | ||
| Fig. 1 STM images of Au electrodes in 0.1 M HClO4 + 1 mM HCl (a) (1,1,29) at −0.17 V SCE, (b) (1,1,17) at +0.45 V SCE and in 0.1 M HClO4 + 1 mM KBr (c) (1,1,29) at −0.25 V SCE, (d) (1,1,29) at +0.31 V SCE. | ||
At low potentials, large terraces are reconstructed. Smaller terraces in regions of high step density remain unreconstructed (Fig. 1(c)). This observation is in accordance with previous data for Au(11n) in sulfuric acid.28 The fact that smaller terraces remain unreconstructed offers the opportunity to measure step fluctuations for the unreconstructed (1 × 1)-surface even at low potentials. Therefore, the data for step fluctuations and the kink energy in the potential range between −0.25 and +0.55 V SCE as presented in this work are for the unreconstructed Au(001) surface exclusively.
Cyclic voltammograms and surface charge density vs. potential plots are presented in Fig. 2 for Cl−- and Br−-containing solutions as solid and dot-dashed curves, respectively. For comparison, we have plotted the data for the bare surface (without specifically adsorbing halides in the solution) as dotted lines. As expected, the surface charge density increases with potential and the higher the adsorption strength of the anion, the larger the surface charge density.
 | ||
| Fig. 2 Cyclic voltammograms (a) and surface charge density vs. potential plots (b) for nominally flat Au(001) electrodes in 9 mM KClO4 + 1 mM X, X = KCl (solid) and X = KBr (dot-dashed). For comparison, data for Au(001) in pure 10 mM KClO4 are plotted as dotted lines (“Bare”) in both graphs. | ||
As demonstrated in ref. 29, capacitance curves on flat Au(001) in pure perchloric acid reveal only minor shifts compared to the respective capacitance of stepped Au(11n). Since we deal with electrolytes where the anion strongly specifically adsorbs onto the Au electrode we expect no considerable influence of the step density on the surface charge density. We therefore conclude that the experimental results for the surface charge density obtained on flat Au(001) are appropriate to determine the kink dipole moment from step fluctuation data as measured on stepped Au electrodes.
Fig. 3 shows two examples of G(y) as measured for Cl− and Br− containing electrolytes at different electrode potentials. For long distances |y − y0|, G(y) is linear. For short distances, the correlation function is obviously curved. This curvature is due to the remaining time contribution to the correlation function. The time contribution originates from the finite scanning speed of the STM tip compared to kink diffusion on the surface. The fast kink movement becomes obvious in the frizzy appearance of the steps30,31 in the STM images in Fig. 1, in particular at high potentials where diffusion is fast. The time contribution in STM images and to the spatial correlation function has been extensively considered in ref. 1, 22 and 30 and is not further discussed here. We emphasize though that G(y) as plotted in Fig. 3 must undergo careful inspection prior to evaluating the slope and the kink energy. In the studies reported here, we have analyzed G(y) only in the range of large distances |y − y0| where the correlation function is linear (solid lines in Fig. 3 are linear fits to G(y) as used for further analysis). From those linear fits one then directly (eqn (1)) obtains the kink energy (Fig. 4). The kink energies for the two electrolytes fall apparently on the same line when plotted vs. electrode potential (Fig. 4(a)). However, when plotted vs. surface charge density, the kink energy data obtained in the two electrolytes fall on different curves with apparently different slopes (Fig. 4(b)). According to eqn (2) the kink dipole moment is determined from the slopes of the linear fits in Fig. 4(b) (solid lines). We find
| μCl = (6.0 ± 0.7) × 10−3 eÅ |
| μBr = (10.1 ± 0.6) × 10−3 eÅ | (3) |
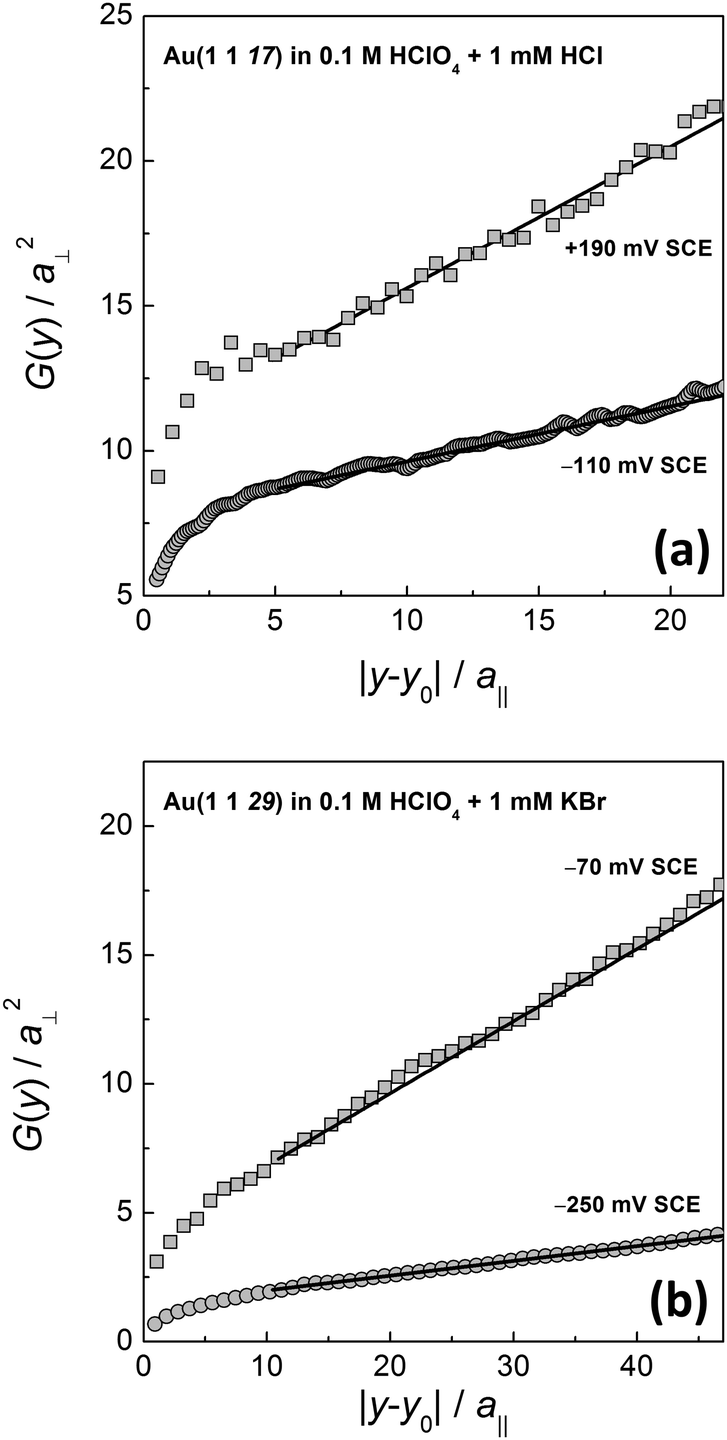 | ||
| Fig. 3 Spatial correlation function G(y) (eqn (1)) for (a) Cl−- and (b) Br−-containing electrolytes at different potentials. Solid lines are linear fits to the curves in the range of large distances (see the text for further discussion). | ||
 | ||
| Fig. 4 (a) Kink energy as determined from experimental data for G(y) (eqn (1)) similar to those presented in Fig. 3 for Cl−-(dark grey squares) and Br−-(light grey circles) containing electrolytes vs. electrode potential. (b) The same data plotted vs. surface charge density taken from Fig. 2(b). Solid lines are linear fits to the data (see the text for further discussion). Error bars are determined as the standard deviation from the mean for independent measurements at the same electrode potential. | ||
Experimental studies of the kink energy on Au(001) electrodes have been published previously for H2SO4 and HCl solutions.32–34 There, kink energies were determined from the curvature of island edges rather than by the analysis of the step correlation function. Fig. 5 shows the previous data (open symbols)32–34 in comparison with the new data reported here (grey symbols).
 | ||
| Fig. 5 Comparison of experimentally determined values for the kink energy on Au(100) as obtained previously from the analysis of the island edge curvature32,34 (open symbols) with the data as measured in this work from the analysis of the spatial correlation function (grey symbols). See the text for discussion. | ||
The data for sulfuric acid (open circles)32,34 are in excellent agreement with the results we obtained for Cl− (grey squares) and Br− (grey circles). The previous data for the HCl electrolyte,33 however, seem to strongly deviate from the results reported here. The reason for this deviation is not fully clear. We assume though that the deviation is typical for island perimeter curvature analysis in cases of limited database: the curvature is determined by calculating the second derivative of the perimeter radius with respect to the polar angle. Hence, low, nevertheless obvious noise in the island perimeter may lead to larger errors in the curvature. These uncertainties are reflected by the relatively large error bars of the previous data in HCl solution (open triangles in Fig. 5). In ultra-high vacuum experiments where the statistical database is generally much higher than in electrochemical studies, the noise in the perimeter of the island equilibrium shape is negligibly low. Here, the curvature analysis is a very reliable tool to measure the kink energy.35 In electrochemical studies, however, the statistical database is generally lower due to finite time where the electrolyte remains clean in the open STM cell. Here, the curvature analysis seems not to be an appropriate tool to obtain exact values of the kink energy, merely an order of magnitude can be determined. We conclude therefore that the more direct method of measuring spatial step fluctuations is superior to obtain precise values of the kink energy in electrolytes.
Several years ago, we measured a kink energy of 0.074 eV on Au(111) in 0.5 mM HCl at −0.1 V SCE using spatial correlation functions.36 This value is in excellent agreement with our data for Au(001) in HCl reported here. Effective medium calculations of kink energies on Au(001) and (111) surfaces in vacuum yield values of comparable size.37 This means that at low potentials where the specific adsorption of halide anions is low, the kink energy in an electrolyte is comparable to that in vacuum.
Theoretical values of kink energies in electrolytes were published by Pötting et al. for Ag(100).11 Using the embedded atom method, these authors calculated the kink energy as a function of the double-layer field. They showed that the kink energy increases with decreasing electric field (i.e. with decreasing potential) in accordance with our results for Au(001). Furthermore, their kink energy of 0.095 eV for a double-layer field of 1 × 1010 V m−1 is of comparable order of magnitude to our findings on Au(001).
Our data for the kink dipole moment determined from the step correlation function as reported here may be compared with previous data on defect dipole moments available so far: several years ago our group presented experimental data on step dipole moments on vicinal Au(11n) and Ag(11n) electrodes in various electrolytes.13,14 We found  and
and  for stepped Au(001) in SO42−, ClO4− and F−, respectively. These values are of comparable order of magnitude to what we find for Au kinks on (001) with co-adsorbed Cl− and Br−, though slightly larger – in particular in the case of Br−. This is reasonable since the adsorption site at a kink is electronically quite similar to step sites. Furthermore, previous studies13,14 considered steps with an equilibrium concentration of kinks. Hence, the values previously reported for steps are not for entirely straight steps but include the contribution of the equilibrium number of kink sites.
for stepped Au(001) in SO42−, ClO4− and F−, respectively. These values are of comparable order of magnitude to what we find for Au kinks on (001) with co-adsorbed Cl− and Br−, though slightly larger – in particular in the case of Br−. This is reasonable since the adsorption site at a kink is electronically quite similar to step sites. Furthermore, previous studies13,14 considered steps with an equilibrium concentration of kinks. Hence, the values previously reported for steps are not for entirely straight steps but include the contribution of the equilibrium number of kink sites.
Tansel and Magnussen recently published data on the dipole moment of hopping Cl adatoms adsorbed on surface sites on Cu(100).10 They found a dipole moment of μClCu(100) = 0.081 eÅ, a value about an order of magnitude larger than our results for Au(001) kink sites with co-adsorbed chloride. The difference between our values for the kink dipole moments and their result for the dipole moment of a free surface adatom is reasonable. The charge redistribution is expected to be larger for freely migrating surface defects compared to step adatoms or kinks. Hence, the dipole moment of free defects should be larger than that of strongly bound defects. A similar trend was found in embedded atom calculations for Ag(001) where the dipole moment increases when the number of broken bonds of a defect site increases.11
The contribution of co-adsorbed halides to the kink dipole moment should be large. In general, co-adsorbed halide anions retain a small negative charge that reduces the dipole moments of defects: as discussed in detail in ref. 6, the work function of a surface is reduced when dipole moments are oriented with their positive end towards the electrolyte. This situation is found for defects such as adatoms, kinks, steps and adatom islands on metal surfaces. Partially negatively charged ad-ions reduce the dipole moment of a defect. This is in accordance with the observation of a lower adatom dipole moment of a chloride covered adatom on Cu(100) of μClCu(100) = 0.081 eÅ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 compared to μCu(100) = 0.195 eÅ for an adsorbate-free adatom on Cu(001).6,8 We therefore expect that the dipole moments as found for kinks on Au(100) in chloride and bromide containing electrolytes are smaller than kink dipole moments in vacuum. Unfortunately, no further experimental or theoretical data on kink dipole moments exist to the best of our knowledge and cannot be compared here.
10 compared to μCu(100) = 0.195 eÅ for an adsorbate-free adatom on Cu(001).6,8 We therefore expect that the dipole moments as found for kinks on Au(100) in chloride and bromide containing electrolytes are smaller than kink dipole moments in vacuum. Unfortunately, no further experimental or theoretical data on kink dipole moments exist to the best of our knowledge and cannot be compared here.
The various data available on dipole moments seem to follow a general trend: free metal adatoms in an excited transition state are expected to have a considerably larger dipole moment than a defect in a more strongly bound state or in the ground state. Here, more quantitative analyses would be of great help. In order to understand transport, migration and degradation processes of electrode materials in more detail in future more experimental and in particular theoretical data on defect dipole moments are desirable.
Conclusions
We presented for the first time experimental data on the dipole moment of kinks upon co-adsorption of Cl− and Br− by measuring the potential dependence of the kink energy and the surface charge. We find kink dipole moments, μCl = (6.0 ± 0.7) × 10−3 eÅ and μBr = (10.1 ± 0.6) × 10−3 eÅ, respectively. Here the indices ‘Cl’ and ‘Br’ indicate that the kink dipole moment is that of an Au kink with co-adsorbed chloride and bromide rather than that of an uncovered Au kink atom.As previously demonstrated by our group,6,7 dipole moments of defects play a significant role in surface transport on metal electrodes in an electrolyte. This is because defects with their associated dipole moments form and migrate within the strong electric field at the solid/liquid interface. Based on the method described here, one would be able to gather the dipole moment of kink sites at steps for a large number of systems since the method is in principle applicable to all electrode–electrolyte systems. Therefore, the presented method could help to acquire quantitative information on migration phenomena at the solid/liquid interface.
Acknowledgements
We acknowledge the skillful sample preparation by Udo Linke and Claudia Steufmehl ICS-7, FZ Jülich. Furthermore, we are grateful for helpful discussions with Wolfgang Schmickler, Ulm and Harald Ibach, PGI-3, FZ Jülich. Financial support provided by the Deutsche Forschungsgemeinschaft and by the Fond der Chemischen Industrie is greatly appreciated.Notes and references
- M. Giesen, Prog. Surf. Sci., 2001, 68, 1 CrossRef CAS.
- M. R. Vogt, A. Lachenwitzer, O. M. Magnussen and R. J. Behm, Surf. Sci., 1998, 399, 49–69 CrossRef CAS.
- O. M. Magnussen and M. R. Vogt, Phys. Rev. Lett., 2000, 84, 357 CrossRef.
- C. Punckt, M. A. Pope, J. Liu, Y. Lin and I. A. Aksay, Electroanalysis, 2010, 22, 2834–2841 CrossRef CAS.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085–5092 CrossRef CAS.
- M. Giesen, G. Beltramo, S. Dieluweit, J. Muller, H. Ibach and W. Schmickler, Surf. Sci., 2005, 595, 127–137 CrossRef CAS.
- E. Pichardo-Pedrero, G. Beltramo and M. Giesen, Appl. Phys., 2007, A87, 461–467 Search PubMed.
- J. E. Müller and H. Ibach, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 085408 CrossRef.
- N. Hirai, K. Watanabe and S. Hara, Surf. Sci., 2001, 493, 568 CrossRef CAS.
- T. Tansel and O. M. Magnussen, Phys. Rev. Lett., 2006, 96, 026101 CrossRef CAS PubMed.
- K. Pötting, N. B. Luque, P. M. Quaino, H. Ibach and W. Schmickler, Electrochim. Acta, 2009, 54, 4494–4500 CrossRef.
- E. Cockayne and B. P. Burton, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 144116 CrossRef.
- G. L. Beltramo, H. Ibach and M. Giesen, Surf. Sci., 2007, 601, 1876–1885 CrossRef CAS.
- G. L. Beltramo, H. Ibach, U. Linke and M. Giesen, Electrochim. Acta, 2008, 53, 6818–6823 CrossRef CAS.
- Y.-C. Yang and O. M. Magnussen, Phys. Chem. Chem. Phys., 2013, 15, 12480–12487 RSC.
- S. Baier and M. Giesen, Phys. Chem. Chem. Phys., 2000, 2, 3675 RSC.
- M. Giesen and S. Baier, J. Phys.: Condens. Matter, 2001, 13, 5009 CrossRef CAS.
- D. M. Kolb, Prog. Surf. Sci., 1996, 51, 109–173 CrossRef CAS.
- H.-W. Fink, IBM J. Res. Dev., 1986, 30, 460–465 CrossRef CAS.
- V. T. Binh, J. Microsc., 1988, 152, 355–361 CrossRef.
- A. J. Melmed, J. Vac. Sci. Technol., B, 1991, 9, 601–608 CAS.
- M. Poensgen, J. F. Wolf, J. Frohn, M. Giesen and H. Ibach, Surf. Sci., 1992, 274, 430–440 CrossRef CAS.
- J. Clavilier, D. Armand, S. G. Sun and M. Petit, J. Electroanal. Chem., 1986, 205, 267–277 CrossRef CAS.
- A. Hamelin, J. Electroanal. Chem., 1995, 386, 1–10 CrossRef.
- W. Schmickler, Interfacial Electrochemistry, Oxford University Press, New York, 1996 Search PubMed.
- G. Beltramo and E. Santos, J. Electroanal. Chem., 2003, 556, 127–136 CrossRef CAS.
- H.-C. Jeong and E. D. Williams, Surf. Sci. Rep., 1999, 34, 171 CrossRef CAS.
- M. Moiseeva, E. Pichardo-Pedrero, G. Beltramo, H. Ibach and M. Giesen, Surf. Sci., 2009, 603, 670–675 CrossRef CAS.
- G. Beltramo, M. Giesen and H. Ibach, Electrochim. Acta, 2009, 54, 4305 CrossRef CAS.
- M. Giesen, J. Frohn, M. Poensgen, J. F. Wolf and H. Ibach, J. Vac. Sci. Technol., 1992, A10, 2597 CrossRef.
- J. F. Wolf, B. Vicenzi and H. Ibach, Surf. Sci., 1991, 249, 233 CrossRef CAS.
- S. Dieluweit, H. Ibach and M. Giesen, Faraday Discuss., 2002, 121, 27 RSC.
- E. Pichardo-Pedrero and M. Giesen, Electrochim. Acta, 2007, 52, 5659–5668 CrossRef CAS.
- S. Dieluweit and M. Giesen, J. Electroanal. Chem., 2002, 524–525, 194 CrossRef CAS.
- M. Giesen, C. Steimer and H. Ibach, Surf. Sci., 2001, 471, 80–100 CrossRef CAS.
- M. Giesen and D. M. Kolb, Surf. Sci., 2000, 468, 149–164 CrossRef CAS.
- P. Stoltze, J. Phys.: Condens. Matter, 1994, 6, 9495 CrossRef.
| This journal is © the Owner Societies 2014 |