 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial charge separation in Cu2O/RuOx as a visible light driven CO2 reduction catalyst†
Ernest
Pastor
a,
Federico M.
Pesci
a,
Anna
Reynal
*a,
Albertus D.
Handoko‡
b,
Mingjia
Guo
b,
Xiaoqiang
An
b,
Alexander J.
Cowan§
a,
David R.
Klug
a,
James R.
Durrant
a and
Junwang
Tang
*b
aDepartment of Chemistry, Imperial College London, Exhibition Road, SW7 2AZ, London, UK. E-mail: a.reynal@imperial.ac.uk
bDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK. E-mail: junwang.tang@ucl.ac.uk
First published on 11th February 2014
Abstract
We employ transient absorption spectroscopy to record the absorption spectrum of photogenerated charge carriers in Cu2O. We have found that CO2 reduction in Cu2O is limited by fast electron–hole recombination. The deposition of RuOx nanoparticles on Cu2O results in a twofold increased yield of long-lived electrons, indicating partially reduced electron–hole recombination losses. This observation correlates with an approximately sixfold increase in the yield of CO2 reduction to CO.
Photochemical reduction of CO2 has the potential to convert this greenhouse gas into clean fuels or value-added chemicals. As such, it can contribute significantly to both renewable energy generation and CO2 mitigation.1 Photoreduction of CO2 in heterogeneous systems was first reported using optical excitation of large bandgap semiconductor materials such as TiO2, SrTiO3, ZnO, or SiC with UV light irradiation.2 Since this initial study, many reports have focused on the development of materials with improved light absorption properties in the visible region, such as narrow bandgap non-metal oxide semiconductors (e.g. ZnS, CdS, and GaP), or the functionalization or doping of wide bandgap semiconductors,3 although overall efficiencies reported for CO2 photoreduction remain limited. These low efficiencies have often been attributed to fast charge recombination, requirements of high over-potentials and competition between CO2 photoreduction reaction with H2 evolution, as well as issues associated with catalyst/co-catalyst degradation or deactivation.4 Identifying the parameters that restrain the performance of catalysts towards CO2 reduction and designing a system capable of overcoming these limitations is therefore a key challenge. In this context, transient absorption spectroscopy (TAS) is a technique that allows the assessment of competing kinetic processes in a semiconductor, namely charge separation and recombination, by monitoring the dynamics of charge carriers. We have, for example previously reported the signals corresponding to electrons and holes in Fe2O3 and other photoanodes for the related reaction of photochemical water-splitting.5
Herein, we report the dynamics of charge carriers in Cu2O. To the best of our knowledge, the absorption spectrum and dynamics of photoexcited electrons in Cu2O have not been identified. This narrow bandgap (ca. 2.2 eV) p-type semiconductor has been shown to be photocatalytically active for reduction of water6 and degradation of inorganic waste.7 Since the conduction band of Cu2O lies above the reduction potential of CO2 to CO,8 it is attracting increasing interest for the photoreduction of CO2.9 As a p-type semiconductor, interfacial band-bending of Cu2O, where present, is likely to favour interfacial reduction reactions but hinder oxidation reactions by presenting an energetic barrier for holes to approach the semiconductor surface. These surface reduction/oxidation reactions will be in kinetic competition with electron–hole recombination loss pathways (Fig. 1a). We have recently reported that the deposition of a charge acceptor (RuOx, x ≤ 2) on top of Cu2O improves its photocatalytic activity towards CO2 reduction.9b Herein, we focus on the study of the dynamics of photogenerated electrons in Cu2O when forming an inorganic heterojunction with a material capable of (1) increasing the accessibility of photogenerated holes to the electrolyte, (2) having appropriate reaction sites to drive photo-oxidation reactions and (3) reducing the electron–hole recombination reaction on the Cu2O surface (Fig. 1b). Ruthenium oxide (RuOx) was selected as the overlayer material because its appropriate work function level can facilitate transfer of holes from Cu2O, thereby potentially increasing the yield of long-lived electrons in Cu2O to enable the reduction of CO2. RuOx is also less likely to react with Cu2O to form an unwanted interfacial layer and has been shown to have the ability to oxidize many hole scavengers, including water.10
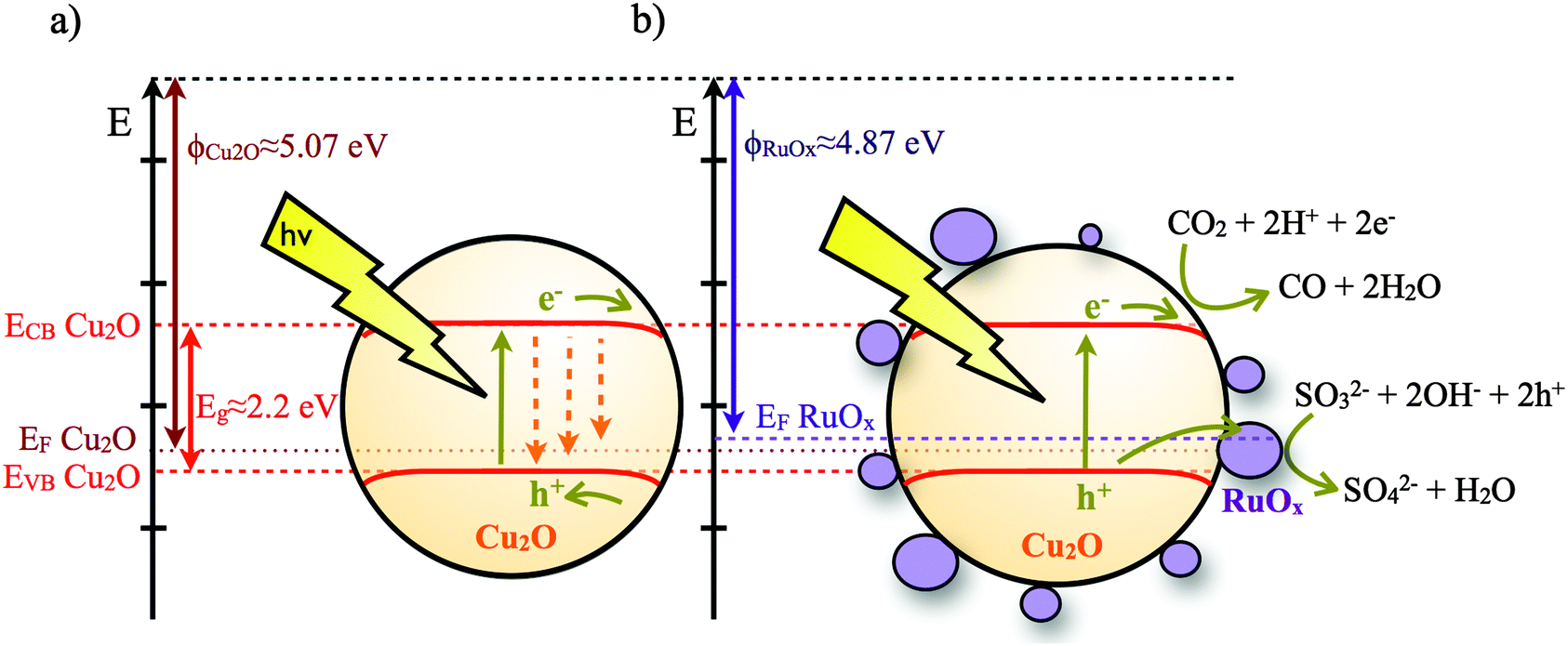 | ||
| Fig. 1 Schematic of Cu2O and the Cu2O/RuOx band diagram and electron and hole transfer reactions for the photocatalytic CO2 reduction. Energy values were taken from ref. 8b and 12. EF stands for Fermi level, ECB and EVB indicate the energy levels of the conduction band and valence band, respectively. | ||
Spectroscopic measurements were performed using Cu2O films deposited onto FTO glass. Cu2O cuboid films with a nanoparticle size of ca. 100 nm were prepared by a chemical solution method using CuSO4, Na2S2O3 and NaOH, as reported previously.11 RuOx was subsequently deposited in the form of nanoparticles <10 nm on the Cu2O surface.13 The RuOx deposition did not alter the bandgap absorption of Cu2O of approx. 2.2 eV. Only Cu2O diffraction peaks were detected in Cu2O–RuOx samples, and Cu(I) species were confirmed by XPS analysis, albeit coexisting with small amounts of Cu(II) (Fig. S4, ESI†). Trace amounts of Ru were detected on the Cu2O–RuOx heterojunction by elemental analysis during TEM and XPS investigations, but not X-ray diffraction due to the low loading amount and small crystallite size of RuOx. TAS spectra were recorded after UV excitation (355 nm) of Cu2O and Cu2O/RuOx, using an experimental setup described previously.14 Details of synthetic procedures, material characterisation and experimental setup for transient absorption measurements can be found in the ESI† (Fig. S1–S4).
The absorption spectrum of charge carriers in a semiconductor can be characterized by TAS, by employing relevant fast hole and electron scavengers.15 Thus, the transient absorption spectrum of Cu2O films deposited onto the FTO glass was obtained (i) in a N2 purged aqueous solution (ii) following the addition of Na2SO3 as a hole scavenger or (iii) AgNO3 as an electron scavenger. Photoexcitation of the Cu2O semiconductor in the absence of charge carrier scavengers results in the appearance of a ground state bleach signal between 475 and 750 nm and two positive photoinduced absorption peaks at wavelengths <475 nm and >800 nm (Fig. 2). The signals at wavelengths >850 nm are significantly enhanced by the addition of Na2SO3 and weakened by the addition of AgNO3. Given that Na2SO3 can effectively scavenge holes, while Ag+ scavenges electrons, we can assign the transient signals above 850 nm to Cu2O photoexcited electrons. On the other hand, we observed only the signal corresponding to photoexcited holes in Cu2O at wavelengths <475 nm when using AgNO3 as an electron scavenger. Thus, the direct probing of holes under the current experimental photocatalytic conditions is hindered by the partial overlap with the bleach signal of the ground state and cannot be directly monitored. Since these signals corresponding to photoexcited electrons and holes in the Cu2O are already present in the 10 μs timescale, our measurements indicate that electron–hole scavenging by the relevant chemicals are very fast (<10 μs), beyond the instrument's limit of response.
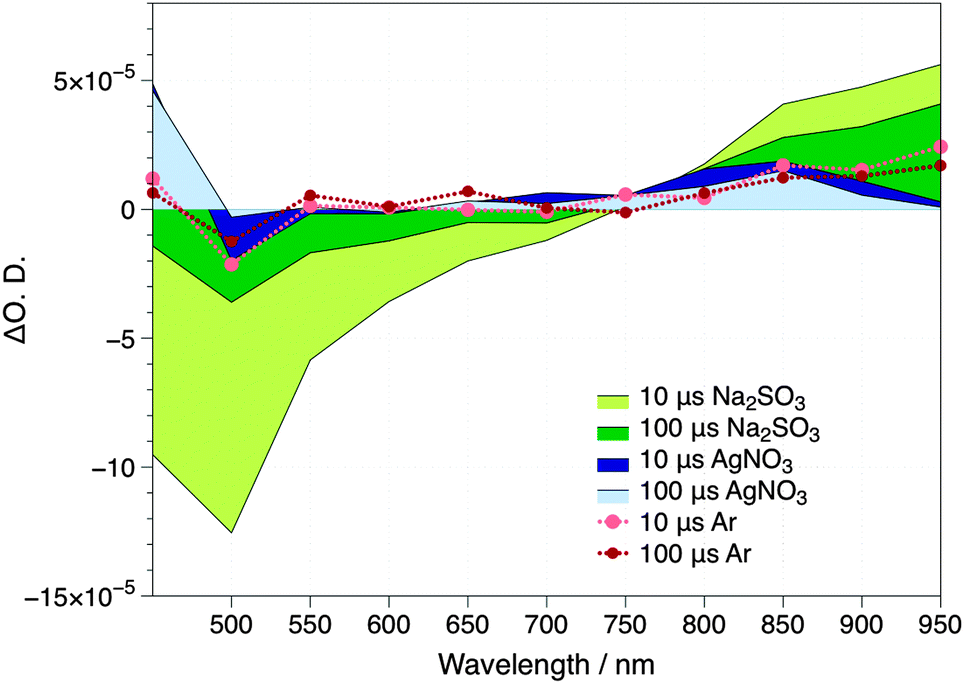 | ||
| Fig. 2 Transient absorption spectrum of Cu2O recorded under N2, in the presence of a 0.1 M Na2SO3 and 0.01 M AgNO3 aqueous solutions. The samples were excited at λex = 355 nm, with a laser intensity of ∼1.2 mJ cm−2. | ||
The formation of inorganic heterojunctions or the addition of sacrificial agents which are able to scavenge charge carriers have been reported to improve the performance of the photocatalytic systems for organic waste decomposition or water splitting reactions by decreasing the recombination of photogenerated electrons and holes in a semiconductor.4b,16 Thus, transient absorption spectroscopy experiments were also employed to assay the effect of RuOx deposition on the Cu2O charge carrier dynamics. Typical kinetic data of the photoinduced electron absorption signals taken at 950 nm are plotted in Fig. 3, showing the decay dynamics on the micro- to second timescales. We note that we only observed significant long-lived signal amplitudes with relatively intense excitation densities, indicative of the presence of significantly fast (<10 μs) electron–hole recombination in these films. The presence of Na2SO3 resulted in an increase in the amplitude of this Cu2O electron signal (Fig. 2), although this effect is relatively minor, indicative of inefficient hole scavenging/fast recombination, and consistent with the expected band bending impeding access of photogenerated holes to the Cu2O surface (Fig. 1a). The deposition of RuOx on the Cu2O surface results in a significant increase in the amplitude of the long-lived electron signal, independent of the presence of Na2SO3, and indicative of a more efficient reduction in fast electron–hole recombination losses. These results qualitatively suggest that RuOx deposition results in a significant increase in the yield of long-lived (>100 μs) Cu2O electrons. This is attributed to a reduction in fast electron–hole recombination losses due to formation of the inorganic junction, most probably due to hole transfer from Cu2O to RuOx, thereby increasing the spatial separation of electrons and holes and facilitating the photooxidation reaction by holes. The lack of absorption of RuO in the near IR region of the spectrum indicates that the increase in absorbance observed at 950 nm is not due to the reduction of RuOx by Cu2O photoexcited electrons, and therefore confirms our peak assignation.17 We want to note that a Cu2O–Al:ZnO–TiO2–RuOx heterojunction for H+ reduction, where the Cu2O and RuOx interfaces are connected through an interfacial (AZO–TiO2) layer, has been reported by Tilley et al.6d Contrary to our approach, where RuOx accepts the holes from Cu2O, charge separation in Cu2O–AZO–TiO2–RuOx takes place through electron transfer from Cu2O to RuOx. We believe that, while in the Cu2O–AZO–TiO2–RuOx there is a favorable downhill electron transfer cascade from Cu2O to RuOx, the AZO–TiO2 layer blocks the hole transfer to RuOx.
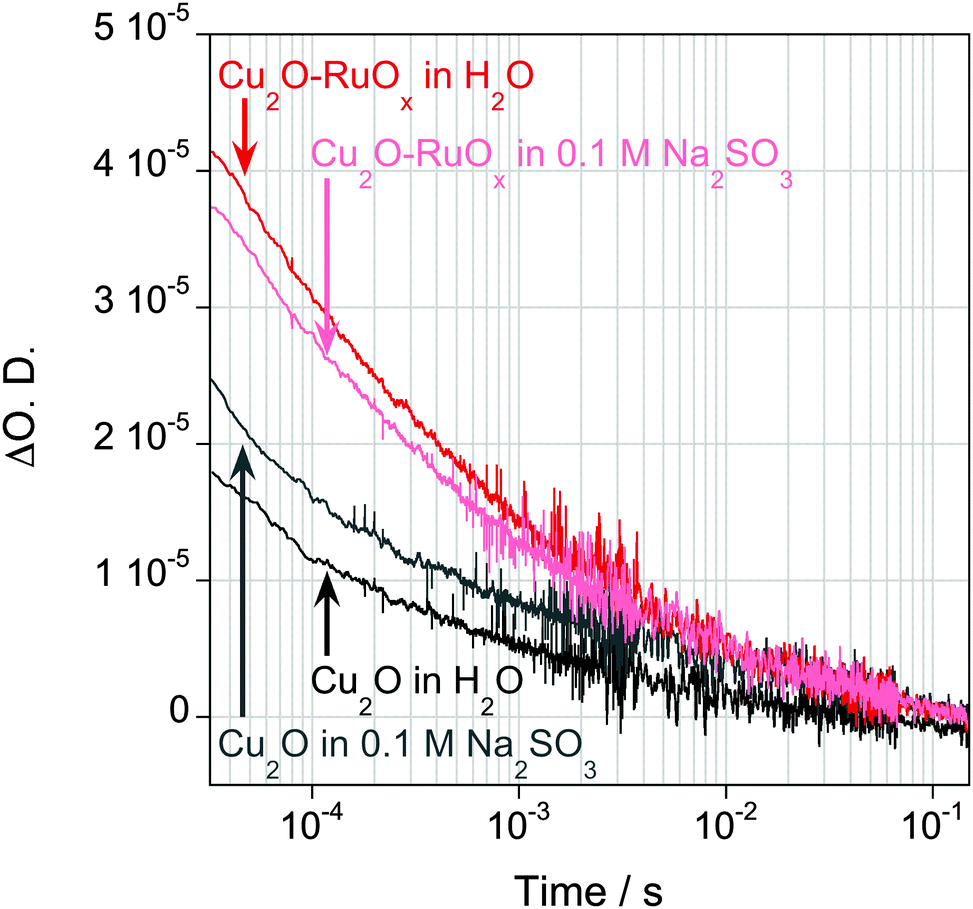 | ||
| Fig. 3 TAS decays of photo-excited electrons of Cu2O films (black trace) and Cu2O–RuOx films (red trace) in N2 purged water, and in the presence of a N2 purged 0.1 M Na2SO3 aqueous solution (Cu2O films: grey trace, Cu2O–RuOx films: pink trace). Data obtained at λprobe = 950 nm with an excitation intensity of 1.6 mJ cm−2 at λex = 355 nm. | ||
The effect of enhanced charge separation by the Cu2O–RuOx junction strategy on CO2 photoreduction was tested using analogous bare Cu2O and Cu2O–RuOx nanoparticulate suspensions in CO2-saturated deionised H2O in the presence of a Na2SO3 hole scavenger upon irradiating under the full arc of a 150 W Xe lamp (Fig. 4). In agreement with the results observed upon irradiating the samples with visible light λ ≥ 420 nm,9b the coupling of Cu2O with RuOx results in a nearly sixfold increase of the initial rate of CO production, from 0.16 μmol g−1 on bare Cu2O to around 0.88 μmol g−1 on the Cu2O–RuOx junction after 1 h irradiation using a 150 W Xe lamp. Trace amounts of methanol (0.01 μmol g−1 h−1) and methane (<0.001 μmol g−1 h−1) were also observed in the Cu2O–RuOx sample during the reaction. Quantum yield measurements were carried out using a near monochromatic visible light 400 nm bandpass filter (Δλ < 10 nm, filtered light output ca. 600 ± 40 μW cm−2). The apparent initial quantum yield for CO generation (20 min) for the Cu2O–RuOx heterojunction was determined to be ca. 1.6%. It is also noted that the CO evolution rate is reduced after the first hour. These results can be explained in terms of the instability of Cu2O towards the presence of increasing amounts of SO42− formed upon scavenging the holes by Na2SO3.18 Another possible explanation for non-linear CO evolution could be the strong interaction between CO and Cu2O, which will result in lowered catalytic activity over time.19 Studies to address this instability by using alternative hole scavengers and further modifying material protection layers are ongoing.
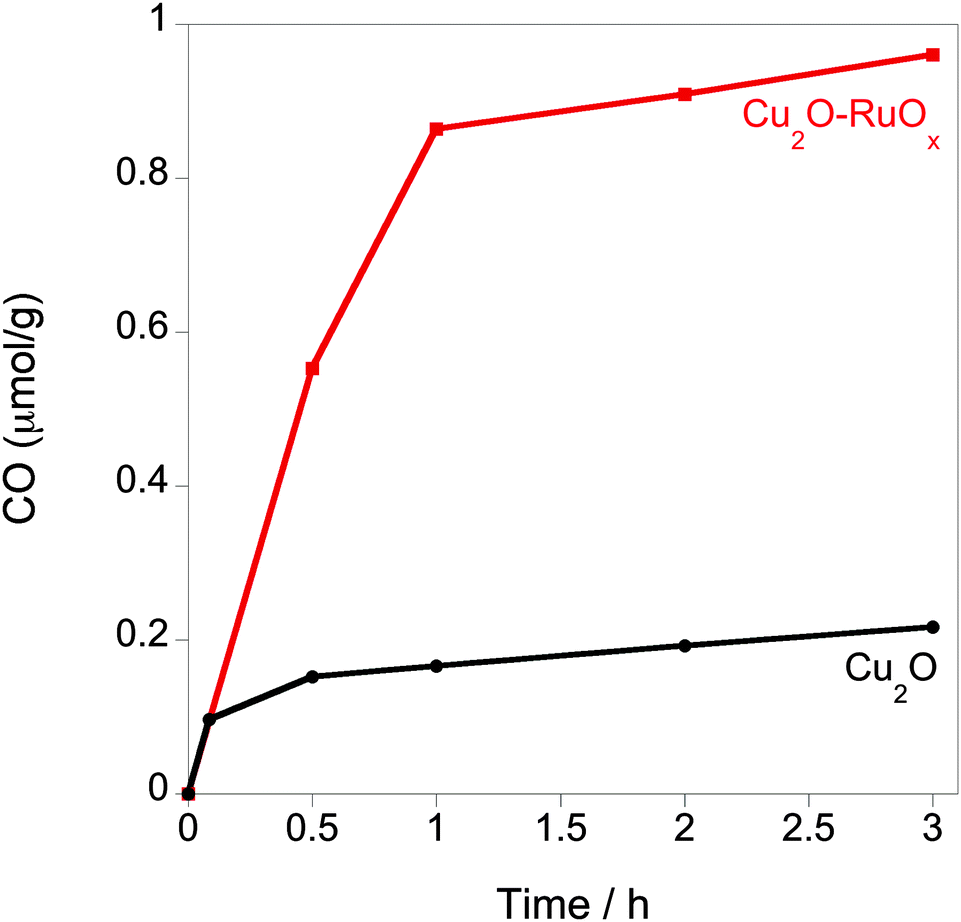 | ||
| Fig. 4 CO2 reduction to CO on bare and heterojunction based Cu2O core photocatalysts under full arc 150 W Xe lamp irradiation. Photoreduction reactions were undertaken in 3 mL of CO2 saturated H2O in the presence of a hole scavenger (ca. 0.7 M Na2SO3). | ||
RuOx has been reported to be able to either reduce or oxidize water,6d,20 but is unable to reduce CO2 due to its work function being more positive than the Ered(CO2/CO) reduction potential. We did not observe any water oxidation and oxygen evolution upon the addition of RuOx, as holes are preferentially scavenged by Na2SO3. Thus, the increase in the CO2 photoreduction yield is consistent with the efficient charge separation due to the inorganic heterojunction, where holes are transferred to RuOx and CO2 reduction takes place at the surface of Cu2O.
In order to provide evidence that the formation of CO is due to the photoreduction of CO2 by Cu2O, three different control experiments were carried out: (1) traces of organic contaminants were removed by treating the aqueous suspension system under strong light irradiation without CO2 until there was no increase in the CO or CO2 amount as detected by gas chromatography before the photocatalytic measurements. (2) Control experiments under similar conditions but in the absence of photocatalyst, CO2 or light, and irradiating the Cu2O with longer wavelength light (λ > 668 nm, much lower than Cu2O bandgap requirement) demonstrated that surface carbon gasification is not a major contributing factor (Fig. S5, ESI†). (3) Gas chromatography – Mass spectrometry isotope labelling analysis showed the sole formation of 13CO (m/z 29) (Fig. S6, ESI†). These results strongly indicate that the CO observed by GC is due to photochemical reduction of CO2 rather than from the reduction of organic contaminants present on the surface of Cu2O–RuOx.
Conclusions
In summary, we have assigned the transient absorption spectrum of Cu2O photoexcited electrons to appear at wavelength λ > 800 nm, and holes at λ < 475 nm. In bare Cu2O films, the major loss pathway limiting their photocatalytic activity is the fast electron–hole recombination. Our results demonstrate that the Cu2O–RuOx heterojunction strategy is effective in partially suppressing this electron–hole recombination loss through the transfer of photogenerated holes from Cu2O to RuOx. This is shown by a significant increase in the yield of long-lived Cu2O electrons as observed by transient absorption spectroscopy. This electron yield and a lifetime increase is translated into a sixfold photocatalytic reaction increase by Cu2O–RuOx coupling in favour of CO2 reduction.Acknowledgements
Financial support from the EPSRC (EP/H046380/1) and ERC (project Intersolar to J. D.) is gratefully acknowledged. A.R. thanks the European Commission Marie Curie CIG (PhotoCO2) and the Spanish Ministry of Education for the EX2010-0479 postdoctoral fellowship. E. P. also thanks the Spanish Ministry of Education, the University of Valencia and the EU for the Erasmus scholarship. The authors thank Dr M. Ardakani (Imperial College London) for TEM and elemental analyses and Prof. P. Cumpson (NEXUS, Newcastle University) for XPS analyses.Notes and references
- (a) E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112 RSC; (b) S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259 CrossRef CAS PubMed; (c) G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 RSC; (d) Y. Izumi, Coord. Chem. Rev., 2013, 257, 171 CrossRef CAS PubMed.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637 CrossRef CAS.
- (a) B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541 CrossRef CAS PubMed; (b) S. Navalón, A. Dhakshinamoorthy, M. Álvaro and H. Garcia, ChemSusChem, 2013, 6, 562 CrossRef PubMed; (c) S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed; (d) T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132 CrossRef CAS PubMed; (e) S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240 CrossRef CAS PubMed; (f) K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596 CrossRef CAS PubMed.
- (a) A. Corma and H. Garcia, J. Catal., 2013, 308, 168 CrossRef CAS PubMed; (b) A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281 RSC.
- M. Barroso, S. R. Pendlebury, A. J. Cowan and J. R. Durrant, Chem. Sci., 2013, 4, 2724 RSC.
- (a) C.-C. Hu, J.-N. Nian and H. Teng, Sol. Energy Mater. Sol. Cells, 2008, 92, 1071 CrossRef CAS PubMed; (b) P. E. de Jongh, D. Vanmaekelbergh and J. J. Kelly, Chem. Commun., 1999, 1069 RSC; (c) A. Paracchino, V. Laporte, K. Sivula, M. Gratzel and E. Thimsen, Nat. Mater., 2011, 10, 456 CrossRef CAS PubMed; (d) S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Graetzel, Adv. Funct. Mater., 2013, 24, 303 CrossRef; (e) C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482 RSC.
- (a) Y. Zhang, B. Deng, T. Zhang, D. Gao and A.-W. Xu, J. Phys. Chem. C, 2010, 114, 5073 CrossRef CAS; (b) C. H. Kuo, C. H. Chen and M. H. Huang, Adv. Funct. Mater., 2007, 17, 3773 CrossRef CAS.
- (a) E. Ruiz, S. Alvarez, P. Alemany and R. A. Evarestov, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 7189 CrossRef CAS; (b) H. Raebiger, S. Lany and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 045209 CrossRef.
- (a) K. Tennakone, A. H. Jayatissa and S. Punchihewa, J. Photochem. Photobiol., A, 1989, 49, 369 CrossRef CAS; (b) A. D. Handoko and J. Tang, Int. J. Hydrogen Energy, 2013, 38, 13017 CrossRef CAS PubMed; (c) K. Rajeshwar, N. R. de Tacconi, G. Ghadimkhani, W. Chanmanee and C. Janáky, ChemPhysChem, 2013, 14, 2251 CrossRef CAS PubMed.
- (a) M. Neumann-Spallart, K. Kalyanasundaram, C. Grätzel and M. Grätzel, Helv. Chim. Acta, 1980, 63, 1111 CrossRef CAS; (b) J. Kiwi, M. Grätzel and G. Blondeel, J. Chem. Soc., Dalton Trans., 1983, 2215 RSC.
- M. Ristov, G. Sinadinovski and I. Grozdanov, Thin Solid Films, 1985, 123, 63 CrossRef CAS.
- (a) J. Deuermeier, J. Gassmann, J. Brotz and A. Klein, J. Appl. Phys., 2011, 109, 113704 CrossRef PubMed; (b) L.-S. Wang, H. Wu, S. R. Desai and L. Lou, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 8028 CrossRef CAS; (c) A. Klein, Thin Solid Films, 2012, 520, 3721 CrossRef CAS PubMed; (d) Y. L. Chueh, C. H. Hsieh, M. T. Chang, L. J. Chou, C. S. Lao, J. H. Song, J. Y. Gan and Z. L. Wang, Adv. Mater., 2007, 19, 143 CrossRef CAS.
- M. Kohno, T. Kaneko, S. Ogura, K. Sato and A. Yasunobu Inoue, J. Chem. Soc., Faraday Trans., 1998, 94, 89 RSC.
- A. Reynal, F. Lakadamyali, M. A. Gross, E. Reisner and J. R. Durrant, Energy Environ. Sci., 2013, 6, 3291 CAS.
- (a) J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885 CrossRef CAS PubMed; (b) M. Barroso, C. A. Mesa, S. R. Pendlebury, A. J. Cowan, T. Hisatomi, K. Sivula, M. Grätzel, D. R. Klug and J. R. Durrant, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15640 CrossRef CAS PubMed.
- (a) S.-I. In, D. D. Vaughn and R. E. Schaak, Angew. Chem., Int. Ed., 2012, 51, 3915 CrossRef CAS PubMed; (b) H. G. Kim, P. H. Borse, J. S. Jang, E. D. Jeong, O.-S. Jung, Y. J. Suh and J. S. Lee, Chem. Commun., 2009, 5889 RSC; (c) S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400 CrossRef CAS PubMed.
- G. L. Zimmerman, S. J. Riviello, T. A. Glauser and J. G. Kay, J. Phys. Chem., 1990, 94, 2399 CrossRef CAS.
- M. J. Siegfried and K.-S. Choi, J. Am. Chem. Soc., 2006, 128, 10356 CrossRef CAS PubMed.
- (a) A. V. Larin, Langmuir, 1987, 3, 318 CrossRef CAS; (b) H. Yamashita, M. Matsuoka, K. Tsuji, Y. Shioya, M. Anpo and M. Che, J. Phys. Chem., 1996, 100, 397 CrossRef CAS; (c) X. Wang, J. C. Hanson, A. I. Frenkel, J.-Y. Kim and J. A. Rodriguez, J. Phys. Chem. B, 2004, 108, 13667 CrossRef CAS.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterisation of Cu2O and Cu2O–RuOx powder and films, and control experimental data. See DOI: 10.1039/c4cp00102h |
| ‡ Present address: Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. |
| § Present address: Stephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. |
| This journal is © the Owner Societies 2014 |