 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of a fibrous carbon envelope on the formation of CoFe nanoparticles for durable electrocatalytic oxygen evolution†
Beomgyun
Jeong‡
a,
Dongyoon
Shin‡
a,
Jae Kwang
Lee
b,
Dae Han
Kim
c,
Young Dok
Kim
c and
Jaeyoung
Lee
*ab
aElectrochemical Reaction and Technology Laboratory (ERTL), School of Environmental Science and Engineering, GIST, Gwangju 500-712, Republic of Korea
bErtl Center for Electrochemistry and Catalysis, RISE, GIST, Gwangju 500-712, Republic of Korea. E-mail: jaeyoung@gist.ac.kr
cDepartment of Chemistry, Sungkyunkwan University, Suwon 440-746, Republic of Korea
First published on 16th April 2014
Abstract
Co oxides are known to be active and stable alternative anode electrocatalysts possessing the potential to replace the best performing but most expensive Ir and Ru oxides in alkaline water electrolysis. Of late, Co oxides loaded on various carbon supports have been reported as a way to outperform Ir or Ru catalysts by improving the utilization efficiency. In this study, we introduce Co and Fe nanoparticles embedded carbon nanofibers (CoFe-CNFs), fabricated through electrospinning and pyrolysis of a polymer mixed with Co and Fe precursors. This method is a facile route for simultaneously making Co and Fe nanoparticles as well as the stable accommodation of the CoFe nanoparticles in the carbon support. We demonstrate the potential of the CoFe-CNFs as active and stable electrocatalysts for the oxygen evolution reaction (OER) in alkaline media. We conducted detailed physico-chemical characterizations to elucidate the effect of the CNFs on the OER activity and stability of the CoFe-CNFs. It is suggested that the CNFs are a medium in which OER-active CoFe alloy nanoparticles are formed homogeneously, and that carbon layers surrounding the nanoparticles are beneficial to the stability of the CoFe-CNFs in the OER.
1. Introduction
The oxygen evolution reaction (OER) has been well studied in the electrochemistry field because it is involved as a rate determining reaction in major electrochemical processes, such as electrometallurgy, water electrolysis, regenerative fuel cells and metal–air batteries.1,2 Along with fundamental studies to understand the electrocatalysis in the OER,3,4 an electrode lowering the degree of polarization required to evolve the same level of oxygen at the same electrical current has been researched. Some of the discovered materials show excellent OER activity approaching the fundamental activation energy limit, such as Beer's dimensionally stable anode (DSA) comprising Ru and Ir oxides, developed in 1969.5 This electrode has been sufficient for use in specialized processes for obtaining high-purity metal, hydrogen, and oxygen. However, as needs for storing renewable energy and also batteries with higher energy densities increase, it is necessary to find a more cost-efficient OER electrocatalyst possessing similar performance and stability to the expensive Ru and Ir oxides based electrocatalyst. This is another challenge for making metal–air batteries and regenerative fuel cells a viable option for storing renewable energy.From a practical point of view, it is extremely difficult to find alternative catalysts that can replace the Ru and Ir oxides in acidic media, in which non-noble metals are chemically unstable and show a worse OER activity. In contrast, Fe, Co, and Ni (iron triad) oxides with amorphous or spinel structures are chemically stable in alkaline media and they show a relatively comparable OER performance to the Ru and Ir oxides.1,6–9 Thus, there are studies on improving the performance of iron triad element based electrocatalysts by introducing novel carbon supports such as carbon nanotubes and graphene.10–13 There are also studies to determine the optimal oxidation state or surface oxide structure of Fe, Co and Ni based electrocatalysts.6,14 These optimal oxidation state and appropriate carbon support electrocatalysts produced in this research resulted in a similar activity toward the OER and also superior stability in alkaline media. In the synthesis of these electrocatalysts, metal salts dissolved and nano-carbon materials dispersed in water or an organic solvent with a high dielectric constant are heated to around 150–200 °C for several hours to make a metal oxide loaded on the carbon nanomaterials. Although the metal oxide loading process on the nanocarbon support is relatively facile, it is not easy to synthesize carbon nanotubes and graphenes and to adjust the graphitic surface to accept the metal precursor easily through the wet process.
We introduced Co and Fe modified electrospun carbon nanofibers (CoFe-CNFs) as a facile one-step synthesis of electrocatalysts toward the oxygen reduction reaction (ORR).15,16 In the ORR, the metal particles embedded in the carbon nanofibers (CNFs) facilitate the formation of ORR active sites and graphitization. Moreover, they contribute to the improvement in the conductivity of the CNF network by enhancing the graphitization of CNFs. In this work, we extended the application of the developed CoFe-CNFs to the OER from the expectation that the metal particles embedded in the CNFs can act as electrocatalysts toward the OER.15,17,18 The metal particles formed inside the carbon nanofibers are uniform because they are under homogeneous temperature and environmental conditions in the high temperature heat-treatment process. The carbon layer surrounding the metal particles provides a stronger interaction to the CNF support and it prevents chemical degradation of the metal surface leading to better stability. Thus, CoFe-CNFs have the potential to be stable, controllable, and cost-effective OER electrocatalysts as well as bi-functional oxygen electrocatalysts applicable to regenerative fuel cells and metal–air batteries. In this study, we tested the OER performance including the activity and stability of the CoFe-CNFs by comparison with Ir electrocatalysts, and we investigated the effect of the carbon layer of the CoFe-CNFs to the durability of the CoFe catalysts during the OER.
2. Experimental methods
The metal-embedded CNFs (Me-CNFs) were prepared by electrospinning the metal precursor containing polymer to produce a nanofiber web and subsequent high temperature heat-treatments. To prepare the metal precursor containing polymer, 4 g polyacrylonitrile (PAN) solution was dissolved in 36 g N,N-dimethylformamide at 60–80 °C for around 3 h. After complete dissolution of PAN in N,N-dimethylformamide, the polymer solution was mixed with the metal precursors, Co(II) nitrate hexahydrate (Co(NO3)2·6H2O) and Fe(III) nitrate nonahydrate (Fe(NO3)3·9H2O). The total weight of the metal precursors was the same, 1.2 g, in all of the metal precursors containing PAN (Me-PAN), and the weight ratio of Co and Fe precursor was 1 to 1 in case of the CoFe-PAN solution. The resultant weight ratio of the precursor is PAN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF = 1:9 and Co or Fe metal precursor:(PAN + DMF) = 3:100. The solution was supplied at a flow rate of 5 ml h−1 for 6 h via a 5 multi-nozzle equipped syringe pump for the electrospinning. A high voltage of +12 kV was applied to the syringe needles and −12 kV to the drum collector, which are located 15 cm apart. All of the processes were performed at room temperature (∼25 °C) under a controlled relative humidity of 30%. In the stabilizing heat treatment, the electrospun Me-PAN fiber web was heated in air at a heating rate of 1 °C min−1 up to 280 °C and then was held for an hour at that temperature. Then, the environment was changed by flowing N2 at a flow rate of 200 ml min−1 and the temperature was increased at a heating rate of 5 °C min−1 to 1000 °C which was maintained for 1 h in order to carbonize the stabilized Me-PAN fiber web. Finally, we obtain the metal containing carbon nanofibers (Me-CNFs), i.e. Co-CNFs, Fe-CNFs, and CoFe-CNFs. The prepared Me-CNFs were ground with a mortar for the electrochemical experiments with a rotating disk electrode (RDE). For preparation of the intensively pulverized CoFe-CNFs, 0.2 g of CoFe-CNFs was mixed with 20 g of zirconium oxide balls with a diameter of 2 mm and the CoFe-CNFs were ball-milled at 500 rpm for 160 min with 10 min idle time and 30 min processing time.
:DMF = 1:9 and Co or Fe metal precursor:(PAN + DMF) = 3:100. The solution was supplied at a flow rate of 5 ml h−1 for 6 h via a 5 multi-nozzle equipped syringe pump for the electrospinning. A high voltage of +12 kV was applied to the syringe needles and −12 kV to the drum collector, which are located 15 cm apart. All of the processes were performed at room temperature (∼25 °C) under a controlled relative humidity of 30%. In the stabilizing heat treatment, the electrospun Me-PAN fiber web was heated in air at a heating rate of 1 °C min−1 up to 280 °C and then was held for an hour at that temperature. Then, the environment was changed by flowing N2 at a flow rate of 200 ml min−1 and the temperature was increased at a heating rate of 5 °C min−1 to 1000 °C which was maintained for 1 h in order to carbonize the stabilized Me-PAN fiber web. Finally, we obtain the metal containing carbon nanofibers (Me-CNFs), i.e. Co-CNFs, Fe-CNFs, and CoFe-CNFs. The prepared Me-CNFs were ground with a mortar for the electrochemical experiments with a rotating disk electrode (RDE). For preparation of the intensively pulverized CoFe-CNFs, 0.2 g of CoFe-CNFs was mixed with 20 g of zirconium oxide balls with a diameter of 2 mm and the CoFe-CNFs were ball-milled at 500 rpm for 160 min with 10 min idle time and 30 min processing time.
The electrochemical properties and OER activity of the synthesized Co-CNFs, Fe-CNFs, and CoFe-CNFs catalysts were evaluated using a three electrode cell connected to a potentiostat/galvanostat (Biologic, VSP). A smooth Pt wire was used as the counter electrode and Hg/HgO as the reference. All of the potentials initially measured versus (vs.) the Hg/HgO electrode immersed in 1 M NaOH were converted to be with respect to RHE reference by adding 0.9083 V to the potentials.
We prepared a catalyst ink by dispersing 25 mg of the synthesized catalysts in a mixture of 25 μl of 10 wt% Nafion solution (Sigma-Aldrich), 2.5 ml D.I. water and 2.5 ml isopropyl alcohol. After the catalyst ink was ultrasonicated for 30 min, a 10 μl aliquot of the suspension was dropped onto a glassy carbon disk electrode (0.2475 cm2) using a micropipette, so that the amount of the catalyst on the electrode was 200 μg cm−2. The reference Ir catalyst ink was prepared by ultrasonically dispersing 5 mg of Ir black (99.9% trace metals basis, Sigma-Aldrich) for 30 min in 5 ml solution containing N-propyl alcohol and deionized water, 1:1 by volume, and the same amount of the suspension was dropped onto the glassy carbon disk electrode.
Cyclic voltammograms (CVs) were obtained in an aqueous solution of 0.1 M potassium hydroxide (KOH) as the electrolyte that was saturated with ultrapure N2 at room temperature. The scan rate was 50 mV s−1 and the potential range was from 0.6 V to −1.2 V vs. Hg/HgO. The OER activities of the synthesized catalysts were measured with the RDE at a rotating rate of 1600 rpm while nitrogen was constantly bubbled through the electrolyte at a flow rate of 10 ml min−1. The potential was scanned from 0 V to 1.5 V vs. Hg/HgO at a scan rate of 100 mV s−1. Chronoamperometry was also performed to obtain long term stability of catalysts at 0.9 V vs. Hg/HgO.
The crystal structures of the embedded metals were investigated using X-ray diffraction (XRD; Rigaku Miniflex II) equipped with a Cu Kα (λ = 0.15406 nm) X-ray source. The morphology and elemental composition of the catalysts were investigated through scanning transmission/transmission electron microscopy (STEM/TEM; Philips TECNAI F20) and energy-dispersive X-ray spectroscopy (EDS) in Gwangju center of Korea Basic Science Institute (KBSI). The X-ray photoelectron spectroscopy (XPS) was performed to investigate the surface functionality and oxidation state of the metal in an ultrahigh vacuum (UHV) chamber with a base pressure ≤2.0 × 10−10 Torr and a concentric hemispherical analyzer (CHA; PHOIBOS-Has 3500, SPECS). All XPS spectra were obtained at room temperature using the Mg Kα-line (1253.6 eV) as the photon source and the binding energy of the measured peaks was calibrated with respect to the C 1s peak position (284.6 eV). The specific surface area and pore size distribution of the Me-CNFs were also determined by N2 adsorption using the BET method and BJH method respectively with the automatic gas adsorption–desorption apparatus (Belsorp-max, Japan).
3. Results and discussion
The TEM images of the Me-CNFs show a similar morphology, most of the metal nanoparticles are homogeneously embedded in the CNFs (Fig. S1, ESI†). The size of the metal particles ranged 10–50 nm as shown in Fig. S1 (ESI†), and the diameter of the CNFs is approximately in the range 100–500 nm from our previous studies.15,16,18 For CoFe-CNFs, Co particles are homogeneously distributed together with the Fe particles as seen in Fig. 1a–d. From the EDS elemental composition shown in Table 1, a slightly larger amount of Co is detected in the CoFe-CNFs, which is expected from the molar ratio of Co and Fe calculated from the precursors’ molecular weights. The relative amount of metal particles in the Me-CNFs measured with point EDS analysis is around 13 wt%. However, according to the ICP-AES analysis conducted in the previous study, approximately 20 wt% of metal is included in the Me-CNFs, which is more representative and conclusive than the EDS elemental ratio for several strands of the Me-CNFs. In the EDS line mapping result for a metal particle in the CoFe-CNFs (Fig. 1f), a molar ratio of Co:Fe = 1:1 is observed, implying the formation of a CoFe alloy.
 | ||
| Fig. 1 (a) TEM image, (b) STEM image of the CoFe-CNFs, EDS maps for (c) Fe and (d) Co, in the same STEM image. (e) Magnified STEM image and (f) linear EDS profile of the carbon layer enclosed metal particles with a roughly Co:Fe = 1:1 ratio. | ||
| Element | wt% | at% |
|---|---|---|
| C | 81.4 | 92.2 |
| N | 0 | 0 |
| O | 5.5 | 4.7 |
| Fe | 5.7 | 1.4 |
| Co | 7.3 | 1.7 |
This implication corresponds with the XRD result showing CoFe as the main crystal component (Fig. 2), and the slight excess of Co seems to comprise CoCx. The crystal structure of the Me-CNFs shows different features according to the types of metal precursor used in the synthesis of the Me-CNFs. In the XRD result of the Co-CNFs, metallic Co is the main crystal component. In contrast, for the Fe-CNFs, Fe carbide (Fe3C) is the main component rather than forming metallic Fe separated from the carbon medium. The crystallite size of the embedded metal particles in Fe-CNFs, estimated from the Debye–Scherrer equation, is around 13 nm, which is smaller than the values for the Co-CNFs and CoFe-CNFs which are around 24–25 nm size (Table 2). The smaller Fe particle size is likely to be related to the formation of Fe carbide where Fe atoms are well-dispersed in the carbon medium. In the case of the CoFe-CNFs, Fe atoms look to form alloys with Co atoms, so that the Fe atoms are less dispersed in the carbon and would not form smaller Fe carbide particles. In the previous study that we conducted with Co-CNFs,16 the FWHM of the graphite peak (002) in the XRD of the Co-CNFs becomes narrower than those of the Fe-CNFs and CoFe-CNFs. Co atoms mixed with a polymer facilitate the graphitization of the carbon fibers under heat-treatment, which can be beneficial to the electrical conductivity.19
 | ||
| Fig. 2 XRD results of the (a) CoFe-CNFs, (b) Co-CNFs, and (c) Fe-CNFs. | ||
| FWHM (°) | Peak location (°) | Lower size limit (nm) | FWHM of graphite (002) peak (°) | |
|---|---|---|---|---|
| CoFe-CNF | 0.55 | 44.9 | 24 | 3.7 |
| Co-CNF | 0.32 | 44.2 | 25 | 3.4 |
| Fe-CNF | 1.01 | 44.9 | 13 | 1.6 |
The cyclic voltammograms (CVs) of the Me-CNFs (Fig. 3) show characteristic redox peaks closely related to the metal components and surface oxide species although most of the metal particles visible in the TEM images are buried inside the CNFs. It suggests that the electrolyte is penetrating through the carbon layers surrounding the metal nanoparticles to reach the important OER active sites. The CV of the Fe-CNFs shows broad peaks appearing around −0.06 V on the cathodic scan and 0.28 V on the anodic scan, which is a characteristic feature of the Fe(II)/Fe(III) redox surface reaction.14,20,21 The CV of the Co-CNFs shows Co(II)/Co(III) redox peaks in the range of 0.95–1.25 V corresponding to CoO or Co(OH)2 → Co3O4, Co2O3 or CoOOH. Co(0)/Co(II) redox peaks corresponding to Co → Co(OH)2, CoO, or CoO·OHaq are also located at around 0.30 V on the anodic scan and 0.40 V on the cathodic scan.14,22 The CoFe-CNFs have an intermediate CV shape between Co-CNFs and Fe-CNFs; the CV shape of the CoFe-CNFs in the lower potential range is similar to that of the Fe-CNFs and there is seemingly a Co-related redox peak in the higher potential range. However, the Co redox peak of the CoFe-CNFs looks different to that of the Co-CNFs, the Co redox peak centered at 1.07 V becomes substantially suppressed and a new Co redox peak appears at around 1.30 V which was identified in CV measurements for Co oxide films in Doyle et al.’s study.14 This Co redox peak at 1.30 V vs. RHE is clearly observed in the CV of the ball-milled CoFe-CNFs and is likely from the more exposed Co and Fe metal surface. Integrated charge and equivalent capacitance of these metal-modified CNFs is in the range roughly between 50–100 F g−1, which is difficult to achieve with the electric double layer of the carbon material having a BET surface area of less than 500 m2 g−1 (Fig. S2, ESI†).
 | ||
| Fig. 3 Cyclic voltammograms of the CoFe-CNFs, Co-CNFs, Fe-CNFs, and ball-milled CoFe-CNFs in 0.1 M KOH aqueous solution at a scan rate of 50 mV s−1. | ||
We tested the OER activity of the Me-CNFs through 50 scans of cyclic voltammetry in the range 0.9–2.4 V vs. RHE in 0.1 M KOH aqueous solution using a RDE at a rotating speed of 1600 rpm to exclude oxygen bubbles blocking the coated catalyst surface (Fig. S3, ESI†). We compared the OER activity of the Me-CNFs (200 μg cm−2 loading on GC) at the fiftieth scan with Ir black catalyst with the same metal loading amount of 40 μg cm−2 considering that the metal content in the Me-CNFs is about 20 wt% as investigated with ICP-AES analysis. The CoFe-CNFs displays a fairly comparable OER current to the Ir black catalyst over 2.0 V vs. RHE (Fig. 4). The OER performance was also maintained over 50 scans even though it went through a highly anodic potential up to 2.4 V vs. RHE in every scan (Fig. 5). The Co-CNFs show a similar trend in their OER stability but with a lower OER activity than the CoFe-CNFs with the same metal loading amount. Thus, the Me-CNFs including Co metal like Co-CNFs and CoFe-CNFs might have advantageous properties for OER stability. Note that the Fe-CNFs show a poor OER activity and stability but the CoFe-CNFs show enhanced activity and stability even compared to Co-CNFs with double the amount of Co metal. The improvement in the OER activity of CoFe is in accordance with the result reported from Smith et al., in which a CoFe oxide layer demonstrates a significantly low Tafel slope close to 40 mV dec−1 and 0.25 V of overpotential at j = 0.5 mA cm−2.8,9 The ball-milled CoFe-CNFs show more enhanced metal redox peaks in the cyclic voltammetry result representing the more exposed metal surface in the ball-milled CoFe-CNFs. The higher OER activity compared to the CoFe-CNFs in the potential range over 1.75 V, implies the disadvantage of the carbon layers around the metal particles with regard to mass transport.
 | ||
| Fig. 4 The OER activity of the CoFe-CNFs, Co-CNFs, Fe-CNFs, ball-milled CoFe-CNFs, and Ir black. The metal loading amount of the Me-CNFs and Ir black on GC is the same, 40 μg cm−2. The arrow mark in this figure indicates the equilibrium potential of the ORR/OER. | ||
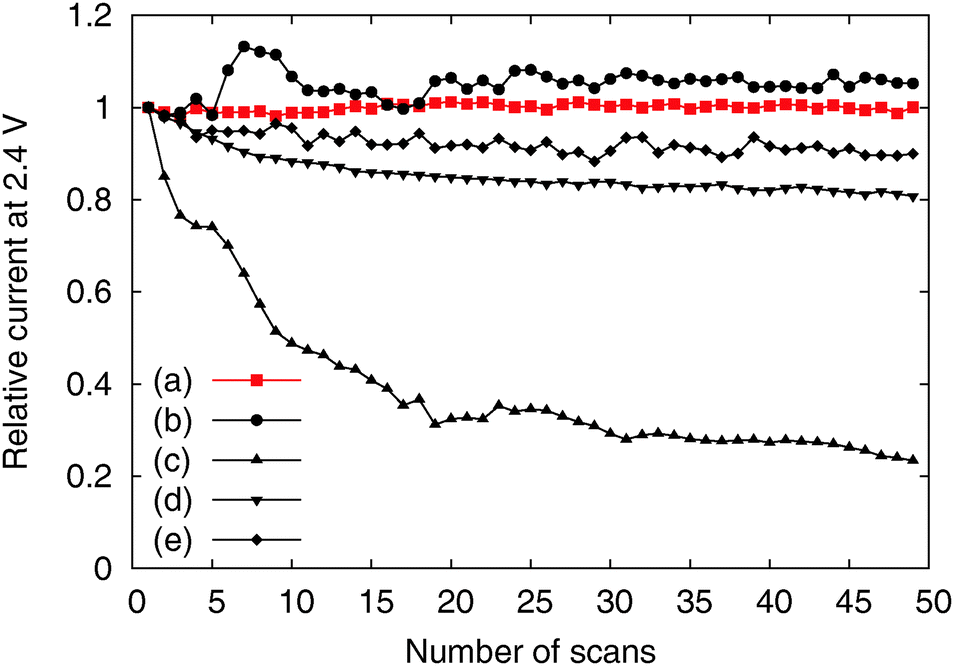 | ||
| Fig. 5 Relative activity change according to number of scans of the (a) CoFe-CNFs, (b) Co-CNFs, (c) Fe-CNFs, (d) ball-milled CoFe-CNFs, and (e) Ir black. | ||
However, the CoFe-CNFs show a similar OER activity to ball-milled CoFe-CNFs at potentials below 1.75 V, and they are relatively more stable than the ball-milled CoFe-CNFs in the OER. Thus, the carbon layers seem to provide a protective layer preventing possible detachment or dissolution of catalyst particles from the CNF support. This means that the CoFe-CNFs have an improved stability without compromising the OER activity performance within a certain range of potential although the metallic nanoparticles themselves do not have this stability under harsh conditions. The concept of enveloping the catalyst surface with thin carbon layers is reported in research papers on the ORR, and the patent documents on bi-functional OER/ORR electrocatalysts.23–25 In this study, however, we could achieve the configuration beneficial to the catalytic stability by simply electrospinning and carbonizing the mixture of metal and polymer precursors. Moreover, the formation of the optimum carbon layer for protecting the catalyst surface seems to depend on the type of the enclosed metal particles, as can be seen from the comparison between Fe-CNFs and Co-CNFs in Fig. 5. We also conducted chronoamperometry (CA) to evaluate the durability of the CoFe-CNF catalysts, with the same metal amount of Ir black for comparison, at 1.8 V vs. RHE (Fig. 6). The durability shown in the CA results is the same trend as in the cyclic durability result in Fig. 5, CoFe-CNFs > ball-milled CoFe-CNFs > Ir black. The CoFe-CNFs suffered little degradation in their OER activity during the measurement time. Furthermore, we did additional experiments to confirm whether the carbon layers around the metal particles prevent the oxidation of the metal surface (Fig. S4 and S5, ESI†). The Co oxidation state does not change before and after the OER experiment and the metal particles are still well dispersed and do not agglomerate. Even though the peak intensity around 288 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) slightly increases in the C 1s XPS spectra, it is not related to the carbon corrosion which can significantly affect the dimensional stability of the catalytic electrode. There is no indication of carbon corrosion, such as substantial development of sp3 bonding (C–C) or C–O functional group. Therefore, we can conclude that carbon layer is fairly stable and it can act as a protective layer to prevent oxidation of the metal particles during the OER.
O) slightly increases in the C 1s XPS spectra, it is not related to the carbon corrosion which can significantly affect the dimensional stability of the catalytic electrode. There is no indication of carbon corrosion, such as substantial development of sp3 bonding (C–C) or C–O functional group. Therefore, we can conclude that carbon layer is fairly stable and it can act as a protective layer to prevent oxidation of the metal particles during the OER.
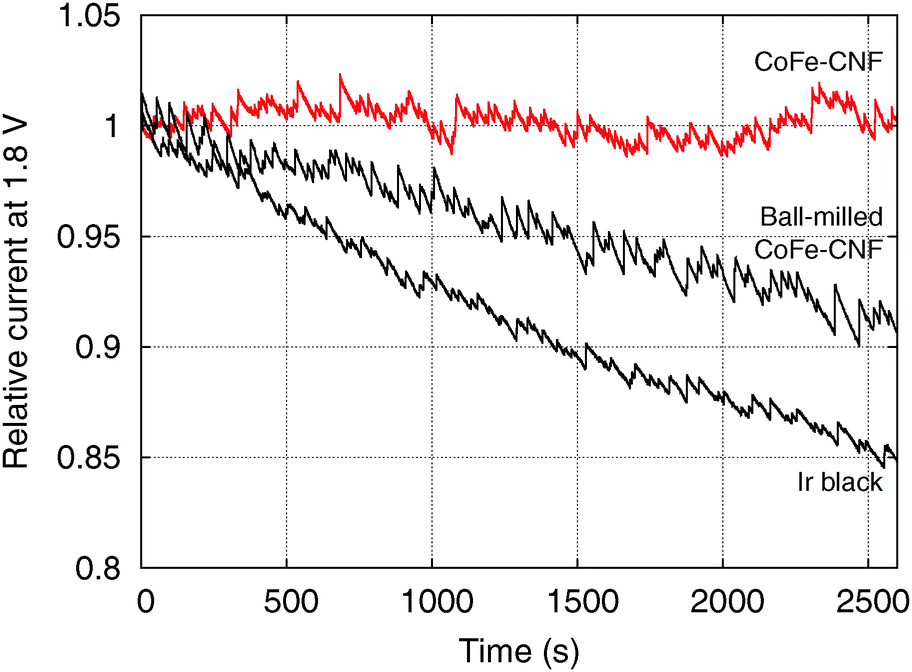 | ||
| Fig. 6 Chronoamperometry of the CoFe-CNFs, ball-milled CoFe-CNFs, and Ir black. | ||
Like other XPS results measured in C 1s, N 1s, and O 1s regions (Fig. S6, ESI†), XPS spectra of the CoFe-CNFs and Co-CNFs measured in the Co 2p region do not show a significant difference in the shape of the profile; the intensity is slightly different due to the higher Co metal content in the Co-CNFs than in the CoFe-CNFs (Fig. 7b and c). The XPS peak shape of the Co-CNFs and CoFe-CNFs resembles the known XPS results of Co(II) oxides, CoO and Co(OH)2.26,27 Although CoO and Co(OH)2 show very similar XPS peak profiles, the XPS peak of CoO in the Co 2p1/2 region has a slight shoulder at a binding energy of 794.5 eV, which is also quite clearly seen in the XPS spectra of the ball-milled CoFe-CNFs having more exposed metal particles (Fig. 7a). The oxidation state of the Co surface observed in the XPS spectra explains the CVs of the CoFe-CNFs and Co-CNFs possessing a predominant Co(II)/Co(III) redox peak. However, despite the similar Co 2p XPS profiles for the CoFe-CNFs and Co-CNFs, the potential location and the shape of the Co(II)/Co(III) peak in the CV are different. Considering the great differences between the ability of XPS and cyclic voltammetry to analyze a sample deeply, the electrochemically influential catalytic sites related to OER activity seem to reside mainly inside the CNFs. This result is due to the synthesis process of the metal nanoparticles; metal precursors are homogeneously mixed with a polymer precursor, then the metal particles form when the polymer becomes carbonized at a high temperature. Then the generated metal nanoparticles are distributed mostly inside the CNFs rather than exposed to the surface of the CNFs. XPS has been a standard and powerful technique for investigating the physicochemical properties of conventional catalysts in which metal nanoparticles are dispersed on the support surface. However, the Me-CNFs possess metal nanoparticles which are enveloped by carbon that is more than several nm thick. A surface analysis method with the capability to analyze a sample more deeply, such as cyclic voltammetry, would be a more reasonable choice for investigating the surface properties of the embedded electrocatalysts.
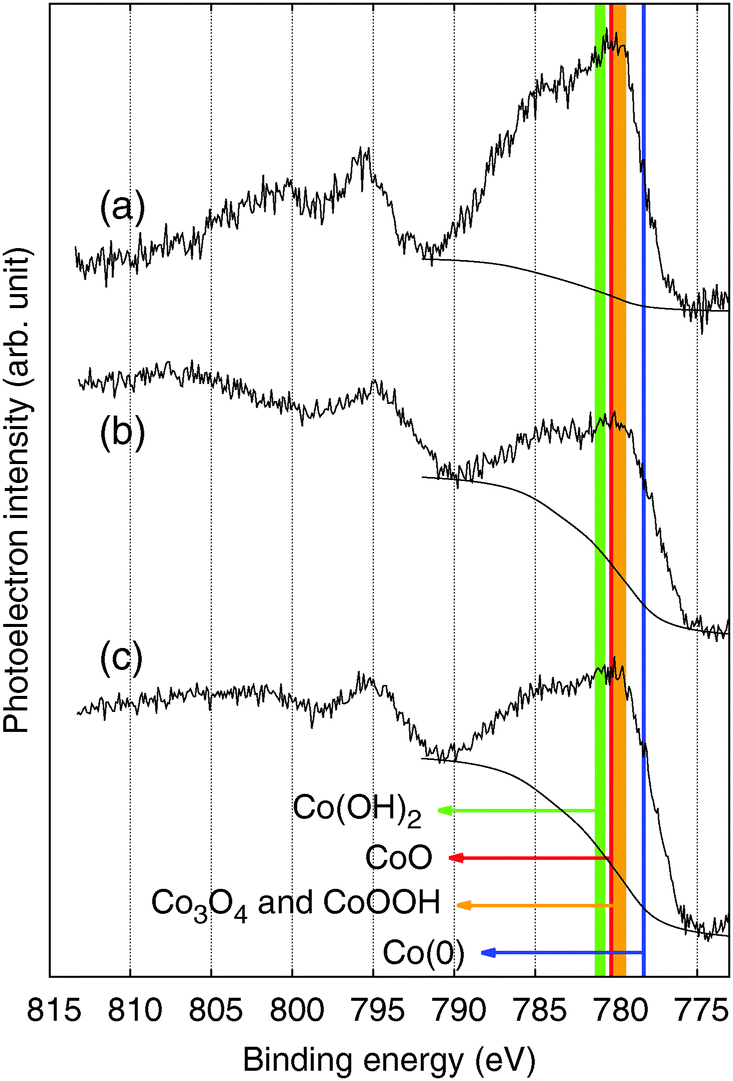 | ||
| Fig. 7 XPS spectra in the Co 2p region for the (a) ball-milled CoFe-CNFs, (b) CoFe-CNFs, and (c) Co-CNFs. | ||
4. Conclusion
We developed a simple synthesis process to produce OER catalysts by carbonizing the electrospun polymer fibers mixed with Co or Fe-based precursors. The CoFe-CNFs showed an OER performance comparable to the Ir black catalyst and a better performance was achieved than the Co-CNFs containing double the amount of Co metal. From the surface characterizations with cyclic voltammetry, we observed that the CoFe alloy shows a Co(III)/Co(II) redox peak of a different potential, which might be related to the improvement in the OER activity performance. In addition, a superior stability to the Ir black catalyst is observed, likely due to the nanofibrous carbon envelopes surrounding the metal nanoparticles. The carbon layers around the nanoparticles do not hinder the electrolyte contact with the active sites as intuitively expected but are helpful in preserving the catalyst surface under highly oxidizing OER conditions. Therefore, the developed CoFe-CNFs have the potential to be bi-functional oxygen electrocatalysts applicable to metal–air batteries or regenerative fuel cells under various potential conditions by utilizing the nanofibrous carbon structure not only as a support for homogeneous dispersion of the catalysts, but also as a protective envelope for enhancing the catalyst stability.Acknowledgements
This work is supported by the Core Technology Development Program of the Research Institute for Solar and Sustainable Energies (RISE), Gwangju Institute of Science and Technology. The authors are greatly thankful to Prof. Dangsheng Su and Prof. Bingsen Zhang for the assistance of STEM analysis. J. Lee is deeply indebted to the Alexander von Humboldt Foundation fellowship for experienced researchers (1141065).References
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley-Interscience, 1st edn, 1992 Search PubMed.
- D.-W. Park, J. W. Kim, J. K. Lee and J. Lee, Appl. Chem. Eng., 2012, 23, 359–366 CAS.
- S. Trasatti, J. Electroanal. Chem., 1980, 111, 125–131 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- H. Beer, Br. Pat., 1 147 442, 1969 Search PubMed.
- M. E. G. Lyons and M. P. Brandon, J. Electroanal. Chem., 2010, 641, 119–130 CrossRef CAS PubMed.
- S. M. Jasem and A. C. C. Tseung, J. Electrochem. Soc., 1979, 126, 1353–1360 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- K. Mette, A. Bergmann, J.-P. Tessonnier, M. Hävecker, L. Yao, T. Ressler, R. Schlögl, P. Strasser and M. Behrens, ChemCatChem, 2012, 4, 851–862 CrossRef CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- S. Uhm, B. Jeong and J. Lee, Electrochim. Acta, 2011, 56, 9186–9190 CrossRef CAS PubMed.
- D. Shin, B. Jeong, B. S. Mun, H. Jeon, H.-J. Shin, J. Baik and J. Lee, J. Phys. Chem. C, 2013, 117, 11619–11624 CAS.
- B. Jeong, S. Uhm and J. Lee, ECS Trans., 2010, 33, 1757–1767 CAS.
- J. Lee, B. Jeong and J. D. Ocon, Curr. Appl. Phys., 2013, 13, 309–321 CrossRef PubMed.
- N. A. M. Barakat, B. Kim, S. J. Park, Y. Jo, M.-H. Jung and H. Y. Kim, J. Mater. Chem., 2009, 19, 7371 RSC.
- C. Abreu, M. Cristóbal, X. Nóvoa, G. Pena, M. Pérez and C. Serra, Electrochim. Acta, 2004, 49, 3057–3065 CrossRef CAS PubMed.
- B. T. Hang, T. Watanabe, M. Eashira, S. Okada, J. Yamaki, S. Hata, S.-H. Yoon and I. Mochida, J. Power Sources, 2005, 150, 261–271 CrossRef CAS PubMed.
- A. Foelske and H.-H. Strehblow, Surf. Interface Anal., 2002, 34, 125–129 CrossRef CAS.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- J. A. Read, US Pat., 6 127 060, 2000 Search PubMed.
- M. N. Golovin and I. Kuznetsov, US Pat., 6 069 107, 2007 Search PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS PubMed.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp00385c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2014 |