 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thieno[3,4-b]pyrazine-based oligothiophenes: simple models of donor–acceptor polymeric materials†
Li
Wen
,
Christopher L.
Heth
and
Seth C.
Rasmussen
*
Department of Chemistry and Biochemistry, North Dakota State University, NDSU Dept. 2735, P.O. Box 6050, Fargo, ND 58108, USA. E-mail: seth.rasmussen@ndsu.edu; Fax: +1 701 231-8831; Tel: +1 701 231-8747
First published on 10th March 2014
Abstract
A series of thieno[3,4-b]pyrazine-based oligomers were synthesized via Stille cross-coupling as models of electronic structure–function relationships in thieno[3,4-b]pyrazine-based conjugated materials. The prepared oligomers include two oligothieno[3,4-b]pyrazine series from monomer to trimer, as well as a series of mixed terthienyls in which the ratio of thieno[3,4-b]pyrazine to either thiophene or 3,4-ethylene-dioxythiophene has been varied. The full oligomeric series was then thoroughly investigated via photo-physical and electrochemical studies, along with theoretical calculations, in order to correlate the effect of conjugation length and oligomer composition with the resulting electronic and optical properties. The corresponding relationships revealed should then provide more advanced models for the elucidation of donor–acceptor interactions in both homopolymeric and copolymeric materials of thieno[3,4-b]pyrazines.
Introduction
Since the initial studies of conjugated organic polymers in the 1960s,1,2 these materials have garnered considerable fundamental and technological interest due to their combination of the electronic and optical properties of classical inorganic semiconductors with many of the desirable properties of organic plastics such as mechanical flexibility and low production costs.3–5 Continued development of conjugated materials has since resulted in the current field of organic electronics. Thus, significant efforts have focused on utilization of conjugated materials in technological applications such as sensors, electrochromic devices, organic photovoltaics (OPVs), organic light-emitting diodes (OLEDs), and field effect transistors (FETs).3–5Within this greater scope of conjugated materials, the study of oligomeric species has found interest as both active materials for device applications and models of the larger polymeric analogues. The structurally defined and monodisperse nature of such oligomers allows correlation of the electronic properties with the conjugation length and overall chemical structure.4–8 Thus the correlation of electronic and optical properties of oligomeric series allows the establishment of valuable structure–function relationships, as well as extrapolation to predict the corresponding properties of defect-free polymer analogues.7 As a result, model oligomeric series of a number of important classes of conjugated polymers have been reported, as illustrated by the various oligothiophene series shown in Fig. 1.4–20
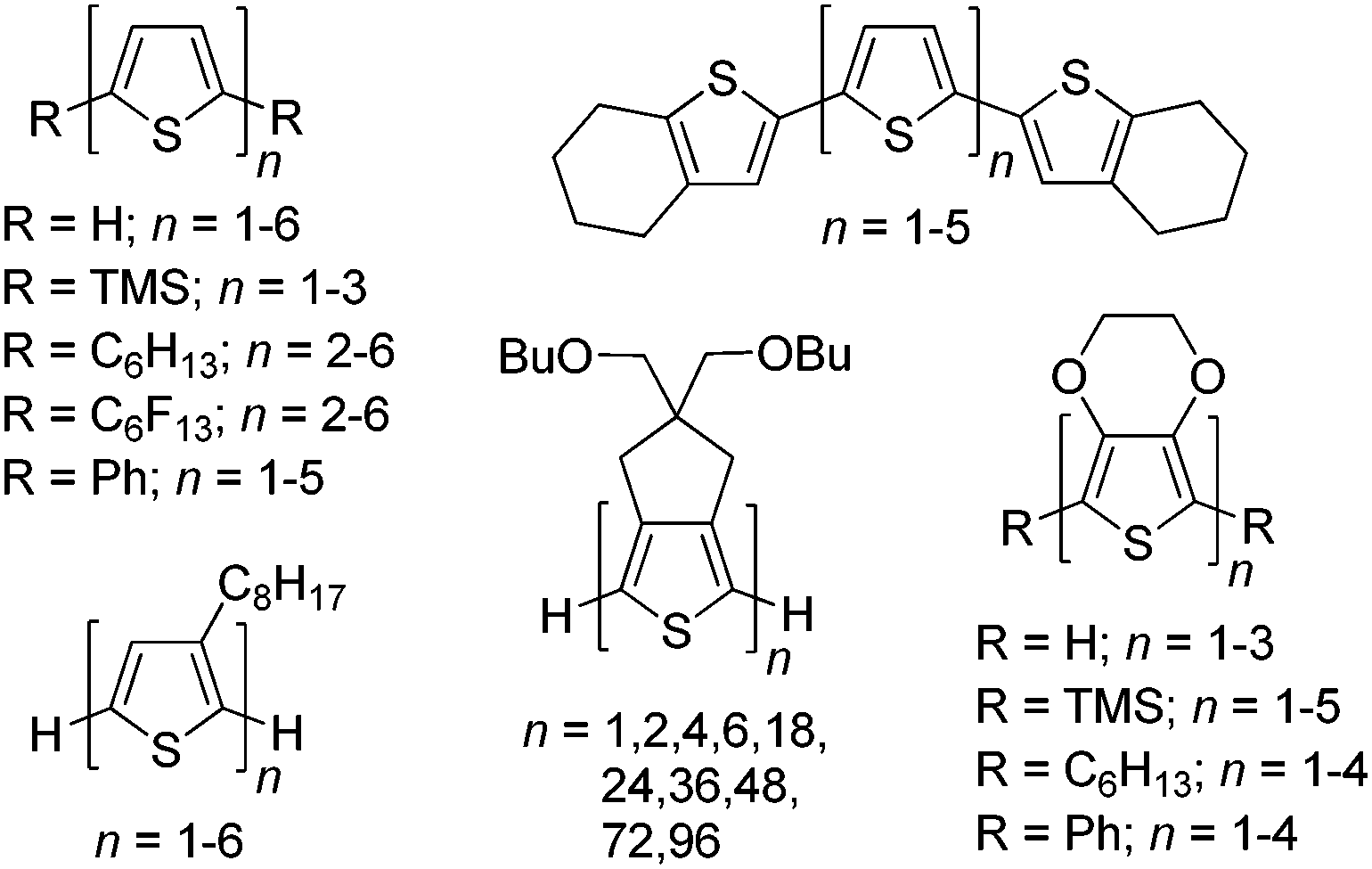 | ||
| Fig. 1 Representative oligothiophene series. | ||
Although numerous studies have been reported for traditional oligothiophene series, analogous studies of fused-ring thiophenes such as the benzo[b]thiophenes, thieno[3,4-b]pyrazines (TPs, 1, Fig. 2),21,22 and thieno[3,4-b]thiophenes are severely limited, with the only currently known examples consisting of two reports of TP oligomer series (Fig. 2).23,24 Such fused-ring thiophenes have been shown to be excellent building blocks for the production of reduced band gap (Eg = 1.5–2.0 eV) and low band gap (Eg < 1.5 eV) conjugated polymers and TPs in particular have been quite popular building blocks for the production of low band gap materials.22 Due to their success and popularity, it is important to continue studies of oligomeric models of TP materials in order to better reveal critical structure–function relationships, thus increasing the ability of researchers to generate materials with improved characteristics for device applications. As such, this current study builds on our preliminary report23 of TP oligomers capped with stabilizing trimethylsilyl (TMS) groups (Fig. 2, 3a–c, 4a–c).
 | ||
| Fig. 2 Thieno[3,4-b]pyrazine oligomeric series. | ||
The low band gaps observed in homopolymeric TP materials are typically attributed to the quinoidal nature of the backbone and the oligomeric series illustrated in Fig. 2 represent appropriate models of such TP homopolymers. However, it is currently far more common for TPs to be utilized as an ‘acceptor’ unit in a ‘donor–acceptor’ framework.25–30 This ‘donor–acceptor’ approach to low band gap conjugated materials was introduced in 1992 by Havinga and co-workers,25 in which it was proposed that alternating electron-rich and electron-deficient moieties along the same backbone could result in a hybrid material with HOMO levels characteristic of the donor and LUMO levels characteristic of the acceptor. While examples of mixed TP oligomers have been reported, particularly in the wealth of known terthienyls containing a central TP unit,21,31 these oligomers have served as precursors to TP-based copolymeric materials, rather than utilized as models of such proposed ‘donor–acceptor’ effects. An exception to this has been two studies on mixed TP oligomers reported by Janssen and coworkers.32,33
In order to probe the extent and nature of ‘donor–acceptor’ properties in TP systems, as well as to better understand the structure–function relationships within all TP-based materials, series of terthienyls with varying ratios of TP to either thiophene or 3,4-ethylenedioxythiophene (EDOT) have also been prepared (Fig. 3). This current study will utilize both TP homo- and mixed-oligomeric series in an attempt to correlate the effects of TP content on the resulting electronic and optical properties. The corresponding relationships revealed should then provide more advanced models for the elucidation of such relationships in the analogous polymeric materials.
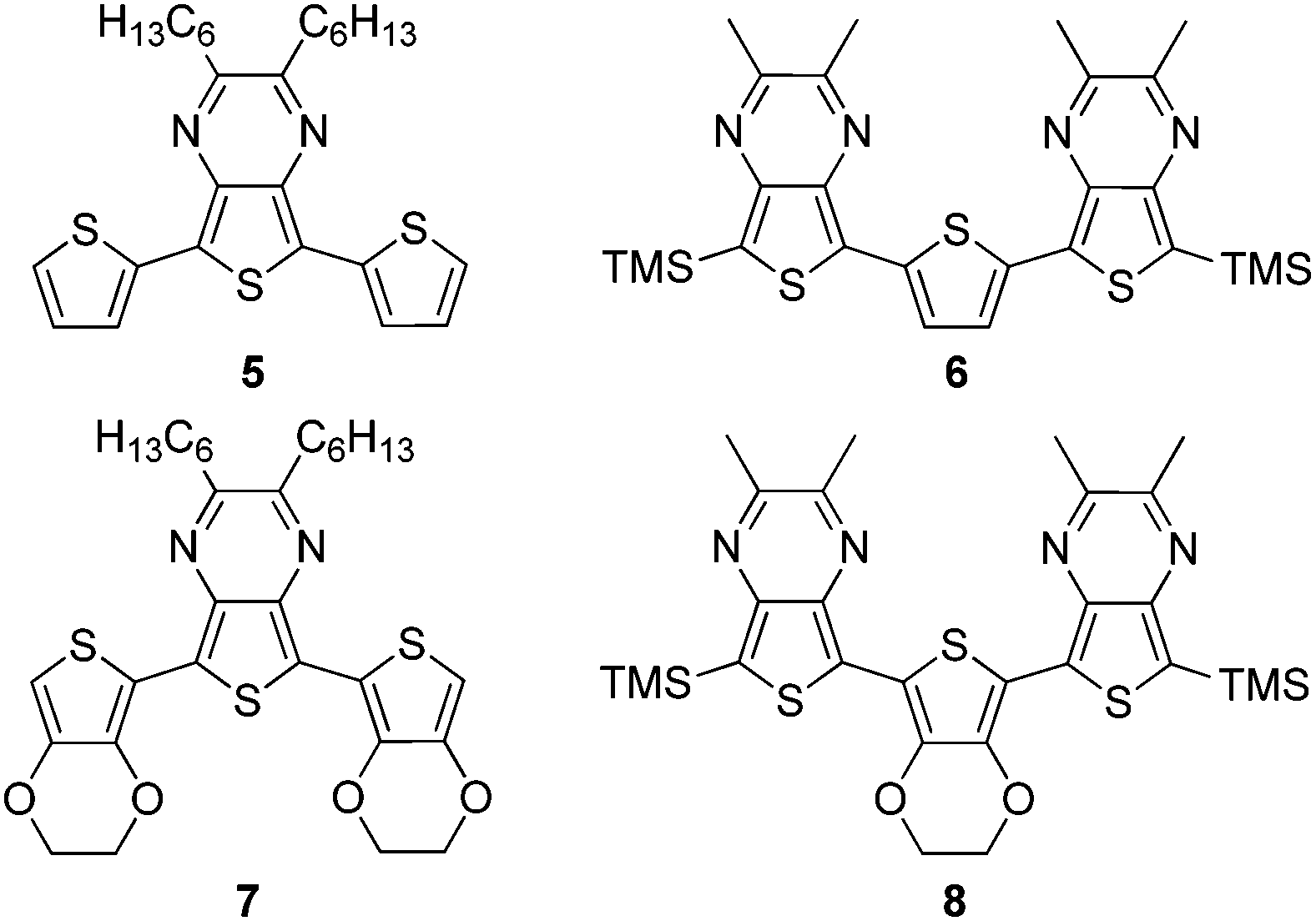 | ||
| Fig. 3 Mixed thieno[3,4-b]pyrazine-based terthienyls. | ||
Results and discussion
Synthesis
Two series of TMS-capped TP oligomers from monomer to trimer were successfully synthesized via Stille cross-coupling34 as shown in Scheme 1. As can be seen in Fig. 1, it is common to utilize substituents in the terminal α-positions of the oligothiophenes of interest. These α-substituents can serve as solubilizing groups to more readily allow solution measurements, as well as to block the reactive terminal positions, thus increasing the stability of reactive oligomers sensitive to oxidative coupling.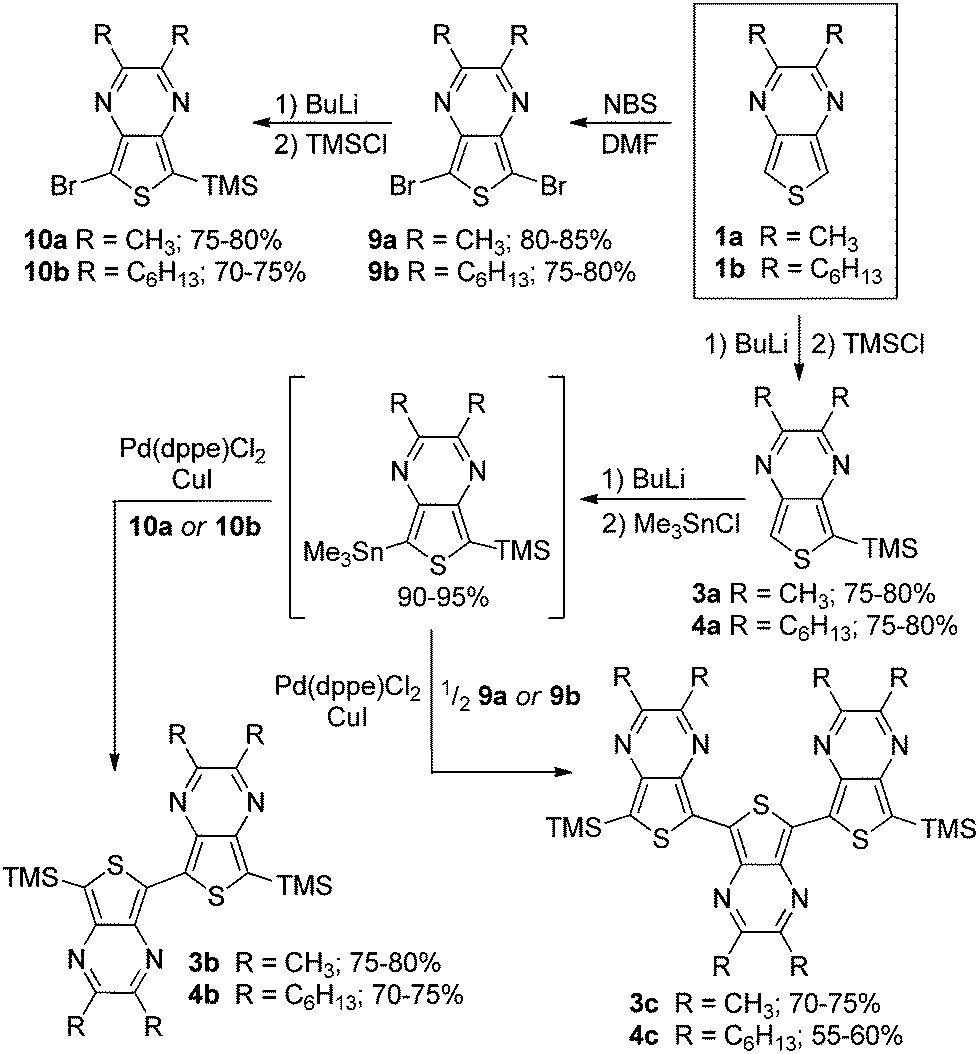 | ||
| Scheme 1 Synthesis of TMS-capped oligothieno[3,4-b]pyrazines. | ||
Due to the reactivity of the TP oligomers, particularly to oxidation, terminal capping groups were indeed necessary in order to allow successful isolation and study of the desired oligomeric products. However, knowing that the inclusion of such terminal α-substituents would also contribute added effects to the final oligomer electronic and optical properties, the choice of capping group was important in order to limit these effects and provide the clearest relationship between oligomer structure and electronic properties. As such, it was decided to utilize TMS groups as these exhibit the smallest electronic effects of the commonly applied capping groups (somewhere between that of a proton and an alkyl group).13,20,35 It should be pointed out that the use of terminal TMS protecting groups does not exclude polymerization via oxidative desilylation,16b but it does stabilize the oligomers enough to allow successful isolation and purification.
The desired stannyl precursors needed for the Stille cross-coupling were successfully produced through sequential treatment of the TMS-functionalized TPs (3a, 4a) with butyl lithium and chlorotrimethylstannane (Me3SnCl) as shown in Scheme 1. Attempts to isolate and purify these intermediates, however, failed due to a lack of sufficient stability. As NMR analysis of the crude intermediate after simple solvent removal indicated efficient conversion with no significant TP-based by-products (see NMR in ESI†), it was determined that full isolation was not necessary and the residue obtained after solvent removal was used without further workup. The desired products could thus be produced via a modified one-pot method in which the stannyl intermediate was redissolved in toluene and the remaining reagents added for the palladium-catalyzed cross-coupling.
The TP oligomers could be produced in this fashion in moderate to good yields, providing the additional use of a CuI co-catalyst.36,37 In the case of the trimer reactions, coupling attempts without the co-catalyst resulted in a single aryl–aryl couple to produce the bromobithienyl intermediate in low yield. According to previous research,37 the addition of CuI can result in a 100-fold increase in the reaction rate, thus accounting for the increased reactivity in the current application. It is believed that this rate increase is due to reaction between the copper salt and the stannyl intermediate to form an organocopper species which can undergo transmetallation with the palladium catalyst much faster than the original stannyl species. In order to further probe the contribution of TP units in ‘donor–acceptor’ materials, a series of mixed terthienyls were also prepared via similar methods (Scheme 2).
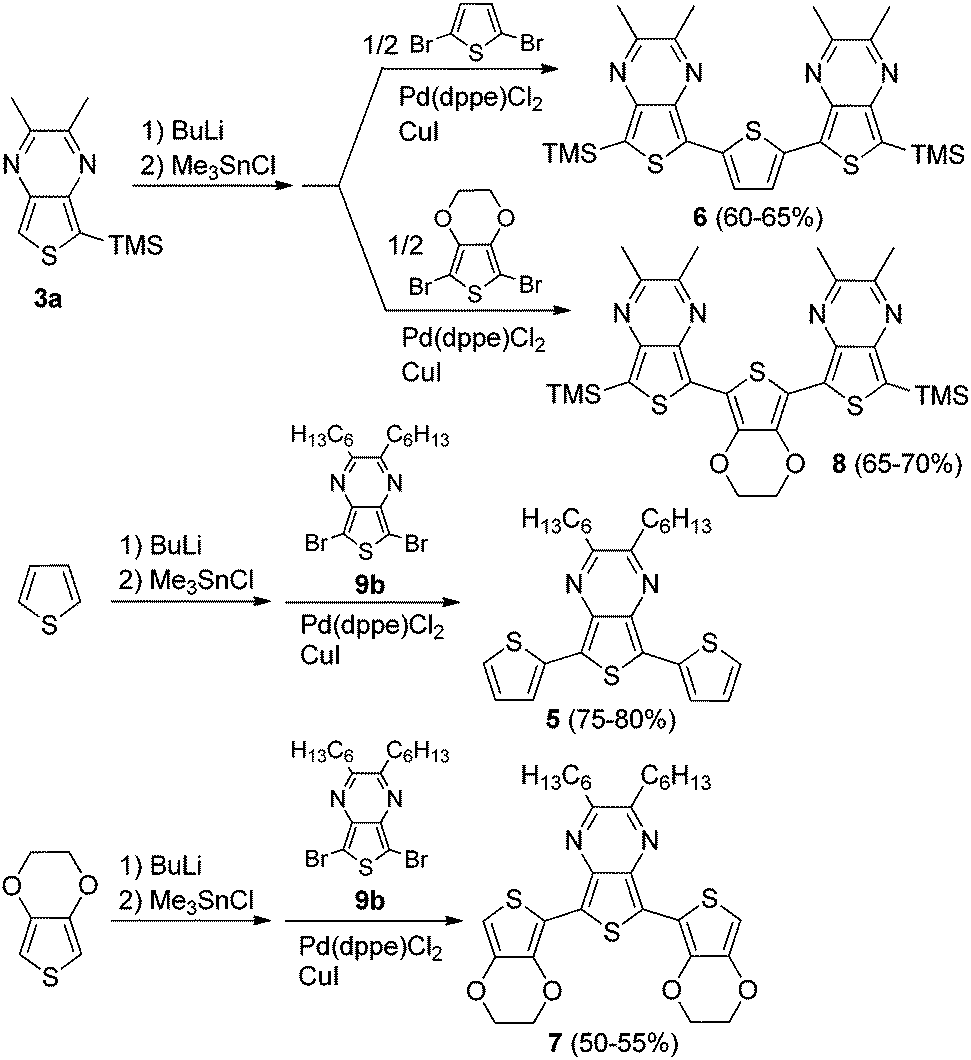 | ||
| Scheme 2 Synthesis of mixed TP-containing terthienyls. | ||
Absorption properties of oligothieno[3,4-b]pyrazines
The full absorption data for both series of oligothieno[3,4-b]-pyrazines are given in Table 1 and representative UV-vis spectra of 3a–c are shown in Fig. 4. As is typical of oligothiophene series, increasing the conjugation length in both sets of oligomers results in lower energy transitions and increased extinction coefficients. In the case of the series 4a–c, the longer hexyl side chains results in more conformational flexibility, resulting in a loss of vibrational structure and broadening of the spectra.| Oligomer | λ max (nm) | ε (M−1 cm−1) | f | Oligomer | λ max (nm) | ε (M−1 cm−1) | f |
|---|---|---|---|---|---|---|---|
| a Measured from dilute CHCl3 solutions in 1 cm quartz cuvettes. (sh) = shoulder. | |||||||
| 3a | 236 | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
0.34 | 4a | 234 | 19600 |
0.31 |
| 294 (sh) | 6400 | 0.32 | 294 (sh) | 6800 | 0.32 | ||
| 306 | 10700 |
307 | 10600 |
||||
| 318 | 11200 |
316 | 11000 |
||||
| 350 | 3190 | 0.05 | 350 | 3100 | 0.05 | ||
| 3b | 257 | 22000 |
0.67 | 4b | 257 | 21800 |
0.57 |
| 303 | 18000 |
0.31 | 304 | 17000 |
0.32 | ||
| 333 (sh) | 9400 | 331 (sh) | 9500 | ||||
| 504 | 7400 | 0.15 | 503 | 7300 | 0.15 | ||
| 539 (sh) | 4700 | 534 (sh) | 4500 | ||||
| 3c | 272 | 23700 |
0.83 | 4c | 274 | 22700 |
0.79 |
| 340 | 11800 |
0.73 | 340 (sh) | 12000 |
0.63 | ||
| 573 (sh) | 7600 | 640 (sh) | 8500 | ||||
| 625 | 10000 |
0.44 | 674 | 9000 | 0.46 | ||
| 668 (sh) | 8800 | ||||||
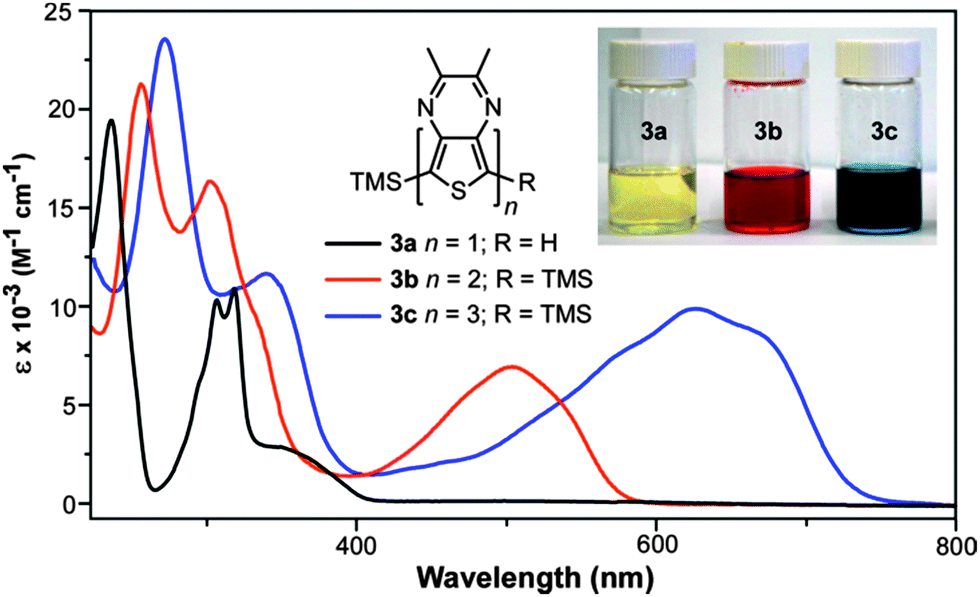 | ||
| Fig. 4 Solution UV-visible spectra for oligomer series 3a–c. | ||
Previous photophysical studies have shown that the lowest energy absorption of monomeric TPs is a broad intramolecular charge transfer (ICT) band resulting from a transition between a predominately thiophene-localized HOMO and a LUMO of greater pyrazine contribution.21,38,39 As can be seen in 3a, this broad band occurs at 350 nm and overlaps with higher energy absorptions assigned as conventional π–π* transitions. All of the higher oligomers exhibit a similar broad, low energy transition, although significantly red-shifted by approximately 120–150 nm with each additional TP unit. As with the monomeric species, this transition is formally assigned as an ICT band, which is also consistent with the assignments given to the first transition of the related TP-based terthienyls such as compound 5.31
To further investigate the optical properties of the oligothieno-[3,4-b]pyrazine series, the electronic density contours for the frontier molecular orbitals and the lowest-energy electronic transition energies were calculated using a combination of HF and ZINDO methods. As can be seen in Fig. 5, the HOMO resides predominately on the thiophene backbone and becomes increasingly localized with increasing oligomer length. In contrast, the pyrazine rings of the TP units fully contribute to the LUMO thus resulting in a HOMO–LUMO transition with significant ICT nature. As shown in Table 2, the ZINDO calculated transition energies are all red-shifted in comparison to the experimental λmax values, falling between the corresponding λmax and onset energies. The general trends both in calculated energies and oscillator strengths, however, are in good agreement with the experimentally measured values.
 | ||
| Fig. 5 Electronic density contours, calculated at HF/6-31G* level, for the HOMO and LUMO of 3b and 3c (TMS groups omitted for clarity). | ||
| Oligomer | Observed (in CHCl3) | ZINDO | |||
|---|---|---|---|---|---|
| λ max (nm) | λ onset (nm) | f | E calc (nm) | f | |
| 1a | 350 | 405 | 0.05 | 396 | 0.01 |
| 3b | 504 | 575 | 0.15 | 551 | 0.56 |
| 3c | 625 | 729 | 0.44 | 697 | 0.80 |
As can be seen in Table 3, the absorptions of TP oligomers are significantly red-shifted in comparison to analogous thiophene or EDOT oligomers, with the TP monomers exhibiting transition energies similar to either dimers or trimers in the other series. This drastic difference can be attributed to the donor–acceptor nature of the TP-based oligomers, thus resulting in low energy ICT transitions. Comparing the two TP-based series, the lowest energy transitions of the TP oligomers reported here are all blue-shifted by approximately 120 nm relative to the analogous thiophene-capped oligomer series reported by Janssen and co-workers (2a–c, Fig. 2, Table 3).24 Thus, in the series 2a–c, each thiophene end-capping unit results in a 60 nm red-shift as a result of their contributions to the conjugation length. In contrast, the TMS endcaps utilized in the series 3a–c and 4a–c appear to contribute very little to the corresponding optical properties.
| Oligomer series | n | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| a In CHCl3; th = thiophene. b Ref. 6. c Ref. 13. d Ref. 20 in CH2Cl2. e Value for parent EDOT; H. J. Spencer, P. J. Skabara, M. Giles, I. McCulloch, S. J. Coles, and M. B. Hursthouse. J. Mater. Chem., 2005, 15, 4783. f Compounds 3a–c reported herein. g Ref. 24 in CH2Cl2. | |||
| H–[th]n–Hb | 231 | 305 | 360 |
| TMS–[th]n–TMSc | 248 | 320 | 368 |
| TMS–[EDOT]n–TMSd | 258e | 350 | 412 |
| TMS–[TP]n–TMSf | 350 | 504 | 625 |
| th–[TP]n–thg | 479 | 620 | 745 |
The contribution of the TMS groups can be evaluated by direct comparison of known TMS-capped oligothiophenes13 to the unfunctionalized parents. As shown in Table 3, the TMS groups result in a red-shift of 8–17 nm, which is believed to be due to conjugation between thiophene and the terminal silicon units via dπ–pπ overlap.13 However, it should also be noted that the observed red-shift decreases with oligomer size and thus conjugation length. To further probe the effect on the TP-based oligomers studied here, the absorption spectra of 1a and 3a were directly compared to reveal no observable shifts in either the onset or λmax of the ICT transition at 350 nm (Fig. 6). However, the addition of the TMS group does increase the intensity of this transition. Small shifts in the higher energy π–π* transitions can be observed, but these shifts are still only 1–3 nm. This is comparable to the magnitude of red shift found for terthiophene (Table 3), which exhibits transitions of similar energy to that of the TP monomers. If the trend observed in the oligothiophene series holds true, one could expect the effect of the TMS group to be even further diminished in the larger TP oligomers. As such, the TMS-end-capped series reported here are as close to the parent, unfunctionalized oligomers as possible while still retaining enough chemical stability to allow significant characterization.
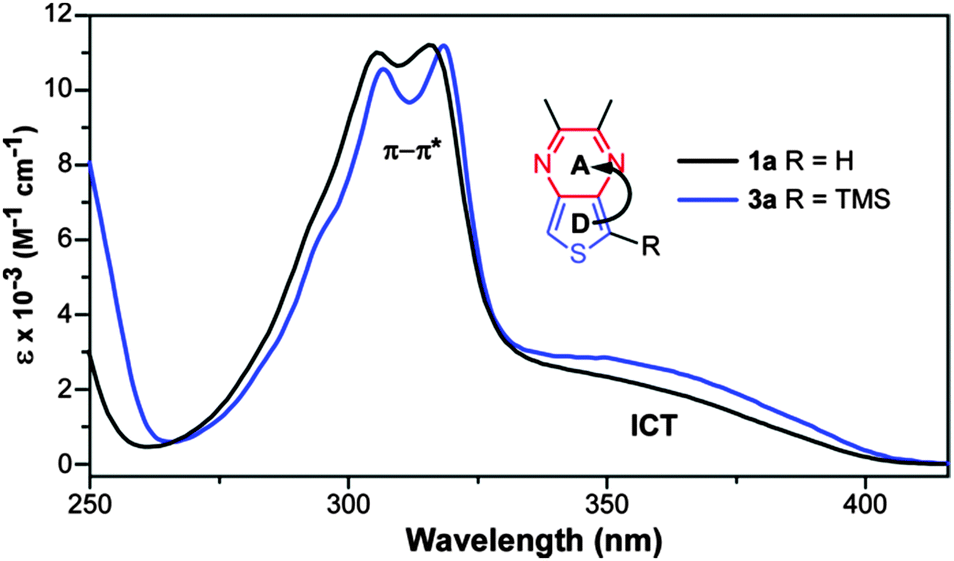 | ||
| Fig. 6 Comparison of the solution UV-vis spectra of 1a and 3a in order to determine the effect of TMS group on the optical transitions. | ||
Finally, in order to predict the optical properties of an infinite polythieno[3,4-b]pyrazine, the absorption onsets of the oligomeric series were extrapolated to predict a value of 1.07 eV for the polymeric solution onset. In comparison, Karsten and Janssen report the slightly higher value of 1.13 eV from the oligomeric series 2a–c.24 Both predicted values agree well with the experimental solution onsets for either FeCl3-polymerized or GRIM-polymerized polythieno[3,4-b]pyrazines (∼1.15 eV).40 However, these materials are of relatively low molecular weights (Mn of 4300–4900).40 In contrast, insoluble electropolymerized materials have produced significantly lower band gaps (∼0.7 vs. 0.93–0.95 eV) and are assumed to be of much higher molecular weights.22,40
Electrochemistry of oligothieno[3,4-b]pyrazines
The collected electrochemical data for both series of oligothieno[3,4-b]pyrazines are given in Table 4 and representative cyclic voltammograms of 4a–c are shown in Fig. 7. In all cases, the oligomers exhibit a well-defined, irreversible oxidation assigned to the oxidation of the oligothiophene backbone, as well as a quasireversible pyrazine-based reduction. As for the majority of thiophene species, the oxidation here is due to the formation of thiophene-based radical cations. Normally, such oxidations in oligothiophenes without endcapping groups are irreversible due to rapid coupling of these radical cations to produce higher oligomeric and polymeric species. The fact that the oxidations observed here are still irreversible, even with the use of the TMS end-groups, strongly suggests the occurrence of electrochemical desilylation. Electrodesilylation has been previously reported with a resulting dimerization rate constant of 104 M−1 s−1 for the TMS-capped terthiophene in CH2Cl2.41 As such, electrodesilylation has been successfully applied to the production of polythiophenes via the electrochemical oxidation of various thienylsilanes.16,41,42| Oligomer | Oxidation | Reduction | HOMOb (eV) | LUMOc (eV) | |
|---|---|---|---|---|---|
| E pa (V) | E 1/2 (V) | ΔE (mV) | |||
|
a Measured in CH3CN with 0.10 M TBAPF6. All potentials vs. Ag/Ag+.
b
E
HOMO = −(E[onset,oxvs.Fc+/Fc] + 5.1) (eV).
c
E
LUMO = −(E[onset,redvs.Fc+/Fc] + 5.1) (eV).
d
Ref. 24, values converted from Fc/Fc+ to Ag/Ag+.
e Reversible, Epa listed for comparison.
|
|||||
| 2a | 0.46 | −2.01 | 260 | −5.34 | −3.17 |
| 2b | 0.13e | −1.76 | 120 | −5.00 | −3.38 |
| 2c | −0.08e | −1.60 | 90 | −4.85 | −3.51 |
| 3a | 1.33 | −2.03 | 90 | −6.31 | −3.15 |
| 3b | 0.50 | −1.83 | 80 | −5.45 | −3.37 |
| 3c | 0.15 | −1.64 | 100 | −5.07 | −3.55 |
| 4a | 1.34 | −2.01 | 110 | −6.31 | −3.15 |
| 4b | 0.50 | −1.83 | 110 | −5.45 | −3.37 |
| 4c | 0.15 | −1.64 | 110 | −5.07 | −3.55 |
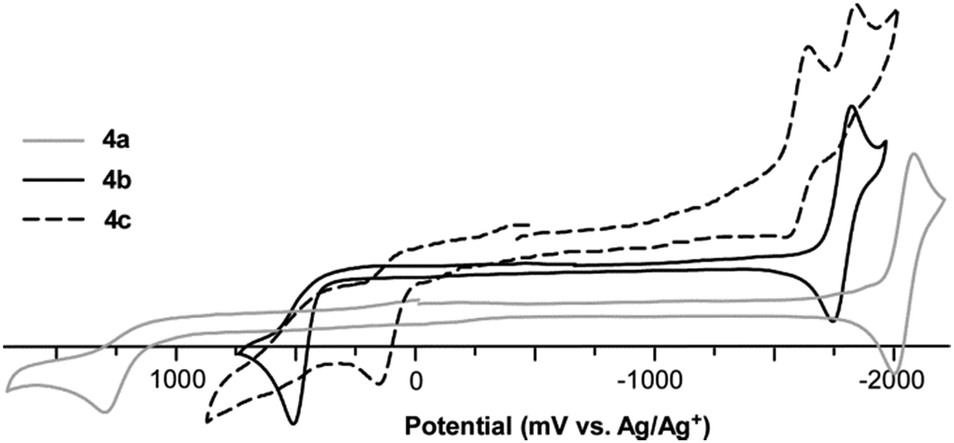 | ||
| Fig. 7 Cyclic voltammograms for oligomer series 4a–c. | ||
In comparison to the previously reported thiophene-capped oligomer series reported by Janssen and co-workers (2a–c, Fig. 2, Table 4),24 the TMS-capped oligothieno[3,4-b]pyrazines exhibit higher potentials of oxidation. This is not surprising as the thiophene endcaps in 2a–c contribute significantly to the oligomer backbones, resulting in destabilization of the corresponding HOMO energies. In contrast, the potentials for the oligomer reductions are very similar between the TMS- and thiophene-capped series. As the LUMO of these TP-based oligomers has more significant contribution from the pyrazine ring of the TP unit,21,22,31,38 the application of thiophene endgroups has relatively little effect on the corresponding LUMO.
Optical and electronic properties of mixed terthienyls
In order to better understand the extent and nature of ‘donor–acceptor’ properties in TP systems, series of mixed terthienyls with varying ratios of TP to either thiophene or EDOT (5–8, Fig. 3 and Scheme 2) were also investigated and their absorption and electrochemical data are given in Tables 5 and 6. As with the oligothieno[3,4-b]pyrazines discussed above, as well as previously reported TP-based terthienyls analogous to terthienyl 5,31 all of the mixed terthienyls exhibit a broad, low energy ICT transition, followed by higher energy transitions of a more conventional π–π* nature. The energy of the ICT transitions in terthienyls 5 and 7 are in good agreement with previously reported values for these two oligomers.43,44| Oligomer | λ max (nm) | ε (M−1 cm−1) | f | Oligomer | λ max (nm) | ε (M−1 cm−1) | f |
|---|---|---|---|---|---|---|---|
| a Measured from dilute CHCl3 solutions in 1 cm quartz cuvettes. (sh) = shoulder. | |||||||
| 5 | 307 | 16800 |
0.30 | 6 | 252 | 18400 |
0.24 |
| 330 (sh) | 11200 |
0.16 | 313 | 13000 |
0.22 | ||
| 342 | 11900 |
0.12 | 334 (sh) | 11600 |
0.30 | ||
| 357 (sh) | 9400 | 0.16 | 456 | 11700 |
0.52 | ||
| 505 | 6800 | 0.28 | 527 (sh) | 5800 | 0.22 | ||
| 7 | 250 | 13500 |
0.74 | 8 | 261 | 22800 |
0.32 |
| 316 | 22000 |
0.58 | 330 | 14500 |
0.10 | ||
| 338 (sh) | 13100 |
0.14 | 357 (sh) | 10800 |
0.22 | ||
| 355 | 16800 |
0.36 | 479 | 9300 | 0.50 | ||
| 373 | 14700 |
0.16 | 548 (sh) | 7900 | 0.24 | ||
| 537 | 7800 | 0.28 | |||||
| Oligomer | Oxidation | Reduction | HOMOb (eV) | LUMOc (eV) | |
|---|---|---|---|---|---|
| E pa (V) | E 1/2 (V) | ΔE (mV) | |||
|
a Measured in CH3CN with 0.10 M TBAPF6. All potentials vs. Ag/Ag+.
b
E
HOMO = −(E[onset,oxvs.Fc+/Fc] + 5.1) (eV).
c
E
LUMO = −(E[onset,redvs.Fc+/Fc] + 5.1) (eV).
d Irreversible, Epc reported.
|
|||||
| 5 | 0.50 | −1.75 | 70 | −5.46 | −3.39 |
| 6 | 0.42 | −1.91d | — | −5.34 | −3.32 |
| 7 | 0.19 | −1.82 | 90 | −5.11 | −3.27 |
| 8 | 0.18 | −2.05d | — | −5.09 | −3.20 |
In comparison to these more conventional terthienyls, both of the ‘inverted’ terthienyls 6 and 8 (containing a central thiophene or EDOT with external TP units) exhibit two broad, overlapping low energy transitions. Due to this overlap, the lower energy transition appears as a shoulder of the higher energy peak. However, due to the large energetic separation of the corresponding λmax, these have been assigned as separate transitions rather than vibrational components of the same transition. The lower energy transitions are red-shifted by ∼25 nm in comparison to their analogous conventional terthienyls 5 and 7.
In terms of electrochemistry, the mixed terthienyls exhibit redox characteristics similar to the oligothieno[3,4-b]pyrazines described above, consisting primarily of irreversible oxidations and quasireversible reductions, and the electrochemical data for terthienyls 5 and 7 are in good agreement with previously reported values for these two oligomers.43,44 The only significant deviation from these general trends is that both terthienyls containing external TP units (i.e.6 and 8), exhibit irreversible reductions. The reason for this is unclear as the terthieno[3,4-b]-pyrazines 3c and 4c containing both external TP units and TMS capping units both showed quasireversible reductions.
Across the series, the EDOT-containing oligomers are both easier to oxidize and harder to reduce in comparison to the thiophene analogues, both of which are consistent with the more electron-rich nature of EDOT in comparison to thiophene. As can be seen in Table 6, however, the effect of thiophene vs. EDOT is much more significant on the corresponding HOMO energies, with only minor effects on the resulting LUMO energies. This observed effect would be consistent with the reductions being primarily pyrazine-based as described previously above.
Effects of TP content on optical and electronic properties
In order to better understand the extent and nature of ‘donor–acceptor’ interactions in TP-based materials, terthienyl series differing only in the ratio of TP to either thiophene or EDOT were directly compared as shown in Tables 7 and 8. Comparing first the thiophene series in Table 7, it can be seen that an increase in TP content results in a significant red-shift of the lowest energy absorption, along with destabilization of the HOMO. This shift in HOMO energies suggests that TP acts as a more electron-rich ‘donor’ than thiophene, which is consistent with the fact that the TP monomer undergoes oxidation at much lower potentials than thiophene (∼1.3 V vs. Ag/Ag+ for TP21,38,45 in comparison to ∼2.0 V vs. Ag/Ag+ for thiophene46). In terms of the LUMO energies, the addition of the first TP unit results in a very large stabilization of the LUMO, consistent with the ‘acceptor’ nature of this unit. The addition of a second TP unit results in only moderate stabilization of the LUMO, potentially due to the fact that the TP units in 6 are separated by the central thiophene. The addition of the third TP unit then provides more significant stabilization and the large red-shift for the absorbance of 3c is thus due to both the high HOMO and low LUMO resulting from the high TP content. This conclusion agrees well with the previous statement of Janssen and coworkers that the TP unit is both a better donor and a better acceptor than thiophene.32| Oligomer | TP:tha |
%TP | λ max (nm) | HOMOc (eV) | LUMOd (eV) |
|---|---|---|---|---|---|
|
a th = thiophene.
b In CHCl3.
c
E
HOMO = −(E[onset,oxvs.Fc+/Fc] + 5.1) (eV).
d
E
LUMO = −(E[onset,redvs.Fc+/Fc] + 5.1) (eV).
e
Ref. 13.
f
Ref. 41.
g Estimated from EHOMO and λmax.
|
|||||
| 3c | 3:0 |
100 | 625 | −5.07 | −3.55 |
| 6 | 2:1 |
66.6 | 527 | −5.34 | −3.32 |
| 5 | 1:2 |
33.3 | 505 | −5.46 | −3.39 |
| TMS–[th]3–TMS | 0:3 |
0.0 | 368e | −5.79f | −2.42g |
| Oligomer | TP:EDOT |
%TP | λ max (nm) | HOMOb (eV) | LUMOc (eV) |
|---|---|---|---|---|---|
|
a In CHCl3.
b
E
HOMO = −(E[onset,oxvs.Fc+/Fc] + 5.1) (eV).
c
E
LUMO = −(E[onset,redvs.Fc+/Fc] + 5.1) (eV).
d R = TMS, ref. 20.
e R = H, ref. 16a.
f Estimated from EHOMO and λmax.
|
|||||
| 3c | 3:0 |
100 | 625 | −5.07 | −3.55 |
| 8 | 2:1 |
66.6 | 548 | −5.09 | −3.20 |
| 7 | 1:2 |
33.3 | 537 | −5.11 | −3.27 |
| R–[EDOT]3–R | 0:3 |
0.0 | 412d | −5.19e | −2.18f |
Comparison of the EDOT series in Table 8 reveals that the replacement of TP with EDOT results in a slight stabilization of the HOMO, suggesting that TP is even a slightly better ‘donor’ than the very electron-rich EDOT. Again, comparing the potentials of oxidation of the monomeric units shows that they are quite similar, although the reported oxidation of EDOT is slightly less than that of TP (∼1.3 V vs. Ag/Ag+ for TP21,38,45 and ∼1.2 V vs. Ag/Ag+ for EDOT16). The remaining trends in the EDOT series mirror that of the thiophene series. However, as the effects on the HOMO are relatively small here, the majority of the red shift in absorption in this case is due to the stabilization of the LUMO by the TP units.
Due to the dual ‘donor’ and ‘acceptor’ nature of the TP unit, we have proposed calling such units ambipolar building blocks and the observed trends in ‘donor–acceptor’ systems utilizing TP as the ‘acceptor’ can now be understood as a consequence of this ambipolar nature. The combination of TPs with various ‘donor’ units is expected to result in a destabilization of the HOMO as a result of the electron-rich ‘donor’. However, in nearly all cases the TP is actually the stronger donor of the two and thus the combination instead reduces the donor character of the backbone, resulting in a stabilized HOMO. This stabilization then leads to an increase in the energy of the ICT transition and thus a larger Eg. This overall effect can be easily seen by the fact that in over 100 materials reviewed, the general trend is that the material band gap increases as the TP content decreases.22
Experimental methods
Unless noted, all materials were reagent grade and used without further purification. 2,3-Dimethylthieno[3,4-b]pyrazine (1a),38,45 2,3-dihexylthieno[3,4-b]pyrazine (1b),38,45 5,7-dibromo-2,3-dihexylthieno[3,4-b]pyrazine (9b),40 and 2,5-dibromo-3,4-ethylene-dioxythiophene47 were prepared according to previously reported literature procedures. Dry THF and toluene were obtained via distillation over sodium/benzophenone. DMF was dried by passing through a silica plug. All glassware was oven-dried, assembled hot, and cooled under a dry nitrogen stream before use. Transfer of liquids was carried out using standard syringe techniques and all reactions were performed under dry N2. Chromatographic separations were performed using standard column chromatography methods with silica gel (230–400 mesh), unless otherwise stated. Melting points were determined using a digital thermocouple with 0.1 °C resolution. 1H and 13C NMR spectra were recorded in CDCl3 on a 400 MHz spectrometer and referenced to the chloroform signal. HRMS (ESI) was performed in house.5,7-Dibromo-2,3-dimethylthieno[3,4-b]pyrazine (9a)
Precursor 1a (4.0 mmol) was dissolved in DMF (120 mL) and the solution cooled to −40 °C. A solution of NBS (1.78 g, 10.0 mmol) in DMF (40 mL) was then added dropwise, after which the combined mixture was stirred at −25 °C for 3.5 h. The mixture was poured onto ice and the precipitated product isolated by filtration. The isolated solid was washed with H2O, dried under vacuum for 6 h, and used without any further purification. 80–85% yield. mp 112.6 °C dec. 1H NMR: δ 2.66 (s, 6H); 13C NMR: δ 155.4, 139.6, 103.4, 23.7. NMR values agree with previously reported values.48General procedure for synthesis of 2,3-dialkyl-5-(trimethyl-silyl)thieno[3,4-b]pyrazines
The desired TP (2.0 mmol) was dissolved in dry THF (80 mL) and cooled to −78 °C. BuLi (2.5 M in hexanes, 0.9 mL, 2.2 mmol) was added and the mixture stirred for 30 min. Chlorotrimethylsilane (0.24 g, 2.2 mmol) was then added and stirring continued for 30 min. The mixture was allowed to warm to room temperature and stirred for 3 h. Water was added and the mixture extracted with CHCl3. The organic layers were combined, dried with Na2SO4, and concentrated by rotary evaporation.2,3-Dimethyl-5-(trimethylsilyl)thieno[3,4-b]pyrazine (3a)
The crude product was purified by column chromatography (15% EtOAc in hexane). 75–80% yield. mp 91.2–92.9 °C. 1H NMR: δ 7.97 (s, 1H), 2.60 (s, 3H), 2.59 (s, 3H), 0.47 (s, 9H); 13C NMR: δ 152.7, 151.8, 148.3, 143.8, 130.9, 121.0, 24.0, 23.8, 0.1; HRMS: m/z 237.0885 [MH+] (calcd for C11H17N2SSi 237.0876).2,3-Dihexyl-5-(trimethylsilyl)thieno[3,4-b]pyrazine (4a)
The crude product was purified by column chromatography (5% EtOAc in hexane). 75–80% yield. 1H NMR: δ 7.99 (s, 1H), 2.89 (m, 4H), 1.88 (m, 2H), 1.78 (m, 2H), 1.47 (m, 4H), 1.36 (m, 8H), 0.91 (m, 6H), 0.48 (s, 9H); 13C NMR: δ 156.1, 154.4, 148.1, 143.5, 130.7, 121.1, 35.9, 35.2, 32.1, 31.9, 29.7, 29.3, 28.6, 27.1, 22.9, 22.8, 14.3, 14.3, 0.1.General procedure for synthesis of 2,3-dialkyl-5-bromo-7-(trimethylsilyl)thieno[3,4-b]pyrazines
The desired 5,7-dibromothieno[3,4-b]pyrazine (2.0 mmol) was dissolved in dry THF (80 mL) and cooled to −78 °C. BuLi (2.5 M in hexanes, 0.9 mL, 2.2 mmol) was then added and the mixture stirred for 30 min. Chlorotrimethylsilane (0.24 g, 2.2 mmol) was then added and stirring continued for 30 min. The mixture was allowed to warm to room temperature and stirred for 3 h. Water was added and the mixture extracted with CHCl3. The organic layers were combined, dried with Na2SO4, and concentrated by rotary evaporation. The crude product was then purified by column chromatography (10% EtOAc in hexanes).5-Bromo-2,3-dimethyl-7-(trimethylsilyl)thieno[3,4-b]pyrazine (10a)
75–80% yield. 1H NMR: δ 2.64 (s, 3H), 2.61 (s, 3H), 0.46 (s, 9H); 13C NMR: δ 154.3, 152.3, 147.6, 145.5, 137.7, 107.4, 23.9, 23.6, 0.1.5-Bromo-2,3-dihexyl-7-(trimethylsilyl)thieno[3,4-b]pyrazine (10b)
70–75% yield. 1H NMR: δ 2.96 (t, J = 7.2 Hz, 2H), 2.89 (t, J = 7.2 Hz, 2H), 1.86 (p, J = 7.2 Hz, 2H), 1.80 (p, J = 7.2 Hz, 2H), 1.46 (m, 4H), 1.36 (m, 8H), 0.89 (m, 6H), 0.49 (s, 9H); 13C NMR: δ 157.3, 155.1, 147.3, 141.0, 131.2, 108.7, 35.8, 35.0, 32.1, 31.9, 29.6, 29.3, 28.3, 27.1, 22.9, 22.8, 14.3, 0.2.General procedure for synthesis of trimethylsilyl-capped thieno[3,4-b]pyrazine dimers
Precursor 3a or 4a (2.0 mmol) was dissolved in dry THF (80 mL) and cooled to −78 °C. BuLi (2.5 M in hexanes, 0.9 mL, 2.2 mmol) was added and the mixture stirred for 30 min. Me3SnCl (1.0 M in THF, 2.2 mL, 2.2 mmol) was then added and stirred for another 30 min. The mixture was allowed to warm to room temperature and stirring continued for an additional 2 h. Pd(dppe)Cl2 (0.058 g, 0.10 mmol), CuI (0.019 g, 0.10 mmol), and either 10a or 10b (2.0 mmol) were then added and the mixture heated at reflux for 20 h. After cooling to room temperature, H2O was added and the mixture extracted with CHCl3. The combined organic layers were then dried with Na2SO4 and concentrated by rotary evaporation.2,2′,3,3′-Tetramethyl-7,7′-bis(trimethylsilyl)-5,5′-bis-(thieno[3,4-b]pyrazine) (3b)
The crude product was purified by column chromatography (10% EtOAc in hexanes). 75–80% yield. mp 158.3 °C dec. 1H NMR: δ 2.72 (s, 6H), 2.62 (s, 6H), 0.52 (s, 18H); 13C NMR: δ 152.2, 152.0, 148.6, 140.2, 130.6, 129.5, 24.0, 23.9, 0.2; HRMS: m/z 471.1524 [MH+] (calcd for C22H31N4S2Si2 471.1523).2,2′,3,3′-Tetrahexyl-7,7′-bis(trimethylsilyl)-5,5′-bis(thieno[3,4-b]pyrazine) (4b)
The crude product was purified by column chromatography (2% EtOAc in hexanes). 70–75% yield. mp 106.5–107.4 °C. 1H NMR: δ 2.97 (t, J = 7.2 Hz, 4H), 2.90 (t, J = 7.2 Hz, 4H), 2.12 (p, J = 7.2 Hz, 4H), 1.89 (p, J = 7.2 Hz, 4H), 1.48 (m, 8H), 1.38 (m, 16H), 0.93 (m, 12H), 0.50 (s, 18H); 13C NMR: δ 154.9, 154.6, 148.1, 139.7, 130.9, 129.0, 35.2, 35.1, 32.4, 32.2, 29.4, 29.4, 27.0, 26.9, 22.9, 14.4, 0.2; HRMS: m/z 751.4688 [MH+] (calcd for C42H71N4S2Si2 751.4653).General procedure for synthesis of trimethylsilyl-capped terthienyls
Precursor 3a or 4a (2.0 mmol) was dissolved in dry THF (80 mL) and cooled to −78 °C. BuLi (2.5 M in hexanes, 0.9 mL, 2.2 mmol) was added and the mixture stirred for 30 min. Me3SnCl (1.0 M in THF, 2.2 mL, 2.2 mmol) was then added and stirred for another 30 min. The mixture was then allowed to warm to room temperature and stirring continued for an additional 2 h. The reaction mixture was concentrated by rotary evaporation and then redissolved in toluene (60 mL). Pd(dppe)Cl2 (0.058 g, 0.10 mmol), CuI (0.019 g, 0.10 mmol), and the desired dihalothiophene (0.40 mmol) were then added and the mixture heated at reflux for 20 h. After cooling to room temperature, H2O was added and the mixture extracted with CHCl3. The combined organic layers were dried with Na2SO4 and concentrated by rotary evaporation.2,2′,2′′,3,3′,3′′-Hexamethyl-7,7′′-bis(trimethylsilyl)-5,5′:7′,5′′-ter(thieno[3,4-b]pyrazine) (3c).
Due to the instability of this terthienyl, it was not subjected to chromatography, but purified by hexane and EtOAc washes. 70–75% yield. 1H NMR: δ 2.78 (s, 6H), 2.77 (s, 6H), 2.64 (s, 6H), 0.53 (s, 18H); HRMS: m/z 633.1803 [MH+] (calcd for C30H37N6S3Si2 633.1775).2,2′,2′′,3,3′,3′′-Hexahexyl-7,7′′-bis(trimethylsilyl)-5,5′:7′,5′′-ter(thieno[3,4-b]pyrazine) (4c)
The crude product was purified by column chromatography (alumina, hexanes). 55–60% yield. 1H NMR: δ 3.01–2.82 (m, 12H), 2.13 (p, J = 7.2 Hz, 4H), 1.99 (p, J = 7.2 Hz, 4H), 1.87 (p, J = 7.2 Hz, 4H), 1.46 (m, 12H), 1.36–1.24 (m, 24H), 0.90 (m, 18H), 0.51 (s, 18H); HRMS: m/z 1052.6376 [M+] (calcd for C60H96N6S3Si2 1052.6392).5,5′-(Thiophene-2,5-diyl)bis[2,3-dimethyl-7-(trimethylsilyl)-thieno[3,4-b]pyrazine] (6)
The crude product was purified by column chromatography (2% EtOAc in hexanes). 60–65% yield. mp 179.6 °C dec. 1H NMR: δ 7.73 (s, 2H), 2.69 (s, 6H), 2.60 (s, 6H), 0.49 (s, 18H); 13C NMR: δ 152.4, 152.3, 146.4, 139.4, 135.8, 132.5, 126.6, 125.0, 24.0, 23.9, 0.2; HRMS: m/z 533.1412 [MH+] (calcd for C26H33N4S3Si2 533.1400).5,5′-(3,4-Ethylenedioxythiophene-2,5-diyl)bis[2,3-dimethyl-7-(trimethylsilyl)thieno[3,4-b]pyrazine] (8)
The crude product was purified by column chromatography (hexanes). 65–70% yield. mp 165.2 °C dec. 1H NMR: δ 4.55 (s, 4H), 2.73 (s, 6H), 2.61 (s, 6H), 0.49 (s, 18H); 13C NMR: δ 152.2, 150.8, 148.5, 139.6, 138.2, 130.4, 126.0, 124.6, 65.6, 24.3, 22.9, 0.3; HRMS: m/z 611.1487 [MH+] (calcd for C28H35N4Si2S3 611.1455).General procedure for synthesis of thienyl-capped thieno[3,4-b]pyrazines
Either thiophene or EDOT (2.0 mmol) was dissolved in dry THF (80 mL) and cooled to −78 °C. BuLi (2.5 M in hexanes, 0.9 mL, 2.2 mmol) was added and the mixture stirred for 30 min. Me3SnCl (1.0 M in THF, 2.2 mL, 2.2 mmol) was then added and stirred for another 30 min. The mixture was then allowed to warm to room temperature and stirring continued for an additional 2 h. The reaction mixture was concentrated by rotary evaporation and then redissolved in toluene (60 mL). Pd(dppe)Cl2 (0.058 g, 0.10 mmol), CuI (0.019 g, 0.10 mmol), and 9b (0.18 g, 0.40 mmol) were then added and the mixture heated at reflux for 20 h. After cooling to room temperature, H2O was added and the mixture extracted with CHCl3. The combined organic layers were dried with Na2SO4 and concentrated by rotary evaporation.2,3-Dihexyl-5,7-bis(2-thienyl)thieno[3,4-b]pyrazine (5)
The crude product was purified by column chromatography (2% EtOAc in hexanes). 75–80% yield. mp 116.2–116.8 °C (lit.43 116–116.5 °C). 1H NMR: δ 7.62 (dd, J = 1.2, 3.6 Hz, 2H), 7.35 (dd, J = 1.2, 5.2 Hz, 2H), 7.10 (dd, J = 3.6, 5.2 Hz, 2H), 2.92 (t, J = 7.2 Hz, 4H), 1.97 (p, J = 7.6 Hz, 4H), 1.51 (m, 4H), 1.41 (m, 8H), 0.94, (t, J = 7.2 Hz, 6H); 13C NMR: δ 156.6, 138.0, 135.3, 127.3, 126.2, 124.3, 124.0, 35.3, 32.1, 29.4, 27.2, 22.9, 14.3. All NMR agrees with previously reported values;42 HRMS: m/z 469.1813 [MH+] (calcd for C26H33N2S3: 469.1800).5,7-Bis(3,4-ethylenedioxythiophen-2-yl)-2,3-dihexylthieno[3,4-b]pyrazine (7)
The crude product was purified by column chromatography (5% EtOAc in hexanes). 50–55% yield. mp 120.3–121.9 °C. 1H NMR: δ 6.41 (s, 2H), 4.47 (m, 4H), 4.30 (m, 4H), 2.91 (t, J = 7.2 Hz, 4H), 1.98 (p, J = 7.4 Hz, 4H), 1.51 (m, 4H), 1.40 (m, 8H), 0.92 (t, J = 7.2 Hz, 6H); 13C NMR: δ 155.3, 141.4, 137.8, 137.0, 121.6, 111.9, 100.6, 65.6, 65.0, 35.1, 32.2, 29.4, 27.0, 23.0, 14.4. All NMR agrees with previously reported values;44 HRMS: m/z 584.1834 [M+] (calcd for C30H36N2O4S3 584.1832).Absorption spectroscopy
UV-vis spectroscopy was performed on a dual beam scanning UV-vis-NIR spectrophotometer using samples prepared as dilute CHCl3 solutions in 1 cm quartz cuvettes. The CHCl3 used in the spectroscopy measurements was dried over molecular sieves prior to use. Oscillator strengths were determined from the visible spectra via spectral fitting to accurately quantify the area of each transition and then calculated using literature methods.49Electrochemistry
All electrochemical methods were performed utilizing a three-electrode cell consisting of Pt disc working electrode, a Pt wire auxiliary electrode, and a Ag/Ag+ reference electrode (0.1 M AgNO3 and 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in CH3CN; 0.251 V vs. SCE).50 Supporting electrolyte consisted of 0.10 M TBAPF6 in spectrochemical grade CH3CN dried over molecular sieves. Solutions were deoxygenated by sparging with argon prior to each scan and blanketed with argon during the measurements. All measurements were collected at a scan rate of 100 mV s−1. EHOMO values were estimated from the onset of oxidation in relation to ferrocene (50 mV vs. Ag/Ag+), using the value of 5.1 eV vs. vacuum for ferrocene.51Theoretical methodology
Calculations were performed with Gaussian 03.52 Optimized geometries were calculated using HF methods and the 6-31G* basis set. A single point energy calculation using methods matching the geometry optimization followed. Molecular orbital diagrams were generated using GaussView 3.09 from the checkpoint file generated during the single point energy calculation and an isovalue of 0.022. In all calculations, very tight SCF convergence criteria were used and molecular symmetry was ignored. When stepwise calculations were performed, the trailing step used only the output geometry from the previous step and did not glean a guess from a previous checkpoint file. Excited state transitions and oscillator strengths were calculated using ZINDO.53Conclusions
A series of TP-based oligomers were synthesized via Stille cross-coupling as models of electronic structure–function relationships in TP-based conjugated materials. In addition to oligothieno[3,4-b]pyrazine series from monomer to trimer, series of mixed terthienyls were analysed in which the ratio of TP to either thiophene or EDOT has been varied. Characterization of the complete oligomeric series via photophysical, electrochemical, and theoretical calculations allowed correlation of the effect of conjugation length and oligomer composition with the resulting electronic and optical properties. Perhaps the most critical aspect revealed by this study is that while TP is commonly viewed as an ‘acceptor’ unit, it also acts as a quite strong ‘donor’ and is on par or slightly more electron-rich than EDOT. This ambipolar nature of the TP unit can now be used to fully understand observed structure–function relationships in both homopolymeric and copolymeric materials of thieno[3,4-b]pyrazines.Acknowledgements
The authors wish to thank the National Science Foundation (CHE-0132886, DMR-0907043) and North Dakota State University for support of this research.Notes and references
- S. C. Rasmussen, in 100+ Years of Plastics. Leo Baekeland and Beyond, ed. E. T. Strom and S. C. Rasmussen, ACS Symposium Series, American Chemical Society, Washington, DC, 2011, ch. 10 Search PubMed.
- A. Elschner, S. Kirchmeyer, W. Lovenich, U. Merker and K. Reuter, PEDOT, Principles and Applications of an Intrinsically Conductive Polymer, CRC Press, Boca Raton, FL, 2011, ch. 1 Search PubMed.
- Handbook of Conducting Polymers, ed. T. A. Skotheim and J. R. Reynolds, CRC Press, Boca Raton, FL, 3rd edn, 2007 Search PubMed.
- Handbook of Thiophene-based Materials, ed. I. F. Perepichka and D. F. Perepichka, John Wiley & Sons, Hoboken, NJ, 2009 Search PubMed.
- Handbook of Oligo- and Polythiophenes, ed. D. Fichou, Wiley-VCH, Weinheim, 1999 Search PubMed.
- J. Nakayama, T. Konishi and M. Hoshino, Heterocycles, 1988, 27, 1731 CrossRef CAS PubMed.
- P. Bäuerle, in The Handbook of Oligo- and Polythiophenes, ed. D. Fichou, Wiley-VCH, Weinheim, 1999, ch. 3 Search PubMed.
- (a) A. Mishra, C.-Q. Ma, J. L. Segura and P. Bäuerle, in The Handbook of Thiophene-based Materials, ed. I. F. Perepichka and D. F. Perepichka, John Wiley & Sons, Hoboken, NJ, 2009, ch. 1 Search PubMed; (b) A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141 CrossRef CAS PubMed.
- Z. Xu, D. Fichou, G. Horowitz and F. Garnier, J. Electroanal. Chem., 1989, 267, 339 CrossRef CAS.
- A. Facchetti, M.-H. Yoon, C. L. Stern, G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 13480 CrossRef CAS PubMed.
- (a) S. A. Lee, Y. Yoshida, M. Fukuyama and S. Hotta, Synth. Met., 1999, 106, 39 CrossRef CAS; (b) S. Hotta, S. A. Lee and T. Tamaki, J. Heterocycl. Chem., 2000, 37, 25 CrossRef CAS.
- G. Bidan, A. De Nicola, V. Enee and S. Guillerez, Chem. Mater., 1998, 10, 1052 CrossRef CAS.
- J. M. Tour and R. Wu, Macromolecules, 1992, 25, 1901 CrossRef CAS.
- (a) N. Sumi, H. Nakanishi, S. Ueno, K. Takimiya, Y. Aso and T. Otsubo, Bull. Chem. Soc. Jpn., 2001, 74, 979 CrossRef CAS; (b) T. Izumi, S. Kobashi, K. Takimiya, Y. Aso and T. Otsubo, J. Am. Chem. Soc., 2003, 125, 5286 CrossRef PubMed.
- P. Bäuerle, U. Segelbacker, A. Maier and M. Mehring, J. Am. Chem. Soc., 1993, 115, 10217 CrossRef.
- (a) G. A. Sotzing, J. R. Reynolds and P. J. Steel, Chem. Mater., 1996, 8, 882 CrossRef CAS; (b) G. A. Sotzing, J. R. Reynolds and P. J. Steel, Adv. Mater., 1997, 9, 795 CrossRef CAS.
- S. Akoudad and J. Roncali, Synth. Met., 1998, 93, 111 CrossRef CAS.
- M. Turbiez, P. Frère and J. Roncali, J. Org. Chem., 2003, 68, 5357 CrossRef CAS PubMed.
- J. J. Apperloo, L. B. Groenendaal, H. Verheyen, M. Jayakannan, R. A. J. Janssen, A. Dkhissi, D. Beljonne, R. Lazzaroni and J. L. Brédas, Chem.–Eur. J., 2002, 8, 2384 CrossRef CAS.
- D. Wasserberg, S. C. J. Meskers, R. A. J. Janssen, E. Mena-Osteritz and P. Bäuerle, J. Am. Chem. Soc., 2006, 128, 17007 CrossRef CAS PubMed.
- S. C. Rasmussen, M. E. Mulholland, R. L. Schwiderski and C. A. Larsen, J. Heterocycl. Chem., 2012, 49, 479 CrossRef CAS.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394 RSC.
- L. Wen and S. C. Rasmussen, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2007, 48(1), 132 CAS.
- B. P. Karsten and R. A. J. Janssen, Org. Lett., 2008, 10, 3513 CrossRef CAS PubMed.
- (a) E. E. Havinga, W. ten Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119 CrossRef CAS; (b) E. E. Havinga, W. ten Hoeve and H. Wynberg, Synth. Met., 1993, 55, 299 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396 CrossRef CAS PubMed.
- M. Kertesz, C. H. Choi and S. Yang, Chem. Rev., 2005, 105, 3448 CrossRef CAS PubMed.
- U. Salzner, O. Karalti and S. Durdagi, J. Mol. Model., 2006, 12, 687 CrossRef CAS PubMed.
- M. Kertesz, S. Yang and Y. Tian, in The Handbook of Thiophene-based Materials, ed. I. F. Perepichka and D. F. Perepichka, John Wiley & Sons, Hoboken, NJ, 2009, ch. 7 Search PubMed.
- P. Ou, W. Shen, R. He, X. Xie, C. Zeng and M. Li, Polym. Int., 2011, 60, 1408 CAS.
- R. L. Schwiderski and S. C. Rasmussen, J. Org. Chem., 2013, 78, 5453 CrossRef CAS PubMed.
- B. P. Karsten, L. Viani, J. Gierschner, J. Cornil and R. A. J. Janssen, J. Phys. Chem. A, 2009, 113, 10343 CrossRef CAS PubMed.
- B. P. Karsten, J. C. Bijleveld, L. Viani, J. Cornil, J. Gierschner and R. A. J. Janssen, J. Mater. Chem., 2009, 19, 5343 RSC.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 CAS.
- R. L. Wells, A. T. McPhail and T. M. Speer, Organometallics, 1992, 11, 960 CrossRef CAS.
- K. Osakada, R. Sakata and T. Yamamoto, Organometallics, 1997, 16, 5354 CrossRef CAS.
- V. Farina, S. Kapadia, B. Krishnan, C. Wang and L. S. Liebeskind, J. Org. Chem., 1994, 59, 5905 CrossRef CAS.
- L. Wen, J. P. Nietfeld, C. M. Amb and S. C. Rasmussen, J. Org. Chem., 2008, 73, 8529 CrossRef CAS PubMed.
- S. C. Rasmussen, D. J. Sattler, K. A. Mitchell and J. Maxwell, J. Lumin., 2004, 190, 111 CrossRef PubMed.
- L. Wen, B. C. Duck, P. C. Dastoor and S. C. Rasmussen, Macromolecules, 2008, 41, 4576 CrossRef CAS.
- P. Hapiot, L. Gaillon, P. Audebert, J. J. E. Moreau, J.-P. Lère-Porte and M. Wong Chi Man, J. Electroanal. Chem., 1997, 435, 85 CrossRef CAS.
- (a) M. Lemaire, W. Buchner, R. Gareau, H. A. Hoa, A. Guy and J. Roncali, J. Electroanal. Chem., 1990, 281, 293 CrossRef CAS; (b) H. Masuda, Y. Taniki and K. Kaeriyama, Synth. Met., 1993, 55–57, 1246 CrossRef.
- (a) C. Kitamura, S. Tanaka and Y. Yamashita, J. Chem. Soc., Chem. Commun., 1994, 1585 RSC; (b) C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570 CrossRef CAS.
- J. Casado, R. P. Ortiz, M. C. R. Delgado, V. Hernández, J. T. L. Navarrete, J.-M. Raimundo, P. Blanchard, M. Allain and J. Roncali, J. Phys. Chem. B, 2005, 109, 16616 CrossRef CAS PubMed.
- D. D. Kenning, K. A. Mitchell, T. R. Calhoun, M. R. Funfar, D. J. Sattler and S. C. Rasmussen, J. Org. Chem., 2002, 67, 9073 CrossRef CAS PubMed.
- S. C. Rasmussen, J. C. Pickens and J. E. Hutchison, Chem. Mater., 1998, 10, 1990 CrossRef CAS.
- Y. Zhu and M. O. Wolf, J. Am. Chem. Soc., 2000, 122, 10121 CrossRef CAS.
- J.-C. Li, E.-O. Seo, S.-H. Lee and Y.-S. Lee, Macromol. Res., 2010, 18, 304 CrossRef CAS PubMed.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991, pp. 86–90 Search PubMed.
- R. C. Larson, R. T. Iwamoto and R. N. Adams, Anal. Chim. Acta, 1961, 25, 371 CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- M. J. Frisch, et al.Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) J. Ridley and M. Zerner, Theor. Chim. Acta, 1973, 32, 111 CrossRef CAS; (b) J. Ridley and M. Zerner, J. Mol. Spectrosc., 1974, 50, 457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, extrapolation of absorption data to infinite chain length, and electronic density contours of additional oligomers. See DOI: 10.1039/c4cp00312h |
| This journal is © the Owner Societies 2014 |