 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnergy transfer in PPV-based conjugated polymers: a defocused widefield fluorescence microscopy study
E. N.
Hooley†
a,
A. J.
Tilley‡
a,
J. M.
White
a,
K. P.
Ghiggino
*a and
T. D. M.
Bell
*b
aSchool of Chemistry and Bio21 Institute, The University of Melbourne, Parkville, Vic. 3010, Australia. E-mail: ghiggino@unimelb.edu.au; Fax: +61 3 9347 5180; Tel: +61 3 8344 8939
bSchool of Chemistry, Monash University, Clayton, Vic. 3800, Australia. E-mail: toby.bell@monash.edu; Fax: +61 3 9905 4597; Tel: +61 3 9905 4566
First published on 4th March 2014
Abstract
Both pendant and main chain conjugated MEH-PPV based polymers have been studied at the level of single chains using confocal and widefield fluorescence microscopy techniques. In particular, defocused widefield fluorescence is applied to reveal the extent of energy transfer in these polymers by identifying whether they act as single emitters. For main chain conjugated MEH-PPV, molecular weight and the surrounding matrix play a primary role in determining energy transport processes and whether single emitter behaviour is observed. Surprisingly in polymers with a saturated backbone but containing the same pendant MEH-PPV oligomer on each repeating unit, intra-chain energy transfer to a single emitter is also apparent. The results imply there is chromophore heterogeneity that can facilitate energy funneling to the emitting site. Both main chain conjugated and pendant MEH-PPV polymers exhibit changes in orientation of the emission dipole during a fluorescence trajectory of many seconds, whereas a model MEH-PPV oligomer does not. The results suggest that, in the polymers, the nature of the emitting chromophores can change during the time trajectory.
Introduction
Single molecule spectroscopy has been used to investigate the complex behaviour of conjugated polymers since the late 1990's, with many reports on poly(2-methoxy-5-(2′-ethylhexyloxy)-1,4-phenylene vinylene) (MEH-PPV). The first single molecule studies on conjugated polymers1 revealed that despite the multi-chromophoric nature of conjugated polymer chains, the fluorescence/time trajectories showed discrete on–off behaviour, as well as distinct fluorescence intensity levels. This was a surprising result, given the established understanding of polymers as a collection of quasi-localized chromophores. A new model was developed, based on the idea that following photoexcitation, energy ‘hopped’ among the constituent chromophores until a local energy minimum was reached from which emission occurred.2 While it had been proposed that conjugated polymers must undergo some degree of energy transfer,3 these early observations raised new questions regarding the mechanism and extent of energy transfer in a single polymer chain. These questions included: does energy migration occur along the chain, or via larger hops through space?4 And, is the final emitter simply the longest conjugation length chromophore or does it depend on the shape of the chromophore5 and local environment?6The important influence of chain conformation on energy transport processes was identified early. Simulations showed that polymer chains can adopt a variety of conformations,7 which are heavily dependent on solvent and the host matrix. However detailed insights into the effects of the local environment and excitation conditions on the behaviour of conjugated polymers have remained elusive.
The inherently complex nature of polymers with distributions of chain and chromophore length, combined with the ill-defined conformation and chromophore microenvironment, limit what can be learned from bulk studies. Single molecule spectroscopy can provide insights beyond the ensemble average by accessing the distributions of behaviour that make up bulk properties.1,2,6,7 Nevertheless energy transfer processes in conjugated polymers are subject to many influences, including the distribution of conjugated polymer backbone lengths that make up the chromophores, the pathways of energy migration, (through space or along the backbone) and the conformation of the polymer chain. By keeping one or more of these factors constant, a more accurate picture of these processes can be constructed.
Synthesis can allow some manner of control over the overall chain length of the polymer, however, it cannot solve the problem of distributed chromophore lengths for conventional polymers with conjugated backbones. Pendant polymers, however, having a saturated backbone with conjugated chromophores of fixed length attached as pendant groups, can provide polymers of the same chromophore type.8 These polymers have a saturated backbone spacer between the pendant chromophore units, effectively restricting the energy migration to the “through space” pathway.
In this work, both fully conjugated polymer chains of MEH-PPV of varying molecular weight and pendant polymers containing MEH-PPV pentamer units as side-chains have been studied by single molecule fluorescence spectroscopy. The role of polymer structure and environment on energy transfer mechanisms in these polymer systems is reported.
Experimental
Fig. 1a shows the molecular structure of MEH-PPV. The pendant polymer used in this study is shown in Fig. 1c and it consists of a saturated backbone with pentamer MEH-PPV oligomer units attached as pendants (Mn = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 516, Mw/Mn = 1.08). The model pentamer oligomer was also studied and is shown in Fig. 1b. The synthesis of the pendant polymer and oligomer are described elsewhere.8,9
516, Mw/Mn = 1.08). The model pentamer oligomer was also studied and is shown in Fig. 1b. The synthesis of the pendant polymer and oligomer are described elsewhere.8,9
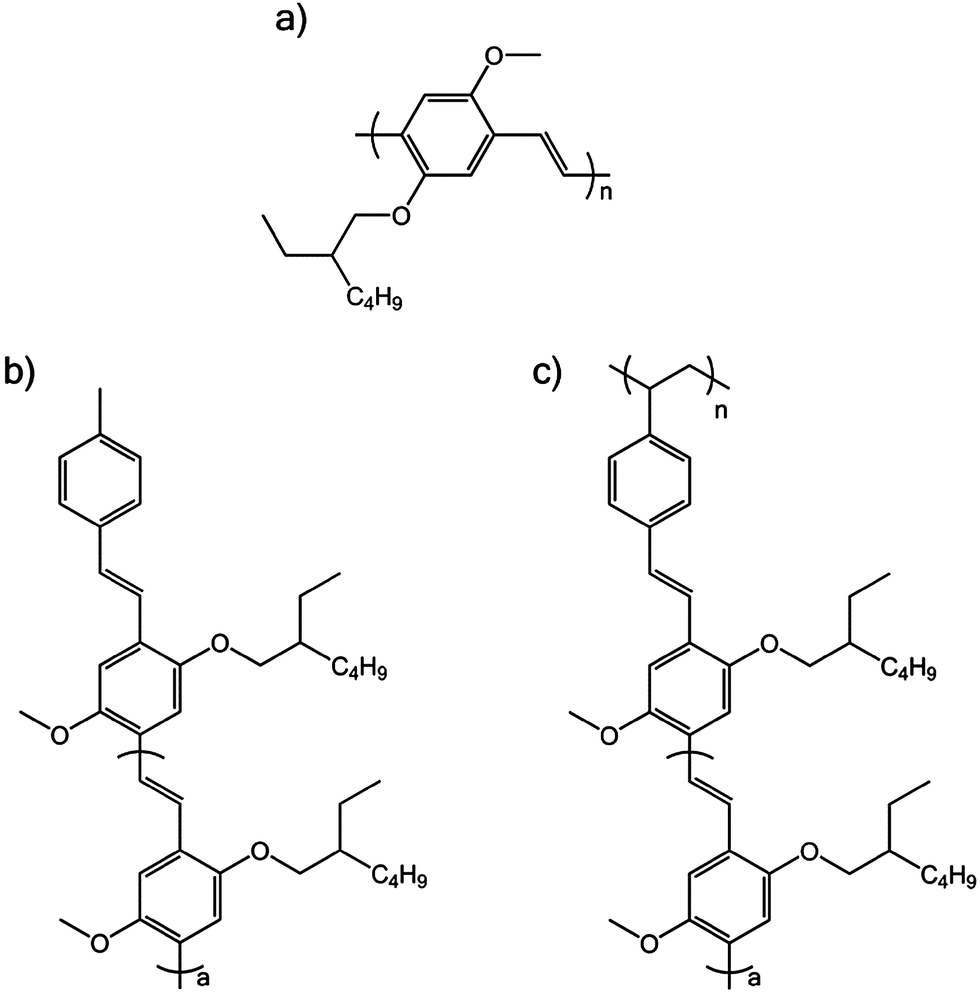 | ||
| Fig. 1 Molecular structure of (a) MEH-PPV, (b) oligomer of MEH-PPV, where a = 3 and (c) pendant MEH-PPV polymer, where a = 3. | ||
Samples for single molecule spectroscopy were diluted to approximately 1 × 10−9 mol dm−3 by serial dilution into chloroform (Aldrich). The final dilution was into an approximately 0.5% (w/w) solution of poly(methyl methacrylate) (PMMA) (Aldrich) or polystyrene (PS) (Fluka) in chloroform. The solution was spin-coated onto a clean glass coverslip using a Specialty Coating System (SCS) P6700 Series spin coater to produce a thin polymer film, approximately 100 nm thick.
The widefield microscope consists of an inverted optical microscope (IX71, Olympus), an air-cooled CCD camera (Photonmax 512, Princeton Instruments) with excitation provided by a 150 mW, 473 nm continuous wave diode laser (Shanghai Laser and Optics Century Co. BL4737-150). The maximum power can be attenuated by a graded neutral density filter (Thorlabs), before passing through a half wave plate (Thorlabs) and into a beam expander to give a beam diameter of approximately 2.5 cm. Koehler illumination is achieved by focusing the expanded excitation beam through a lens (f = 50 cm) onto the back of the objective lens (100×, 1.4 NA oil objective, Olympus). This provides an illumination area of approximately 0.1 mm2 with a flux on the order of 0.5–1 kW cm−2. The emitted light is collected by the objective, separated from the excitation light by a dichroic mirror (Chroma Technology) and a further long pass filter (Chroma Technology) then passed through a 3.3× beam expander (Olympus) and collected by the CCD camera. The CCD chip is 512 × 512 pixels with each pixel being 16 μm × 16 μm. The image mostly fills the chip following expansion, implying a field of view of 24.6 μm × 24.6 μm. All measurements were recorded under ambient atmosphere and at room temperature.
For defocused widefield images, the focused image was degraded by defocusing approximately 1 μm, by moving the objective towards the sample. The defocused pattern maps the angular distribution of emitted light intensity.10,11 The patterns observed are a function of the emission dipole orientation and thus molecular orientation. The defocused patterns were matched to theoretical patterns calculated as described elsewhere10,12 using a non-linear least-squares algorithm.13
Molecules were selected for analysis using a program written in MATLAB13 with the following criteria: the molecule must be separated from those surrounding it such that the outer rings of the emission pattern do not overlap, (preferably a separation of 1.5 μm or greater), and the molecule must be bright enough to be distinguished from the background with a suitable signal to noise ratio, (greater than 2:1).
Confocal single molecule microscopy measurements were recorded on a system described elsewhere.14 Single molecule trajectories were determined using BIFL 1.4 software package (Scientific Software Technologies Center). Further details of the instrument, data collection and analysis procedures have been outlined previously15 with 470 nm excitation.
Results and discussion
MEH-PPV
Conjugated polymer fluorescence is known to be strongly affected by the solvent or host matrix for the polymer. In poor solvents, the chains coil up into compact, “collapsed” conformations, while in a good solvent, the chains extend into a more relaxed conformation. The changes in conformation have a significant effect on the photophysics of the polymer, and can be observed as changes to the emission spectrum,16,17 photobleaching behaviour, and the number of emitting chromophores.18
Fig. 2 shows two widefield images of high molecular weight MEH-PPV (2600000 Da) in PS (Fig. 2a) and PMMA (Fig. 2b), recorded under the same illumination conditions. The images are shown in reverse grey scale where high intensity (molecules) is dark and low intensity (background) is bright. There is a distinct difference in fluorescence intensity of the polymer chains in the two host matrices. While the background levels are comparable, the reverse grey scale clearly shows there is a much lower signal/background ratio for the MEH-PPV chains in PMMA indicating the polymer chains in PS are significantly brighter.
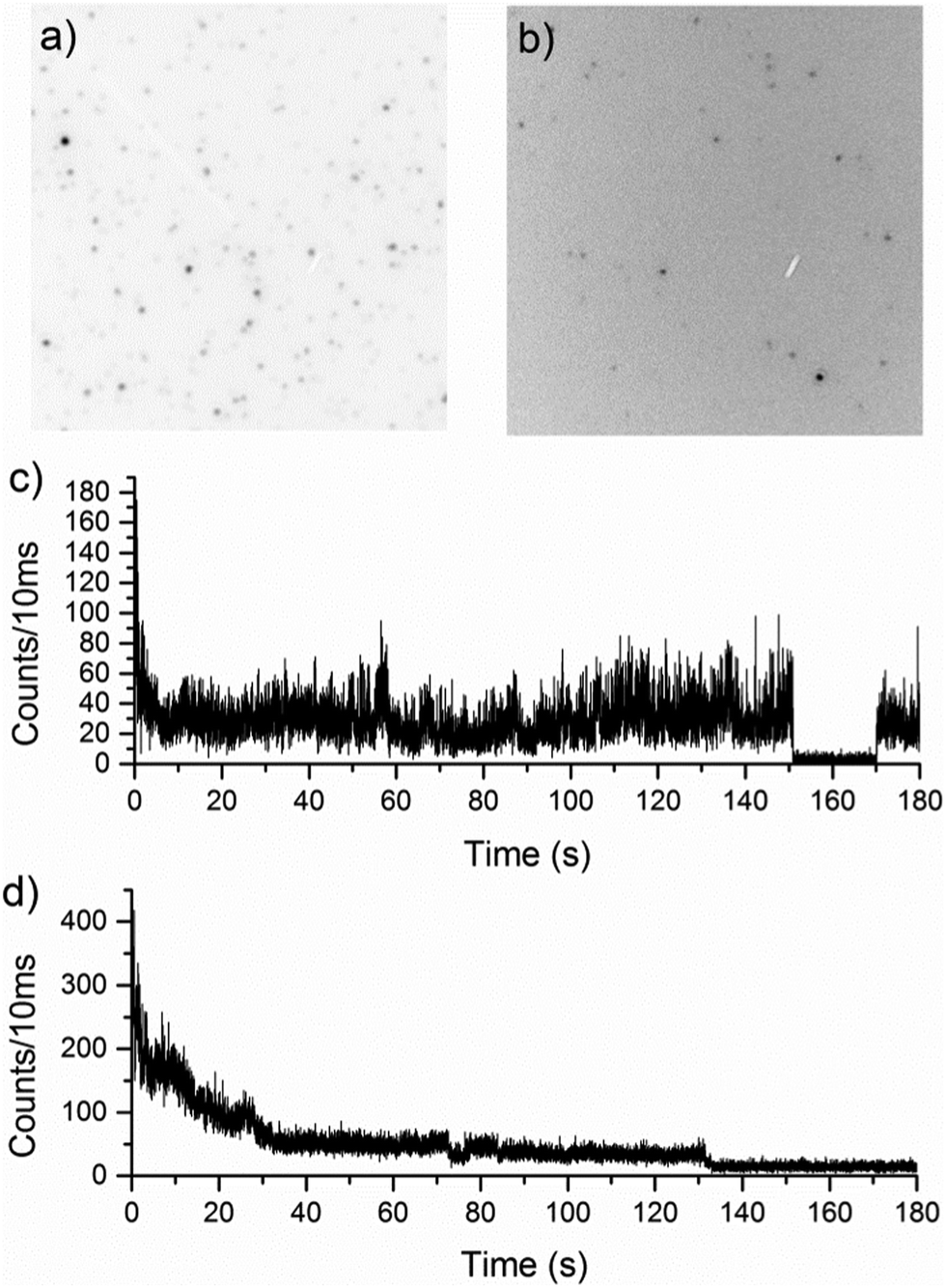 | ||
| Fig. 2 Focused widefield microscopy images of high molecular weight MEH-PPV in (a) PMMA and (b) PS; and confocal fluorescence traces in (c) PMMA and (d) PS. | ||
Fig. 2c and d clearly shows the previously reported19,20 difference in photobleaching behaviour in the two polymer matrices investigated using confocal fluorescence microscopy. Polymer chains in PMMA have discrete intensity levels, with distinct changes between them, whereas in PS, a more gradual ‘exponential-like’ photobleaching of the chromophores is seen. The latter photobleaching behaviour is more typical of multi-chromophoric species, and can be attributed to individual emitters photobleaching, one after the other. Discrete on–off type behaviour in fluorescence trajectories of single conjugated polymers is associated with efficient energy transfer from the initially excited polymer chromophores to the lowest energy chromophore in the polymer from which emission occurs. In extended conformations, the increased distances between chromophoric units, means energy transfer is less efficient, leading to multi-chromophoric type emission behaviour. For an ensemble of independent emitters, emission intensity depends on their number and the decay due to photobleaching of the intensity from this population over time can appear as an exponential decay. This type of behaviour is apparent for the first 30 seconds of the polymer fluorescence in Fig. 2d suggesting that initially in PS, MEH-PPV behaves like a small ensemble. After most of the emitters have decayed, the emission is steady and shows the discrete emission levels associated with single emitters.
In addition to the difference in photobleaching, there is a clear difference in the emission intensity of MEH-PPV in the two matrices. 50 molecules of MEH-PPV in PMMA and PS were plotted in 100 ms bins, and a histogram made of the counts in each bin. The histograms are shown in Fig. 3.
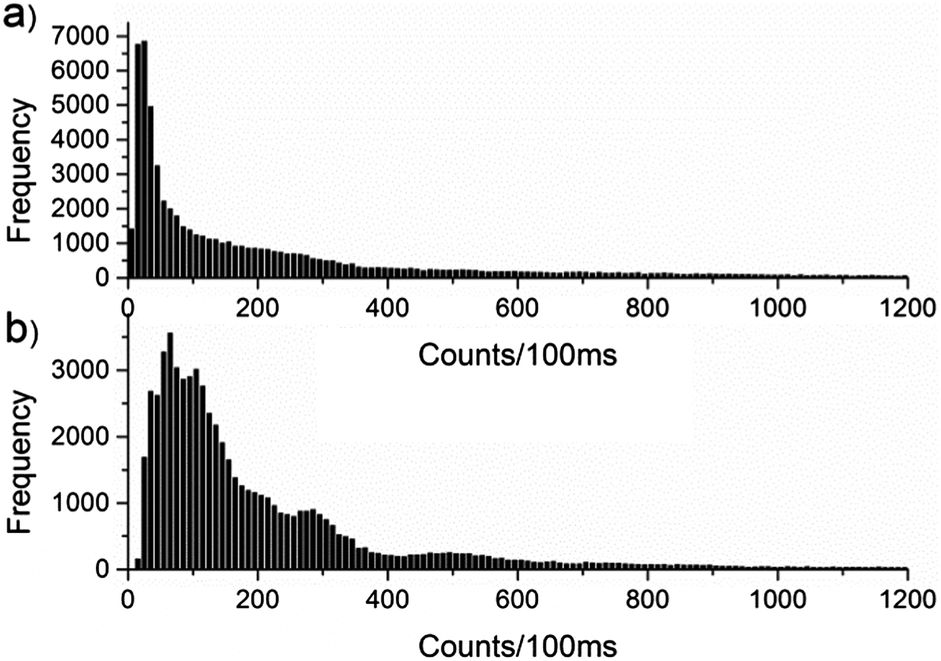 | ||
| Fig. 3 Histogram of counts per 100 ms for MEH-PPV in (a) PMMA and (b) PS. | ||
The difference in intensity may be accounted for by considering that in PS, MEH-PPV adopts an extended conformation that restricts the efficiency of the energy transfer, allowing more than one chromophore to emit simultaneously. In the more compact, coiled conformation in PMMA, possible singlet–singlet and singlet–triplet annihilation events and efficient energy transfer result in emission from a single chromophore and an overall loss of intensity. Photon antibunching experiments18 have shown that in PMMA, MEH-PPV forms collapsed conformations that favour efficient energy transfer, resulting in emission from a single chromophore. In PS, where the MEH-PPV exists in a more extended conformation, simultaneous emission from multiple chromophores is evident.
One method for examining the relative efficiencies of energy transfer in conjugated polymers is to count the number of simultaneously emitting chromophores. Energy transfer is known to be efficient1,7 in conjugated polymer chains, but is also thought to be conformation dependent.20,21 In any given chain, energy transfer can occur along the chain, hopping between neighbouring chromophores (Fig. 4a) or through-space, where the excitation moves between two nearby, but not directly connected, sections of the chain (Fig. 4b). In polymers, through-space energy transfer is reported to be much faster than along-chain energy hopping, and is the dominant energy transfer mechanism in collapsed sections of the chain.22 This leads to an increased likelihood of single chromophore emission occurring in shorter, collapsed polymer chains, compared to those in extended conformations.
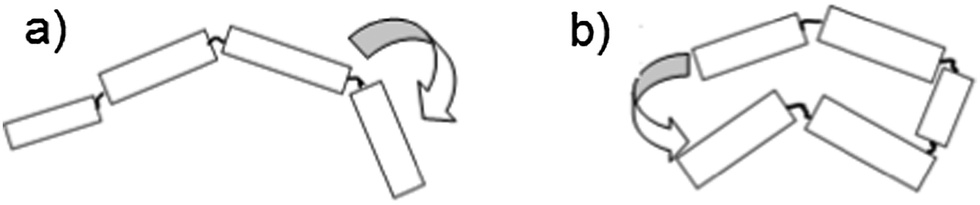 | ||
| Fig. 4 Energy transfer from an excited chromophore to a neighbouring one (a) along the chain or (b) through space. | ||
Defocused widefield imaging is a powerful method for determining whether or not a molecule is a single emitter. By defocusing the fluorescence from a molecule, the spatial distribution of intensity from the single emitting dipole can be resolved.10 Emission from a single dipole shows an uneven distribution of intensity (i.e. a bi-lobed shape), and depends on the orientation of the transition dipole. These patterns can be compared to theoretical patterns,10,12 and the orientation of the dipole deduced.
Providing the emission comes from a single transition dipole, the pattern can be fitted to one theoretically derived using methods described.10,12 However, if more than one emitter is present, the defocused image will show the sum of the emission patterns, and appear as a ring or doughnut shape. These patterns are distinguishable from out-of-plane emission from a single transition dipole by the radius of the ring. Single emitters oriented perpendicular to the excitation plane (in line with the optical axis) show a far narrower ring than the sum of many, in-plane emitters.
To investigate the influence of chain length and host polymer on the efficiency of energy transfer in MEH-PPV, three molecular weight polymers (2600, 24000 and 2600000 Da, Polymer Source) were diluted into both PMMA (a poor solvent matrix) and PS (a good solvent matrix) and examined using the defocused widefield microscope. Fig. 5 shows examples of defocused widefield images from the three molecular weight polymer chains in both PMMA and PS.
 | ||
| Fig. 5 Defocused widefield images of isolated chains of high (a and b), medium (c and d) and low (e and f) molecular weight MEH-PPV in PS (a, c and e) and PMMA (b, d and f). | ||
Each molecule was tracked over the length of the widefield movie, and any changes to the defocused patterns could be observed. Any molecule that showed an emission spatial pattern corresponding to more than one emission was assigned as a multiple emitter (i.e. defocused patterns that were indicative of superimposed single transition dipoles patterns as discussed above) even if it reverted to a single emitter during the experiment. The total number of single and multiple emitters in each data set are shown in Table 1. A clear trend can be observed towards multiple chromophore emission from single chains at higher molecular weights and in extended conformations (i.e. in PS). These observations agree with the photon anti-bunching experiments performed by Masuo et al.18
| M w (kDa) | PMMA | PS | ||||
|---|---|---|---|---|---|---|
| Multia (%) | Δφb (%) |
Total | Multia (%) | Δφb (%) |
Total | |
| a % of total molecules measured for that condition showing emission patterns indicative of multiple chromophore emission. b % of molecules measured for that condition showing at least one change of angle of greater than 20° in the orientation of the emission dipole, φ. | ||||||
| 2.6 | 1 | 1 | 103 | 2 | 2 | 85 |
| 24 | 11 | 18 | 114 | 25 | 24 | 108 |
| 2600 | 48 | 23 | 108 | 51 | 21 | 107 |
The relationship between conformation and number of emitting chromophores can be explained by considering the relative efficiencies of the exciton migration in the two matrices. The energy transfer is more efficient in the collapsed conformation (i.e. in PMMA), as in addition to along-chain energy hopping, the energy can move through space between two sections of the same chain, whereas in extended conformations, the exciton migration is limited to along-chain hopping.23–25 Additionally, in collapsed conformations, exciton–exciton annihilation can occur if more than one chromophore is excited simultaneously. This, in addition to the well-documented energy funnelling to the lowest energy chromophore, leads to a single emission pattern recorded in defocused widefield microscopy. In extended conformations, exciton–exciton annihilation is less efficient, leading to emission from several chromophores – the local energy minimum for that section of chain.
For short polymer chains, along-chain energy migration and exciton–exciton annihilation are efficient along the entire chain length, regardless of conformation. As a result, most low molecular weight chains showed single chromophore emission. Medium chain length polymers show an increased dependence on conformation, with 25% of chains in an extended conformation (i.e. PS matrix) showing multiple emitting chromophores, compared to 10% in the collapsed conformation. The proportion of multiple emitters increases further for the high molecular weight chains, with 51% in PS and 48% in PMMA showing multiple emissions.
It appears that there is less of a conformation effect for single versus multiple emitters at high molecular weights, however it should be noted that there is a difference between the total number of emitters. In PMMA, high molecular weight chains tend to show fewer emitting chromophores, that photobleach quickly to leave a single emitter. The extended chains show multiple chromophore emission for longer, and photobleaching leaves many chromophores still emitting.
Defocused widefield imaging is also useful for observing changes to the emitting chromophore. Quenching events, or the photobleaching of the emitting chromophore can lead to the next-lowest energy chromophore becoming the emitter. In confocal single molecule experiments, chromophore switching has been used to explain sudden shifts in the emission spectrum26 and fluorescence intensity.27 In defocused widefield experiments, this is seen as a sudden change of the angle of the emission dipole. The orientation of the emission dipole is conveniently described by two angles, phi, the rotation of the dipole in the sample (or x, y) plane, and theta, the tilt away from the sample plane towards the optical (or z) axis. Example images for a single chain of medium molecular weight MEH-PPV are shown in Fig. 6.
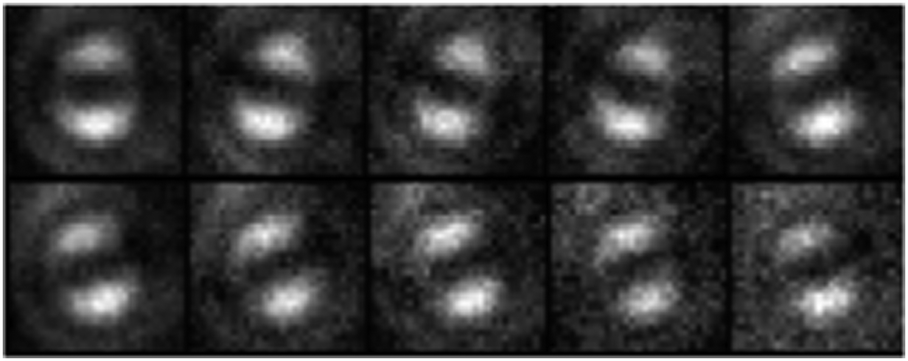 | ||
| Fig. 6 Defocused widefield images taken every 2 s of the same single chain for medium molecular weight MEH-PPV in PS, showing three distinct emission dipoles (φ = 180°, 160° and 20°, θ = ∼0°). | ||
The number of individual emitters increases with polymer chain length, as more chromophores are available for emission. Table 1 also gives the number of molecules in each condition that show changes, as defined by an abrupt change in the emitter phi angle of greater than 20°.
Changes in the emitting chromophore may be accompanied by a change in the emission intensity, fluorescence lifetime or emission spectrum. Fig. 7 shows the fluorescence trajectory, theta angle and defocused images for a MEH-PPV chain in PS, which undergoes a change in emitting chromophore.
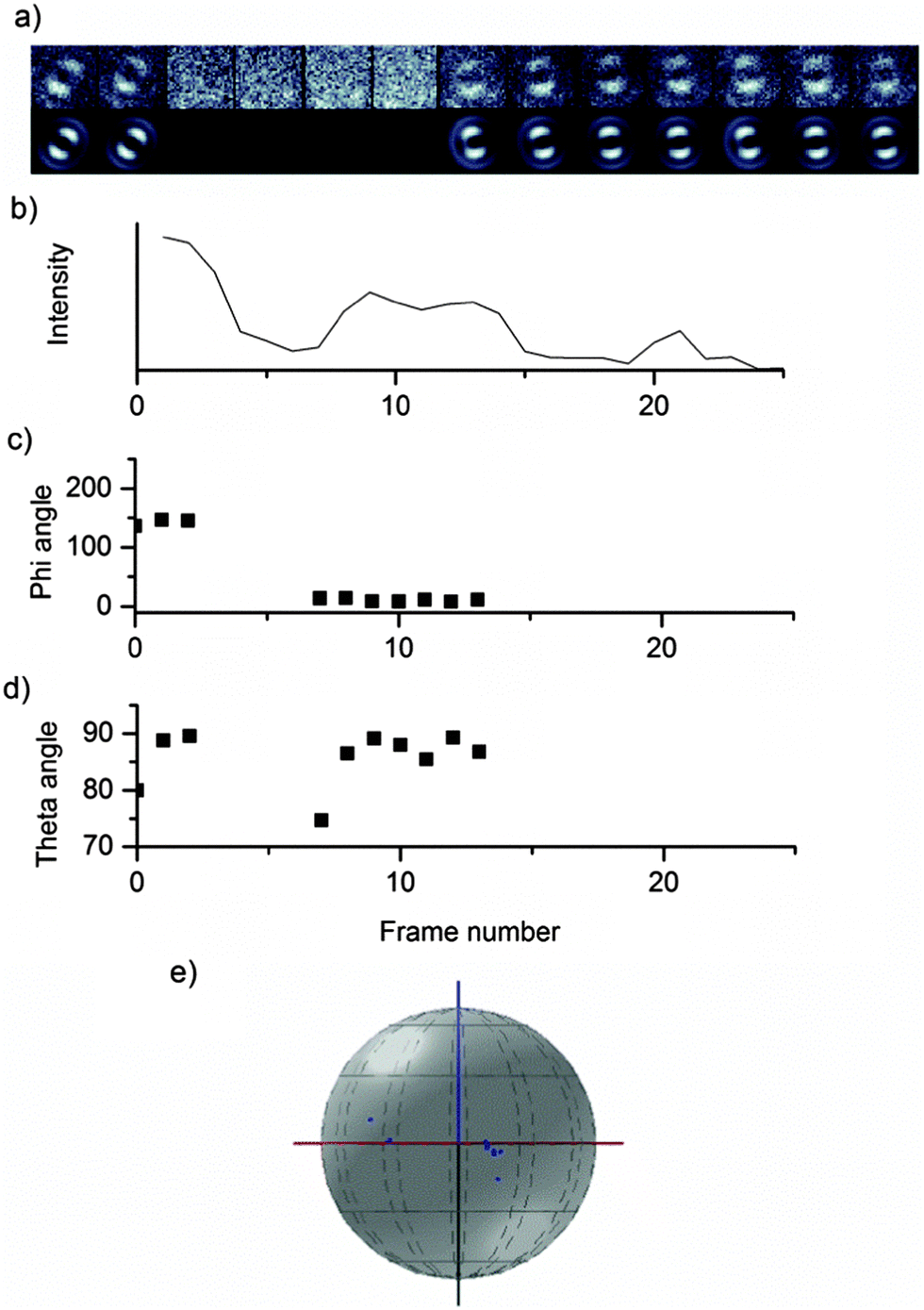 | ||
| Fig. 7 Defocused widefield images of a single chain of medium molecular weight MEH-PPV in polystyrene. The defocused images (a) have been fitted using the MATLAB fitting routine and the phi (c) and theta (d) angles plotted against the fluorescence intensity (b); (d) shows the transition dipole angles (φ and θ) plotted in 3D space. | ||
Pendant MEH-PPV polymers
Defocused widefield microscopy is an effective method for determining whether or not a multi-chromophoric molecule is emitting from a single or multiple emitters. When applied to pendant polymers, it can be used to investigate the efficiency of energy transfer when the size and distance of the chromophores is fixed.The defocused fluorescence emission behaviour of a model MEH-PPV pentamer oligomer (Fig. 1b) is compared here to single polymers that have the MEH-PPV pentamer oligomers appended as side chains along a saturated polymer backbone (Fig. 1c). Fig. 8 shows defocused widefield images of a MEH-PPV pentamer oligomer in PS.
 | ||
| Fig. 8 Defocused images of an isolated pentamer oligomer in PS (top row), along with the fitted theoretical patterns (bottom row). | ||
Of the 100 oligomer molecules fitted, 100% showed single emitter patterns with no variation in emission dipole orientation over time. This is to be expected as each pentamer is a single chromophore embedded in a solid matrix. Defocused widefield movies of the pendant polymer show that the majority of single chains of this system display defocused patterns consistent with a single emitter (93%) and only 10 of the 140 molecules measured showed multiple emitter behaviour. Furthermore, most of the molecules imaged (89%) had no variation in emission dipole orientation with only 15 molecules showing change in this orientation over time.
Given there is no such movement in the emission dipole for the oligomers, shifts in the emission dipole of the pendant polymer molecules may be attributed to a change in the emitting chromophore, rather than movement within the host matrix. One concern with defocused widefield microscopy is that rotation of the molecules in pockets of solvent within the otherwise rigid host matrix may be misinterpreted as a change in the emitting chromophore. As no change is seen in the smaller, less hindered oligomer patterns, it may be assumed that rotation in polystyrene films is minimal.
The tendency of pendant polymer chains to show emission from only one chromophore indicates that energy transport within a polymer chain remains efficient and that some nominally similar chromophores are of lower energy than others. We have previously reported9 that the MEH-PPV oligomers in solution consist of a range of torsional isomers in the ground state due to rotation about the single bonds in the vinylene linker that accounts for their broadened absorption spectra. In solution rapid conformational relaxation following photoexcitation leads to emission from a more planar configuration. In the solid films where such excited state conformational relaxation is inhibited, one could expect the pendant chromophores will exist in a range of energetically distinct conformational species. Under these conditions funnelling of excitation energy could be expected among the gradient of energetically distinct species along the chain until it resides on the pendant chromophores of lowest energy. This is consistent with previous conclusions – that the ‘shape’ of a single chromophore may affect its electronic energy.5
An additional effect could arise from the heterogeneity in the environment of the host matrix that may impose a range of configurations for the pendant groups, providing some lower energy chromophores where the excitation finally resides. The close proximity of the chromophores could also be conducive to interactions between adjacent chromophores, increasing or decreasing their electronic energy. We have reported fluorescence polarization studies for the pendant polymers in frozen 2-methyltetrahydrofuran solutions8 that demonstrate that energy migration occurs and that emission arises from lower energy planar chromophores. In room temperature polymer films where the range of torsional isomer configurations will be ‘frozen-in’ energy transfer to the lowest energy configurations would be favourable. Based on a simple Förster energy transfer model, rates of inter-chromophore energy transfer between two similar pendant MEH-PPV pentamer oligomers, assuming a separation of adjacent pendant chromophores along the polymer chain of 6.6 Å is calculated to be 8 × 1012 s−1 demonstrating the potential high efficiency of this process.8 Other studies have also concluded that through-space energy transfer in conjugated polymers can be quite efficient.4,28
The small number of pendant polymer chains that do show multiple emission can be understood by considering the variety of conformations the polymer chains can adopt. In a large sample, there may be some chains that exist in conformations that make energy migration from one section of the chain to another less likely. In these cases it is possible the orientation of adjacent chromophores is such that the energy transfer probability is zero and the funnelling of excitation energy is confined to non-interacting clusters of pendant chromophores. This splitting of a polymer chain into smaller, tightly packed domains separated by more extended sections of chain has also been proposed in non-pendant polymers, such as MEH-PPV.22,29,30 The multiple emitting nature of some of the polymer chains may also be due to several chromophores very close in energy. Fluctuations in the microenvironments may cause the energies to fluctuate, leading to two (or more) energy minima and this is seen as emission from multiple chromophores over the long exposure time of the defocused widefield instrument.
Another phenomenon observed using defocused widefield imaging is the switching between different emitting chromophores. Fig. 9 shows the change in emission dipole over time for a single, isolated pendant polymer chain. This change in emitting chromophore may be due to the photobleaching of the initially emitting chromophore, and emission then arises from the next lowest energy one that has a different relative orientation. These observations confirm that the single emitter behaviour observed cannot fortuitously arise from all pendant chromophores having the same alignment (i.e. a semi-crystalline structure) since individual chromophores in the chain appear to have a different orientation.
 | ||
| Fig. 9 Defocused widefield patterns (top row) with fitted theoretical patterns (bottom row) for an isolated chain of the pendant polymer in PS. A change in emitting chromophore can be seen after a temporary “off” state. | ||
Conclusions
Energy transfer between the chromophores of a conjugated polymer is not well understood, but is thought to be influenced by chain conformation and the relative distances between the chromophores. Defocused widefield imaging has been used to determine the number of emitting chromophores from single chains and found to be an effective and direct way to examine energy transfer pathways. In MEH-PPV, chain conformation plays an important role in the efficiency of energy transfer. Collapsed conformations allow for effective energy transfer and emission from single, low energy chromophores. Environments that favour extended conformations, however, show multiple emitting chromophores due to poorer energy transfer.Pendant polymers that comprise chromophores of nominally fixed chromophore lengths allow additional insights into energy transfer pathways. Single oligomer units show uniformly single and unchanging emission whereas polymers containing these oligomers as pendant groups show more complex behaviour. Surprisingly energy funnelling to single emitting sites is observed in most polymer chains. The results suggest that the configuration of each individual chromophore and its interaction with the surrounding environment play significant roles in determining the energy transfer processes.
Acknowledgements
We thank the Australian Research Council (ARC) for financial support of this research through grants DP0986166, LE100100131 and LE110100161.Notes and references
- D. A. Vanden Bout, W. Yip, D. Hu, D. Fu, T. M. Swager and P. F. Barbara, Science, 1997, 277, 1074 CrossRef CAS.
- W. Yip, D. Hu, J. Yu, D. A. Vanden Bout and P. F. Barbara, J. Phys. Chem. A, 1998, 102, 7564 CrossRef CAS.
- L. Rothberg, M. Yan, F. Papadimitrakopoulos, M. Galvin, E. Kwock and T. Miller, Synth. Met., 1996, 80, 41 CrossRef CAS.
- B. Van Averbeke, D. Beljonne and E. Hennebicq, Adv. Funct. Mater., 2008, 18, 492 CrossRef CAS.
- K. Becker, E. Da Como, J. Feldmann, F. Scheliga, E. Thorn Csányi, S. Tretiak and J. M. Lupton, J. Phys. Chem. B, 2008, 112, 4859 CrossRef CAS PubMed.
- S. Sarzi Sartori, S. De Feyter, J. Hofkens, M. Van der Auweraer, F. De Schryver, K. Brunner and J. W. Hofstraat, Macromolecules, 2003, 36, 500 CrossRef.
- D. Hu, J. Yu, K. Wong, B. Bagchi, P. J. Rossky and P. F. Barbara, Nature, 2000, 405, 1030 CrossRef CAS PubMed.
- A. J. Tilley, M. Chen, S. M. Danczak, K. P. Ghiggino and J. M. White, Polym. Chem., 2012, 3, 892 RSC.
- A. J. Tilley, S. M. Danczak, C. Browne, T. Young, T. Tan, K. P. Ghiggino, T. A. Smith and J. M. White, J. Org. Chem., 2011, 76, 3372 CrossRef CAS PubMed.
- D. Patra, I. Gregor and J. Enderlein, J. Phys. Chem. A, 2004, 108, 6836 CrossRef CAS.
- M. Böhmer and J. Enderlein, J. Opt. Soc. Am. B, 2003, 20, 554 CrossRef.
- P. Dedecker, B. Muls, A. Deres, H. Uji-i, J. Hotta, M. Sliwa, J. Soumillion, K. Müllen, J. Enderlein and J. Hofkens, Adv. Mater., 2009, 21, 1079 CrossRef CAS.
- H. Uji-i, S. M. Melnikov, A. Deres, G. Bergamini, F. De Schryver, A. Herrmann, K. Müllen, J. Enderlein and J. Hofkens, Polymer, 2006, 47, 2511 CrossRef CAS.
- D. Gómez, T. A. T. Tan, J. M. White, T. D. M. Bell and K. P. Ghiggino, Aust. J. Chem., 2009, 62, 1577 CrossRef.
- T. D. M. Bell, S. Yap, C. H. Jani, S. Bhosale, J. Hofkens, F. C. De Schryver, S. J. Langford and K. P. Ghiggino, Chem. –Asian J., 2009, 4, 1542 CrossRef CAS PubMed.
- T. Pullerits, O. Mirzov and I. G. Scheblykin, J. Phys. Chem. B, 2005, 109, 19099 CrossRef CAS PubMed.
- Y. Ebihara, S. Habuchi and M. Vacha, Chem. Lett., 2009, 38, 1094 CrossRef CAS.
- S. Masuo, T. Tanaka, S. Machida and A. Itaya, Appl. Phys. Lett., 2008, 92, 233114 CrossRef.
- J. Liang, J. D. White, Y. C. Chen, C. F. Wang, J. C. Hsiang, T. S. Lim, W. Y. Sun, J. H. Hsu, C. P. Hsu, M. Hayashi, W. S. Fann, K. Y. Peng and S. A. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 085209 CrossRef.
- Y. Ebihara and M. Vacha, J. Phys. Chem. B, 2008, 112, 12575 CrossRef CAS PubMed.
- T. Sugimoto, S. Habuchi, K. Ogino and M. Vacha, J. Phys. Chem. B, 2009, 113, 12220 CrossRef CAS PubMed.
- H. Lin, S. R. Tabaei, D. Thomsson, O. Mirzov, P. Larsson and I. G. Scheblykin, J. Am. Chem. Soc., 2008, 130, 7042 CrossRef CAS PubMed.
- P. F. Barbara, A. J. Gesquiere, S. Park and Y. Lee, Acc. Chem. Res., 2005, 38, 602 CrossRef CAS PubMed.
- C. Hollars, S. M. Lane and T. Huser, Chem. Phys. Lett., 2003, 370, 393 CrossRef CAS.
- T. Q. Nguyen, J. Wu, V. Doan, B. J. Schwartz and S. H. Tolbert, Science, 2000, 288, 652 CrossRef CAS PubMed.
- K. P. Ghiggino, T. D. M. Bell and E. N. Hooley, Faraday Discuss., 2012, 155, 79 RSC.
- S. Habuchi, S. Onda and M. Vacha, Chem. Commun., 2009, 4868 RSC.
- J. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef CAS PubMed.
- H. Lin, Y. Tian, K. Zapadka, G. Persson, D. Thomsson, O. Mirzov, P. O. Larsson, J. Widengren and I. G. Scheblykin, Nano Lett., 2009, 9, 4456 CrossRef CAS PubMed.
- S. Habuchi, S. Onda and M. Vacha, Phys. Chem. Chem. Phys., 2010, 13, 1743 RSC.
Footnotes |
| † Current address: Nano-Science Center/Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark |
| ‡ Current address: Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON M5S 3H6, Canada |
| This journal is © the Owner Societies 2014 |