 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence spectroscopy of individual semiconductor nanoparticles in different ethylene glycols
Sandra
Flessau
,
Christopher
Wolter
,
Elmar
Pöselt
,
Elvira
Kröger
,
Alf
Mews
and
Tobias
Kipp
*
Institute of Physical Chemistry, University of Hamburg, Grindelallee 117, 20146 Hamburg, Germany. E-mail: kipp@chemie.uni-hamburg.de
First published on 15th April 2014
Abstract
The optical properties of single colloidal semiconductor nanoparticles (NPs) are considerably influenced by the direct environment of the NPs. Here, the influence of different liquid and solid glycol matrices on CdSe-based NPs is investigated. Since the fluorescence of individual NPs varies from one NP to another, it is highly desirable to study the very same individual NPs in different matrices. This was accomplished by immobilizing NPs in a liquid cell sample holder or in microfluidic devices. The samples have been investigated by space-resolved wide-field fluorescence microscopy and energy- and time-resolved confocal scanning fluorescence microscopy with respect to fluorescence intensities, emission energies, blinking behavior, and fluorescence decay dynamics of individual NPs. During the measurements the NPs were exposed to air, to liquid ethylene glycols H(OCH2CH2)nOH (also called EGn) with different chain lengths (1 ≤ n ≤ 7), to liquid 2-methylpentane-2,3-diol, or to solid polyethylene oxide. It was found that EG6–7 (also known as PEG 300) is very well suited as a liquid matrix or solvent for experiments that correlate chemical and physical modifications of the surface and of the immediate environment of individual NPs to their fluorescence properties since it leads to intense and stable fluorescence emission of the NPs.
1 Introduction
Colloidal II–VI semiconductor nanoparticles (NPs) are crystalline fragments of the corresponding bulk material with sizes of 1–10 nm. In this range, the surface-to-volume ratio is considerably large, so that for a CdSe NP with a diameter of 3 nm, already one-third of its atoms are at the surface. Electronic surface states influence charge carriers which are three-dimensionally confined within this volume and thus are important in determining many of the NP fluorescence properties. Therefore, the environment, which is naturally located directly adjacent to the surface, plays a crucial role as well.One of the main applications of NPs is their use as bio-compatible fluorescent markers1–3 with high optical stability in medical applications. On the one hand, NPs can be used to track cellular transport processes by measuring the fluorescence with spatial resolution.4 On the other hand, the analysis of their fluorescence properties can be simultaneously used to probe their local environment, e.g., the pH value,5,6 or to show the presence of certain molecules,7 using fluorescence resonance energy transfer8 or charge transfer processes.9–11 The interactions of NPs at the cellular level and with biochemical substances are very complex and by far not fully understood. To utilize NPs for biomedical tasks, modifying them via colloidal chemistry is of great importance. In this context, poly(ethylene glycol) (PEG)-based ligands are of special interest12,13 as they render NPs hydrophilic, are nontoxic, minimize unspecific interactions,14 and preserve the fluorescence of the NPs.15,16 The molecule endings of ethylene glycols are nearly arbitrarily functionalizable and therefore offer various opportunities to further process these systems, on the one hand by tailoring the bonds between NPs and ligands to achieve well-defined coatings, and on the other hand by making use of the outward-positioned terminal groups that determine chemical interactions with the environment. For example, complex hybrids of fluorescent semiconductor NPs and other nanostructured components like magnetic iron nanoparticles or plasmonic gold nanoparticles were developed to enable bimodal probing.17–20 Detailed knowledge of the influence of ligand shells on the fluorescence properties of the NPs is crucial, first, to optimize the encapsulation, and second, to be able to separate the influence of the biological environment from the impact of the ligand shell itself.
Despite highly developed and well controllable synthesis routines, it is still not possible to obtain perfectly monodisperse, identical NPs in solution. Each individual NP emits in a relative small wavelength range, but due to different sizes and consequently different quantization energies they do not luminesce with exactly the same energy so that ensemble spectra are broadened to a Gaussian shape. Also, it is difficult to determine an absolute intensity since single semiconductor NPs show fluorescence blinking.21,22 These intensity fluctuations are accompanied by fluctuating fluorescence lifetimes that arise from fluctuating non-radiative decay channels, which lead to a multi-exponential decay even for single NPs.23
Since characteristic features of individual NPs are masked through ensemble averaging, single-particle experiments are crucial to unravel the fundamental interactions between the environment and the NP.24,25 There are elaborate investigations on, e.g., the influence of dielectric properties of surrounding polymers26 and of different ligands27 on the fluorescence intermittency. Such examinations are mainly based on a set of different single NPs. However, for even deeper investigations of the impact of different environments on the optical properties of NPs, it is actually necessary to follow the change in the fluorescence behavior of one and the same particle before and after a change of the environment.
Time-resolved confocal single-nanocrystal fluorescence spectroscopy is a powerful tool to explore the details of photophysical changes when individual CdSe NPs are, e.g., exposed to different gases like oxygen and nitrogen28 or when they are electrochemically charged.29 The investigation of individual NPs in their native situation, in which they are diffusing in solution, is experimentally challenging. One can approximate this situation by immobilizing NPs in microfluidic devices to study the fluorescence behavior of the same individual nanocrystal under systematic manipulation.30 For instance, the fluorescence intensity and blinking time traces have been observed via wide-field fluorescence microscopy to quantify the presence of β-mercaptoethanol31 and via total internal reflection fluorescence microscopy to detect maltose32 and potassium permanganate.33 In another study, confocal time-tagged time-resolved (TTTR) methods are used to correlate fluorescence intensity time traces with fluorescence lifetime trajectories to compare radiative and non-radiative decay rates under the influence of propyl gallate.34 To date, no survey has combined both spectral and time-resolved information of one and the same NP in different environments.
This paper provides extensive investigations at a fundamental level of the basic influence of ethylene glycols on the fluorescence properties of semiconductor NPs. The aim of this study is to correlate chemical and physical modifications of the surface and of the immediate environment of individual CdSe-based NPs to their optical properties in order to gain deeper insight into the interactions that are occurring and to better understand the underlying physical processes. In particular, the impact of liquid and solid ethylene glycol oligomers with different chain lengths and of chemically similar 2-methylpentane-2,4-diol is investigated with respect to fluorescence intensities, emission energies, blinking behavior, and fluorescence decay times. Measurement techniques are space-resolved wide-field fluorescence microscopy and energy- or time-resolved confocal scanning fluorescence microscopy. The work can be divided into three parts, with each part representing a certain kind of experiment. The first experiment is about the measurement of the very same individual NPs, once in air and once surrounded by a glycol matrix. For this purpose, the NPs were immobilized on the bottom of a liquid cell and were investigated before and after the cell was filled with a liquid glycol matrix or polymer solution. The second experiment is about the measurement of the very same NPs subsequently covered by different glycols. Here, the NPs were immobilized within microfluidic channels, which were fabricated by soft-lithography and enabled the exchange of the liquid glycols during the optical measurements. The third experiment is about the changes of the optical properties of the very same NPs upon mixing different glycol liquids. These measurements were conducted using again the liquid cell as the sample holder.
2 Experimental
2.1 Experimental setup
The optical measurements were performed using a homebuilt inverted confocal scanning microscope setup. For excitation, a 485 nm diode laser (PDL800-D, LDH-D-C-485, PicoQuant GmbH) was used. For confocal measurements, the laser beam was focused by a 100× oil objective with 1.25 NA (Zeiss Achroplan) onto the sample. For wide-field fluorescence imaging, a widening lens was inserted into the beam path in front of the objective. Confocal scanning measurements and detailed confocal investigations of single particles were performed with the pulsed laser (repetition rate of 5 MHz) having powers of 1.27 μW and 45 nW, respectively, measured in front of the objective. Wide-field measurements were conducted with the laser in its continuous wave mode. The sample was positioned and scanned using a computer-controlled piezo stage. Light from the sample was collected by the objective and scattered laser light was filtered out using a longpass filter with 522 nm edge wavelength (FF01-515/LP-25, Semrock). Fluorescence light was then guided either to an avalanche photo diode (PDM Series, Micro Photon Devices) for scanned images, fluorescence lifetime measurements, and blinking time traces (time-tagged time-resolved (TTTR) measurements with PicoHarp 300, PicoQuant GmbH), or to a spectrograph (Acton SP2500, Princeton Instruments) equipped with a charge-coupled device (CCD) camera (ProEm 512B, Princeton Instruments) for spectra or for wide-field images.Two of the three experiments presented here involved a liquid cell (CoverslipHolder from JPK Instruments) whose bottom was formed by a glass slide on which NPs were immobilized. The other experiment involved microfluidic devices. They consisted of a glass slide with immobilized NPs on top, covered by a polydimethylsiloxane (PDMS) chip that exhibited a channel structure prepared by soft lithography techniques. Both ends of the channel were attached to polyethylene (PE) tubes. For the experiment reported here, a syringe pump (NE500 Programmable OEM Syringe Pump, New Era Pump Systems) filled the inlet tube and the channel with different solutions that were separated from each other by gaseous nitrogen sections as follows: one measurement cycle started with injection of the respective chemical into the channel. After taking a confocal scan to locate the NCs, they were investigated in terms of single-particle spectra, fluorescence lifetimes and blinking time traces. Then, a nitrogen bubble extruded the first solution which was subsequently replaced by the second one to enter the second measurement cycle. Nitrogen was used to minimize the effect of aerial oxygen within a measurement series. Three cycles were conducted. During confocal scans and single-particle measurements, the syringe pump was switched off to avoid vibrations. This also means that the flow velocity was set to zero.
2.2 Nanoparticle samples
The samples used for the first two experiments presented here were semiconductor nanoparticles with a spherical CdSe core and an elongated CdS shell, so-called DotRods, which were synthesized following the seeded-growth approach by Carbone and coworkers.35 These nanoparticles are labeled as DRs in the following. For the third experiment, spherical CdSe-core–CdS shell nanocrystals were used that were synthesized by common methods.36,37 They are labeled as CS NCs throughout this work. Both kinds of NPs were capped with trioctylphosphine (TOP) and trioctylphosphine oxide (TOPO) ligands, and were dispersed in chloroform.Fig. 1a and b show typical transmission electron microscope (TEM) (JEM-1011 by JEOL) images of the elongated DRs and the spherical CS NCs, respectively. The diameter distributions, shown in Fig. 1c and d as derived from the corresponding TEM image, correspond to narrow Gaussian distributions with mean values in the range of about 4–5 nm. The small relative standard deviations of <18% in the case of DRs and <15% for CS NCs prove the high quality of the samples. Fig. 1e and f show emission spectra (red lines) measured with an excitation wavelength of 460 nm using a FluoroMax-4 spectrophotometer (Horiba Jobin Yvon), as well as absorption spectra (black lines) measured using a Cary 5000 UV-Vis spectrophotometer (Varian). The ensemble center emission wavelengths are approximately at 600 nm, i.e., they luminesce orange-red. From the absorption and emission spectra, quantum yields are calculated in relation to the dye Rhodamine 6G.38 Quantum yields are measures of the number of emitted photons per number of absorbed photons and are QYDRs = 75% and QYCSNCs = 16%. The DRs show strong and stable fluorescence and thus are suitable for extensive single-particle investigations. The thinness of the CS NC shell is responsible for the relatively low QY and is expected to render the NCs sensible against the surroundings.25 On the one hand, a high optical quality of the sample is desirable to ease measurement procedures, but on the other hand, this insensibility makes the detection of the environmental influence difficult, such that the choice of the sample is always a compromise.
 | ||
| Fig. 1 (a and b) TEM images of the DR and CS NC nanoparticle samples. (c and d) Diameter (short axis in the case of DRs) distributions (red) evaluated from (a and b). Black curves are fitted Gaussian distributions to determine the mean diameters which are specified below the sample name in the diagrams. (e and f) UV/vis spectra of the DRs and CS NCs. The mean emission wavelengths are specified in red ink. | ||
2.3 Glycol matrices
Table 1 lists the different glycols to which the NPs were exposed during our studies. The color coding accords with that used in all diagrams throughout this paper. Primarily, ethylene glycols (EGs) with different chain lengths from one to seven ethylene units were used. At room temperature, they are liquid with viscosities of 18.8 mPa s to 95 mPa s. Their refractive indices have been specified to be between 1.431 and 1.463, while their relative static dielectric constants range from 41.3 to 19.2.39,40 In addition, a solid polymer matrix consisting of long-chained poly(ethylene glycols) (for which the conventional nomenclature is poly(ethylene oxide) (PEO)) with an average molecular weight of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and a refractive index of 1.4539 was applied. For comparison, 2-methylpentane-2,4-diol (MPD, C6H14O2) that has a similar chemical composition as triethylene glycol (C6H14O4) but a different geometry was employed. Monoethylene glycol and di(ethylene glycol) were purchased from Merck, and 2-methyl-2,4-pentanediol, tri(ethylene glycol), poly(ethylene glycol) 200, poly(ethylene glycol) 300 and poly(ethylene oxide) 100000 were purchased from Sigma-Aldrich.
000 and a refractive index of 1.4539 was applied. For comparison, 2-methylpentane-2,4-diol (MPD, C6H14O2) that has a similar chemical composition as triethylene glycol (C6H14O4) but a different geometry was employed. Monoethylene glycol and di(ethylene glycol) were purchased from Merck, and 2-methyl-2,4-pentanediol, tri(ethylene glycol), poly(ethylene glycol) 200, poly(ethylene glycol) 300 and poly(ethylene oxide) 100000 were purchased from Sigma-Aldrich.

|
3 Results and discussion
3.1 The very same NPs in air and different glycols
In a first experiment we investigated the influence of different glycols on the optical properties of individual DRs by comparing the fluorescence intensities of one and the same DRs surrounded by air and by a particular glycol.For these investigations, diluted solutions of the NPs were spin-coated on glass slides such that the area density of NPs is low enough to allow for single-particle spectroscopy. These glass slides were then inserted into the liquid cell and served as its bottom. Fig. 2 shows wide-field fluorescence image sections of different samples. Each panel a to g compares one and the same sample region exposed to air and subsequently to one of the glycols listed in Table 1. In the case of liquid glycols (a–f), in each measurement, a drop of 30 μl was dropped into the liquid cell. Panel g belongs to a measurement in which the NPs were covered by a solid glycol matrix. Here, a 0.05 wt% solution of PEO 100000 in a 2:1 toluene–chloroform mixture was dropped on the glass slide and was allowed to dry before the second wide-field image was taken. In all these measurements, the exposure time of the CCD camera was set to 0.3 s and the excitation intensity applied to the sample was kept constant. The intensity color scaling is same in all images.
 | ||
| Fig. 2 Wide-field fluorescence image sections of DRs that are exposed to (a) EG1, (b) EG2, (c) EG3, (d) EG4, (e) EG6–7, (f) MPD, and (g) PEO. Length and intensity scales are valid for all images. | ||
The bright spots in the wide-field fluorescence images indicate the fluorescence of DotRods. Simply the visual inspection of Fig. 2 already gives some valuable information about the influence of the different glycols. First of all, one can state that for liquid matrices, i.e., in panels a–f, the number of emitting DRs is larger in the first measurement in air than in the second measurement in a glycol. Also the intensity of the emission seems to be generally larger in air than in the glycol. The situation is changed for the solid glycol, as shown in panel g, where both the number of emitting DRs and the intensity are more balanced. Comparing the wide-field images for different glycols in Fig. 2 one can conclude that the number of emitting DRs as well as their emission intensity are by trend increasing with increasing chain lengths of the EGs.
For a more detailed analysis of the wide-field fluorescence images in Fig. 2, for each bright spot in every image the intensity of its brightest pixel was read out. Then, for each DR the ratio of the intensity in medium to the intensity in air was determined. Histograms of the intensity ratios are shown in Fig. 3. Here, the occurrences have been normalized, i.e., they have been divided by the total number of investigated DRs. The bars representing intmed/intair ≤ 0 correspond to the situation that the DR was emitting in air but was not found back when covered by a glycol. All occurrences of ratios intmed/intair > 2 are summarized in the bar at the right-hand side of each histogram marked by the dashed area.
 | ||
| Fig. 3 Distribution of fluorescence intensity development of DRs in medium compared to the fluorescence intensity of the same DR in air. | ||
The histograms support the trends that were already identified by visual inspection of Fig. 2. The number of DRs that emit in air but are dark when covered by a glycol (intmed/intair ≤ 0) is generally decreasing with increasing chain length of the EGs (only EG1 is slightly out of the ordinary). Accordingly, there is a significant number of DRs emitting more intense in a matrix than in air only for EG4 and longer EGs. Furthermore, also the center of mass of histograms with 0 < intmed/intair ≤ 2 generally seems to shift from left to right with increasing chain length (again EG1 is slightly out of the ordinary). The histogram for MPD nicely fits between the histograms for EG3 and EG4. This result is in line with the fact that MPD resembles EG3 in chemical composition.
From the experiments above it is obvious that the nature of the surrounding matrix has an impact on the emission intensity of the DRs. In order to investigate the influence of electrodynamic effects, i.e., the change of the dielectric surrounding of the individual DR emitters, on the measured emission intensity, we performed two-dimensional finite-difference time-domain (FDTD) simulations. We modeled both the excitation and emission processes of a NP lying on a glass surface covered by media of different refractive indices. These simulations were done using the software Lumerical FDTD Solutions, which solves Maxwell's equations to calculate the time evolution of electromagnetic fields.
The wide-field laser excitation was simulated by a plane wave of a fixed amplitude impinging from below on a glass–medium interface. We analyzed the electromagnetic field intensities at the interface for media with different refractive indices n, in particular for the glycols (with n in the range of 1.431 to 1.463) and air (n = 1). We found that the higher the refractive index of the medium is the smaller is the excitation intensity. Applying the values for the glycols used in this study, we obtained excitation intensities in glycol matrices of only about 70% as in air. In order to simulate the emission process, we assumed a point dipole positioned at the glass–medium interface with its dipole axis lying within the glass surface plane. Fig. 4 shows the field distributions of a point dipole with its axis perpendicular to the image plane, located on a glass slide (n = 1.5255) either in (a) air (n = 1) or (b) a medium with n = 1.463 (EG6–7). The figure demonstrates that the emission of a dipole into the half-space with a high refractive index is enhanced when the other half-space has a lower refractive index. The collection cone of the microscope objective is denoted by the dashed lines in the glass half-space. We analyzed the emission and subsequent collection of light for a dipole surrounded by media of different refractive indices. For values of the employed glycols we found the collected intensity to be only about 70% as compared to the case of air.
 | ||
| Fig. 4 FDTD simulation of the emission process of a dipole on a glass substrate surrounded by either (a) air (n = 1) or (b) EG6–7 (n = 1.463). The color scale encodes the field intensity. Diagonal lines in the glass half-space represent the collection cone of the microscope objective. | ||
Summarizing the FDTD simulations, considering just electrodynamics, an increase of the refractive index of the medium that surrounds a NP leads to a decrease of the measurable fluorescence intensity. On the one hand, comparing air and glycols, the simulations rationalize a decrease of the measured intensity of nanoparticles in glycols to values of about 50% of the intensity in air just because of the different refractive indices. This explains and correctly classifies, at least for the liquid glycols, the general result of the wide-field fluorescence-image analysis in Fig. 3 that the measured fluorescence intensity is on average considerably lower in the matrix than in air.
On the other hand, comparing the different glycols among each other with their slightly increasing refractive index with increasing chain length, the FDTD simulations would let one expect a slight monotonic decrease of the average intensity ratio intmed/intair from EG1 to EG6–7. Interestingly, this is in contrast to the measurements. Thus, the measured solvent-dependent intensities of the DRs cannot be explained by changes in excitation and emission efficiency, i.e., by electrodynamics, solely. Instead, it is proved that the solvents directly impact the optical properties in chemical or quantum mechanical ways. As an example of a chemical modification, oxygen that might be dissolved in the glycol matrices in different amounts influences the optical properties of the nanoparticles.28 To ascertain these interactions, more detailed investigations are demonstrated in the following.
3.2 The very same NPs under subsequent exchange of ethylene glycols
The first experiment dealt with the comparison of the intensity of individual NPs in air and in different glycols. From that, one can deduce the general result that long-chained EGs lead to more fluorescence light; however, the conclusions about the different impact of the different EGs on the DR fluorescence are limited. This is mainly because the covering glycol matrix was not changed from one to another for a particular DR during the experiment. Furthermore, the intensity alone is not the only measure that characterizes the fluorescence. It is well known that individual NPs exhibit a blinking behavior. This can on the one hand tamper the conclusions drawn from an experiment like the first one. On the other hand the blinking, if properly investigated, can be utilized to learn something about the interaction of the surrounding glycol with the NP emitter. Time-resolved measurements of the fluorescence decay dynamics can yield further information on the matrix–emitter interaction.Better but experimentally much more challenging than the first experiment would be the detailed investigation of individual DRs (including measurements of intensity time traces, spectra, and decay curves) that are subsequently covered by different liquid glycols. Such an experiment requires a very stable setup to have enough time to conduct all measurements within several matrices to gain statistics. Also the NPs themselves have to be stable enough without undergoing degradation during the measurements, for example induced by illumination.
We realized such an experiment using microfluidic devices. The NPs under investigation were again DRs because of their relative high optical long-term stability. A diluted solution of DRs was spin-coated on a glass slide. It was then covered by a PDMS microchannel chip that was attached to inlet and outlet tubes. In order to restrict the measuring time, only three liquid ethylene glycols were subsequently channeled above the NPs under investigation: first EG1, then EG4, and at last EG6–7.
Fig. 5a shows a confocal scan of a sample area within a microflow channel filled with EG1. The bright spots are (presumably) single DRs. Such scans were used to localize individual DRs for further confocal measurements. 14 of the bright spots were further investigated by recording their spectra and by recording spectrally integrated fluorescence time traces in the time-tagged time-resolved mode that also allows for determination of the fluorescence decay dynamics. Considering also the alignment, both kinds of measurements took at least 6 min for each DR under investigation. After all measurements in EG1 had been completed, the matrix was changed to EG4. Then a fluorescence image of the same sample area was taken, as shown in Fig. 5b. Several of the bright spots occurring in panel a can be found back. However, also some new spots have arisen and other spots are missing. This is mainly due to the blinking of nanoparticles which causes the nanoparticles to appear bright or dark depending on whether they are in an on-state or in an off-state during the recording of the confocal scan. In total 12 of the 14 spots investigated in EG1 could be found back in Fig. 5b and were also investigated with respect to their spectra, their lifetimes, and their blinking behavior. Again, such measurements took about 6 min for each DR. Finally, the matrix was changed to EG6–7 and again, a fluorescence image was scanned, which is depicted in Fig. 5c. Here, in total 6 of the 12 spots investigated in EG1 and EG4 could be relocated. These spots were again probed concerning their spectra, blinking, and lifetimes. After sorting out data sets that were either incomplete or arose most likely not from a single DR, the whole measuring process delivered a set of five individual DRs that have been each thoroughly investigated in three different liquid EGs. These five DRs are encircled and numbered in each of the fluorescence scans in Fig. 5. In the following we will discuss and compare the spectra, the blinking dynamics, and the fluorescence decay dynamics of these five DRs in the three different environments.
 | ||
| Fig. 5 Confocal scans of one and the same sample area that was subsequently exposed to (a) EG1, (b) EG4, and (c) EG6–7. | ||
Comparing Fig. 5a–c it is obvious that the total number of emitting DRs is decreasing with increasing duration of the experiment. This irreversible disappearance is most likely due to the chemical degradation of the respective nanoparticles. A washing-off should be inhibited since the TOPO ligands do not render the nanoparticles soluble in the glycols.
 | ||
| Fig. 6 Fluorescence spectra of DR No. 3 (cf.Fig. 5) in EG1, EG4 and EG6–7 with Lorentz fits. | ||
It is known that spectral diffusion occurs in the temporal evolution of the fluorescence emission of individual NPs.41 Spectral diffusion broadens the fluorescence spectra when it happens on time scales smaller than the integration time, i.e., the single-particle line shapes result from rapid spectral shifting of the emission peak.24 When spectral diffusion occurs on time scales larger than the integration time, it is visible as jumps of the whole spectrum. To estimate the spectral diffusion for our DRs, we performed a control experiment in which single DRs were embedded in a Zeonex matrix and the spectral diffusion was monitored over several minutes. Here, spontaneous jumps in the maximum emission wavelengths of up to 10 nm occurred. These jumps will mask any matrix-induced shifting of the fluorescence wavelength. In order to clearly observe such shiftings, they should be at least in the range of the above mentioned 10 nm.
A matrix-induced shifting, also called the solvatochromatic effect, is caused by polarization interactions:42 charge carriers in a NP, which are introduced through optical excitation, polarize the dielectric environment and form image charge distributions. These in turn generate electrostatic potentials at the positions of the charge carriers that perturb the energy of the confined exciton and shift the emission wavelength of the NP. The magnitude of the effect can be estimated quasi-analytically for spherical NPs in a dielectric medium without and with an additional capping ligand layer on the NP surface.42,43 If we for a moment assume spherical NPs without ligands, calculations reveal that a change of the matrix from EG1 to EG6–7 should lead to a blue shift of the emission of only about 3 nm, assuming static dielectric constants given in Section 2.3. A capping layer of TOPO ligands, as present in our experiments, however decreases the shift to only very small values far below 1 nm. Even though our experiments deal with DotRods in which the solvatochromatic effect should be larger than in spherical NPs because of the reduced symmetry,44–46 the above estimation leads one to assume also a rather small solvatochromatic shift for our system. This is in line with the absence of any experimental evidence in our measurements and we conclude that the shifting of the spectra shown in Fig. 6 is essentially due to large spectral diffusion.
As a final remark concerning the fluorescence spectra, despite their above mentioned weak dependence on changing EGs, they deliver valuable information during our study. In particular the width and the shape of an emission peak in a fluorescence spectrum can help to make sure that the nanostructure under investigation is indeed an individual DR and not an aggregation of two or more DRs.
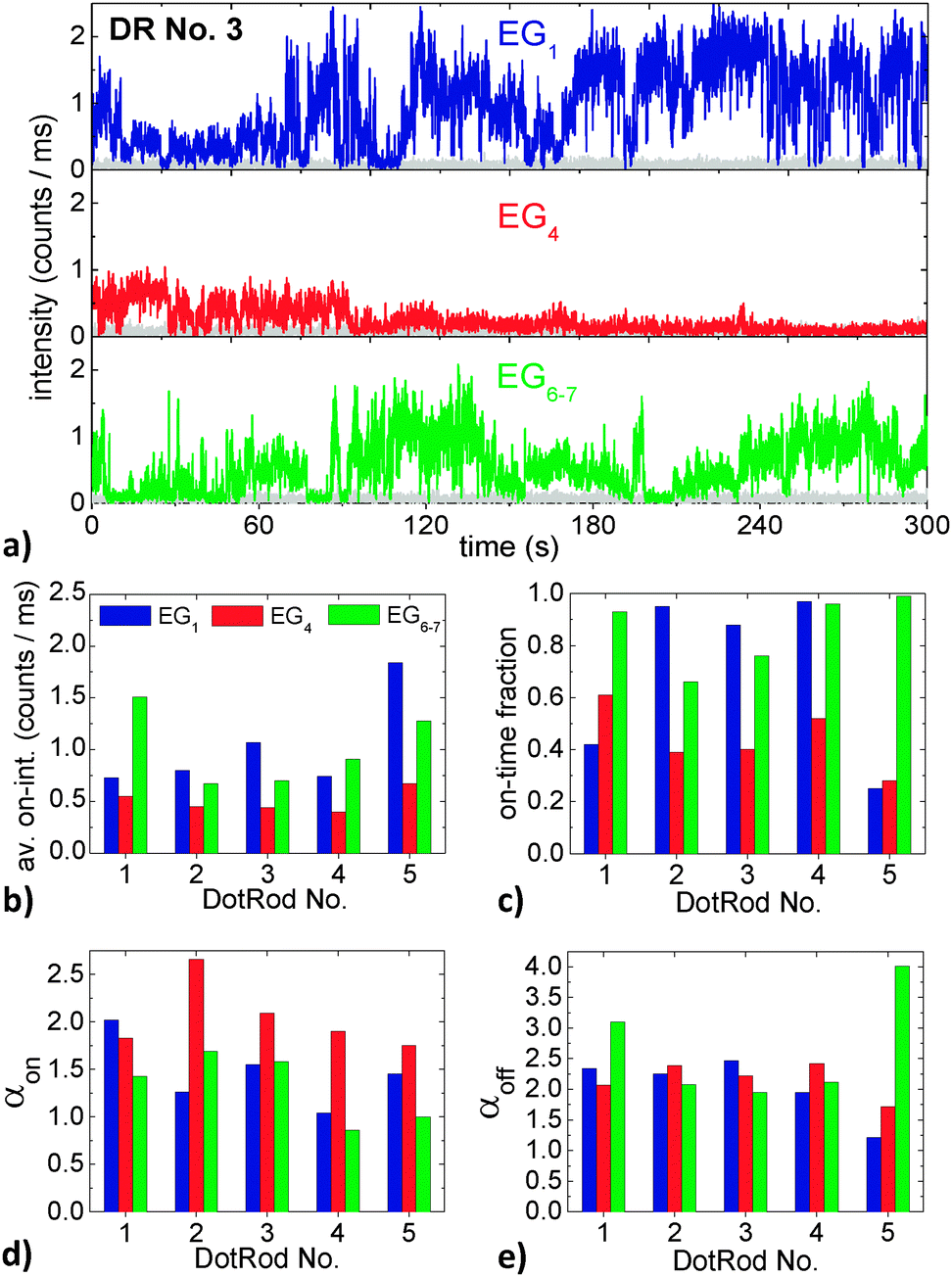 | ||
| Fig. 7 (a) Fluorescence intensity time trace of DR No. 3 in EG1, EG4 and EG6–7. The gray traces denote the background intensity. (b) Average on-intensity, (c) on-time fraction, (d) on-time power-law exponents, and (e) off-time power-law exponents of the five DRs marked in the confocal scans in Fig. 5 in EG1, EG4 and EG6–7. | ||
Regarding first the time trace of DotRod No. 3 in EG1 (Fig. 7a, top panel), it is obvious that the intensity is strongly fluctuating. The DR does not show a clear binary blinking with one well-defined on-state and another off-state. Instead, when in an on-state, various intensity values (up to about 2 counts per ms, i.e., 100 counts per time bin) have been measured. After having changed the matrix to EG4, the intensity time trace has drastically changed (Fig. 7a, center panel). The maximum intensity is much lower (<1 counts per ms) and the fluorescence seems to fade out with increasing time. Interestingly, the fluorescence returns after having changed the matrix to EG6–7 (Fig. 7a, bottom panel).
The strong fluctuations in the fluorescence intensity time traces complicate an exact analysis of the impact of the surrounding matrix on the optical properties of DRs. From Fig. 7a one might deduce that EG1 is the best glycol with respect to a high fluorescence yield, followed by EG6–7 and finally EG4. However, one has to keep in mind that the intensity time traces could have been significantly different for another temporal window. In order to find some measure of the individual time traces and to compare time traces of different DRs, we analyzed them following the method of Kuno et al.:47 after defining an intensity threshold that discriminates off-states from on-states, histograms of on-state durations and of off-state durations were generated. Each data point of a histogram was weighted by a value representing the average time to nearest neighbor events to account for the low event density at long times, yielding probability densities P(ton/off) for on- and off-times. It has been shown that both probability densities typically follow inverse power laws P(ton/off) ∝ (ton/off)−αon/off over several decades in probability and time. For our experimental data, we plotted P(ton) and P(toff) double-logarithmically against ton and toff and fitted the graphs by straight lines with their slopes being the power-law exponents αon and αoff, respectively. The results of the analysis depend to some degree on the choice of the binning time, the duration of the time trace and the threshold level.48 The intensity threshold was set to twice the average background value, as proposed by Kuno et al.,49 plus once the standard deviation of the background intensity distribution to take additionally the noise fluctuations into account. Having defined a threshold level it is also possible to define other measures of the blinking behavior, like the average on-intensity (which was calculated by averaging the intensity of every time bin that had fluorescence intensity above the threshold intensity) and the on-time fraction (which was calculated by the number of time bins that showed fluorescence intensity above the background intensity divided by the total number of time bins).
The above described measures of the blinking behavior of the five different DRs each within the three different liquid EGs are compiled in Fig. 7b–e. The average on-intensity (Fig. 7b) is always lowest in EG4, while the highest values occur sometimes for EG1 and sometimes for EG6–7. By trend, the smallest on-time fractions (Fig. 7c) occur again in EG4. This suggests a correlation between the average on-intensity and the on-time fraction; however such a correlation cannot unambiguously be deduced from the measurements in EG1 and EG6–7. The on-time power-law exponents (Fig. 7d) are, except for DR No. 1, highest in EG4, which suggests a correlation of αon with the average on-intensity and the on-time fraction. The off-time exponents (Fig. 7e) however are strongly varying and do not show distinct trends.
From the above discussion it is obvious that it is difficult to conclude unambiguous findings from the experiment. This is mainly because the statistics based on five different DRs is too poor. The comparatively small number of investigated DRs on the other hand is a consequence of the extremely difficult experiment with its particular requirements as discussed above. In this sense, our experiment can be seen as a proof of principle; it shows that it is indeed possible to investigate the blinking behavior of one and the same individual NPs in different matrices.
The discussion of the blinking behavior in dependence of the medium that is surrounding the emitter is particularly interesting, because it might help to finally understand the fundamental processes underlying the blinking. It is commonly assumed that the blinking occurs because of the changeover from the NP being neutral and charged. It is argued that after the photoexcitation of an electron–hole pair in a NP, one of the charge carriers gets trapped, leaving behind a delocalized excess charge inside the NP. This excess charge gives rise to the off-state of the NP because it quenches the radiative recombination of further photogenerated electron–hole pairs by a very fast non-radiative Auger process.22 Until now, the exact processes of charge trapping and the subsequent neutralization of the NP, returning it into an on-state, have not been fully understood. They are surely influenced by the material that is surrounding the NP, as can already be seen in the experiments presented here. In particular, the molecular dynamics at the NP surface is of interest, for which the viscosity is a measure. One might expect a systematic variation of the on- and off-time power-law exponents α with the viscosity of the surrounding medium, but further experiments are necessary.
 | ||
| Fig. 8 (a) Fluorescence decay curves of DR No. 3 in EG1, EG4, and EG6–7. (b) Stretched-exponential fluorescence lifetimes 〈τ〉 of the five DRs that are marked in the confocal scan in Fig. 5 in EG1, EG4, and EG6–7, respectively. (c) 1/e lifetime and stretching parameter resulting from the fits of the fluorescence decay curves from the five DRs in the different EGs. (d) Dependence of fluorescence intensity and lifetime of single DRs in the different EGs. The cipher depicts the number of the DR. | ||
Fig. 8b summarizes the measured decay curves by comparing the average lifetimes of the five different DRs each within the three different EGs. These lifetimes range between 1.6 ns and 16 ns. Comparing the different matrices, by trend, the shortest lifetimes occur in EG4. For each DR at least one of the lifetimes measured in EG1 or EG6–7 is significantly larger than in EG4. This behavior already suggests a correlation between short lifetimes and low average on-intensities (cf.Fig. 7b) as well as short on-times (cf.Fig. 7c).
Fig. 8c and d allow for a deeper analysis of the fluorescence decay dynamics. In Fig. 8c the fitting parameters β and τe of each decay curve are correlated. Fig. 8d connects the average intensity of the different DRs to the average lifetimes 〈τ〉 (as shown in Fig. 8b). In both panels, the color of the data points represents the particular EG while the cipher names the DR number.
Overall, it can be seen that larger β values correspond to larger 1/e lifetimes and that larger average intensities come along with longer average lifetimes. This is congruent with reports in the literature that found that high fluorescence intensities of nanocrystals come along with long fluorescence decay times,23 and that maximum fluorescence intensities yield nearly single-exponential decays.51 A more detailed interpretation of the different fluorescence dynamics of DRs in different EGs is difficult because of the small number of DRs that were investigated. For example, it would be very interesting to accurately investigate the influence of the matrices on the β parameter.
The analyses of the blinking behavior and of the fluorescence dynamics both are based on the same measurement, i.e., the photon-counting over a time period of 5 min using the TTTR mode. Both analyses revealed that the measurement of a larger number of NPs is highly desirable. This, however, is difficult to achieve since it requires a very stable system. Here, the stability of the system not only includes the experimental setup itself, but also the NPs under investigation. For example, it is not sufficient that the experimental setup is stable such that the optical pathways do not change over time and upon frequently changing the liquid matrix surrounding the NPs. It should also be ensured that the NPs under investigation are stable enough during matrix exchange and optical measurements, such that their optical properties are not permanently changed by fundamental and irreversible changes of the NPs themselves. Even though the accomplishment of such a stable experiment is challenging, our study shows the principle feasibility.
3.3 The very same NPs in different mixtures of ethylene glycols
The second experiment described above is the most sophisticated one in this work, which can principally deliver the deepest insight into the systems under investigation, but which is by far also the most difficult one. The third experiment described here is much simpler. It aims to investigate reversibility of changes in the optical properties of NPs upon exchange of their surrounding medium from one to another and back to the first. Such an experiment can be conducted within microfluidic channels, but here we chose an easier approach using again the liquid cell. The idea is to start with the investigation of NPs with the liquid cell being filled by the first liquid EG. Then a further glycol is added into the cell, such that a liquid mixture of different EGs covers the NPs during the next optical investigation. Finally, a large excess of the first EG is added to the cell, such that a situation similar to that in the beginning of the experiment is created before the final optical investigation is performed.Unlike in the first and second experiments, where DotRod samples were used that offer high quantum yields and a good stability against environmental influences, here, we used the simpler and less stable spherical core–shell nanocrystals (CS NCs) (see Section 2.2). As argued above, on the one hand, a high optical stability of the sample is desirable to ease long-lasting experiments. On the other hand, a high optical quality involves certain insensibility against the environment, which makes the detection of environmental influences themselves difficult. Thus, one generally has to deal with a trade-off between stability against the environment and changeability by the environment.
The CS NCs were immobilized on a glass slide that formed the bottom of the liquid cell. The cell was filled with 200 μl of EG1. The sample was then imaged by confocal scanning microscopy. The left column of Fig. 9a shows three scanned sample areas; the bright spots arise from CS NCs. Most of the NCs were stable enough to outlast recordings of their fluorescence emission over a time period of 30 s without obvious degradation. The fluorescence was measured by single-photon counting in TTTR mode; thus both intensity time traces and fluorescence decay curves could subsequently be constructed from the measurement. In total 28 individual CS NCs were investigated in such a way. For the next measurement cycle, 200 μl of EG6–7 was dropped into the liquid cell which then contained a 1:1 mixture of EG1 and EG6–7. After waiting for more than one hour so that the glycols could mix evenly upon diffusion, the sample was scanned again (see the center column of Fig. 9a). 26 of the 28 CS NCs investigated in the previous measurement cycle could be identified and were again measured by TTTR single-photon counting. For the third and last cycle, 150 μl of the EG mixture was removed from the liquid cell before 600 μl of EG1 was added. This resulted in a 5.8:1 mixture of EG1:EG6–7 in the liquid cell, in which the NCs were investigated again. The scanned images are shown in the right column of Fig. 9a. In total 13 of the CS NCs that were investigated in both previous cycles could be again identified and were subsequently again measured by time-correlated single-photon counting. These CS NCs are marked and numbered in the scanned images in Fig. 9a.
 | ||
| Fig. 9 (a) Confocal scans of the same areas of individual CS NCs which were subsequently exposed to pure EG1 (left), to a 1:1 mixture (center) and a 5.8:1 mixture of EG1 and EG6–7 (right). (b) Fluorescence intensity time traces (bin time 50 ms) and (c) fluorescence decay curves of CS NC No. 13 also marked in (a) in the three different mixing ratios of EGs. (d) Average on-intensities and (e) stretched-exponential fluorescence lifetimes of the 13 CS NCs marked in the confocal scans in (a) in the different mixtures of EG1 and EG6–7. | ||
Fig. 9b shows exemplarily intensity time traces of CS NC No. 13 in the three different EG mixtures recorded during the three measurement cycles (from top to bottom). Because of the short duration of only 30 s, the time traces delivered no sufficient statistics for an elaborate blinking analysis. However, the average on-intensity can be analyzed. Results are depicted in Fig. 9d. Only with one exception, the average on-intensities are always highest for the solution that contained the highest relative amount of EG6–7, i.e., during the second measurement cycle. We tested this result in an additional experiment in which we changed the order of EGs added, starting with pure EG6–7. The outcome was the same: the average on-intensities were high for pure EG6–7 and for EG6–7 in large excess. Remarkably, this observation is fully congruent with the findings of the first experiment described above.
Fig. 9c presents fluorescence decay curves of, again, CS NC No. 13 in the three different EG mixtures. The curves are clearly multi-exponential, like the decay curves discussed above. We again fitted each decay curve with a stretched-exponential function and calculated the average lifetime 〈τ〉. These lifetimes are compiled in Fig. 9e. It is found that 〈τ〉 in the second measurement cycle (EG1:EG6–7 = 1:1) is always larger than in the first cycle (pure EG1) and in most cases larger than in the third cycle (EG1:EG6–7 = 5.8:1). This finding, together with the finding on the average on-intensities, is again congruent with the above mentioned correlation between high fluorescence intensities and long lifetimes.23
4 Conclusions
In this work, we investigated the influence of glycols on the optical properties of CdSe-based nanoparticles. In particular, liquid and solid ethylene glycol oligomers with different chain lengths (EG1, EG2, EG3, EG4, EG6–7 and PEO) and 2-methylpentane-2,4-diol (MPD) were probed with respect to their impact on the fluorescence intensities, the emission energies, the blinking characteristics, and the fluorescence lifetimes of elongated DotRods (DRs) and spherical core–shell nanocrystals (CS NCs). The study concentrates on the examination of individual NPs that were immobilized either within a liquid cell or within microfluidic devices. This allowed us to follow changes in the optical properties of the very same NPs under the influence of different matrices using space-resolved wide-field fluorescence microscopy and energy- and time-resolved confocal scanning microscopy.Comparison of wide-field microscopy images of the very same individual DRs that were immobilized on a glass slide and exposed to air and subsequently to one of the above-mentioned matrices revealed that their fluorescence intensity generally increased with increasing chain length of the liquid EGs, with EG1 being an exception from this sequence. The measured average intensities were larger in air than in all investigated glycols, except for PEO. The solid PEO matrix led to by far the highest fluorescence intensities and long-term stabilities of the NPs.
To estimate the effect of a changed dielectric environment of individual NPs on their excitation and emission efficiency, electromagnetic finite-difference time-domain calculations were performed. They revealed that the measurable fluorescence intensity of an emitter decreases with the increasing refractive index n of the matrix covering the emitter. This finding is in line with the observed differences in intensity for liquid glycols (n = 1.431 − 1.463) as compared to air (n = 1). Interestingly, the slight monotonic increase of the refractive index of liquid ethylene glycols from EG1 to EG6–7 should in theory lead to a decrease of the fluorescence intensity from short-chained EGs to long-chained EGs, which is in contrast to the measurements. This discrepancy and also the high fluorescence intensity of NPs in solid PEO prove that other effects of the matrix impact the fluorescence of NPs. These effects are of chemical or quantum mechanical nature. They should be also affected by the molecular dynamics of the covering matrices, as suggested by the general trend of increasing fluorescence for glycol matrices with increasing viscosity, i.e., chain length, that culminates in the very stable and intense fluorescence for the solid PEO matrix.
For a better insight into the underlying processes, it is inevitable to investigate the very same NPs under exchange of the covering matrix and to correlate changes in the optical properties to physical and chemical changes of the NP surface and its environment. Such experiments are difficult to accomplish because they demand a high long-term stability of both the experimental setup and the NPs under investigation.
In this work, such an experiment was realized, in which individual NPs immobilized in a microfluidic channel were subsequently covered by EG1, EG4, and EG6–7. The optical properties of the very same NPs were extensively investigated in each matrix. It is shown that solvatochromatic effects resulting from polarization interactions are masked by spectral diffusion of the NP emission. However, time-tagged time-resolved measurements allowed for the analysis of fluorescence intensities, time traces, blinking characteristics, and fluorescence decay dynamics. It was found that EG6–7 is an excellent liquid glycol matrix promoting more intense and stable fluorescence than EG4. A further experiment on individual CS NCs in a liquid cell subsequently filled with pure EG1, with a 1:1 mixture of EG1 and EG6–7, and finally with a EG1:EG6–7 mixture with a large excess of EG1 demonstrated the principle reversibility of changes in the optical properties of NPs upon exchange of their surrounding matrix. Again it was found that the existence of EG6–7 inside the matrix covering a NP increases its fluorescence intensity.
For further measurements of this kind, the finding that EG6–7, i.e., polyethylene glycol 300, is a fluorescence-supporting medium for CdSe nanoparticles can be of great value. PEG 300 is an excellent choice of solvent when working with PEG-encapsulated NPs since it does not interfere with the NP capping being of the very same material. It may for example be used as a solvent for different quenching agents to investigate their interaction with NPs. Its high viscosity allows for long diffusion times, such that the quenching processes happen on longer time scales which eases their investigation.
References
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- W. R. Algar, K. Susumu, J. B. Delehanty and I. L. Medintz, Anal. Chem., 2011, 83, 8826–8837 CrossRef CAS PubMed.
- F. Pinaud, S. Clarke, A. Sittner and M. Dahan, Nat. Methods, 2010, 7, 275–285 CrossRef CAS PubMed.
- X. Ji, G. Palui, T. Avellini, H. B. Na, C. Yi, K. L. Knappenberger and H. Mattoussi, J. Am. Chem. Soc., 2012, 134, 6006–6017 CrossRef CAS PubMed.
- X.-d. Wang, J. A. Stolwijk, T. Lang, M. Sperber, R. J. Meier, J. Wegener and O. S. Wolfbeis, J. Am. Chem. Soc., 2012, 134, 17011–17014 CrossRef CAS PubMed.
- S. J. Clarke, C. A. Hollmann, Z. Zhang, D. Suffern, S. E. Bradforth, N. M. Dimitrijevic, W. G. Minarik and J. L. Nadeau, Nat. Mater., 2006, 5, 409–417 CrossRef CAS PubMed.
- R. C. Somers, M. G. Bawendi and D. G. Nocera, Chem. Soc. Rev., 2007, 36, 579–591 RSC.
- S. F. Wuister, C. de Mello Donegá and A. Meijerink, J. Phys. Chem. B, 2004, 108, 17393–17397 CrossRef CAS.
- J. Völker, X. Zhou, X. Ma, S. Flessau, H. Lin, M. Schmittel and A. Mews, Angew. Chem., Int. Ed., 2010, 49, 6865–6868 CrossRef PubMed.
- A. A. Cordones, M. Scheele, A. P. Alivisatos and S. R. Leone, J. Am. Chem. Soc., 2012, 134, 18366–18373 CrossRef CAS PubMed.
- F. Zhang, E. Lees, F. Amin, P. Rivera-Gil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113–3127 CrossRef CAS PubMed.
- J. Ostermann, J.-P. Merkl, S. Flessau, C. Wolter, A. Kornowksi, C. Schmidtke, A. Pietsch, H. Kloust, A. Feld and H. Weller, ACS Nano, 2013, 7, 9156–9167 CrossRef CAS PubMed.
- H. Kloust, E. Pöselt, S. Kappen, C. Schmidtke, A. Kornowski, W. Pauer, H.-U. Moritz and H. Weller, Langmuir, 2012, 28, 7276–7281 CrossRef CAS PubMed.
- K. Boldt, S. Jander, K. Hoppe and H. Weller, ACS Nano, 2011, 5, 8115–8123 CrossRef CAS PubMed.
- G. Chilla, T. Kipp, T. Menke, D. Heitmann, M. Nikolic, A. Frömsdorf, A. Kornowski, S. Förster and H. Weller, Phys. Rev. Lett., 2008, 100, 057403 CrossRef.
- W. J. M. Mulder, R. Koole, R. J. Brandwijk, G. Storm, P. T. K. Chin, G. J. Strijkers, C. de Mello Donegá, K. Nicolay and A. W. Griffioen, Nano Lett., 2006, 6, 1–6 CrossRef CAS PubMed.
- E. Pöselt, C. Schmidtke, S. Fischer, K. Peldschus, J. Salamon, H. Kloust, H. Tran, A. Pietsch, M. Heine, G. Adam, U. Schumacher, C. Wagener, S. Förster and H. Weller, ACS Nano, 2012, 6, 3346–3355 CrossRef PubMed.
- C. Schmidtke, H. Kloust, N. G. Bastús, J.-P. Merkl, H. Tran, S. Flessau, A. Feld, T. Schotten and H. Weller, Nanoscale, 2013, 5, 11783–11794 RSC.
- C. Schmidtke, E. Pöselt, J. Ostermann, A. Pietsch, H. Kloust, H. Tran, T. Schotten, N. G. Bastús, R. Eggers and H. Weller, Nanoscale, 2013, 5, 7433–7444 RSC.
- F. D. Stefani, J. P. Hoogenboom and E. Barkai, Phys. Today, 2009, 62, 34–39 CrossRef CAS PubMed.
- P. Frantsuzov, M. Kuno, B. Janko and R. A. Marcus, Nat. Phys., 2008, 4, 519–522 CrossRef.
- G. Schlegel, J. Bohnenberger, I. Potapova and A. Mews, Phys. Rev. Lett., 2002, 88, 137401 CrossRef.
- S. A. Empedocles, R. Neuhauser, K. Shimizu and M. G. Bawendi, Adv. Mater., 1999, 11, 1243–1256 CrossRef CAS.
- D. E. Gómez, J. van Embden, J. Jasieniak, T. A. Smith and P. Mulvaney, Small, 2006, 2, 204–208 CrossRef PubMed.
- A. Issac, C. von Borczyskowski and F. Cichos, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 161302 CrossRef.
- Y. Kim, N. W. Song, H. Yu, D. W. Moon, S. J. Lim, W. Kim, H.-J. Yoon and S. K. Shin, Phys. Chem. Chem. Phys., 2009, 11, 3497–3502 RSC.
- F. Koberling, A. Mews and T. Basché, Adv. Mater., 2001, 13, 672–676 CrossRef CAS.
- C. Galland, Y. Ghosh, A. Steinbrück, M. Sykora, J. A. Hollingsworth, V. I. Klimov and H. Htoon, Nature, 2011, 479, 203–207 CrossRef CAS PubMed.
- X. Shi, X. Meng, L. Sun, J. Liu, J. Zheng, H. Gai, R. Yang and E. S. Yeung, Lab Chip, 2010, 10, 2844–2847 RSC.
- S. Hohng and T. Ha, J. Am. Chem. Soc., 2004, 126, 1324–1325 CrossRef CAS PubMed.
- S. R. Opperwall, A. Divakaran, E. G. Porter, J. A. Christians, A. J. Denhartigh and D. E. Benson, ACS Nano, 2012, 6, 8078–8086 CrossRef CAS PubMed.
- X. X. Yu, J. N. Li, K. C. Kwok, M. C. Paau, M. M. F. Choi, K. K. Shiu, J. Y. Chen and N. H. Cheung, J. Phys. Chem. C, 2012, 116, 18479–18486 CAS.
- V. Fomenko and D. J. Nesbitt, Nano Lett., 2008, 8, 287–293 CrossRef CAS PubMed.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS.
- D. V. Talapin, I. Mekis, S. Götzinger, A. Kornowski, O. Benson and H. Weller, J. Phys. Chem. B, 2004, 108, 18826–18831 CrossRef CAS.
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285–6294 CrossRef CAS.
- R. J. Sengwa, K. Kaur and R. Chaudhary, Polym. Int., 2000, 49, 599–608 CrossRef CAS.
- N. Koizumi and T. Hanai, J. Phys. Chem., 1956, 60, 1496–1500 CrossRef CAS.
- U. T. D. Thuy, N. Q. Liem, D. X. Thanh, M. Protière and P. Reiss, Appl. Phys. Lett., 2007, 91, 241908 CrossRef PubMed.
- C. Leatherdale and M. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 165315 CrossRef.
- M. Iwamatsu, M. Fujiwara, N. Happo and K. Horii, J. Phys.: Condens. Matter, 1997, 9, 9881–9892 CrossRef CAS.
- A. Piryatinski, S. A. Ivanov, S. Tretiak and V. I. Klimov, Nano Lett., 2007, 7, 108–115 CrossRef CAS PubMed.
- Y. Luo and L.-W. Wang, ACS Nano, 2010, 4, 91–98 CrossRef CAS PubMed.
- G. Rainò, T. Stöferle, I. Moreels, R. Gomes, J. S. Kamal, Z. Hens and R. F. Mahrt, ACS Nano, 2011, 5, 4031–4036 CrossRef PubMed.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2001, 115, 1028–1040 CrossRef CAS PubMed.
- X. Ma, K. Fletcher, T. Kipp, M. P. Grzelczak, Z. Wang, I. Pastoriza-Santos, A. Kornowski and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2011, 2, 2466–2471 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2000, 112, 3117 CrossRef CAS PubMed.
- P. Lindsey and G. D. Patterson, J. Chem. Phys., 1980, 73, 3348–3357 CrossRef PubMed.
- B. R. Fisher, N. E. Stott and M. G. Bawendi, J. Phys. Chem. B, 2004, 108, 143–148 CrossRef CAS.
| This journal is © the Owner Societies 2014 |