 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azaporphine guest–host complexes in solution and gas-phase: evidence for partially filled nanoprisms and exchange reactions†
Patrick
Weis
*a,
Ulrike
Schwarz
a,
Frank
Hennrich
b,
Danny
Wagner
c,
Stefan
Bräse
cd and
Manfred
Kappes
ab
aInstitute of Physical Chemistry, Karlsruhe Institute of Technology, Kaiserstr. 12, 76131 Karlsruhe, Germany. E-mail: patrick.weis@kit.edu
bInstitute of Nanotechnology, Karlsruhe Institute of Technology, 76021 Karlsruhe, Germany
cInstitute of Organic Chemistry, Karlsruhe Institute of Technology, Kaiserstr. 12, 76131 Karlsruhe, Germany
dInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology, 76021 Karlsruhe, Germany
First published on 7th February 2014
Abstract
Supramolecular guest–host complexes comprising various azaporphines stacked in a coordination nanoprism consisting of tris(4-pyridyl)triazines as panels, 1,4-bis(pyridyl)benzenes as pillars and (en)Pd as hinges were synthesized according to the procedure of Fujita and coworkers and characterized as ions in the gas-phase by high-resolution electrospray ionization mass spectrometry and collision induced dissociation as well as in solution by analytical ultracentrifugation. Apart from fully filled nanoprisms we have also prepared and observed partially filled as well as empty congeners in aqueous solutions. Upon mixing room temperature solutions of two types of nanoprisms, we observe that azaporphine guest exchange reactions occur on a timescale of minutes, indicating that the formation of the guest–host complexes is reversible.
1. Introduction
Metalloporphyrins play a key role in the biochemistry of basically all organisms, therefore they are well studied in the condensed phase.1 Besides their obvious biological importance, they represent fascinating building blocks in supramolecular chemistry, since they are known to easily self-assemble in solution2–8 with the structures observed depending strongly on the substituents linked to the porphyrin ring. Prominent examples are H- and J-type aggregates, i.e. stacks and chains of porphyrins. The interactions involved were π–π stacking, hydrogen bridges, electrostatic and van der Waals interactions, with varying relative extents. Controlling and tuning the size of the aggregates is desirable for applications which depend on the number of monomeric units. Typically this is very difficult – particularly for bulk scale preparations. One way to solve this problem is to orient the porphyrins along template structures. Along these lines Sugimoto et al. have constructed ladder-type assemblies using dialkylammonium cations as templates.9 Di Costanzo et al. have created calixarene/porphyrin crystals with stoichiometry tunable by pH variations.10 In a particularly elegant approach, Fujita and coworkers have developed a surprisingly simple method to confine (and stack) up to three metal-(aza)-porphine guests in a coordination “nanoprism”. The corresponding prismatic host framework is composed of triazine and bipyridine derivatives which act as “panels” and “pillars”, respectively, with the corners constituted by cis-protected palladium and platinum “hinges”11–13 (see structural schematic in Scheme 1). The guest–host complex self-assembles upon heating an aqueous solution of the four components in roughly stoichiometric ratios. Also, the number of (aza)-porphine guests can be adjusted simply by varying the length of the pillar: with pyrazine pillars the nanoprism can accommodate one, with 4,4′-dipyridine pillars two, and with 1,4-bis(pyridyl)benzenes it can accommodate three (aza)-porphines. | ||
| Scheme 1 A – tris(4-pyridyl)triazine “panels”, B 1,4-bis(pyridyl)benzene “pillars”, (en)Pd “hinges”, H, Cu azaporphine “guests”. | ||
The goal of the present study was to further investigate the influence of the azaporphine guest on the energetics of these fascinating host–guest complexes. For this we have applied a novel combination of analytical ultracentrifugation (AUC) and high resolution electrospray ionization mass spectrometry (ESI-MS) to determine the compositions of solutions containing the host–guest complexes. Furthermore, we have used collision induced dissociation studies to compare the gas-phase stabilities of nanoprism hosts filled with different sets of guests. As part of our effort on completely filled nanoprisms, we have also explored whether partially or even empty nanoprisms can be formed and whether the formation of the guest–host complex is in fact reversible under room temperature solution conditions.
We decided to carry out a joint ESI-MS/AUC study because it was not clear to what extent either method can in fact be used to determine the actual solution compositions. Fujita and coworkers have propagated the use of “cold-spray” ESI-MS to study these systems – specifically so as to provide softer ionization with less fragmentation. AUC on the other hand is a well-established method used in biochemistry and polymer science to study the composition of complex solutions. More recently, it has been rediscovered as a useful means of probing liquid dispersions of size-selected nanoparticles. The interest derives in part from the fact that recent 2D mathematical and computational modelling advancements14 allow sedimentation-velocity analytical ultracentrifugation (SV-AUC) data to be much more rapidly analyzed towards mapping the sedimentation coefficient and diffusion coefficient distributions of the major species present in solution.15,16 While Carney et al. have recently demonstrated that AUC can quickly characterize the size, density and molecular weight distribution of stable ligand-stabilized gold clusters – within one single measurement17 – there are no literature precedents which show that AUC can in fact be used to characterize solutions of charged supramolecular structures like those of interest here.
2. Experimental
Synthesis
The nanoprism-samples with metal-free “1” and copper-tetraazaporphine “2” were prepared according to Ono et al.11 The procedure for sample “1” is as follows: in a capped vial, (ethylenediamine)-palladium(II)nitrate (34.86 mg; 120 μmol; 6 eq.), 1,4-bis(2,6-dimethyl-4-pyridyl)benzene (17.3 mg; 60 μmol; 3 eq.), tris(4-pyridyl)triazine (12.5 mg; 40 μmol; 2 eq.) and tetraazaporphine (25.12 mg; 80 μmol; 4 eq.) were mixed with 2 ml of water and the reaction mixture was stirred at 100 °C for two hours. The mixture was filtered through a small plug of cotton and the filtrate submitted to lyophilization. 72.6 mg (17.39 μmol) of a violet, fluffy powder were obtained. Yield: 90%. The analytical data correspond to literature. For sample “2” we use the same protocol with 80 μmol of Cu-tetraazaporphine (which contained small amounts of metal-free tetraazaporphine, see below). In case of the empty host sample “3” the same typical procedure was followed without addition of any tetraazaporphine. A white powder was obtained.Mass spectrometry
The mass spectrometric and collision induced dissociation (CID) studies were performed with a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) consisting of an electrospray ionization source (ESI), a linear ion trap mass spectrometer and a high resolution orbitrap mass spectrometer. For electrospray, solutions of 4 mg ml−1 (roughly 1 mmol l−1 in H2O/DMF 20/1) of reaction products 1, 2, 3 were used. The spray voltage was typically 3 kV, the temperature of the transfer capillary was kept rather low, at 50 °C, in order to minimize in-source fragmentation. The resolution of the orbitrap analyzer (m/Δm) was on the order of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, allowing us to unequivocally determine the molecular formula of each species based on its exact mass.
000, allowing us to unequivocally determine the molecular formula of each species based on its exact mass.
Collision induced dissociation
For CID, the respective species was isolated in the linear ion trap at much lower mass resolution: in order to minimize collisional excitation upon isolation we worked with an isolation width of typically 10 amu. Subsequently, the isolated ions within this mass range were resonantly excited by a radio-frequency pulse (duration 30 ms, amplitude varied in the range 0–20 internal units) applied to the trap electrodes and simultaneously collided with the helium gas present in the ion trap (1.5 × 10−5 mbar), thereby inducing fragmentation. Note that this CID protocol is comparatively mild since fragment ions go out of resonance with the radio-frequency pulse and are therefore not excited further. The remaining parent ions as well as the fragment ions produced were then analyzed at high mass resolution in the orbitrap.Analytical ultracentrifugation
For all experiments we used a Beckman Coulter (Proteomelab XL-A/XL-I) analytical ultracentrifuge equipped with an AN-50 rotor and an optical detection system. Equal aliquots (440 μl) of the guest–host complexes (1 mg ml−1 of 1 and 2 in H2O/NaCl (∼10 mg ml−1)) and appropriate reference solvents were injected into a two-sector cell (12 mm path length) comprising sapphire windows and an Epon charcoal-filled centerpiece. The optical path length of these cells is 1.2 cm. Data were collected in sedimentation velocity mode with both the interference optics and absorption optics set to a detection wavelength of 440 nm. All experiments were performed at 50000 rpm with a radial resolution of 0.001 cm; scans were recorded every 20 min for 12–24 h depending on the experiment. The numerical fitting software SEDFIT18,19 was used to fit the absorbance profiles with Lamm's equation solutions to calculate the distribution of sedimentation and diffusion coefficients. A typical 2D SEDFIT screen capture is shown in the ESI† (Fig. S1a and b). Size distribution and molar mass of the guest–host complexes were then calculated according to the procedure described by Carney et al.17 (see ESI† 2). The sedimentation process is monitored by a scanning UV/VIS optical detection system that records the concentration profile, c(r, t), with respect to the radial distance from the rotor (r) and time (t). The c(r, t) profile is subsequently numerically modelled and transformed into a sedimentation coefficient (s) and diffusion coefficient (D) distribution, c(s, D).
3. Results and discussion
Centrifugation of 1 and 2
Solutions of ∼1 mg ml−1 of 1 and 2 in H2O/NaCl (∼10 mg ml−1) were ultra-centrifuged as described in the previous section. During centrifugation, UV/VIS spectra were also recorded. The spectra obtained are consistent with the data presented by Ono et al.,11 see ESI,† Fig. S3. We have analyzed our sedimentation velocity data to yield distributions of diffusion (D) and sedimentation (s) coefficients – using the method recently applied by Carney et al.17 to characterize dissolved gold nanoparticles. This method numerically fits the experimental data using Lamm's equation and a two-dimensional (2D) model for s and D coefficient distributions (which takes into account that the diffusion coefficient contains additional information on the particle size). The approach does not require a priori knowledge of particle density but instead offers an independent way of estimating it in addition to the particle molecular mass and its hydrodynamic radius. This is particularly useful for the characterization of core–shell nanoparticles because particle composition (i.e. shell thickness) can be estimated from these three numbers by mass conservation. The specific assumptions inherent in this approach are: (i) diffusion and sedimentation coefficient distributions are independent and (ii) diffusion contributions are fully resolved experimentally.Fig. 1 shows the resulting best fits to the AUC data of diffusion (D) and sedimentation (s) coefficient distributions. From these we have determined the average mass of the species present in solution. In both cases, the mass distribution is dominated by a single peak. For 1 it is centered at 4400 amu, for 2 at 4600 amu (see Fig. 1a and b). These numbers are within 10% of the values obtained by ESI-MS (see below), thus proving that the assumptions made in analyzing the AUC data are reasonable even for such small size guest–host complexes. Equally interesting is the fact that SV-AUC allows a first order structural characterization of the guest–host complexes in solution (yielding a hydrodynamic radius of 2.14 nm under the assumption of uniform density). This is not possible with ESI-MS.
 | ||
| Fig. 1 2-D sedimentation and diffusion coefficient distributions for aqueous solutions (1 mg ml−1 in H2O/NaCl 10 mg ml−1) of 1 (left, predominantly A2B3H3Pd6(NO3)12) and 2 (right, predominantly A2B3Cu3−xHxPd6(NO3)12) resulting from a fit of the corresponding sedimentation velocity data obtained by analytical ultracentrifugation (AUC). From these we obtain molecular masses as indicated in the ESI† 2. The traces below the large spots correspond to impurities. | ||
Mass spectrum of 1
For the following we first refer the reader to the experimental section which provides an explanation of the abbreviations used below to describe the composition of parent and fragment ions. The mass spectrum obtained by electrospray-ionisation of a 4 mg ml−1 solution of 1 (nominally 1 mmol l−1 [A2B3H3Pd6 + 12NO3]) in H2O/DMF (20/1) is dominated by [B1Pd1 + NO3]+ (516 m/z), [A1Pd1 + NO3]+ (542 m/z), [A1B1Pd1 − H]+ (767 m/z), and [A1H1Pd1 − H]+ (855 m/z), see Fig. 2, left inset. The observation of [A1H1Pd1 − H]+ is quite unexpected, it implies that the azaporphine – triazine interaction is rather strong and might help to stabilize the coordination complex. | ||
| Fig. 2 ESI mass spectrum of a 4 mg ml−1 solution of 1 (nominally 1 mmol l−1 of [A2B3H3Pd6(NO3)12] in H2O/DMF (20/1)). The mass accuracy allows us to unambiguously assign the molecular formula for each species, see inset. | ||
In the range 1100–1500 m/z the mass spectrum consists of a series of peaks corresponding to the isotopic distribution of the expected [A2B3Pd6]–host complex filled with three azaporphine guests, [A2B3H3Pd6 + 9NO3]3+ (1330 m/z). Furthermore, we observe a smaller signal (50% of the [A2B3H3Pd6 + 9NO3]3+ intensity) at 1115 m/z corresponding to [A2B2H3Pd5 + 6NO3 − H]3+, which can be regarded as a disrupted host nanoprism – with one pillar and hinge missing – but still containing three azaporphines. The peak at 1225 m/z [A2B3H2Pd6 + 9NO3]3+ corresponds in some respects to the opposite: the host nanoprism is intact but one azaporphine is missing (see Fig. 2). The question we will address below is whether this represents a consequence of the electrospray ionization process, i.e. in-source fragmentation, or whether the partially filled nanoprisms are present in solution already.
We attribute the finding that the mass spectrum is dominated by low mass ions and that the intensity of the guest–host nanoprism complex [A2B3H3Pd6 + 9NO3]3+ (1330 m/z) is only on the order of 1% of the dominating [A1Pd1 + NO3]+ (516 m/z) signal to both in-source fragmentation and to the fact that low mass ions are formed preferentially in the electrospray process. We do not expect that the latter species are prevalent in solution. Based on the centrifugation results it is clear that a species with roughly 4000 amu, i.e. the filled box [A2B3H3Pd6 + 12NO3] is the dominating species in solution (see above).
Mass spectrum of 2
The mass spectrum of 2, the copper-derivative, was recorded under the same conditions as for 1, i.e. we use again a 4 mg ml−1 solution. As can be seen from Fig. 3, the mass spectrum of 2 is significantly more complicated than that of 1. We observe an essentially binomial distribution of nanoprisms filled with three azaporphines having either hydrogens or copper as central atoms [A2B3CuxH3−xPd6 + (12 − y)NO3]y+, (x = 0–3, y = 4, 3) with x = 2 dominating (relative intensities 5:30:45:20 for x = 0–3), see Fig. 3, red labels. This is most likely a consequence of incomplete exchange of hydrogen by copper upon preparation of the constituting azaporphine.
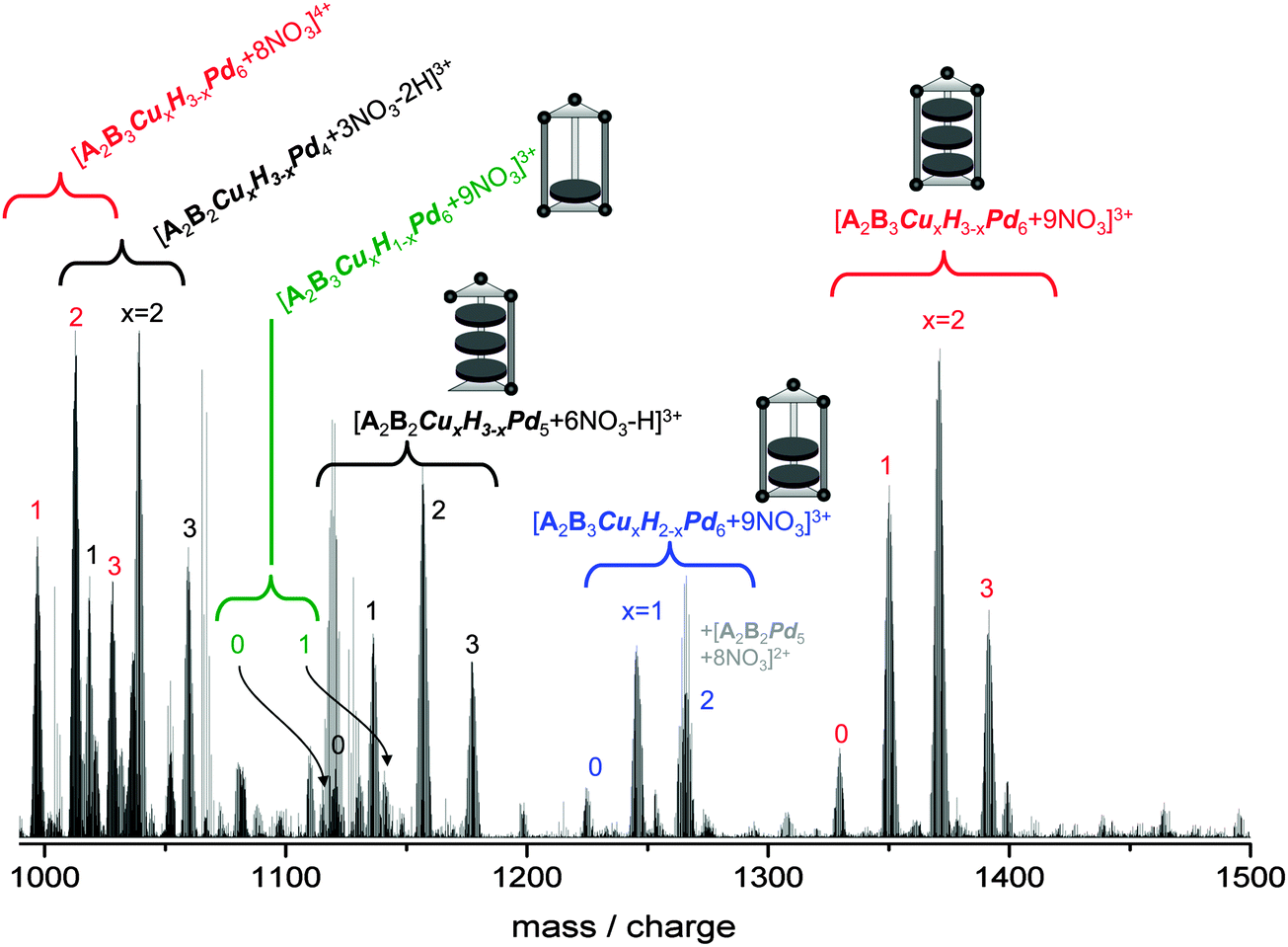 | ||
| Fig. 3 ESI mass spectrum of a 4 mg ml−1 solution of 2 in H2O/DMF (20/1). The color code of the labels distinguishes the different series we observe in the 1000–1500 mass/charge range: red – [A2B3CuxH3−xPd6 + (12 − y)NO3]y+x = 3, 2, 1, 0; y = 3, 4; blue – [A2B3CuxH2−xPd6 + 9NO3]3+x = 2, 1, 0; green – [A2B3CuxH1−xPd6 + 9NO3]3+x = 1, 0; black – [A2B2CuxH3−xPd5 + 6NO3 − H]3+ and [A2B2CuxH3−xPd4 + 3NO3 − 2H]3+, x = 3, 2, 1, 0. | ||
As for 1 (see above) we observe a series of nanoprisms filled with only two azaporphines [A2B3CuxH2−xPd6 + 9NO3]3+, (x = 0–2, Fig. 3, blue labels) and even, at a significantly smaller intensity, but clearly resolvable, a series of nanoprisms with only one azaporphine center [A2B3CuxH1−xPd6 + 9NO3]3+, (x = 0–1, green labels). As before, the question arises whether these partially filled nanoprisms are a consequence of the ionization process or are already present in solution.
Furthermore we observe in the range between 1100 and 1200 m/z a series with the molecular formula [A2B2CuxH3−xPd5 + 6NO3 − H]3+, (x = 0–3) and around 1050 a series that can be regarded as [A2B2CuxH3−xPd4 + 3NO3 − 2H]3+ (Fig. 3, black labels), i.e. nanoprisms with three azaporphyrins but missing pillar and hinge(s).
Mass spectrum of 3
The mass spectrum of 3 was recorded under the same conditions as for 1, and 2, however with a 3 mg ml−1 solution. We can clearly identify the empty nanoprism in two different charge states, i.e. [A2B3Pd6 + 9NO3]3+ and [A2B3Pd6 + 10NO3]2+, respectively (see Fig. 4, red labels). The relative intensities of these peaks are however much smaller than what we observe for 1 and 2. Here the spectrum in the 900–1600 amu range is dominated by species which can be regarded as fragments of the nanoprism with one (blue labels) or two (green labels) missing pillars. Furthermore we observe a strong peak centered at 1435 m/z (doubly charged) which cannot be explained as a fragment of the empty nanoprism framework. Based on the mass determination we assign the molecular formula [A4Pd6 + 10NO3]2+ corresponding to the highly stable octahedral A4Pd6-box20 and fragments thereof (black labels). Apparently this species is formed as a byproduct in the synthesis of 3. Based on the low abundance of the empty nanoprism it seems likely that it is less stable than the corresponding filled nanoprism, implying that the azaporphine guests help in stabilizing the host nanoprism framework. As before the question arises to what extent the fragments observed in the mass spectra are present in solution (or formed in the electrospray-ionisation). Furthermore we cannot rule out that the topology of the [A2B3Pd6] species differs from the nanoprism structure.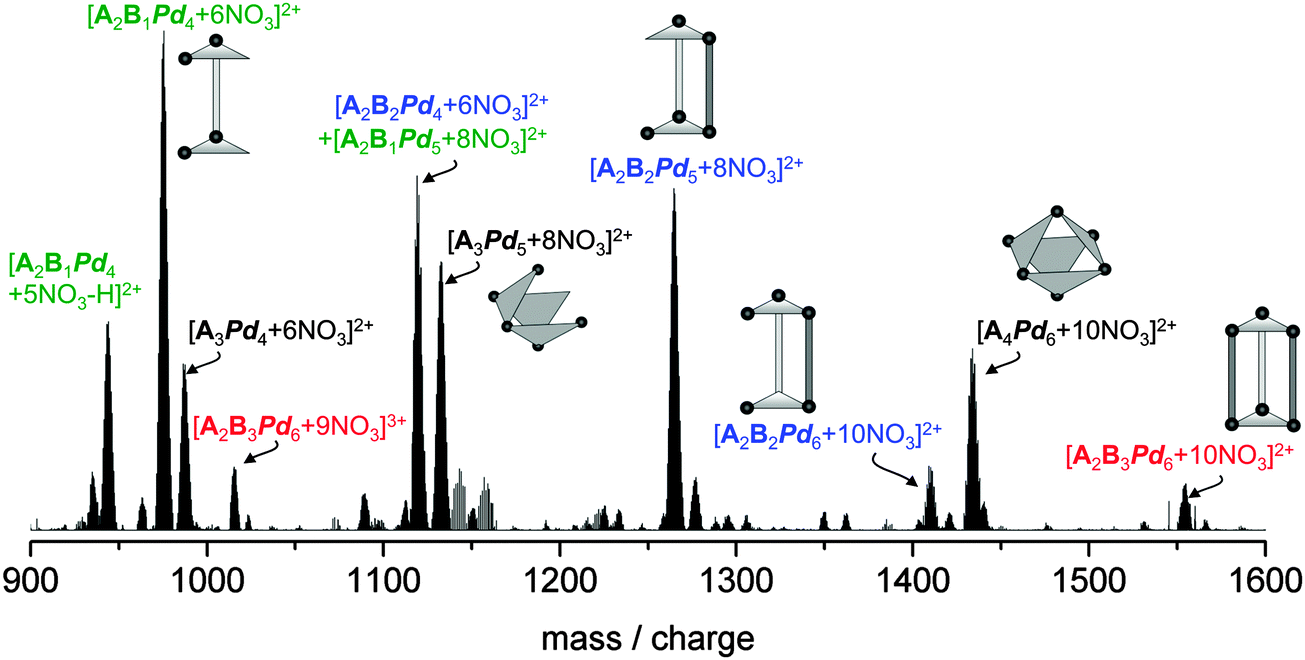 | ||
| Fig. 4 ESI mass spectrum of a 3 mg ml−1 solution of 3 in H2O/DMF (20/1)). The color code of the labels distinguishes the different series we observe in the 900–1600 mass/charge range: red – [A2B3Pd6 + (12 − x)NO3]x+x = 3, 2; blue – one missing pillar “B” [A2B2Pd6−x + (10 − 2x)NO3]2+x = 2…0; green – two missing pillars [A2B1Pd4 + 5NO3 − H]2+ and [A2B1Pd5−x + (8 − 2x)NO3]2+x = 1, 0; black – [A4Pd6 + 10NO3]2+ and fragments thereof. | ||
In order to address some of these questions we have next selectively fragmented the different nanoprism structures (filled and empty) and have studied the corresponding collision energy dependencies.
Collision induced dissociation
We performed collision induced dissociation (CID) measurements according to the following protocol. The respective precursor (parent) ion was isolated in the linear quadrupole ion trap using a relatively large selection window of 10 amu (in order to minimize excitation upon isolation). Subsequently, in order to induce CID (in He, 1.5 × 10−5 mbar) a radio-frequency excitation pulse was applied (duration 30 ms, amplitude varied in the range 0–20 internal units). The fragmentation products were then analyzed in the orbitrap with high mass resolution. A typical CID mass spectrum of a filled nanoprism is shown in Fig. 5a. We present the case of [A2B3Cu3Pd6 + 9NO3]3+ as an example. The spectra of the other filled nanoprisms [A2B3Cu3−xHxPd6 + 9NO3]3+ (see ESI,† Fig. S4) show the same features except that of the hydrogen-only-nanoprism [A2B3H3Pd6 + 9NO3]3+ fragments slightly more easily (see Fig. 5b). | ||
| Fig. 5 (a) CID mass spectrum of [A2B3Cu3Pd6(NO3)9]3+. The peaks marked by (*) correspond to electronic noise. Isolation width 10 amu, excitation 5 internal units. (b) Relative parent ion intensity, calculated as Iparent/ΣIparent+fragments as function of CID energy (in internal units). The lines serve to guide the eye. | ||
The dominant fragmentation channels are loss of a positively charged pillar ([B1Pd1 + NO3]+ and [B1Pd1 − H]+) and loss of a neutral pillar B (indirectly observed by the corresponding fragment ions [A2B2Cu3Pd6 + 9NO3]3+). We do not observe the loss of a triazine panel A as a significant fragmentation channel which can be easily rationalized since each triazine panel is bound within the nanoprism by three coordinative bonds to Pd, while the bipyridine pillars B are held by only two Pd bonds. So, assuming roughly the same Pd–N bond energy, removal of a pillar should require only two thirds of the energy required for removal of a panel and should therefore be the favored fragmentation channel. This finding is in line with the pattern we observe directly in the primary ESI mass scan (see above) and implies that the observed abundant [A2B2(Cu,H)3Pd6−x]-species (see Fig. 2 and 3) are likely the result of in-source fragmentation.
Note, however that in CID we do not observe the loss of an azaporphine center from an otherwise intact nanoprism (i.e. no partially filled nanoprisms like [A2B3Cu2−xHxPd6 + 9NO3]3+ and [A2B3Cu1−xHxPd6 + 9NO3]3+) – although we do observe these species in comparatively high intensity in the primary ESI-mass scans of 1 and 2 (see above). This suggests that these partially filled nanoprisms are most likely present in solution and that they are not the result of in-source fragmentation.
What about the empty nanoprism, 3? Its fragmentation behavior clearly differs from that of the filled nanoprism in two ways: first, the fragmentation threshold is much lower than for the filled nanoprisms, as can be seen from Fig. 5b. While for the filled nanoprisms the radio frequency pulse energy needed for 50% fragmentation is between 5 and 7 internal units (with the 2H-azaporphine-nanoprism being the least stable), the threshold for the empty host framework is only 2 internal units. Second, the fragmentation pattern is completely different: we observe both loss of a panel (triazine) and loss of a pillar (bipyridine) with comparable yields, while for the filled nanoprisms we observe only the loss of one or two pillars, see Fig. 6. This implies a different structure, or at least that some of the bonds of the framework are broken.
 | ||
| Fig. 6 CID of [A2B3Pd6 + 9NO3]3+. (a) Isolation of the parent ion. The situation is slightly complicated by a low mass impurity (singly charged) with an overlapping isotope pattern. (b) CID mass spectrum with an activation energy of 2 internal units. Red: loss of a triazine panel, black: loss of a bipyridine pillar. Note that the parent ion is completely fragmented, only the low mass impurity remains. | ||
Azaporphine exchange reaction in solution
In order to further investigate the stability of the guest–host complex in solution we mixed solutions of both the 1 and 2 (1 mmol l−1 in H2O/DMF 20/1), heated the reaction mixture to 100 °C for 30 min and analyzed the reaction products mass spectrometrically after cooling to room temperature (within another 30 min). Immediately after mixing but prior to heating the solution consists predominantly of [A2B3H3Pd6 + 9NO3]3+ (1330 m/z) and [A2B3Cu2H1Pd6 + 9NO3]3+ (1371 m/z) – the intermediate [A2B3Cu1H2Pd6 + 9NO3]3+ (1350 m/z) is significantly lower in intensity, as expected from the component distributions of the constituting solutions (see Fig. 7, reaction time 0). After heating, the distribution changed to a binomial distribution with [A2B3Cu1H2Pd6 + 9NO3]3+ dominating, indicating that an interchange of azaporphines between the nanoprisms has taken place. | ||
| Fig. 7 Left: intensity ratio of [A2B3Cu1H2Pd6 + 9NO3]3+ (1350 m/z) and [A2B3Cu2H1Pd6 + 9NO3]3+ (1371 m/z) “Cu1H2:Cu2H1” plotted as a function of reaction time. The data points are obtained by integrating the respective mass ranges, the line is a guide to the eye. Right: schematics illustrating the guest exchange. | ||
The question arises whether such interchange already occurs at room temperature. This would support our conclusion based on the fragmentation patterns that partially filled nanoprisms can exist in solution. Therefore and also to get more information on the reaction kinetics, we repeated the exchange experiment at room temperature. As before, we mixed solutions of 1 and 2 and followed the exchange reaction by continuously electrospraying the reaction solution into our mass spectrometer. We monitored the [A2B6Cu1H2Pd6 + 9NO3]3+ to [A2B6Cu2H1Pd6 + 9NO3]3+ intensity ratio by accumulating the ion signal in the respective mass ranges for short intervals (2 minutes at a time). The results are shown in Fig. 7.
Immediately upon mixing of the solutions we observe a bimodal intensity distribution with [A2B3H3Pd6 + 9NO3]3+ (1330 m/z) and [A2B3Cu2H1Pd6 + 9NO3]3+ (1371 m/z) dominating over [A2B3Cu1H2Pd6 + 9NO3]3+ (1350 m/z), reflecting the relative concentrations of the starting solutions. On a timescale of minutes this distribution changes and the relative intensities of the copper rich species [A2B3Cu2H1Pd6 + 9NO3]3+ and [A2B3Cu3Pd6 + 9NO3]3+ decrease. Finally a binomial distribution is obtained with [A2B3Cu1H2Pd6 + 9NO3]3+ dominating (Fig. 7). This clearly proves that aza-porphine exchange in solution occurs at room temperature and furthermore implies that partially filled nanoprisms are probably also present in solution. They are not (only) a consequence of fragmentation upon ionization. How is this consistent with our observation of only a single species upon analytical ultracentrifugation? The boxes interconvert during centrifugation and we observe a time averaged mass.
For weakly bound supramolecular complexes AUC and ESI-mass spectrometry complement each other: while inferior in mass resolution AUC reflects the solution composition more closely since it is not affected by in-source fragmentation as is ESI-MS. This might have applications for the investigation of various weakly bound supramolecular complexes.
4. Conclusion
Supramolecular guest–host complexes, “nanoprisms”, consisting of a prismatically shaped coordination box filled with 2H and Cu-azaporphines were prepared according to the method of Fujita and coworkers.11 Aqueous solutions of these nanoprisms were then analysed by analytical ultracentrifugation confirming that the host–guest-complexes are the dominating species in solution. This is the first time that AUC has been used to characterize solutions of charged supramolecular host–guest structures.Nanoprism solutions were also characterized by high-resolution electrospray ionization mass spectrometry. The expected (parent) product ions were identified in the mass spectra. Also seen in significantly higher abundances were low mass species which we attribute to in-source fragmentation. Partially filled nanoprisms with only two and one azaporphine guests were observed in the mass spectra as well. The stability of both filled and empty nanoprism derived ions was investigated by collision induced dissociation: the azaporphine guests drastically stabilize the nanoprisms.
Based on these observations, we have also prepared empty nanoprisms wet-chemically and have characterized their solution composition by mass spectrometry. Furthermore, we have used mass spectrometry to follow composition ratios upon combining solutions of preformed nanoprisms filled with two different sets of guests. These measurements indicate that the complexation reactions leading to nanoprism formation in solution are reversible (at least in part) and importantly that azaporphine guest exchange reactions take place on timescales of minutes at room temperature.
Acknowledgements
We thank Professor Mathias Senge (Dublin) for the donation of azaporphine-type compounds. We thank the Deutsche Forschungsgemeinschaft (DFG) for support of this work through projects B2 and C6 of the collaborative research centre SFB/TRR 88 “3MET” (Kooperative Effekte in homo- und hetero-metallischen Komplexen). Funding of an orbitrap mass spectrometer by DFG and Land/KIT (Art 91b) is also gratefully acknowledged. U. Schwarz thanks the “Stiftung der deutschen Wirtschaft” for financial support.References
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2003 Search PubMed.
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed.
- V. E. Yushmanov, H. Imasato, T. T. Tominaga and M. Tabak, J. Inorg. Biochem., 1996, 61, 233–250 CrossRef CAS.
- S. C. M. Gandini, V. E. Yushmanov and M. Tabak, J. Inorg. Biochem., 2001, 85, 263–277 CrossRef CAS.
- J. M. Ribó, J. Crusats, J. A. Farrera and M. L. Valero, J. Chem. Soc., Chem. Commun., 1994, 681–682 RSC.
- H.-L. Ma and W.-J. Jin, Spectrochim. Acta, Part A, 2008, 71, 153–160 CrossRef PubMed.
- R. F. Pasternack, K. F. Schafer and P. Hambright, Inorg. Chem., 1994, 33, 2062–2065 CrossRef CAS.
- J. V. Hollingsworth, A. J. Richard, M. G. H. Vicente and P. S. Russo, Biomacromolecules, 2012, 13, 60–72 CrossRef CAS PubMed.
- T. Sugimoto, K. Sada, Y. Tateishi, T. Suzuki, Y. Sei, K. Yamaguchi and S. Shinkai, Tetrahedron Lett., 2005, 46, 5347–5350 CrossRef CAS PubMed.
- L. G. S. Di Costanzo, L. Randaccio, R. Purrello, R. Lauceri, D. Sciotto, F. G. Gulino and V. Pavone, Angew. Chem., Int. Ed., 2001, 40, 4245–4247 CrossRef CAS.
- K. Ono, M. Yoshizawa, T. Kato and M. Fujita, Chem. Commun., 2008, 2328–2330 RSC.
- K. Ono, M. Yoshizawa, T. Kato, K. Watanabe and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 1803–1806 CrossRef CAS PubMed.
- K. Ono, J. K. Klosterman, M. Yoshizawa, K. Sekiguchi, T. Tahara and M. Fujita, J. Am. Chem. Soc., 2009, 131, 12526–12527 CrossRef CAS PubMed.
- P. H. Brown and P. Schuck, Comput. Phys. Commun., 2008, 178, 105–120 CrossRef CAS PubMed.
- P. H. Brown and P. Schuck, Biophys. J., 2006, 90, 4651–4661 CrossRef CAS PubMed.
- E. Brookes, W. M. Cao and B. Demeler, Eur. Biophys. J., 2010, 39, 405 CrossRef PubMed.
- R. P. Carney, J. Y. Kim, H. Qian, R. Jin, H. Mehenni, F. Stellacci and O. M. Bakr, Nat. Commun., 2011, 2, 335 CrossRef PubMed.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS.
- J. Dam and P. Schuck, Methods Enzymol., 2004, 384, 185–212 CAS.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp55486d |
| This journal is © the Owner Societies 2014 |