 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A near ambient pressure XPS study of Au oxidation†
Alexander Yu.
Klyushin
*a,
Tulio C. R.
Rocha
a,
Michael
Hävecker
ab,
Axel
Knop-Gericke
a and
Robert
Schlögl
a
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195, Berlin, Germany. E-mail: klyushin@fhi-berlin.mpg.de
bDivision Solar Energy Research, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Albert-Einstein-Str. 15, 12489 Berlin, Germany
First published on 27th February 2014
Abstract
The surface of a gold foil under ozone oxidation was examined by near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) and scanning electron microscopy (SEM). Our in situ observations show that a surface oxide phase is formed during the exposure to ozone; however this phase decomposes under vacuum and even in the presence of ozone at temperatures higher than 300 °C. Assuming that an oxide overlayer completely covers the Au surface, the thickness of the oxide phase was estimated to be between 0.29 and 0.58 nm by energy-dependent XPS depth profiling. The surface oxidation led to structural modifications of the gold surface. These morphological changes do not disappear even under vacuum. In the Au 4f spectra, an additional component at low binding energy (83.3 eV), which appears during/after O3 treatment, is assigned to the presence of low-coordinated atoms which appear on the Au surface as a result of surface restructuring under oxidation. Ex situ SEM images demonstrate that only the region of the sample that was exposed to O3 shows the presence of ridges on the Au surface.
Introduction
The noble character of bulk Au has been well known for centuries. Corrosion and oxidation resistance are the hallmarks of Au, with these properties being exploited in many technological and everyday applications, such as dentistry, jewelry and electronics. However, gold's noble characteristics spectacularly vanish as the dimensions of the material are reduced to the nanometer scale. Experiments have shown that isolated Au atoms are readily oxidized in air.1 Radiolysis experiments have demonstrated that Au atoms have a redox potential similar to sodium borohydride, one of the most potent reductants in chemistry. Impressive demonstrations of this phenomenon were provided by Haruta and co-workers,2 who found Au nanoparticles (supported on transition metal oxides) to have a high catalytic activity in CO and H2 oxidation, even at low temperatures. This pioneering work has prompted Au catalysis to become a very active research field in recent years.3–5Presently, the catalytic applications of Au concern not only low-temperature CO oxidation,6 but also epoxidation of olefins,7,8 water–gas-shift reactions,9 C–C coupling reactions,10 and liquid-phase oxidation of alcohols,11 among others. Nano-structured Au, supported on transition metal oxides, is a highly efficient oxidation catalyst; however, the mechanism of these reactions is still the focus of intense debate in the literature. For instance, edge sites and changes in the electronic structure of Au, due to its interaction with the support, have been suggested to be important factors in establishing activity.12 Several studies have clearly shown that CO can be readily oxidized to CO2, at any kind of Au surface, if a source of atomic oxygen is present.12 Ozone (O3), N2O, and O2 plasma have been used as sources of atomic oxygen to show that even extended surfaces of Au single-crystals can be catalytically active towards CO oxidation.13 Based on DFT calculations,14 N. Lopez and J. Nørskov proposed that low-coordinated atoms on Au clusters play a central role in molecular oxygen activation. Contrary to the case of extended Au surfaces, adsorbed O on Au clusters is stable relative to O2 in the gas phase. In turn, recent theoretical and experimental investigations show that Au perimeter atoms can be active sites in CO oxidation reaction.15–18 The interpretation of the results combines the role of low coordinated Au atoms and their interaction with the support.19
In this study we investigate the interaction of extended Au surfaces with O3, using a combination of in situ XPS and ex situ SEM. NAP-XPS measurements allow us to characterize the electronic structure of surface oxide, while SEM provides information about morphological changes after oxidation. The combination of these techniques may shed light on understanding the nature of the Au–O phase and clarify issues discussed in some recent surface science reports.
Experimental
The experiments were performed at ISISS beamline at BESSY II/HZB (Berlin, Germany). All measurements were carried out in a stainless steel NAP-XPS chamber described in detail elsewhere.20,21 The sample was a polycrystalline gold foil (>99.95% purity) mounted on a sapphire sample holder between a stainless steel back-plate and a lid with 8 mm hole, shown in the Fig. S1 in the ESI.† The sample was heated from behind by an infrared laser and the temperature was measured by a type-K thermocouple positioned at the sample's surface. The overall spectral resolution was 0.3 eV in O 1s and 0.2 eV Au 4f regions. The spectral intensity was normalized by the incident photon flux, which was measured using Au reference foil. The binding energies (BE) were calibrated using both the Fermi edge and the Au 4f7/2 second-order peak. The accuracy of BE calibration was estimated to be around 0.05 eV.The gold foil was cleaned by standard surface science procedures, with Ar+ sputtering (2 × 10−4 mbar, 1.5 kV) for 30 min at room temperature and subsequent annealing to 400 °C under vacuum. Cycles of cleaning were repeated until carbon was no longer detected by C 1s XPS.
O3 was produced using a commercial ozone generator TC-1KC. Oxygen was passed at a rate of 1 L min−1 through Teflon tubing to the ozone generator. The effluent gas from the generator contained a mixture of approximately 1% ozone and 99% un-reacted oxygen. The O3–O2 mix was dosed through Teflon tubing into the experimental cell using a leak valve with the sample kept at 100 °C. The total pressure in the experimental cell was 0.3 mbar.
We recorded Au 4f7/2 and O 1s spectra at four different kinetic energies: 175 eV, 350 eV, 600 eV and 1000 eV, using a special combination of exit slit width of the monochromator and pass energy of the electron analyzer, which keeps the total resolution almost the same over the relevant interval of kinetic energies. These kinetic energies correspond to inelastic mean free paths (IMFP) of 0.49 nm, 0.67 nm, 0.91 nm and 1.29 nm (assuming predominantly gold in the near-surface region).22 All X-ray photoemission spectra were collected in normal photoemission mode. For quantitative XPS analysis least-squares fitting of the Au 4f7/2 spectra was performed using the commercially available CasaXPS software (www.casa xps.com). A Doniach–Sunjic line shape was used to obtain the best fit. All fitting parameters are given in Table S1 in the ESI.†
Results
The clean Au surface
The first step to study the interaction of O3 with Au is to obtain a clean Au surface. The fresh Au foil was cleaned by repeating cycles of Ar+ sputtering and thermal annealing until no additional elements other than Au were visible in a broad-energy survey XPS spectrum (Fig. S2 in the ESI†). The corresponding Au 4f spectrum of the clean Au foil (Fig. 1A(i)) shows the expected metallic peak at a binding energy (BE) of 83.92 eV23–25 together with an additional component shifted by 0.32 eV towards lower BE, assigned as a surface core-level shift according to the literature.26,27 The surface-sensitive O 1s spectrum (using 150 eV kinetic energy photoelectrons) shows only a small background step but no sign of adsorbed oxygen or oxygenated contaminants. (Fig. 1B(i)).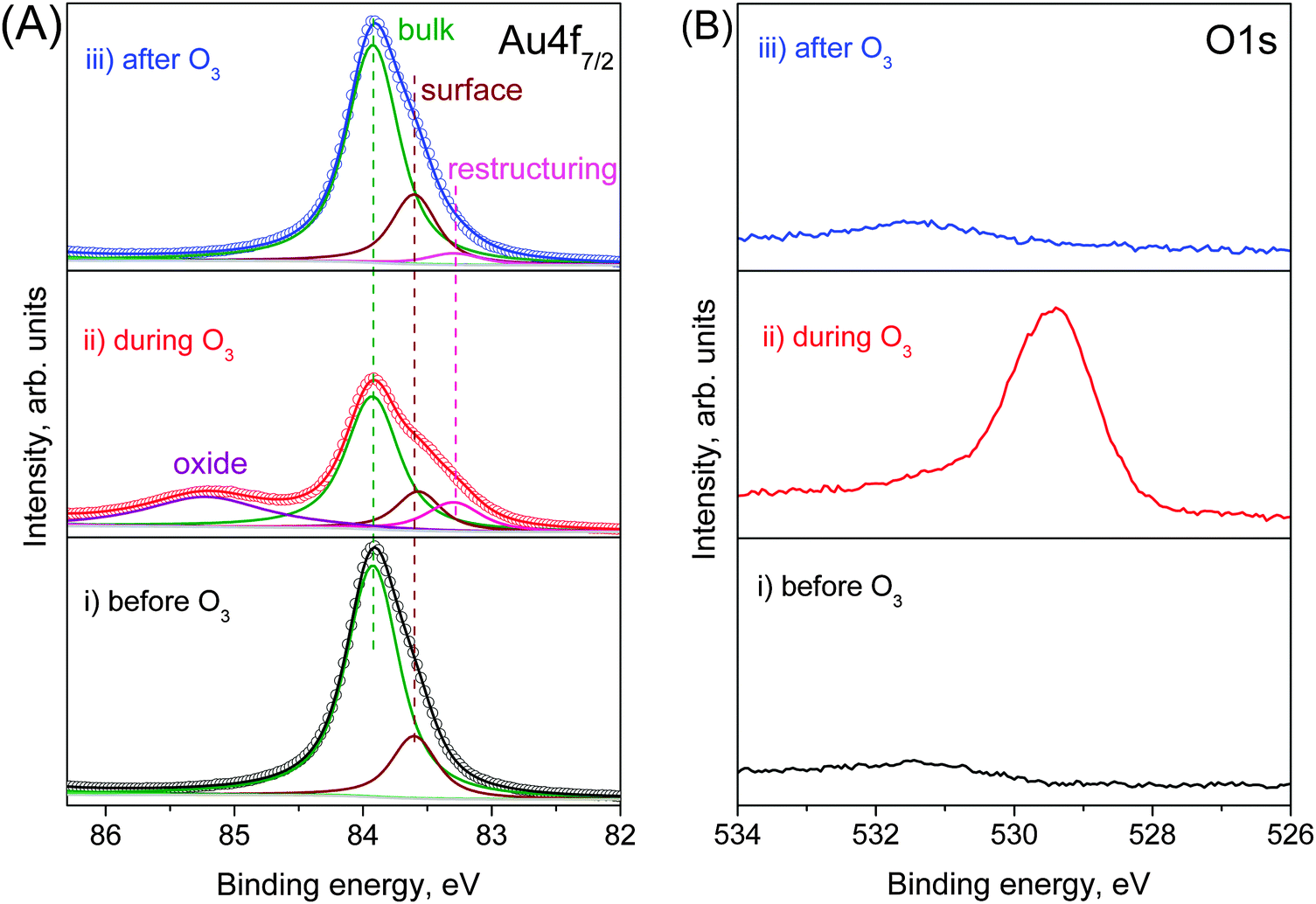 | ||
| Fig. 1 Fitting of NAP (A) Au 4f7/2 and (B) O 1s photoelectron spectra (using 150 eV kinetic energy photoelectrons) from gold foil at 100 °C (i) before (ii) during and (iii) after O3 treatment. | ||
Au oxidation under O3
When we introduce the O2–O3 mixture into the chamber, the O 1s spectrum immediately changes, developing an intense peak at 529.4 eV (Fig. 1B(ii)). The atomic element ratio is O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au = 0.84 ± 0.05 within a depth of 0.49 nm. Since no contaminants, within the detection limits of XPS, appear on the Au surface during oxidation, this peak can be assigned as oxygen bonded to gold. The corresponding Au 4f spectrum during O3 exposure (Fig. 1A(ii)) shows new features in comparison with the clean Au surface. A well-defined peak appears at 85.22 eV, which is assigned to ionic Au species (Auδ+),25,28 together with an additional feature at low BE (83.29 eV). This additional component is assigned to the presence of low-coordinated atoms that appear on the Au surface as a result of surface restructuring during oxidation.29 This assignment is further discussed below.
:Au = 0.84 ± 0.05 within a depth of 0.49 nm. Since no contaminants, within the detection limits of XPS, appear on the Au surface during oxidation, this peak can be assigned as oxygen bonded to gold. The corresponding Au 4f spectrum during O3 exposure (Fig. 1A(ii)) shows new features in comparison with the clean Au surface. A well-defined peak appears at 85.22 eV, which is assigned to ionic Au species (Auδ+),25,28 together with an additional feature at low BE (83.29 eV). This additional component is assigned to the presence of low-coordinated atoms that appear on the Au surface as a result of surface restructuring during oxidation.29 This assignment is further discussed below.
Stability of the oxide phase
Both the Auδ+ component and the O 1s peak disappear when the O2–O3 mixture is removed and the chamber reaches high vacuum (HV) conditions, as shown in the top spectra of Fig. 1A and B. Even the higher chemical potential of 0.3 mbar of O2 at 100 °C instead of HV does not prevent the decomposition of the surface oxide. The same behavior is also observed if O3 is replaced by O2. Under O3, the Au oxide is stable from room temperature up to 250 °C. At higher temperatures the Auδ+ component vanishes even in the presence of O3 as shown in Fig. 2. However, as the sample is cooled from 400 °C to room temperature, the oxide forms on the surface again at temperatures below 300 °C (see Fig. S3 in the ESI†).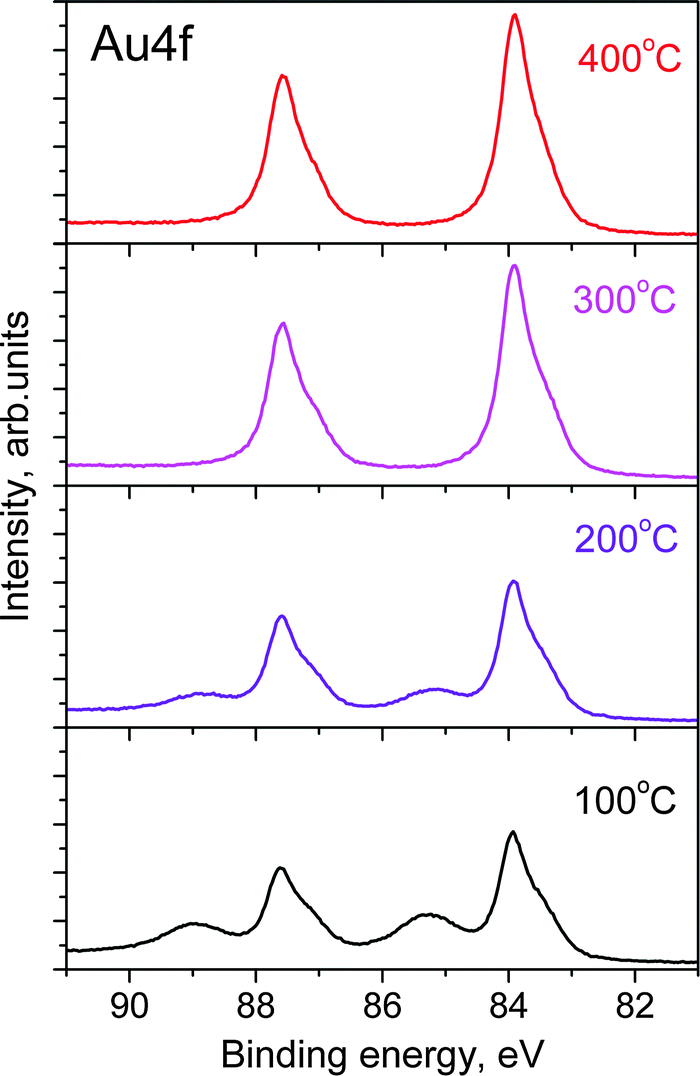 | ||
| Fig. 2 Au 4f7/2 photoelectron spectra (hν = 270 eV) from gold foil during heating at various temperatures. | ||
Interestingly, although the oxide phase totally vanishes after O3 is removed, the Au 4f spectral shape is not exactly the same as before O3 treatment. The Au 4f7/2 peak is slightly broader (FWHM changes from 0.59 eV before O3 to 0.64 eV after) with a more pronounced tail to low BE (Fig. S4 in the ESI†). The difference spectrum before and after ozone oxidation (Fig. S4 in the ESI†) clearly shows the existence of an additional component at 83.29 eV assigned to restructuring. At 100 °C, the initial spectrum is recovered only after 8 h, but at 350 °C the additional low BE features disappear after 10 min (not shown). The persistence of the additional low-BE component even after the decomposition of the oxide phase and its removal by thermal annealing reinforces the assignment as an additional surface core-level shift related to restructuring.
Surface restructuring
The structural modifications of the Au foil after O3 exposure, as suggested by the XPS results, were further confirmed by ex situ SEM characterization. Fig. 3 shows an image of a particular region of the Au surface that was partially covered by the sample holder lid. The nearly vertical line marks the position where the edge of the steel lid was pressed against the Au foil. The region of the sample that was exposed to O3 (region A in Fig. 3) shows pronounced contrast variations that are consistent with the presence of ridges on the Au surface. Conversely, the region that was shadowed by the lid shows a rather uniform contrast suggesting a less-structured surface on this length scale (region B in Fig. 3). | ||
| Fig. 3 Ex situ SEM image after O3 (0.3 mbar) treatment at 100 °C showing ridges in the gold foil only in areas that were exposed to O3 (region B). On the left side, where the surface was covered by the sample holder lid, no ridges are observed (region A). | ||
Extent of oxidation
We exploited the energy tunability of synchrotron radiation to measure the depth profile of the sample. By generating photoelectrons with different kinetic energies of the photoelectrons, the sample could be probed at various depths. Fig. 4 shows Au 4f and O 1s spectra measured using kinetic energies of 175, 350, 600 and 1000 eV. The O 1s spectra intensity is normalized to the area of the corresponding metallic component Au0 in order to emphasize the variations in the total amount of oxygen. It can be clearly seen that the Auδ+ feature and the O 1s peak are consistently less-pronounced for higher-KE photoelectrons, which originates from deeper layers of the Au foil. Moreover, the main peak at 83.95 eV in the Au4f is more symmetric at higher kinetic energies, which confirms the assignment of the low-BE components to surface related features. These measurements suggest that only the near-surface region of the Au foil is oxidized by O3 at mbar pressures; however, quantification of the depth-dependent measurements is required for a better assessment of the thickness of the oxide phase.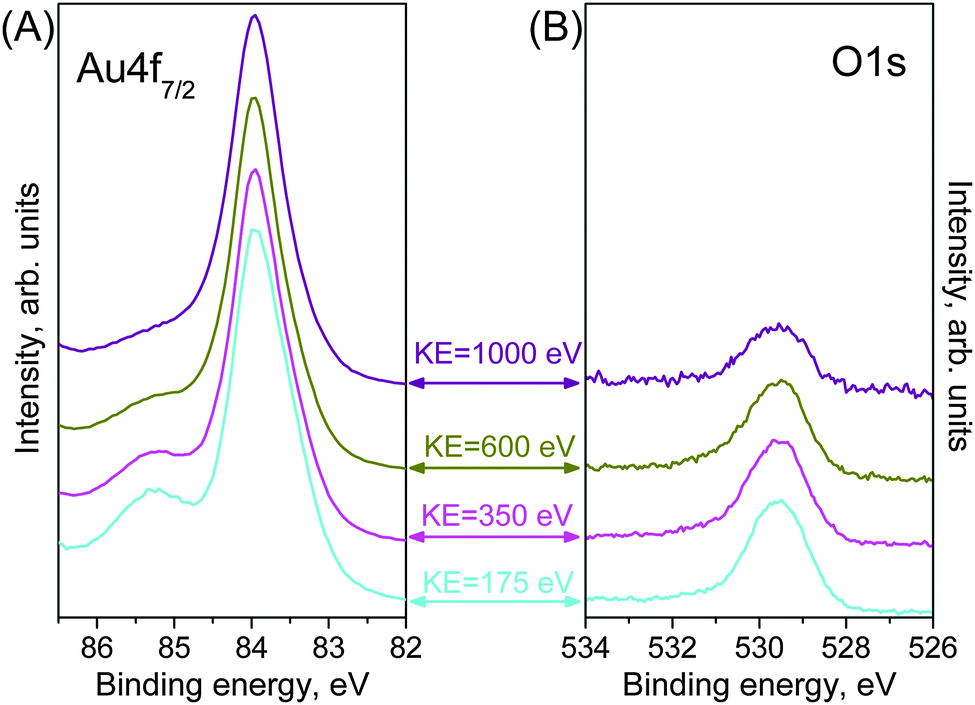 | ||
| Fig. 4 NAP-XPS depth profile (A) Au 4f7/2 and (B) O 1s as obtained from a gold foil during O3 (0.3 mbar) treatment at 100 °C for increasing kinetic energies of photoelectrons, i.e. decreasing surface sensitivity. Spectra were measured at 175 eV (cyan line), 350 eV (magenta line), 600 eV (dark yellow line) and 1000 eV (purple line) kinetic energy of photoelectrons. | ||
Thickness of the oxide phase
The mean escape depth for each measurement at different kinetic energies can be approximated using known values of the inelastic mean free path‡ (IMFP) for Au.22 Depth profiles are constructed from the integrated XPS areas. Fig. 5 shows the integrated peak areas of the O 1s and Auδ+ divided by the Au0 area as a function of the average escape depth. The normalization by Au0 is necessary to remove the influence of elastic scattering in the gas phase that is also energy dependent. The thickness of the oxide phase can be estimated by quantification of the depth profile using a simplified structural model consisting of a uniform oxide layer of thickness t over a semi-infinite substrate.30,31 Using known atomic densities and assuming the same IMFP for the metal and oxide, the unknown thickness t can be obtained by fitting the experimental data. Details about the procedure are given in the ESI.† By assuming that the oxide over-layer completely covers the Au surface, a good fit was obtained (solid line in Fig. 5) and gave a value of t = 0.29 ± 0.02 nm. However, similar models with incomplete coverage of the surface by the oxide phase (patches) also result in reasonable fittings within the experimental errors, resulting in larger values for the thickness. In the extreme case of 60% coverage (dash-dot line in the Fig. 5), the derived over-layer thickness increases to 0.58 nm (lower coverages do not result in reasonable fittings). Comparing these values to the shortest Au–O distance of 0.201 nm in the Au2O3 structure (most stable bulk Au–oxide) or the smaller axis of its unit cell of 0.404 nm,32 it is reasonable to conclude the formation of a surface oxide phase during the oxidation of a Au surface with O3 at mbar pressures.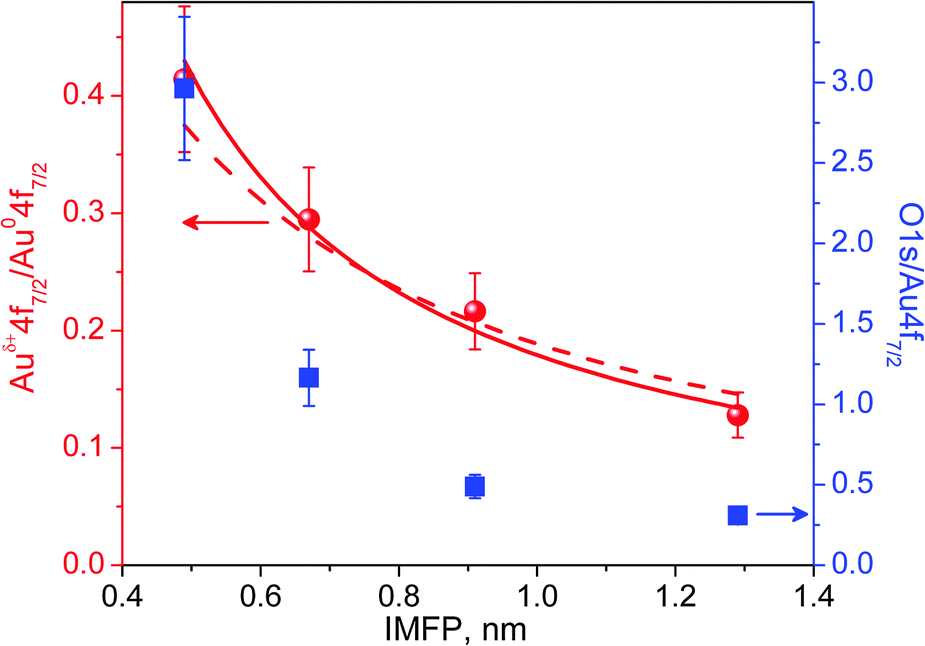 | ||
| Fig. 5 The area ratios between Auδ+ 4f7/2 and Au0 4f7/2 (red dots), O 1s and Au0 4f7/2 (blue squares) XPS peak as a function of inelastic mean free path (IMPF). Fitting results, assuming an oxide over-layer completely (solid line) and 60% (dash-dot line) covering the Au surface. | ||
Oxygen overlayer
The area ratio in both depth profile curves in Fig. 5 decays exponentially with escape depth; however, the oxygen curve (blue squares) decays much faster than the Auδ+ when probing deeper into the sample. This observation suggests the presence of an additional adsorbed oxygen over-layer that totally or partially covers the surface oxide layer/patches.Discussion
The limiting step in the oxidation of Au seems to be the production of atomic oxygen.12 Since the dissociation probability of O2 is very small on Au surfaces,33 atomic oxygen must be supplied by other methods. Recent studies have clearly shown that Au can be readily oxidized when exposed to strong oxidative environments, such as O2-plasma and UV/ozone.24,34 The formation of Au oxide has been reported, not only for nanoparticles,1,6 but also for extended Au surfaces, such as single-crystals35 and polycrystalline thin films.25,28,36 In our case, the ozone treatment at 100 °C resulted in the formation of a surface oxide phase with a thickness of a few atomic layers, instead of the 3–4 nm thick plasma-oxidized thin films reported by others.25,28,37This Au surface oxide is unstable under UHV conditions and in an O2 atmosphere at 100 °C, leaving a clean Au surface when O3 is removed (Fig. 1A and B(iii)), in agreement with a previous study.25 However, under constant O3 exposure, the surface oxide is stable up to 250 °C. In contrast, based on resistivity measurements, Tsai et al.28 reported a decomposition half-life time of approximately 10 min for 4 nm Au–oxide films (Au2O3) at 100 °C in air. The stability of the surface oxide under an O3-containing atmosphere suggests that the surface oxide is in dynamic equilibrium with the gas environment. The constant supply of atomic oxygen, provided in this case by O3 dissociation, counteracts the spontaneous decomposition and the surface oxide that prevails at higher temperatures. The surface-limited oxidation, observed under our conditions, might be related to this competition between oxidation and reduction/decomposition, and would kinetically limit the oxide growth. This model system under dynamic equilibrium is more representative of a practical catalytic system than static phases under UHV. In the real catalyst, atomic O might be supplied by the oxide support or by direct dissociation of O2 at the kinks or steps of nanoparticles. It is worth mentioning that we did not observe any X-ray-induced oxidation of Au, as has been previously reported by P. Jiang et al.,38 most likely due to the lower photon flux density in our case that limits beam-induced chemistry. The broad O 1s and Auδ+ peaks suggest that the surface oxide is most likely not a single, long-range, ordered phase, but might consist of multiple phases. For example, in the Ag–O system, multi-phase co-existence has been predicted theoretically39 and experimentally verified.40,41 Similar conclusions – i.e. that several Au–O species can be generated by kinetic control of the oxidation conditions – were drawn by Ming and Friend after reviewing several studies about the oxidation of Au.12
One of the most controversial aspects of the mechanism of CO oxidation on nano-structured Au catalysts is the source of atomic oxygen. While some authors suggest that a spillover of lattice oxygen from reducible oxide supports is the source of atomic oxygen, others point to the direct dissociation of O2 at low-coordination sites on the surface of Au nano-particles.6,12,41 The additional surface core-level shift component, related to the restructuring that is observed during ozone treatment in our XPS spectra, (Fig. 1A(ii)) indicate the formation of low-coordinated atoms on the Au surface during oxidation. Similar spectral features were previously observed for stepped single-crystal surfaces29 with a clear correlation between the coordination number of atoms at steps and terraces with the measured surface core-level shift for Au(321) and Au(310) crystals. The surface roughening observed by SEM after the O3 treatment (Fig. 2) indicates extensive surface restructuring during oxidation, and might have generated the low-coordinated atoms that we had inferred from the XPS analysis. Surface restructuring induced the formation of Au ad-atoms and clusters on a Au(111) surface were directly observed by STM after atomic oxygen exposure, generated by electron-induced NO2 dissociation.42
Our combined spectroscopy and microscopy results can be interpreted by the restructuring of the Au surface as reported by others, suggesting the presence of low-coordinated atoms on the surface of the Au foil after ozone treatment. However, it should be stressed that the presence of such proposed active sites for molecular oxygen dissociation does not prevent the disappearance of the Au–oxide when O3 is removed at 100 °C. Although the low-coordinated atoms may have an enhanced interaction strength with surrounding molecules, and still might play a role in O2 adsorption at room temperature, our results do not show that they enhance O2 dissociation. This observation suggests that low-coordinated Au sites alone might not dissociate molecular oxygen as efficiently as at room temperature11,42 or the number of active sites was not sufficient to compensate for the thermal decomposition of the oxide, unlike the case of O3 dissociation.
Conclusions
From the results presented we suggest the following picture of the system gold–ozone under a constant chemical potential of O3. The surface consists of an overlayer of atomic oxygen being chemisorbed on a surface oxide by a thickness of 0.29 nm. This layer may be presented as a tri-layer oxygen–gold–oxygen with a region of oxygen atoms diffusing in deeper layers with decreasing probability. The formation of the surface oxide mobilized gold atoms leads to a significant restructuring of the surface and the frequent formation of coordinatively undersaturated species, which leads to shape changes in the Au 4f spectra. These species may be responsible for holding the atomic oxygen at the surface. Whereas the restructuring remains upon lowering the chemical potential of oxygen to vacuum levels, the absorbed oxygen species and also the surface oxide vanish even at 100 °C indicating that the heat of adsorption and heat of formation, respectively, are small even in the presence of the massive restructuring. This sheds light on the energetic consequences of the formation of coordinatively undersaturated gold species.We prefer such a picture of a laterally homogeneous multi-layer architecture over the alternative of laterally inhomogeneous patches of different surface oxide species as the SEM image does not reveal indications for extensive lateral inhomogeneity.
Acknowledgements
We thank HZB for the allocation of synchrotron radiation beamtime and Gisela Weinberg (Fritz Haber Institute, Berlin) for the SEM characterization.References
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240(2), 222–234 CrossRef CAS PubMed.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115(2), 301–309 CrossRef CAS.
- T. Hayashi, T. Inagaki, N. Itayama and H. Baba, Catal. Today, 2006, 117(1–3), 210–213 CrossRef CAS PubMed.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- E. E. Stangland, K. B. Stavens, R. P. Andres and W. N. Delgass, J. Catal., 2000, 191(2), 332–347 CrossRef CAS.
- M. C. Kung, R. J. Davis and H. H. Kung, J. Phys. Chem. C, 2007, 111, 11767–11775 CAS.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178(2), 566–575 CrossRef CAS.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Top. Catal., 2004, 29(3–4), 95–102 CrossRef CAS.
- I. Andreeva, T. Tabakova and A. Andreev, J. Catal., 1996, 158(1), 354–355 CrossRef.
- C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Chem. Commun., 2005, 3451–3453 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20–44 RSC.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107(6), 2709–2724 CrossRef CAS PubMed.
- B. K. Min, A. R. Alemozafar, D. Pinnaduwage, X. Deng and C. M. Friend, J. Phys. Chem. B, 2006, 110(40), 19833–19838 CrossRef CAS PubMed.
- N. Lopez and J. Nørskov, J. Am. Chem. Soc., 2002, 124(38), 11262–11263 CrossRef CAS PubMed.
- Y. Iizuka, T. Tode, T. Takao, K. Yatsu, T. Takeuchi, S. Tsubota and M. Haruta, J. Catal., 1999, 187(1), 50–58 CrossRef CAS.
- L. M. Molina and B. Hammer, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69(15), 155424 CrossRef.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197(1), 113–122 CrossRef CAS.
- M. Okumura, S. Nakamura, S. Tsubota, T. Nakamura, M. Azuma and M. Haruta, Catal. Lett., 1998, 51(1), 53–58 CrossRef CAS.
- S. Chrétien, S. K. Buratto and H. Metiu, Curr. Opin. Solid State Mater. Sci., 2007, 11(3), 62–75 CrossRef PubMed.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, Adv. Catal., 2009, 52, 213–272 CAS.
- H. Bluhm, M. Hävecker, A. Knop-Gericke, E. Kleimenov, R. Schlögl, D. Teschner, V. I. Bukhtiyarov, D. F. Ogletree and M. Salmeron, J. Phys. Chem. B, 2004, 108, 14340–14347 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1991, 17, 911–926 CrossRef CAS PubMed.
- N. D. S. Canning, D. Outka and R. J. Madix, Surf. Sci., 1984, 141, 240–254 CrossRef CAS.
- D. E. King, J. Vac. Sci. Technol., A, 1995, 13(3), 1247–1253 CAS.
- B. Koslowski, H.-G. Boyen, C. Wilderotter, G. Kästle, P. Ziemann, R. Wahrenberg and P. Oelhafen, Surf. Sci., 2001, 475(1–3), 1–10 CrossRef CAS.
- P. H. Citrin, G. K. Wertheim and Y. Baer, Phys. Rev. Lett., 1978, 41, 1425–1428 CrossRef CAS.
- P. Heimann, J. F. van der Veen and D. E. Eastman, Solid State Commun., 1981, 38, 595–598 CrossRef CAS.
- H. Tsai, E. Hu, K. Perng, M. Chen, J.-C. Wu and Y.-S. Chang, Surf. Sci., 2003, 537(1–3), L447–L450 CrossRef CAS.
- C. J. Weststrate, E. Lundgren, J. N. Andersen, E. D. L. Rienks, A. C. Gluhoi, J. W. Bakker, I. M. N. Groot and B. E. Nieuwenhuys, Surf. Sci., 2009, 603(13), 2152–2157 CrossRef CAS PubMed.
- A. Cimino, D. Gazzoli and M. Valigi, J. Electron Spectrosc. Relat. Phenom., 1999, 104(1–3), 1–29 CrossRef CAS.
- C. S. Fadley, in Electron Spectroscopy: Theory, Techniques and Applications, ed. C. R. Brundle and A. D. Baker, Academic Press, New York, 1978, ch. 1, vol. 2, pp. 2–157 Search PubMed.
- H. Shi, R. Asahi and C. Stampfl, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(20), 205125 CrossRef.
- X. Deng, B. K. Min, A. Guloy and C. M. Friend, J. Am. Chem. Soc., 2005, 127(25), 9267–9270 CrossRef CAS PubMed.
- A. Krozer and M. Rodalh, J. Vac. Sci. Technol., A, 1997, 15(3), 1704–1709 CAS.
- B. K. Min, A. R. Alemozafar, M. M. Biener, J. Biener and C. M. Friend, Top. Catal., 2005, 36(1–4), 77–90 CrossRef CAS.
- K. Juodkazis, J. Juodkazyte, V. Jasulaitiene, A. Lukinskas and B. Sebeka, Electrochem. Commun., 2000, 2(7), 503–507 CrossRef CAS.
- H. Ron and I. Rubinstein, Langmuir, 1994, 10(12), 4566–4573 CrossRef CAS.
- P. Jiang, S. Porsgaard, F. Borondics, M. Köber, A. Caballero, H. Bluhm, F. Besenbacher and M. Salmeron, J. Am. Chem. Soc., 2010, 132(9), 2858–2859 CrossRef CAS PubMed.
- W. X. Li, C. Stampfl and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67(4), 045408 CrossRef.
- J. Schnadt, J. Knudsen, X. L. Hu, A. Michaelides, R. T. Vang, K. Reuter, Z. Li, E. Lægsgaard, M. Scheffler and F. Besenbacher, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80(7), 075424 CrossRef.
- T. C. R. Rocha, A. Oestereich, D. V. Demidov, M. Hävecker, S. Zafeiratos, G. Weinberg, V. I. Bukhtiyarov, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2012, 14(13), 4554–4564 RSC.
- B. K. Min, X. Deng, D. Pinnaduwage, R. Schalek and C. M. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(12), 121410 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp00308j |
| ‡ The IMFP is defined as the distance a photoelectron beam can travel inside the solid before its intensity decays to 1/e of its initial value. |
| This journal is © the Owner Societies 2014 |