 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydration of the oxygen-evolving complex of photosystem II probed in the dark-stable S1 state using proton NMR dispersion profiles†
Guangye
Han
ab,
Yang
Huang
a,
Faisal Hammad Mekky
Koua
c,
Jian-Ren
Shen
c,
Per-Olof
Westlund
*a and
Johannes
Messinger
*a
aDepartment of Chemistry, Kemiskt Biologiskt Centrum, Umeå University, Linnaeus väg 6, 901 87 Umeå, Sweden. E-mail: perolof.westlund@chem.umu.se; johannes.messinger@chem.umu.se
bPhysical Biosciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road, MS 66-327, Berkeley, CA 94720, USA
cGraduate School of Natural Science and Technology, Department of Biology, Faculty of Science, Okayama University, 1-1, Naka 3-chome, Tsushima, Okayama 700-8530, Japan
First published on 2nd April 2014
Abstract
The hydration of the oxygen-evolving complex (OEC) was characterized in the dark stable S1 state of photosystem II using water R1(ω) NMR dispersion (NMRD) profiles. The R1(ω) NMRD profiles were recorded over a frequency range from 0.01 MHz to 40 MHz for both intact and Mn-depleted photosystem II core complexes from Thermosynechococcus vulcanus (T. vulcanus). The intact-minus-(Mn)-depleted difference NMRD profiles show a characteristic dispersion from approximately 0.03 MHz to 1 MHz, which is interpreted on the basis of the Solomon–Bloembergen–Morgan (SBM) and the slow motion theories as being due to a paramagnetic enhanced relaxation (PRE) of water protons. Both theories are qualitatively consistent with the ST = 1, g = 4.9 paramagnetic state previously described for the S1 state of the OEC; however, an alternative explanation involving the loss of a separate class of long-lived internal waters due to the Mn-depletion procedure can presently not be ruled out. Using a point-dipole approximation the PRE-NMRD effect can be described as being caused by 1–2 water molecules that are located about 10 Å away from the spin center of the Mn4CaO5 cluster in the OEC. The application of the SBM theory to the dispersion observed for PSII in the S1 state is questionable, because the parameters extracted do not fulfil the presupposed perturbation criterion. In contrast, the slow motion theory gives a consistent picture indicating that the water molecules are in fast chemical exchange with the bulk (τw < 1 μs). The modulation of the zero-field splitting (ZFS) interaction suggests a (restricted) reorientation/structural equilibrium of the Mn4CaO5 cluster with a characteristic time constant of τZFS = 0.6–0.9 μs.
1. Introduction
In oxygenic photosynthesis, light-driven water-splitting is carried out by the oxygen-evolving complex (OEC) of Photosystem II (PSII), a pigment–protein complex embedded in the thylakoid membranes of higher plants, green algae, and cyanobacteria. The OEC consists of an inorganic Mn4O5Ca cluster and its surrounding protein matrix, which includes the redox-active tyrosine residue YZ (D1-Tyr161).1–10 YZ/YZ˙ is a critical redox couple, since it quickly reduces P680˙+ after light-induced charge separation and then retrieves an electron at a much slower rate from the Mn4O5Ca cluster, thus allowing chemistry to take place instead of charge recombination.11 Four consecutive light-induced charge separations drive the Mn4O5Ca cluster through a cycle (Kok cycle) of five intermediate oxidation states denoted as Sn (n = 0–4).8,12,13 The S0 state is the most reduced state while S1 is the dark stable state. The S2 and S3 states are metastable states that decay back to the S1 state within a few minutes at room temperature. S4 is a transient state involved in the formation and release of O2 during the S3 → (S4) → S0 transition.8,12,14EPR and ENDOR, as well as X-ray spectroscopy have been employed to characterize the electronic structure of the Mn4CaO5 cluster.4,5,9,15–23 Most groups agree that the oxidation states of the S0 state are Mn4(III,III,III,IV)24,25 and that the subsequent transitions up to S3 involve MnIII → MnIV oxidations26,27 (see however refs. 28 and 29). Via the μ-oxo bridges the unpaired d-electrons of the four MnIII/IV centers couple to total spins of ST = 1/2, ST = 1, ST = 1/2 (and ST = 5/2), and ST = 3 for the S0, S1, S2 and S3 states, respectively. The corresponding EPR signals have been observed at g = 2 (S0), g = 4.9 (S1), g = 2 and g ≥ 4.1 (S2), and g = 8 and 12 (S3) at liquid helium temperatures using parallel (S1, S3) or perpendicular mode (S0, S2) EPR.17–20,30–36
Several crystal structures of PSII, which crystallizes as a dimer of 700 kDa, have been reported at progressively better resolutions of 3.8 Å to 2.9 Å, and recently 1.9 Å.1,37–40 This latest structure not only revealed a very detailed picture of the protein and its cofactors including the Mn4CaO5 cluster, but also identified about 1300 water molecules per monomer that are associated with the stromal surface of PSII or ‘bound’ within the luminal extensions of the CP43 and CP47 inner light-harvesting proteins and within the three extrinsic proteins of PSII, PsbO (33 kDa), PsbU (12 kDa) and PsbV (cyt c550; 15 kDa).1 This lumenal shield protects and stabilizes the Mn4CaO5 cluster, and it also forms several channels for water access, as well as for proton and oxygen release.1,39,41–51
Studying the access of water to and the escape path of protons from the catalytic site of water oxidation, as well as the binding of substrate water to the Mn4CaO5 cluster are crucial aspects for deriving the mechanism of photosynthetic water oxidation. Substrate water-binding has been mostly studied by time resolved membrane-inlet mass spectrometry6,9,52–54 and FTIR spectroscopy using 18O isotope labelling,9,55,56 but also by advanced EPR spectroscopy using 17O-labelling.57–61 It was concluded that at least one substrate water is bound in the S0 and S1 states, while both substrates are bound in the S2 and S3 states.4,6,52,62,63 Consistent with this conclusion the 1.9 Å crystal structure has identified four water molecules that are ligated to the Mn4CaO5 cluster: two to Ca, and two to one of the four Mn ions.1 In addition, several water molecules were found in their vicinity that are part of an intricate H-bonding network, which is highly important for the function of the OEC.
In proton spin–lattice NMR dispersion experiments (R1-NMRD) one measures the proton R1 (spin–lattice) relaxation rate as a function of the Larmor frequency by varying the static magnetic field B. The experiment reports on the water spin relaxation rate of bulk water, which can change dramatically due to relatively few water molecules which are reorienting slowly due to interactions with the protein.64 Such long-lived water molecules (τw > 10 ns) are usually characterized by restricted access to the bulk, a small Debye–Waller factor (B < 20 Å2) and several strong H-bonds to protein ligands or other long-lived water-molecules.65 When there is a paramagnetic metal ion or some other paramagnetic species present and in contact with exchangeable waters, the water spin relaxation of the bulk water is generally dominated by the electron spin relaxation of the paramagnetic center and the resulting profiles are referred to as PRE-NMRD profiles (Paramagnetic relaxation enhancement = PRE). Parameters that may be derived from such experiments include the number of water molecules/protons near the paramagnetic site, the limit value of their exchange rates with the bulk water, the characteristics of the spin state of the paramagnetic center, i.e. the zero field splitting and the characteristic correlation time describing its modulation, and the distance between the water protons and the electron spin density.66–68 This technique thereby forms a promising tool for studying the mechanism of water oxidation under physiological conditions.
The first PRE-NMRD experiments on PSII were performed by Wydrzynski et al.69–72 They reported flash-induced changes of the proton spin–spin relaxation rate R2 of thylakoid suspensions employing a NMR spectrometer operating at 27 MHz. A period four oscillation was observed upon illuminating PSII preparations using a series of light flashes, which was attributed to MnII → MnIII oxidation state changes in the S0 → S1 → S2 → S3 transitions.69,72 Subsequent investigations by Sharp et al. sparked a debate as to whether or not these first R1 and R2 relaxation rate measurements may have been dominated by the oxidation of free Mn2+ by the superoxide produced on the reducing side of PSI.73,74 Sharp and coworkers added EDTA to remove unspecific Mn2+. Yet, they also found a flash-dependence of R1 at about 20 MHz which they assigned to Mn-oxidation state changes of the Mn4CaO5 cluster.75–80
These early measurements and interpretations were limited by sample preparations and structural information available at the time. Both have dramatically improved over the past 20–30 years due to the development of highly stable, clean and active PSII core complexes (PSIIcc) from Thermosynechococcus vulcanus (T. vulcanus) and Thermosynechococcus elongatus (T. elongatus).1,37 The former not only allowed obtaining the 1.9 Å crystal structure, but also provided practically Mn2+-free preparations of high reaction center (RC) concentration, potentially allowing to obtain artifact-free data with much improved S/N. This prompted us to take up the NMRD and PRE-NMRD experiments again. In this paper, the hydration of the OEC in its dark-stable S1 state and the water exchange with the bulk is monitored for PSIIcc from T. vulcanus by studying the solvent water proton nuclear magnetic relaxation dispersion employing a field cycling approach.
2. Experimental procedures
2.1 Preparation of the PSII core complex
Core complexes of PSII were isolated from the thermophilic cyanobacterium T. vulcanus as described previously.81 The activity of the O2 evolution rate was measured in a buffer with 30 mM Mes (pH 6.0), 20 mM NaCl, 3 mM CaCl2 at 30 °C. Under saturating illumination, the O2 evolution rate exceeded 3000 μmol O2 (mg of Chl)−1 h−1 in the presence of 0.5 mM phenyl-p-benzoquinone and 0.5 mM potassium ferricyanide as electron acceptors. The chlorophyll (Chl) a concentration was employed for calculating the quantity of the PSII centers by using 35 Chl a/PSII as a reference. The Chl a concentration was determined according to ref. 82 by recording the absorbance at 665 nm in methanol according to [Chl a] = A665 × 13.4 × dilution factor.2.2 Manganese depletion of the PSII core complex
To remove Mn from the OEC, the PSII core complexes were treated with 5 mM NH2OH in a buffer containing 20 mM Mes (pH 6.0), 20 mM NaCl, and 3 mM CaCl2 (buffer A) at a chlorophyll concentration of 0.5 mg mL−1 for 60 min in the dark on ice. After NH2OH treatment, the samples were precipitated at 4 °C by adding polyethylene glycol (PEG 1450) to a final concentration of 15% (w/v) followed by centrifugation. The samples were then diluted in buffer A and concentrated in centrifugal concentrators (Vivaspin). For EPR and NMRD measurement the samples were finally resuspended in buffer A containing 5% (w/v) glycerol.2.3 Cyanide and dithionite treatments of the Mn-depleted PSII core
For the cyanide treatment, the Mn-depleted PSII core complexes (c[RC] = 27 μM) were washed once in a buffer containing 60 mM Hepes-NaOH (pH = 8.0), 20 mM NaCl, 3 mM CaCl2, 5% (w/v) glycerol and then resuspended in a small volume of the same buffer. The non-heme iron was converted to its low-spin form (S = 0) by incubating the Mn-depleted sample with 350 mM KCN at pH 8.0 for 3 h at 4 °C, according to Sanakis et al.83 To induce the QA− signal, Mn-depleted PSIIcc were incubated for 20 min with 50 mM dithionite at room temperature (20 °C) in the respective KCN or KCl buffer.2.4 Sample characterization by EPR measurement at liquid helium temperatures
EPR measurements were used to check the integrity of the Mn4CaO5 cluster of the intact PSII core and the Mn content of the Mn-deleted PSII core samples. The measurements were performed using a Bruker ELEXYS E500 spectrometer using a Super X EPR049X microwave bridge and a Bruker SHQ4122 cavity. The system was fitted with a liquid helium cryostat and a temperature controller (ITC 503S, Oxford instrument). Spectrometer settings are given in the figure legends.2.5 The NMRD experiment
The 1H NMRD experiments were carried out by using a fast field cycling NMR relaxometer, SPINMASTER FFC2000-CDC (STELAR Company, Italy) equipped with a STELAR broad brand probe thermostated with a STELAR VTC90 variable temperature controller that kept the sample temperature to within ±0.1 °C. The longitudinal relaxation rate (R1) was measured over a 0.01 to 40 MHz (1H resonance frequency) range by using the pre-programmed basic pre-polarization sequence (at low relaxation fields) and basic non-polarizing sequence (at higher relaxation fields). The switching time (SWT) of filed values between the fixed polarization field Bpol, the variable relaxation field Brlx and the fixed detection field Bacq was 3 ms.In our NMRD experiment, 0.25 mL of the sample in a buffer containing 20 mM Mes-NaOH (pH 6.0), 20 mM NaCl, 3 mM CaCl2, 5% (w/v) glycerol and calibrated NMR tubes (10 mm diameter) were used. The number of blocks used for R1 data was 16. The width of the RF 90° pulse was 7.5 μs. The polarization time and the recycle delay were 5 times longer than R1 at 25 MHz. To subtract the background from the relaxation contribution, NMRD profiles of the buffer were also measured under all conditions. The R1 values were estimated to have an experimental uncertainty of about 2%.
3. Experimental results
The spin–lattice relaxation rate measurements of water protons were performed in the 0.01–40 MHz proton Larmor frequency range using a Stellar fast cycling relaxometer. The reported NMRD profiles give either the net-relaxation rates obtained after subtraction of the buffer-relaxation rates from those of the sample suspensions or, where indicated, the difference between the NMRD profiles of intact and Mn-depleted PSIIcc.The NMRD profiles of intact PSII and Mn-depleted PSII core preparations of T. vulcanus were recorded at 4 various concentrations (27–188 μM PSIIcc) and three temperatures (1, 10, 20 °C) (Fig. 1 and 2). Fig. 1A shows that the relaxation enhancement effect, which is largest at the lowest fields, is approximately proportional to the PSIIcc concentration. However, scaling to the PSIIcc concentration reveals for the two highest concentrations small deviations in the relaxivity in the low-frequency region. We provisionally assign this to increasing inter-complex interactions of the PSIIcc at these higher concentrations that may slow down the dynamics and thus increase the relaxivity. We therefore focus our further analysis on measurements at 60 μM PSII reaction center concentration.
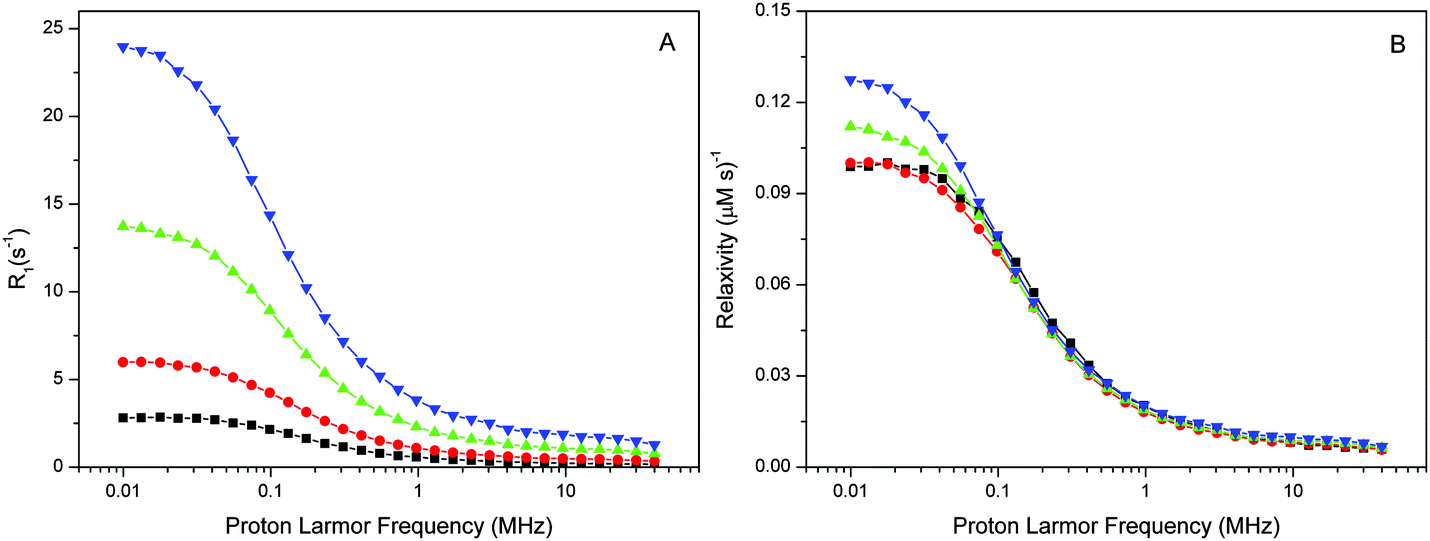 | ||
| Fig. 1 Panel A displays the proton R1 NMRD profiles of the intact PSIIcc of T. vulcanus obtained at T = 10 °C for four different PSII reaction center concentrations (29 μM, black squares; 60 μM, red circles; 123 μM, green triangles; d = 188 μM, blue inverted triangles). Panel B displays the same data after normalization to the reaction center concentrations. | ||
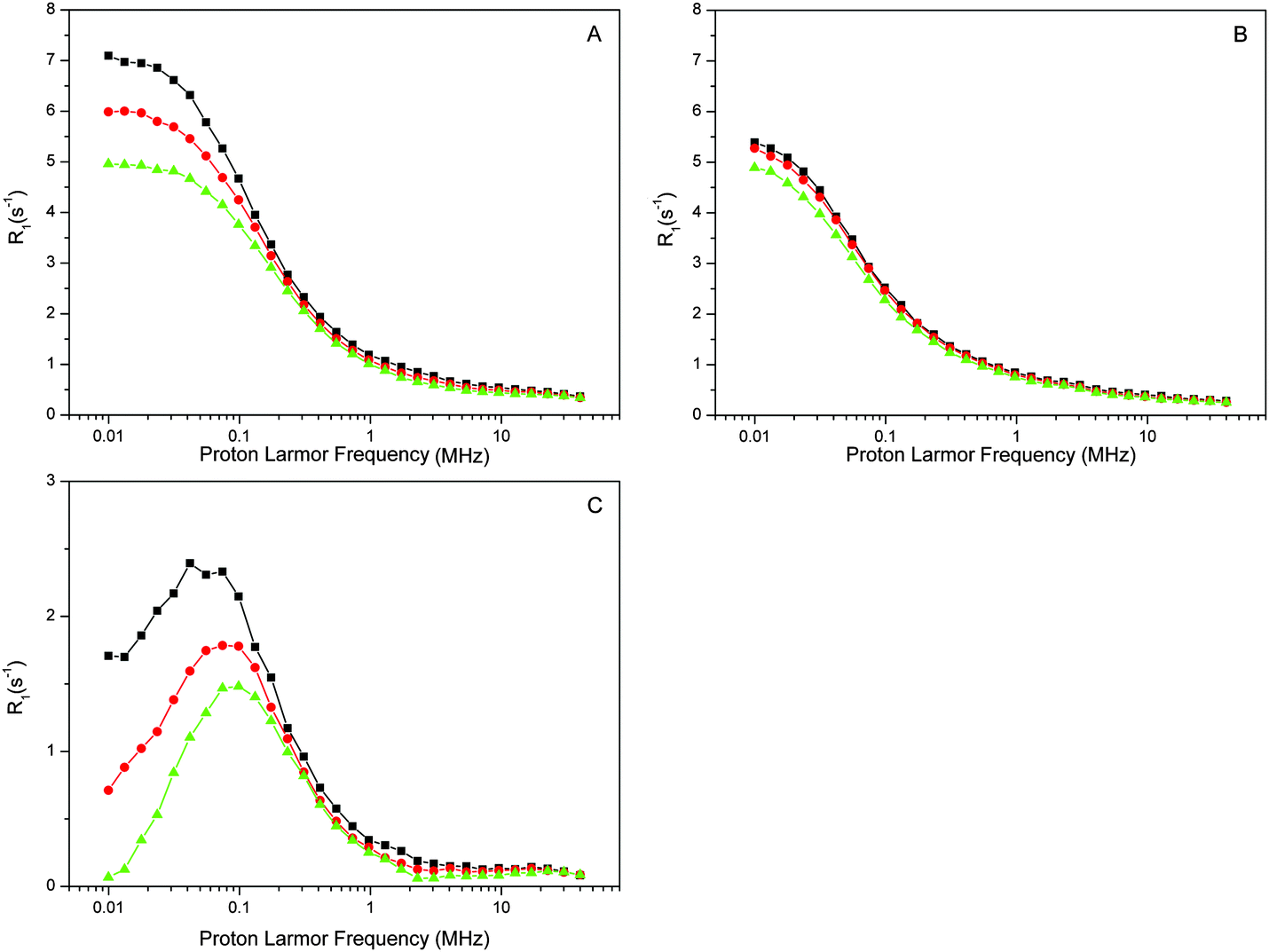 | ||
| Fig. 2 Proton R1-NMRD profiles of PSII core preparations from T. vulcanus ([RC] = 60 μM) at three temperatures: T = 1 °C (black squares); T = 10 °C (red circles); T = 20 °C (green triangles). A: intact samples, B: Mn depleted, C: intact-minus-depleted. | ||
Next, the temperature dependence was measured as this reveals important information about the relaxation process. Fig. 2A shows that the relaxation rate decreases with increasing temperature, implying that the exchange of the bulk protons with those in PSII is fast compared to the characteristic correlation time τc. This means that the ‘fast exchange limit’ can be applied during data analysis (see below).
The proton relaxation effect of PSIIcc is a composite of many factors that includes restriction of water movements by binding to the protein or cofactors. Especially strong effects are caused by paramagnetic centers such as the Mn4CaO5 cluster. In the dark-stable S1 state the Mn4CaO5 cluster is known to attain at liquid helium temperatures largely the first exited S = 1 spin state. To test the contribution of the Mn4CaO5 cluster we removed it by incubation with NH2OH. The Mn2+ released was carefully washed away and the absence of free Mn2+ was confirmed by EPR spectroscopy (see ESI,† Fig. S1). The EPR spectra of the intact and Mn-depleted samples also show that no paramagnetic cofactors that are visible under these conditions at perpendicular mode EPR were lost during Mn removal. The NMRD profiles of the Mn-depleted sample are displayed in Fig. 2B. While the overall shape of the relaxation profile remains similar, it is obvious that the relaxation rates decrease and that the temperature dependence is less pronounced. The relaxation effect of the Mn4CaO5 cluster is then given by the difference intact-minus-(Mn) depleted (Fig. 2C). This difference may have some protein contributions, since the Mn-depletion procedure may also partially release some of the extrinsic proteins.84,85
The relaxation enhancement in the absence of the Mn4CaO5 cluster (Fig. 2B) is dominated by the protein. One additional cofactor that may contribute is the non-heme iron on the acceptor-side of PSII. The non-heme iron is usually in the high-spin form of Fe2+, which is paramagnetic (S = 2). To probe its contribution we added to the Mn-depleted samples CN−, which is known to impose the low spin configuration (S = 0) onto the non-hem iron. This effect, known from spinach BBY-type preparations, was confirmed for the PSIIcc samples from T. vulcanus using low temperature EPR spectroscopy. For this, the Mn-depleted samples were treated for the EPR spectroscopy with dithionite, which reduces the quinone QA on the acceptor side of PSII to its radical form QA−. QA is tightly bound in the vicinity of the non-heme iron. Thus its EPR signal is broadened beyond detection if the non-heme iron is in its usual paramagnetic high spin S = 2 state.83 This situation is seen in the KCl control (the lower trace in Fig. 3B). In contrast, the QA− EPR signal becomes visible at g = 2.0045 in the KCN treated sample, confirming that the strong ligation of the non-heme iron by CN− converts it to the diamagnetic S = 0 state. Fig. 3A demonstrates that the NMRD profiles of the KCl and KCN treated Mn-depleted PSII samples (not treated with dithionite) are very similar, suggesting that the contribution of the non-heme iron to the overall relaxation enhancement effect of the Mn-depleted PSIIcc is small. This may indicate that the water which is 5.5 Å near the non-heme iron is not in the fast chemical exchange regime, and thus does not contribute to the bulk water proton relaxation rate measured here by NMRD. Thus, the dispersion observed for the Mn-depleted sample appears to be mostly due to the protein, although other cofactors may also contribute.
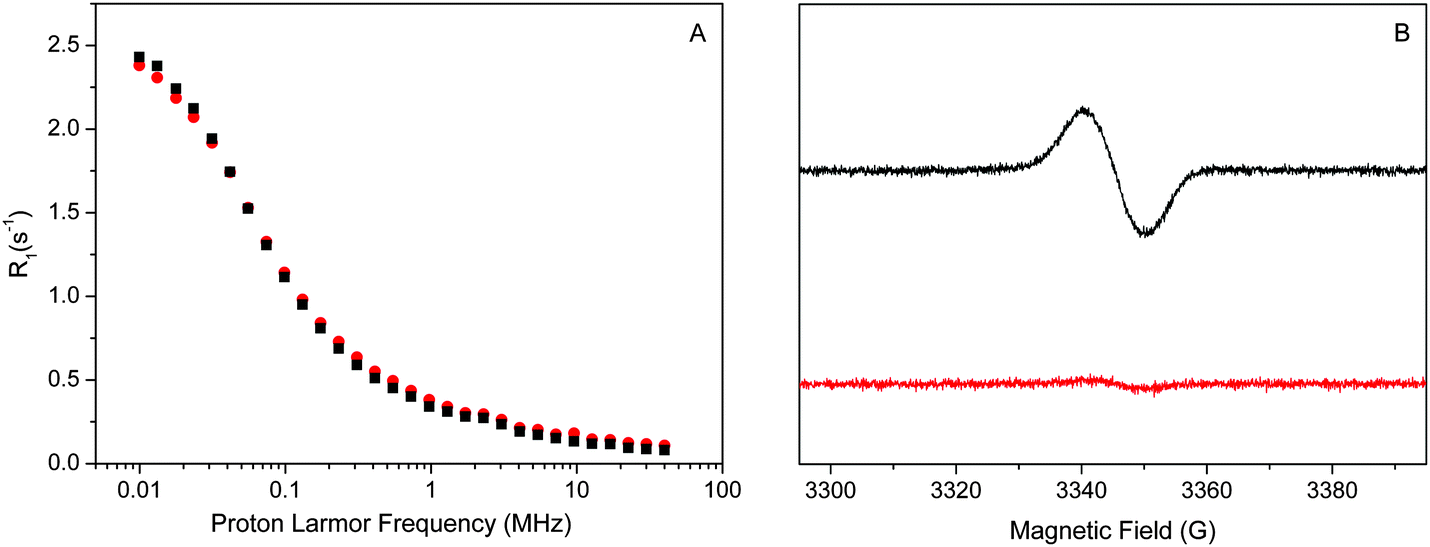 | ||
| Fig. 3 NMRD profiles (A) and the related EPR spectra (B) of the Mn-depleted Photosystem II (from T. vulcanus) treated by high concentration KCN (black squares and line) and KCl (red circles and line). For the EPR spectra the samples were additionally treated with dithionite to reduce the acceptor side quinone QA to QA−. NMRD profiles were recorded at 20 °C. EPR spectra were obtained at 13 K; microwave frequency, 9.39 GHz; modulation amplitude, 4 G; microwave power, 200 nW. | ||
Our experimental results thus suggest that the Mn4CaO5 cluster in its S1 state has a significant proton relaxation enhancement effect at room temperature, and that the solvent waters/protons are in rapid exchange with water protons in the vicinity of the Mn4CaO5 cluster. However, for a final proof, future experiments will need to establish the Sn state dependence of this signal. Below the present data are analyzed by two different theoretical approaches.
4. Theoretical analysis
4.1 Is there a paramagnetic contribution of the Mn4CaO5 cluster to the NMRD profiles?
In this section we evaluate if it is possible to rationalize the measured (PRE)-NMRD profiles of the water proton spin–lattice relaxation rates (proton R1-NMRD profiles) of the Mn-depleted PSIIcc sample and the native PSIIcc sample without including the paramagnetic relaxation enhancement effect due to the paramagnetic Mn4CaO5 cluster of the OEC. This scenario would tacitly imply that there are no fast exchangeable waters/protons in the vicinity of the OEC cluster, and that the changes in relaxivity observed after Mn-depletion are either due to creating an extra water-filled void or by the loss of extrinsic proteins during Mn removal. According to the relaxation theory for solvated proteins the R1-NMRD profile is then given by the relaxation contribution from two types of waters that are either associated with the protein surface or buried inside the protein, namely α and β waters:64,86–94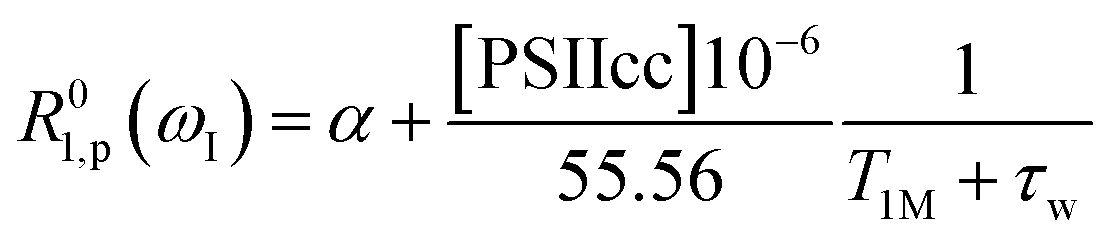 | (1a) |
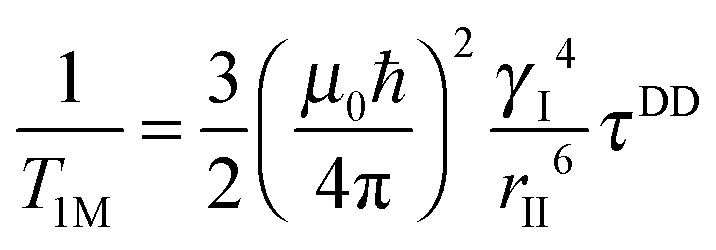 | (1b) |
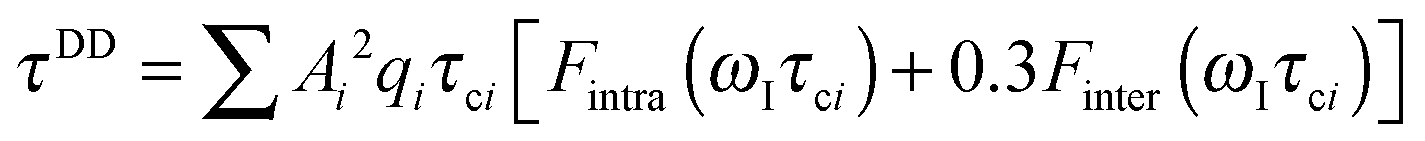 | (1c) |
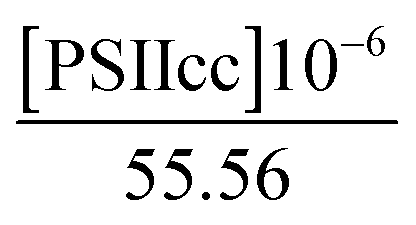 where the concentration of PSIIcc is given in μM. The spin–lattice relaxation rate of water associated with the protein, denoted β waters, is 1/T1M and fast chemical exchange conditions are assumed (see above), meaning that the residence times are short compared to the water proton spin relaxation (T1M ≫ τw). Several classes of β protons may exist (see below). The α relaxation contribution refers to water molecules with a spin–lattice relaxation rate that is frequency independent (extreme narrowing regime). But since we already have subtracted the bulk relaxation contribution the α term is defined as the difference between the measured relaxation rate of water in the extreme narrowing regime and the water proton relaxation rate of the buffer solution. Consequently, this relaxation contribution can be ascribed to short residence time water molecules at the protein surface.
where the concentration of PSIIcc is given in μM. The spin–lattice relaxation rate of water associated with the protein, denoted β waters, is 1/T1M and fast chemical exchange conditions are assumed (see above), meaning that the residence times are short compared to the water proton spin relaxation (T1M ≫ τw). Several classes of β protons may exist (see below). The α relaxation contribution refers to water molecules with a spin–lattice relaxation rate that is frequency independent (extreme narrowing regime). But since we already have subtracted the bulk relaxation contribution the α term is defined as the difference between the measured relaxation rate of water in the extreme narrowing regime and the water proton relaxation rate of the buffer solution. Consequently, this relaxation contribution can be ascribed to short residence time water molecules at the protein surface.
The effective spin dipole–dipole correlation time τDD is field-dependent (eqn (1c)) due to the proton Larmor frequency (ωI) dependence of both its intra- and inter-molecular contributions. The total relaxation contribution of a sample is thus obtained by summing up the relaxation contributions from all groups of waters characterized by an effective correlation time.86–93,95 In eqn (1c) the dipole–dipole interaction strength between the nuclear spin of the water protons and the electron spin of the paramagnetic centre has been approximately expressed as 0.3 times the intramolecular dipole interaction. The number of β waters is qi per PSIIcc, and they are characterized by the effective correlation time τci and the order parameter Ai. Ai is one for completely fixed water molecules that rotate with the speed of the protein. The field dependence of the proton relaxation is described by two functions:
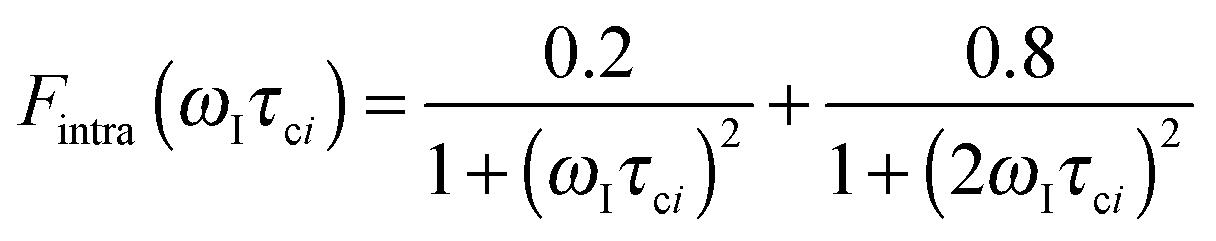 | (2a) |
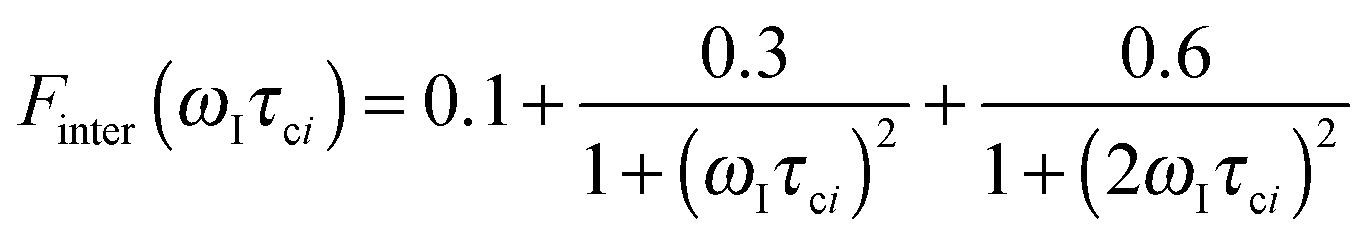 | (2b) |
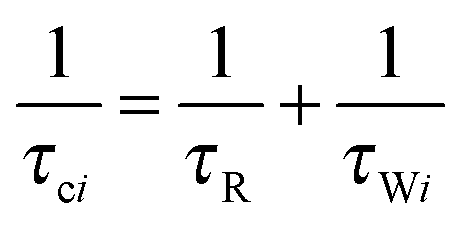 | (3) |
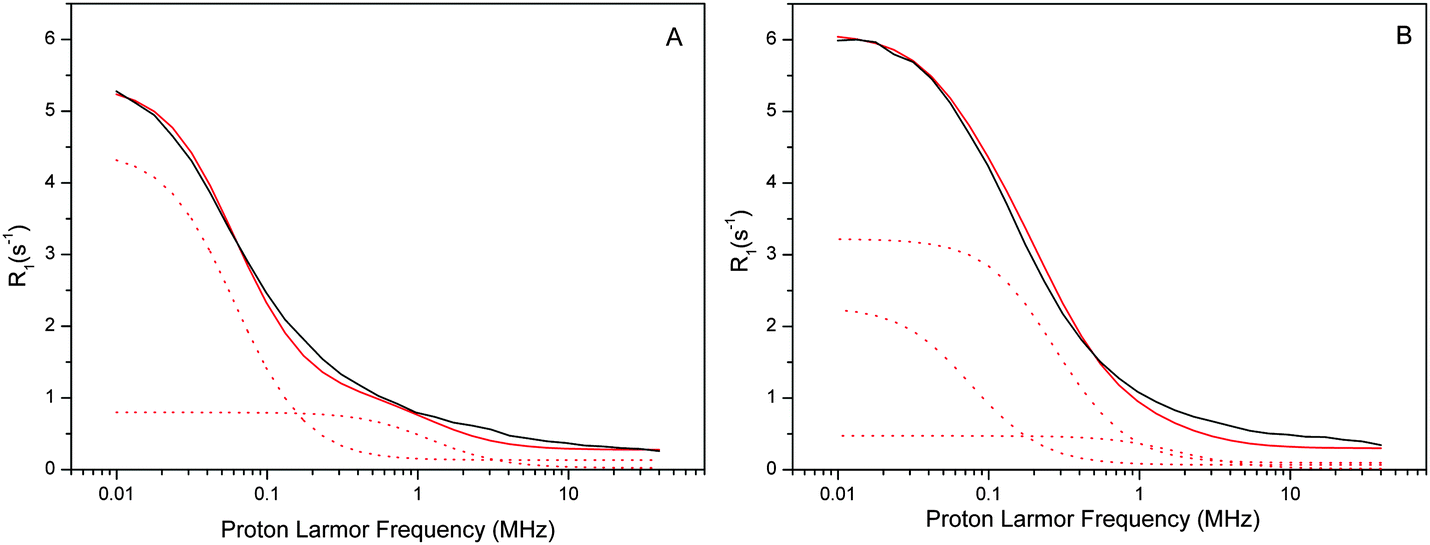 | ||
| Fig. 4 Experimental water proton R01,p(ωI)–NMRD profiles of the Mn-depleted sample (A) and the intact sample (B) at a temperature of 10 °C. Both samples had a [PSIIcc] = 60 μM. The individual β-water components of the fits are indicated by dashed red lines, while the resulting fit is given by the solid red line. All fit parameters are given in Table 1. Note that the amplitude reflects (among several other factors) the number of waters, while the position of the dispersion depends on the effective correlation time. The small frequency independent component (0.06 s−1 and 0.12 s−1, respectively) of the α waters is not shown for clarity. | ||
| Figure | Number of β waters, qi | Effective correlation times τci (ns) | Sample type |
|---|---|---|---|
| 4A | 50 | τ c1 = 1500 | Mn-depleted PSIIcc |
| 180 | τ c2 = 80 | ||
| 4B | 30 | τ c1 = 1200 | Intact PSIIcc |
| 170 | τ c2 = 330 | ||
| 160 | τ c3 = 50 | ||
The water proton NMRD profiles of the Mn-depleted PSIIcc (Fig. 4A) can be reproduced by using two classes of β-waters which are characterized by different effective correlation times τc1 = 1500 ns and τc2 = 75 ns, respectively, plus a small field independent contribution α = 0.06 (s−1). A total of about 230 β waters were identified. For the fit of the native sample containing the Mn4CaO5 cluster significantly more β waters are required, about 360. These are distributed into three classes, of which two (τc1 and τc3) are similar to the two found in Mn-depleted PSIIcc. In addition, a third component with τc2 = 330 ns is required. Similar fits were obtained for the data collected at 1 °C and 20 °C. It is noted that the τc1 phase in the intact PSIIcc is especially temperature sensitive with regard to the number of β waters and the effective correlation time (see Tables S1 and S2 in ESI†).
In order to rationalize these estimates without involving paramagnetic relaxation effects one needs to assume that the chemical process that removed the Mn-cluster also changed the hydration of PSIIcc with β waters. For this we have two options. In the first scenario the Mn-depletion did not alter the protein structure/composition. In that case the hydration with β waters should be larger in the Mn-depleted sample as compared to the native sample since water is expected to fill up the Mn4CaO5-cluster cavity. The opposite trend is observed and therefore this case can be excluded. In the second scenario, the lower number of β waters in Mn-depleted PSIIcc may then be rationalized by the NH2OH-induced loss of extrinsic proteins during the Mn-depletion procedure. Indeed this has been reported previously for spinach samples84,85 and may also happen to some extent for the more stable PSIIcc of T. vulcanus. So while we cannot exclude this possibility, we think that the presence of a large apparent separate class of β waters in the intact PSIIcc indicates that the difference is mainly due to the paramagnetic contribution of the Mn4CaO5 cluster to the R1 NMRD profile of intact PSIIcc. In the following, the NMRD difference profiles of Fig. 2C are therefore analyzed by two different theoretical models using this assumption.
4.2 The theoretical framework for the analysis of the R1 PRE-NMRD difference profiles of the paramagnetic Mn4CaO5 complex
The analysis of the intact-minus-depleted PSIIcc NMRD difference profiles presented in Fig. 2C is performed using either the traditional Solomon–Bloembergen–Morgan (SBM) equations96–98 or the slow-motion theory that is based on the Stochastic Liouville theory.66,67,99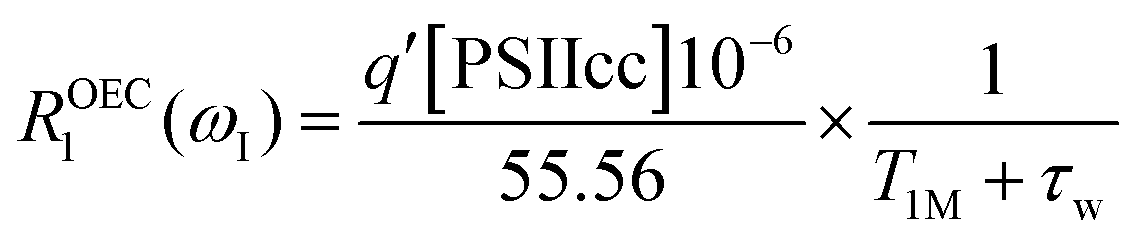 | (4a) |
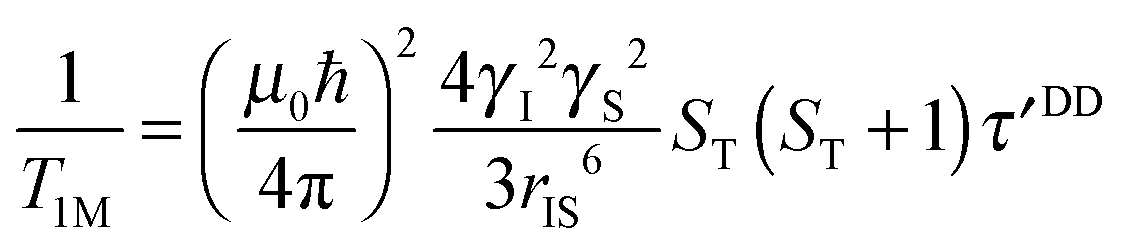 | (4b) |
| τ′DD = 0.7sDD1 + 0.3sDD0 | (5) |
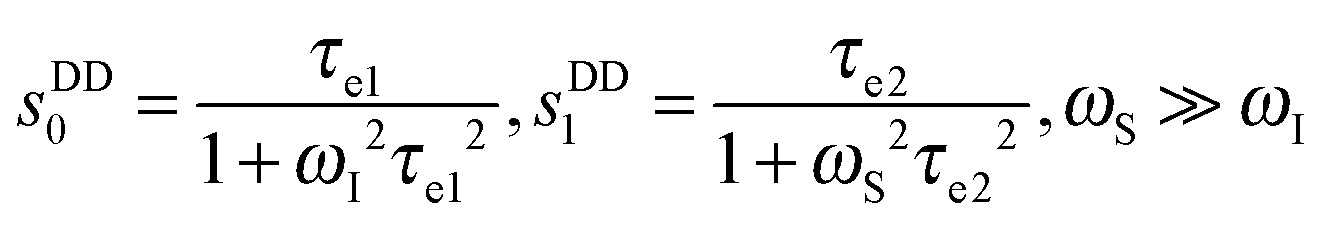 | (6) |
These electron spin spectral density functions depend on the electron and the nuclear Larmor frequencies ωS and ωI, the electron spin–lattice relaxation time T1e and the spin–spin relaxation time T2e through the effective correlation times:
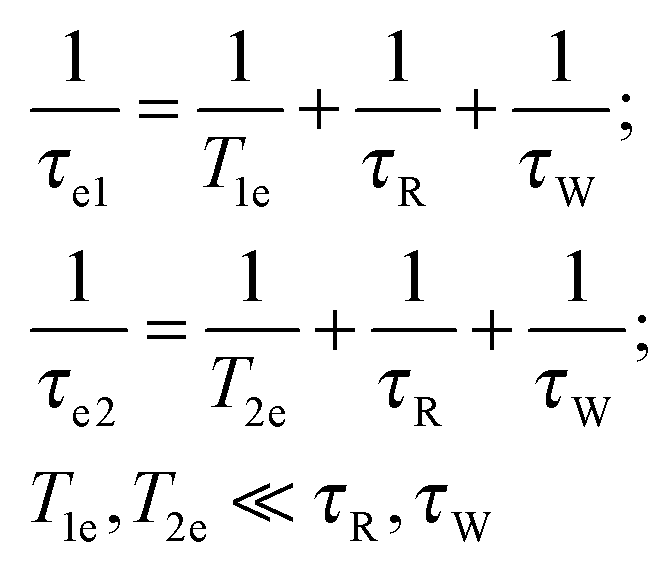 | (7) |
The reorientation correlation time τR thus refers to all sorts of motions in the protein which modulate the water proton–electron dipole–dipole coupling HDD(t) including the reorientation of the PSIIcc. The electron spin relaxation times are given within the Redfield perturbation theory by:66,67,99
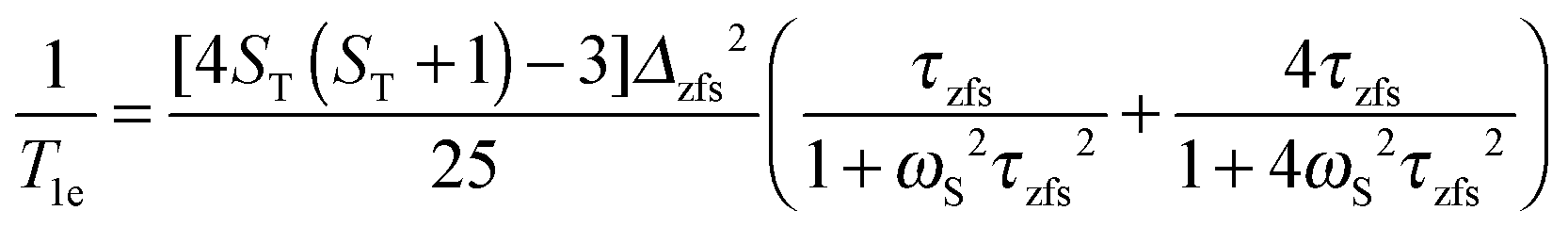 | (8a) |
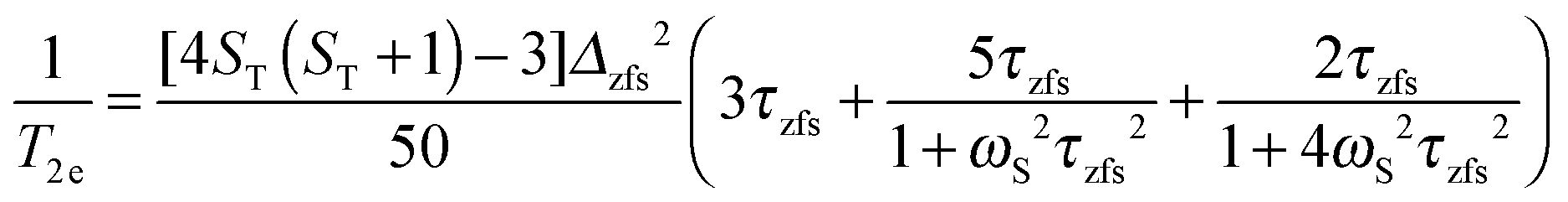 | (8b) |
Here the electron spin relaxation times are determined by the electron spin quantum number ST and the (transient) zero-field splitting (ZFS) interaction Δzfs, which is a stochastic time dependent quantity characterized by the correlation time τzfs.
The SBM theory is valid, if the modulation of the ZFS interaction is due to relatively fast local motions that are much faster than the overall tumbling of the PSIIcc (τzfs ≪ τR). If the term Δzfsτzfs (Kubo term) becomes much smaller than 1 (Δzfsτzfs ≪ 1), then the time dependent perturbation (Redfield) theory is applicable.66,67,99
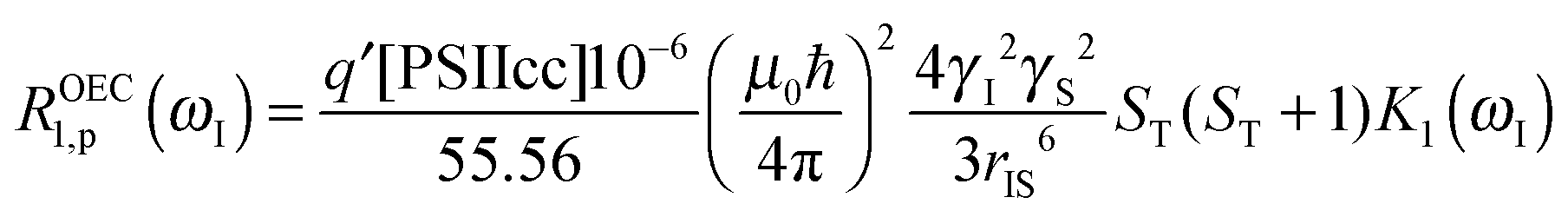 | (9) |
In the analysis of the difference PRE-NMRD profile we have to introduce the decomposition approximation67 assuming that the modulation of the ZFS interaction and the electron spin relaxation is statistically independent of the protein reorientation. This assumption is questionable if the paramagnetic metal cluster is firmly bound to the protein which means that the permanent (residual) ZFS becomes modulated only by the reorientation tumbling of the whole protein. However, for this case the slow motion theory predicts a field dependent paramagnetically enhanced proton spin–lattice relaxation profile which is qualitatively different from the difference NMRD profile observed here. Therefore, we must assume that the reorientational motion of the metal cluster is independent of the reorientational motion of the whole protein complex. In other words, we need to assume that the ZFS causing the electron spin relaxation is not modulated by the reorientation of the whole PSII complex, but is caused by structural fluctuations (in the μs time range) of the metal cluster relative to the PSIIcc complex. Such dynamics were recently suggested for the Mn4CaO5 cluster in the S2 state of the OEC,100–102 and may also occur in the S1 state studied here. However, further quantum chemical calculations will be required to verify that.
4.3 Analysis of the proton relaxation dispersion of the paramagnetic Mn4CaO5 complex
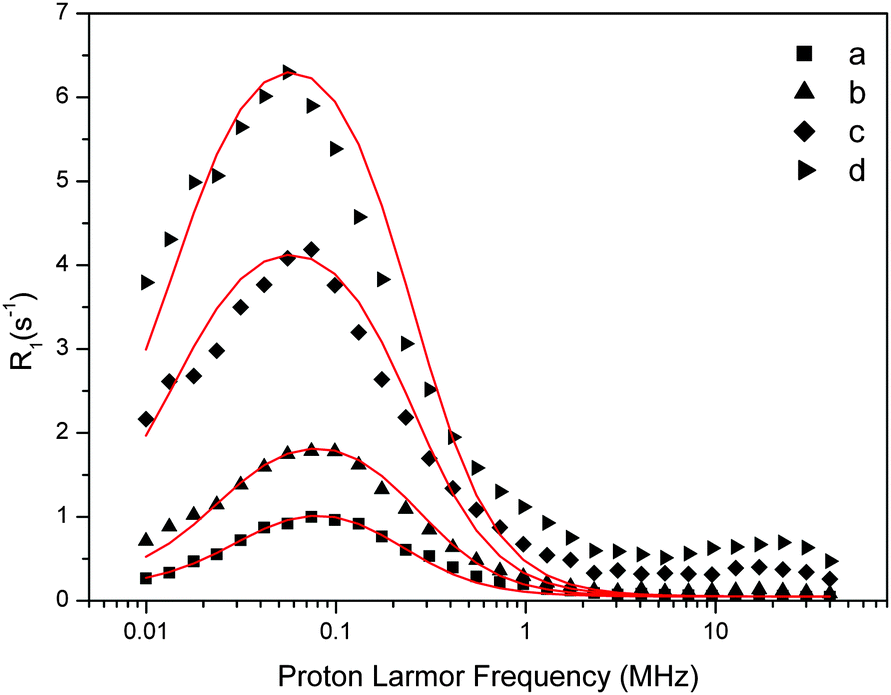 | ||
| Fig. 5 Experimental water proton ROEC1,p(ωI) NMRD difference profiles of OEC (symbols; a = 29 μM, b = 60 μM, c = 123 μM, d = 188 μM) at 10 °C using theoretical simulation (red line; SBM theory). Parameters are given in Table 2. Note that the peak position is determined by the g-value. | ||
| [PSIIcc], μM | Δ ZFS, cm−1 | The Kubo term | τ R, μs | τ zfs, μs | T 1M, μs |
|---|---|---|---|---|---|
| 29 | 1.5 × 10−4 | 0.231 | 0.73 | 0.0035 | 8.83 |
| 60 | 3.5 × 10−4 | 0.310 | 0.59 | 0.0047 | 7.32 |
| 123 | 2.3 × 10−4 | 0.282 | 0.63 | 0.0065 | 7.75 |
| 188 | 2.3 × 10−4 | 0.282 | 0.63 | 0.0065 | 7.75 |
In the fitting procedure using SBM and the slow motion theory (see below) we have to use a value on the amplitude factor in front of the field dependent expressions in eqn (9). This factor contains as unknowns q′ST(ST +1)/r6. Since we assume on the basis of EPR data in the literature that the electron spin quantum number for the intact, dark-adapted Mn4CaO5 cluster in photosystem II is ST = 1,20,36 only the ratio q′/r6 needs to be optimized during the fitting procedure. This number can be interpreted assuming q′ = 1 which gives r = 9.8 Å, or if q′ = 2 then the distance r becomes 10.9 Å and so forth (Table 3).
| q′ | SBM theory | Slow motion theory |
|---|---|---|
| r IS, Å | r IS, Å | |
| 1 | 10.7 | 9.7 |
| 2 | 12.0 | 10.9 |
| 3 | 12.8 | 11.7 |
| 4 | 13.4 | 12.3 |
| 5 | 13.9 | 12.7 |
| 10 | 14.4 | 14.3 |
| 20 | 14.7 | 16.0 |
| 30 | 15.1 | 17.2 |
The results in Table 2 show that the characteristic correlation time of the ZFS interaction (τzfs) is about 100-times shorter than the reorientation correlation time τR, which may indicate that it refers to structural fluctuations/vibrations of the OEC that occur at the ns time scale. These fluctuations/vibrations may also lead to a partial averaging of the ZFS interaction parameter, consistent with its rather small ΔZFS value (Table 2). Using the SBM theory the Kubo factor must be much smaller than 1. This is not quite the case. We therefore analyze our data below using the more general slow motion theory.
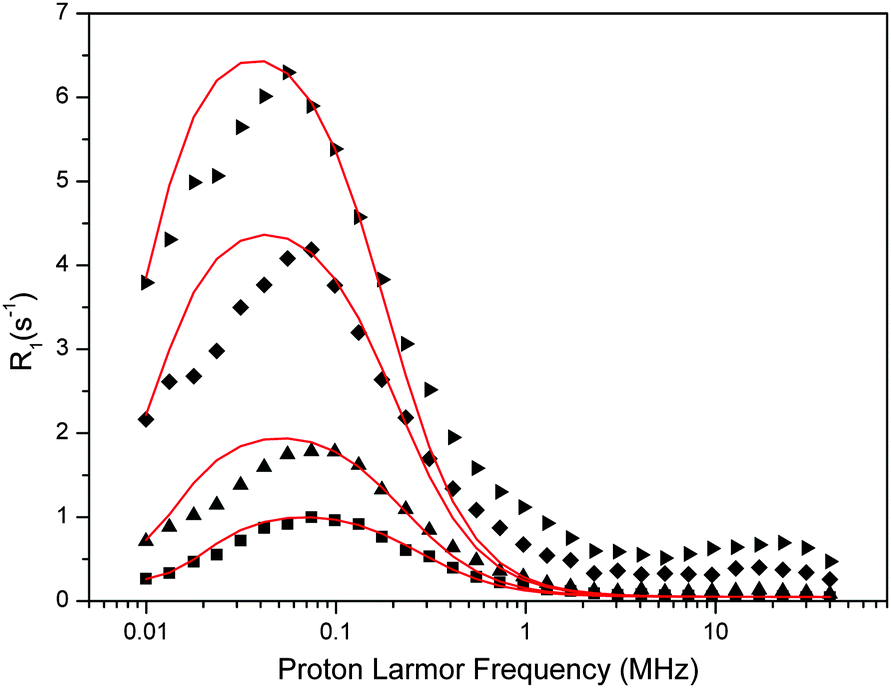 | ||
| Fig. 6 Experimental water proton ROEC1,p(ωI) NMRD difference profiles of OEC (symbols; squares, 29 μM; triangles, 60 μM; diamonds, 123 μM, inverted triangles, 188 μM) at 10 °C using theoretical simulation (red line; slow motion theory, eqn (9)). Parameters are given in Table 4. Note that the peak position is determined by the g-value. | ||
The fit parameters are summarized in Table 4. The Kubo term is always at least 20 times larger than one, confirming that this theoretical approach is applicable to the data. Fluctuations of the ZFS have a time constant of about 0.7 μs, which is about 200-times longer than estimated by the SBM theory. However, the reorientation time τR, which reflects the fluctuations within the OEC that determine the dispersion effect, is rather similar in both cases, about 0.6–0.9 μs.
| [PSIIcc], μM | Δ ZFS, cm−1 | The Kubo term | τ R = τzfs, μs | T 1M, μs | q′ |
|---|---|---|---|---|---|
| 29 | 3.9 × 10−4 | 42.6 | 0.58 | 43.3 | 2.2 |
| 60 | 2.9 × 10−4 | 37.6 | 0.70 | 51.2 | 1.7 |
| 123 | 2.3 × 10−4 | 33.7 | 0.77 | 55.7 | 1.7 |
| 188 | 2.0 × 10−4 | 33.2 | 0.87 | 62.3 | 1.45 |
The slow motion theory indicates a remarkably similar hydration pattern than the SBM theory (Table 3). Assuming integer q′ values, only slightly shorter distances of water molecules to the electron spin center in the OEC are found.
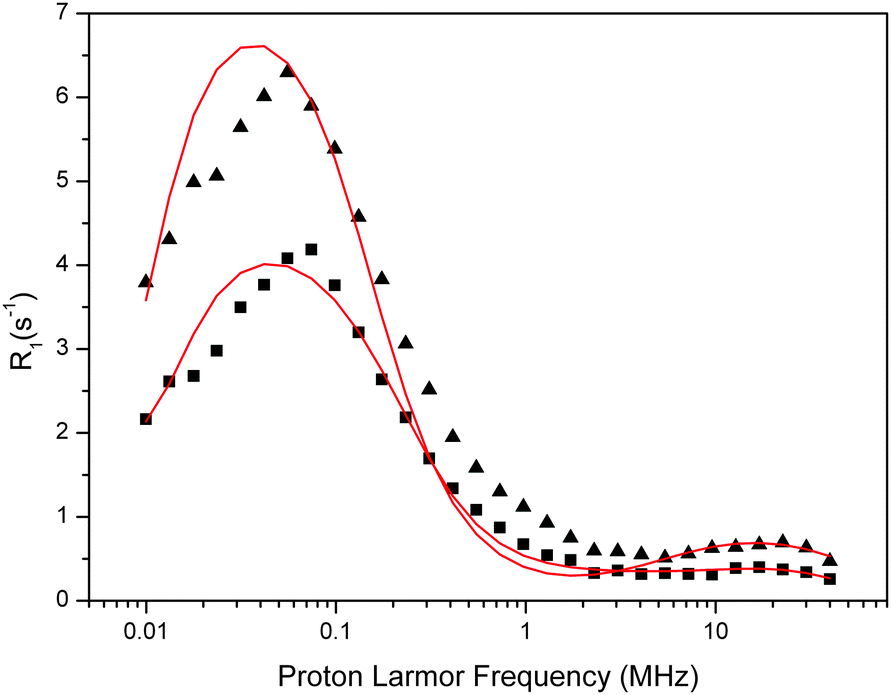 | ||
| Fig. 7 Fit of the experimental water proton ROEC1,p(ωI) NMRD difference profiles of the OEC (squares = 123 μM, triangles = 188 μM) at 10 °C using theoretical simulation (red line) using the slow motion theory and two g-values (g = 4.9 and g = 2.0). Parameters are given in Table 5. Note that the peak positions are determined by the g-values. | ||
| [PSIIcc], μM | Δ ZFS (1 cm−1) | The Kubo term | τ R, μs | τ zfs, μs | T 1M, μs | q′ | g |
|---|---|---|---|---|---|---|---|
| 123 | 3.0 × 10−4 | 36.9 | 0.650 | 0.650 | 47.8 | 1.45 | 4.9 |
| 0.018 | 0.033 | 0.039 | 0.0001 | 744 | 38 | 2 | |
| 188 | 2.1 × 10−4 | 41.5 | 1050 | 1.050 | 74.4 | 1.19 | 4.9 |
| 0.0225 | 0.32 | 0.028 | 0.0008 | 2180 | 52 | 2 |
5. Discussion
In earlier NMR relaxation studies by Wydrzynski et al. and Sharp et al. relaxation features at around 20–27 MHz were reported that were assigned to paramagnetic state changes of the Mn4CaO5 cluster. These signals were found in Sn state difference signals, i.e. for example S2-minus-S1 PRE-NMRD spectra. Here we find by analyzing the intact-minus-(Mn)depleted PRE-NMRD difference profile a strong relaxation effect at around 0.1 MHz. Theoretical simulations based on the SBM and the slow-motion theory both required an electron transition characterized by a g-value of 4.9 for its simulation. This is consistent with the known parallel mode ESR spectrum of the S1 state, which has a first excited state with ST = 1 at liquid helium temperature, and thus suggests the assignment of this relaxation feature to a paramagnetic effect by the Mn4CaO5 cluster. While the analysis appears fully consistent, the final assignment can only be made on the basis of future Sn state dependent PRE-NMRD profiles.However, assuming for the time being the assignment is correct some possible implications regarding the water access to the catalytic site shall be discussed. Since for both the SBM and slow motion approaches the fast chemical exchange condition applies, which is consistent with the temperature dependence observed (Fig. 2), the residence time of the water τw needs to be much shorter than the spin lattice relaxation time of the protons, T1M, which means that τw < 1 μs. Thus, our analysis predicts a sub μs chemical exchange between the bulk and waters near the Mn4CaO5 cluster. Vassiliev et al. calculated that most water channels have barriers of about 10 kcal mol−1 and that this translates into permeabilities of about 5000 water molecules per s at 300 K,44 which corresponds to a residence time of 200 μs. For most channels this barrier was found 10–12 Å away from the OEC, and subsequent barriers would not exceed 5 kcal. This estimate would suggest that we do not observe the waters in the immediate vicinity of the Mn4CaO5 cluster, but rather those directly behind the barrier. The number of waters within the spheres of 10–15 Å from the center of the OEC complex is about 30. This is qualitatively consistent with the estimates in Table 3, if one takes into account that the point dipole approximation for obtaining the hydration numbers in Table 3 is only a very rough estimate. More accurate values may be obtained in future on the basis of detailed quantum chemical calculations that derive the distribution of spin density in the OEC complex in the S1 state. It is noted, however, that the distance of W1 and W2 to Mn1/Mn2 is about 7 Å, so that it presently cannot be fully excluded that they cause the observed PRE-NMRD effect.
6. Conclusion
The experiments and analysis presented here strongly suggest that electron spin relaxation of the S1 state in PSII is an important contribution to the overall correlation time at low field, and also identify a second contribution at around 20 MHz. They thus make NMRD an interesting tool for studying the dynamic features of the Mn4CaO5 cluster in PSII under physiological conditions. The numbers given in the tables have at this early stage to be viewed as our best estimates. The intact-minus-(Mn)depleted difference NMRD profiles of T. vulcanus PSIIcc can be consistently interpreted on the basis of the slow motion theory as being due to a paramagnetic enhanced relaxation (PRE) of 1–2 water molecules residing about 10 Å away from the spin center of the Mn4CaO5, which is in a paramagnetic state characterized by ST = 1, g = 4.9. These water molecules exchange with the bulk water with sub μs exchange times, is consistent with previous theoretical calculations.44 The modulation of the ZFS interaction indicates a (restricted) reorientation or structural equilibrium of the Mn4CaO5 cluster in the S1 state that occurs on the μs time scale. The validity of the SBM theory is however questionable because the parameters extracted do not fulfil the presupposed perturbation criterion. Future Sn state dependent PRE-NMRD experiments are required to confirm these assignments. They may also reveal important information on the role of diffusion barriers within the water channels of PSII.Acknowledgements
The authors thank Tobias Sparrman and Dmitriy Shevela for technical support. The Kempe foundation is acknowledged for supporting the purchase of the NMRD field cycling instrument and for a postdoc stipend to GH. Further financial support was provided by the Artificial Leaf Project Umeå (K&A Wallenberg foundation; JM), the Solar Fuels strong research environment Umeå (Umeå University; JM), VR (YH, POW, JM) and Energimyndigheten (JM). JRS was supported by a Grant-in-Aid for Specially Promoted Research No. 24000018 from MEXT/JSPS of Japan. Author contributions: JM conceived the experiment, FHMK prepared the PSIIcc in the lab of JRS, GH performed all NMRD and EPR experiments, YH and POW performed the theoretical analysis, the manuscript was written by GH, POW and JM, with contributions from all other authors.References
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–61 CrossRef CAS PubMed
.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821–825 CrossRef CAS PubMed
- P. E. M. Siegbahn, Biochim. Biophys. Acta, 2013, 1827, 1003–1019 CrossRef CAS PubMed
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS PubMed
- N. Cox and J. Messinger, Biochim. Biophys. Acta, 2013, 1827, 1020–1030 CrossRef CAS PubMed
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 CAS
-
J. Messinger and G. Renger, in Primary Processes of Photosynthesis – Part 2: Basic Principles and Apparatus, ed. G. Renger, The Royal Society of Chemistry, Cambridge, UK, 2008, pp. 291–349 Search PubMed
-
J. Messinger, T. Noguchi and J. Yano, in Molecular Solar Fuels, ed. T. J. Wydrzynski and W. Hillier, RSC, London, 2012, vol. 5, pp. 163–207 Search PubMed
- N. Nelson and C. F. Yocum, Annu. Rev. Plant Biol., 2006, 57, 521–565 CrossRef CAS PubMed
- C. Tommos and G. T. Babcock, Acc. Chem. Res., 1998, 31, 18–25 CrossRef CAS
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–476 CrossRef CAS
- G. Renger, Biochim. Biophys. Acta, 2012, 1817, 1164–1176 CrossRef CAS PubMed
- B. Forbush, B. Kok and M. P. McGloin, Photochem. Photobiol., 1971, 14, 307–321 CrossRef CAS
- R. D. Britt, K. A. Campbell, J. M. Peloquin, M. L. Gilchrist, C. P. Aznar, M. M. Dicus, J. Robblee and J. Messinger, Biochim. Biophys. Acta, 2004, 1655, 158–171 CrossRef CAS PubMed
- J. M. Peloquin, K. A. Campbell, D. W. Randall, M. A. Evanchik, V. L. Pecoraro, W. H. Armstrong and R. D. Britt, J. Am. Chem. Soc., 2000, 122, 10926–10942 CrossRef CAS
- G. C. Dismukes and Y. Siderer, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 274–278 CrossRef CAS
- J. Messinger, J. H. A. Nugent and M. C. W. Evans, Biochemistry, 1997, 36, 11055–11060 CrossRef CAS PubMed
- K. A. Åhrling, S. Peterson and S. Styring, Biochemistry, 1998, 37, 8115–8120 CrossRef PubMed
- T. Yamauchi, H. Mino, T. Matsukawa, A. Kawamori and T. Ono, Biochemistry, 1997, 36, 7520–7526 CrossRef CAS PubMed
- J. Yano and V. K. Yachandra, Inorg. Chem., 2008, 47, 1711–1726 CrossRef CAS PubMed
- V. K. Yachandra, K. Sauer and M. P. Klein, Chem. Rev., 1996, 96, 2927–2950 CrossRef CAS PubMed
- J. Messinger, J. H. Robblee, U. Bergmann, C. Fernandez, P. Glatzel, H. Visser, R. M. Cinco, K. L. McFarlane, E. Bellacchio, S. A. Pizarro, S. P. Cramer, K. Sauer, M. P. Klein and V. K. Yachandra, J. Am. Chem. Soc., 2001, 123, 7804–7820 CrossRef CAS PubMed
- L. V. Kulik, B. Epel, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2007, 129, 13421–13435 CrossRef CAS PubMed
- L. V. Kulik, B. Epel, W. Lubitz and J. Messinger, J. Am. Chem. Soc., 2005, 127, 2392–2393 CrossRef CAS PubMed
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed
- M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke and H. Dau, Biochemistry, 2005, 44, 1894–1908 CrossRef CAS PubMed
- D. R. J. Kolling, N. Cox, G. M. Ananyev, R. J. Pace and G. C. Dismukes, Biophys. J., 2012, 103, 313–322 CrossRef CAS PubMed
- P. Glatzel, H. Schroeder, Y. Pushkar, T. Boron, S. Mukherjee, G. Christou, V. L. Pecoraro, J. Messinger, V. K. Yachandra, U. Bergmann and J. Yano, Inorg. Chem., 2013, 52, 5642–5644 CrossRef CAS PubMed
- J. Messinger, J. H. Robblee, W. O. Yu, K. Sauer, V. K. Yachandra and M. P. Klein, J. Am. Chem. Soc., 1997, 119, 11349–11350 CrossRef CAS
- K. A. Åhrling, S. Peterson and S. Styring, Biochemistry, 1997, 36, 13148–13152 CrossRef PubMed
- T. Matsukawa, H. Mino, D. Yoneda and A. Kawamori, Biochemistry, 1999, 38, 4072–4077 CrossRef CAS PubMed
- J. L. Zimmermann and A. W. Rutherford, Biochim. Biophys. Acta, 1984, 767, 160–167 CrossRef CAS
- J. L. Casey and K. Sauer, Biochim. Biophys. Acta, 1984, 767, 21–28 CrossRef CAS
- A. Boussac, M. Sugiura, Y. Inoue and A. W. Rutherford, Biochemistry, 2000, 39, 13788–13799 CrossRef CAS PubMed
- S. L. Dexheimer and M. P. Klein, J. Am. Chem. Soc., 1992, 114, 2821–2826 CrossRef CAS
- A. Zouni, H. T. Witt, J. Kern, P. Fromme, N. Krauß, W. Saenger and P. Orth, Nature, 2001, 409, 739–743 CrossRef CAS PubMed
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed
- A. Guskov, J. Kern, A. Gabdulkhakov, M. Broser, A. Zouni and W. Saenger, Nat. Struct. Mol. Biol., 2009, 16, 334–342 CAS
- N. Kamiya and J.-R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 98–103 CrossRef CAS PubMed
- F. M. Ho and S. Styring, Biochim. Biophys. Acta, 2008, 1777, 140–153 CrossRef CAS PubMed
- J. W. Murray, K. Maghlaoui, J. Kargul, M. Sugiura and J. Barber, Photosynth. Res., 2008, 98, 523–527 CrossRef CAS PubMed
- J. W. Murray and J. Barber, J. Struct. Biol., 2007, 159, 228–237 CrossRef CAS PubMed
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, 2012, 1817, 1671–1678 CrossRef CAS PubMed
- S. Vassiliev, T. Zaraiskaya and D. Bruce, Biochim. Biophys. Acta, 2013, 1827, 1148–1155 CrossRef CAS PubMed
- K. Linke and F. M. Ho, Biochim. Biophys. Acta, 2014, 1837, 14–32 CrossRef CAS PubMed
- A. Gabdulkhakov, A. Guskov, M. Broser, J. Kern, F. Mueh, W. Saenger and A. Zouni, Structure, 2009, 17, 1223–1234 CrossRef CAS PubMed
- J. W. Murray and J. Barber, J. Struct. Biol., 2007, 159, 228–237 CrossRef CAS PubMed
- H. Ishikita, W. Saenger, B. Loll, J. Biesiadka and E.-W. Knapp, Biochemistry, 2006, 45, 2063–2071 CrossRef CAS PubMed
- J. M. Anderson, FEBS Lett., 2001, 488, 1–4 CrossRef CAS
- T. Wydrzynski, W. Hillier and J. Messinger, Physiol. Plant., 1996, 96, 342–350 CrossRef CAS
- W. Hillier and T. Wydrzynski, Coord. Chem. Rev., 2008, 252, 306–317 CrossRef CAS PubMed
- J. Messinger, M. Badger and T. Wydrzynski, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3209–3213 CrossRef CAS
-
W. Hillier and J. Messinger, in Photosystem II. The Light-Driven Water*Plastoquinone Oxidoredutase, ed. T. Wydrzynski and K. Satoh, Springer, Dordrecht, 2005, vol. 22, pp. 567–608 Search PubMed
- H. Suzuki, M. Sugiura and T. Noguchi, Biochemistry, 2008, 47, 11024–11030 CrossRef CAS PubMed
- T. Noguchi, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1189–1194 CrossRef CAS PubMed
- M. P. Navarro, W. M. Ames, H. Nilsson, T. Lohmiller, D. A. Pantazis, L. Rapatskiy, M. M. Nowaczyk, F. Neese, A. Boussac, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15561–15566 CrossRef CAS PubMed
- L. Rapatskiy, N. Cox, A. Savitsky, W. M. Ames, J. Sander, M. M. Nowaczyk, M. Rögner, A. Boussac, F. Neese, J. Messinger and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 16619–16634 CrossRef CAS PubMed
- I. L. McConnell, V. M. Grigoryants, C. P. Scholes, W. K. Myers, P. Y. Chen, J. W. Whittaker and G. W. Brudvig, J. Am. Chem. Soc., 2012, 134, 1504–1512 CrossRef CAS PubMed
- Ö. Hansson, L.-E. Andréasson and T. Vänngård, FEBS Lett., 1986, 195, 151–154 CrossRef
- S. Turconi, D. J. MacLachlan, P. J. Bratt, J. H. A. Nugent and M. C. W. Evans, Biochemistry, 1997, 36, 879–885 CrossRef CAS PubMed
- J. Messinger, Phys. Chem. Chem. Phys., 2004, 6, 4764–4771 RSC
- G. Hendry and T. Wydrzynski, Biochemistry, 2002, 41, 13328–13334 CrossRef CAS PubMed
- B. Halle, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1207–1224 CrossRef CAS PubMed
- V. P. Denisov, B.-H. Jonsson and B. Halle, Biochemistry, 1999, 121, 2327–2328 CAS
-
P.-O. Westlund, in Dynamics of solutions and fluid mixtures by NMR, ed. J. J. Delpuech, Wiley, Chichester, 1995, pp. 173–229 Search PubMed
- J. Kowalewski, L. Nordenskiöld, N. Benetis and P. O. Westlund, Prog. Nucl. Magn. Reson. Spectrosc., 1985, 17, 141–185 CrossRef CAS
- L. Helm, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 45–64 CrossRef CAS PubMed
- T. Wydrzynski, N. Zumbulyadis, P. G. Schmidt, H. S. Gutowsky and Govindjee, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1196–1198 CrossRef CAS
- T. Wydrzynski, N. Zumbulyadis, P. G. Schmidt and Govindjee, Biochim. Biophys. Acta, 1975, 408, 349–354 CrossRef CAS
- T. J. Wydrzynski, Photosynth. Res., 2004, 80, 125–135 CrossRef CAS
- T. J. Wydrzynski, S. B. Marks, P. G. Schmidt, Govindjee and H. S. Gutowsky, Biochemistry, 1978, 17, 2155–2162 CrossRef CAS
- T. Wydrzynski and G. Renger, Biochim. Biophys. Acta, 1986, 851, 65–74 CrossRef CAS
- H. H. Robinson, R. R. Sharp and C. F. Yocum, Biochem. Biophys. Res. Commun., 1980, 93, 755–761 CrossRef CAS
- A. N. Srinivasan and R. R. Sharp, Biochim. Biophys. Acta, Bioenerg., 1986, 850, 211–217 CrossRef CAS
- A. N. Srinivasan and R. R. Sharp, Biochim. Biophys. Acta, Bioenerg., 1986, 851, 369–376 CrossRef CAS
- R. R. Sharp and C. F. Yocum, Photobiochem. Photobiophys., 1983, 5, 193–199 CAS
- H. H. Robinson, R. R. Sharp and C. F. Yocum, Arch. Biochem. Biophys., 1981, 207, 1–8 CrossRef CAS
- J. M. Bovet, E. J. Park and R. R. Sharp, Photosynth. Res., 1993, 38, 347–354 CrossRef CAS PubMed
-
R. R. Sharp, in Manganese Redox Enzymes, ed. V. L. Pecoraro, VCH Publishers, New York, 1992, pp. 177–196 Search PubMed
- J. R. Shen and N. Kamiya, Biochemistry, 2000, 39, 14739–14744 CrossRef CAS PubMed
- R. J. Porra, W. A. Thompson and P. E. Kriedemann, Biochim. Biophys. Acta, 1989, 975, 384–394 CrossRef CAS
- Y. Sanakis, V. Petrouleas and B. A. Diner, Biochemistry, 1994, 33, 9922–9928 CrossRef CAS
- N. Tamura and G. Cheniae, Biochim. Biophys. Acta, 1985, 809, 245–259 CrossRef CAS
- R. Mei and C. Yocum, Photosynth. Res., 1993, 38, 449–453 CrossRef CAS PubMed
- V. R. Denisov and B. Halle, J. Mol. Biol., 1995, 245, 682–697 CrossRef CAS PubMed
- V. P. Denisov and B. Halle, J. Mol. Biol., 1995, 245, 698–709 CrossRef CAS PubMed
- V. P. Denisov and B. Halle, Biochemistry, 1995, 117, 8456–8465 CAS
- V. P. Denisov, B. Halle, J. Peters and H. D. Hoerlein, Biochemistry, 1995, 34, 9046–9051 CrossRef CAS
- V. P. Denisov, J. Peters, H. D. Hoerlein and B. Halle, Nat. Struct. Biol., 1996, 3, 505–509 CrossRef CAS PubMed
- K. Venu, V. P. Denisov and B. Halle, Biochemistry, 1997, 119, 3122–3134 CAS
- B. Halle, H. Johannesson and K. Venu, J. Magn. Reson., 1998, 135, 1–13 CrossRef CAS PubMed
-
B. Halle, V. P. Denisov and K. Venu, Biol. Magn. Reson., Kluwer Academic/Plenum Publishers, 1999, vol. 17, pp. 419–484 Search PubMed
- Y. Huang, K. Nam and P.-O. Westlund, Phys. Chem. Chem. Phys., 2013, 15, 14089–14097 RSC
- V. P. Denisov and B. Halle, Biochemistry, 1994, 116, 10324–10325 CAS
- N. Bloembergen and L. O. Morgan, J. Chem. Phys., 1961, 34, 842–850 CrossRef CAS PubMed
- N. Bloembergen, J. Chem. Phys., 1957, 27, 572–573 CrossRef CAS PubMed
- I. Solomon and N. Bloembergen, J. Chem. Phys., 1956, 25, 261–266 CrossRef CAS PubMed
- N. Benetis, J. Kowalewski, L. Nordenskioeld, H. Wennerstroem and P. O. Westlund, Mol. Phys., 1983, 50, 515–530 CrossRef CAS
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed
- D. Bovi, D. Narzi and L. Guidoni, Angew. Chem., 2013, 52, 1–6 Search PubMed
- Y. Kurashige, G. K. L. Chan and T. Yanai, Nat. Chem., 2013, 5, 660–666 CrossRef CAS PubMed
- M. Roncel, A. Boussac, J. L. Zurita, H. Bottin, M. Sugiura, D. Kirilovsky and J. M. Ortega, JBIC, J. Biol. Inorg. Chem., 2003, 8, 206–216 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp55232b |
| This journal is © the Owner Societies 2014 |