 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA theoretical study of the ozonolysis of C60: primary ozonide formation, dissociation, and multiple ozone additions†
Robert C.
Chapleski
Jr.
,
John R.
Morris
and
Diego
Troya
*
Department of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: troya@vt.edu
First published on 28th January 2014
Abstract
We present an investigation of the reaction of ozone with C60 fullerene using electronic structure methods. Motivated by recent experiments of ozone exposure to a C60 film, we have characterized stationary points in the potential energy surface for the reactions of O3 with C60 that include both the formation of primary ozonide and subsequent dissociation reactions of this intermediate that lead to C–C bond cleavage. We have also investigated the addition of multiple O3 molecules to the C60 cage to explore potential reaction pathways under the high ozone flux conditions used in recent experiments. The lowest-energy product of the reaction of a single ozone molecule with C60 that results in C–C bond breakage corresponds to an open-cage C60O3 structure that contains ester and ketone moieties at the seam. This open-cage product is of much lower energy than the C60O + O2 products identified in prior work, and it is consistent with IR experimental spectra. Subsequent reaction of the open-cage C60O3 product with a second ozone molecule opens a low-energy reaction pathway that results in cage degradation via the loss of a CO2 molecule. Our calculations also reveal that, while full ozonation of all bonds between hexagons in C60 is unlikely even under high ozone concentration, the addition of a few ozone molecules to the C60 cage is favorable at room temperature.
Introduction
Pioneering studies of the ozonolysis of unsaturated molecules date back more than a century, with initial investigations focused on the use of ozone to process organics in solution.1 Though studies throughout the years have proposed several alternatives for the reaction mechanism, the mechanism originally put forward by Criegee has been found to predominate.2–4 In the Criegee mechanism,5 the ozone molecule undergoes a concerted cycloaddition to a carbon–carbon double bond. The addition results in the breakage of the π bond between the carbon atoms and the concerted formation of two carbon–oxygen bonds that results in an unstable cyclic molozonide intermediate (the primary ozonide, or POZ). Even though the POZ is an unstable intermediate in the overall reaction mechanism, its thermal stabilization and low-temperature spectroscopic detection have been possible under carefully controlled conditions.6,7Subsequent reactions of the POZ are quite complex and depend on the structure of the unsaturated reactant. Limited spectroscopic evidence of intermediates and their chemistry7–10 has made theoretical studies paramount to the understanding of the later stages of the ozonolysis reaction mechanism. Naturally, the best-studied ozonolysis reaction using theoretical methods is that of ethylene. CCSD(T) single-point calculations using a split-valence triple-zeta basis set with geometries obtained at the CASSCF level provided a complete description of the connections between the POZ and the various products that can be formed.11 Following a number of dissociation, decomposition, recombination, and isomerization pathways involving the POZ, a Criegee intermediate carbonyl oxide, and a secondary ozonide, the simplest carbon-containing products that can be formed include CO2, CO, and formaldehyde. The general mechanism for ozonolysis of small gas-phase alkanes complements what is known for ethylene and has been reviewed in ref. 2, 12, and 13.
In contrast to the wealth of information on the ozonolysis of alkenes, much less is known about the processing of organic aromatic molecules by ozone. Examination of kinetic data indicates that the ozonolysis thermal rate constants decrease from ethylene to aromatic compounds.14 To investigate this trend, Hendrickx and Vinckier15 used calculations performed at the B3LYP and CCSD(T) levels with the 6-31G** basis set that compared the energetics of the formation of the primary ozonides of ethylene and benzene. The calculated barrier for the addition of ozone to benzene (15.8 kcal mol−1) compared favorably to the experimentally determined barrier of 14.6 kcal mol−1,14 and was found to be much larger than that for the reaction with ethylene (3.3 kcal mol−1). This comparison of the barriers agrees with the fact that the thermal rate constant for the ethylene reaction is much larger than for the reaction with benzene. The increase in the barrier in going from ethylene to benzene was attributed to the disruption in benzene aromaticity by the introduction of the ozone moiety. Regarding the reaction energy, the POZ formation with ethylene was found to be about 30 kcal mol−1 more exothermic than for benzene.
Moving beyond benzene toward polycyclic aromatic hydrocarbons (PAH), Chu et al. performed geometry optimizations and vibrational frequency calculations at the B3LYP/6-31G** level of theory for the addition of ozone to surface-adsorbed anthracene, pyrene, and benzo[a]pyrene.16 The loss in planarity upon ozone addition as the C atoms undergoing reaction change hybridization from sp2 to sp3 was found to be an important factor in the determination of the reaction energy. In effect, the loss of planarity in PAHs that are adsorbed on a surface involves liftoff from the surface, which carries an energetic penalty. For the ozone addition to anthracene, two pathways involving the central ring were examined. The first pathway exhibits addition along a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, characteristic of the Criegee mechanism. The second pathway is a 1,4-cycloaddition that involves the formation of a bridge-like structure between the carbon atoms that belong exclusively to the central ring in anthracene. This addition is thermodynamically more favorable than addition to a CC bond.
C bond, characteristic of the Criegee mechanism. The second pathway is a 1,4-cycloaddition that involves the formation of a bridge-like structure between the carbon atoms that belong exclusively to the central ring in anthracene. This addition is thermodynamically more favorable than addition to a CC bond.
For surface-adsorbed pyrene, Criegee addition was considered for ozonolysis along CC bonds in the perimeter of the molecule. For this PAH, the authors went beyond the formation of the primary ozonide and investigated the first steps in the subsequent reaction mechanism. Dissociation of the POZ provided a secondary ozonide of much lower energy than the POZ. Evolution of the secondary ozonide via additional dissociation reactions was not investigated. Finally, the energetics of ozone addition to surface-adsorbed benzo[a]pyrene across selected CC bonds were found to be strongly dependent upon the degree of planarity of the benzo[a]pyrene moiety in the primary ozonide. Generally, POZ formation favors the least reduction in molecular planarity.
In this work, we make progress in the determination of the mechanism of ozonolysis of CC double bonds beyond ethylene and PAHs by investigating the reaction of O3 with the most abundant fullerene, C60. Reactions of ozone with C60 have been studied in various solvents,17–20 and the primary ozonide for C60 has been identified using liquid chromatography. Moreover, UV-VIS and NMR spectra for several fullerenes and their oxidation intermediates in solution are available.21 In certain solvents, ozonolysis of C60 and C70 fullerenes results in polymeric fullerene oxide formation,17,19 and analogies have been drawn between these polymeric oxides and graphitic oxide.18 Beyond the solution phase, the reactivity of fullerenes with ozone has been investigated mostly using electronic structure calculations. Shang et al.22 used the AM1 semiempirical Hamiltonian to calculate the energetics of the stationary points along the reaction coordinate for C60 ozonolysis. The calculations showed that of the two types of CC bonds in C60 (the one between two hexagons, 6,6, and that between a hexagon and a pentagon, 6,5), addition to the 6,6 bonds was thermodynamically favored. The barrier for POZ formation was found to be 20 kcal mol−1, which would indicate a slow process at ambient temperatures. Decomposition of the POZ species ultimately results in an epoxide of C60O stoichiometry after evolution of O2(1Δ). A similar study was reported more recently for C70.23 Sabirov et al.24,25 performed theoretical calculations using the PBE functional with a split-valence triple-zeta basis set for the addition of ozone to a range of fullerenes from C20 to C76. The authors noted a direct correlation between the reaction energy and the radius of fullerene curvature at the site of ozone addition, with POZ formation on high-curvature fullerenes leading to larger exothermicity. Further confirming this trend, the energetics of the ozonolysis reaction of C70 were investigated in a separate study employing plane-wave DFT techniques.26 These calculations traced the reaction through the primary ozonide and the Criegee intermediate to epoxide product formation and O2 release. The most probable sites of ozonolysis were found in this study to also be those with larger curvature in the C70 moiety.
In a recent paper, Davis et al.27 reported the first room-temperature spectroscopic determination of the primary ozonide in O3 reactions with C60 beyond the solution phase. Under ultra-high vacuum (UHV) conditions, a C60 film was exposed to pure ozone and the reaction was monitored using reflection–absorption infrared spectroscopy (RAIRS). Comparison of low-exposure IR spectral signatures to B3LYP/6-31G* harmonic frequency calculations of the POZ unequivocally attributed the experimental spectrum to the 6,6 POZ. The calculations showed that, in contrast to prior work on PAHs,16 ozone addition to a 6,6 bond in C60 is barrierless, which follows the expectation of heightened reactivity of the double bond as a result of cage curvature.
The experiments of Davis et al.27 provided a variety of information about the reaction of O3 with C60 that motivates the work we present in this paper. First, while the measured IR spectrum at low ozone exposures agrees well with the calculated spectrum for a single POZ on a 6,6 bond, the possibility exists that more than one POZ can be formed on the same cage. In this paper, we address whether multiple ozone addition to C60 is thermodynamically possible and whether the calculated IR spectra of C60 containing more than one POZ agrees with the low-exposure IR measurements. Second, it is well known that the POZ is an intermediate in the ozonolysis of alkenes, and further progress along the reaction coordinate yields more stable products. This was captured in the experiments, as the IR spectrum at longer exposure times changes from that of the POZ to one of products that has strong absorption at ∼1800 cm−1, characteristic of CO moieties. Third, at extended exposures, a pronounced IR band develops at approximately 2200 cm−1. However, previously reported computational studies of gas-phase fullerene ozonolysis identified the C60O epoxide22 as the primary product, which is inconsistent with the IR studies of Davis et al. These observations provide additional motivation for the detailed studies of alternative POZ decomposition reactions presented in this paper.
In the following, we first present the details of the calculations. We then show the results of the study of multiple ozone addition to C60 and subsequently detail our investigation of the POZ evolution towards low-energy products. Finally, we offer concluding remarks.
Computational details
Most of the calculations presented in this work have been carried out with the B3LYP method and the 6-31G* basis set as implemented in the Gaussian09 code.28 While one would ideally like to use multireference methods to properly take into consideration the biradical character of ozone and of some of the intermediates presented in this paper, such types of calculations have only been possible for the ozonolysis of the smallest alkenes.11 However, comparison between CASSCF and B3LYP geometries shows that the more inexpensive DFT method provides results that are qualitatively correct.11 Therefore, DFT methods have been the technique of choice for prior calculations of larger molecules, including PAHs16 and fullerenes.22,25,26,29 Additional benchmarks for the performance of the B3LYP/6-31G* method for ozonolysis are provided by studies of the reaction of O3 with benzene, where high-level energy calculations (CCSD(T) using B3LYP geometries) were compared to the B3LYP results.15 B3LYP reasonably captured reaction energies and barriers predicted by CCSD(T).To further test the accuracy of B3LYP/6-31G* calculations, in this work we have carried out single-point hybrid QM:QM calculations at the CCSD(T)/6-311G*:B3LYP/6-31G* level using the ONIOM formalism30 for the primary ozonide formation. The high-level layer of these benchmarks, calculated at the CCSD(T)/6-311G* level, corresponds to the ozone molecule and the naphthalene moiety whose central C–C bond is the 6,6 bond to which ozone adds. The rest of the cage is treated at the B3LYP/6-31G* level, and hydrogen-atom links are employed between the two layers. A schematic of the QM:QM partitioning is depicted in S1 (ESI†). As we show below, the ONIOM benchmarks agree quite well with the B3LYP/6-31G* estimates of the reaction energetics. This validation is important for our work, as gold-standard CCSD(T) calculations on the entire system will continue to be prohibitive in years to come for some of the molecules calculated in this work, which possess up to 150 non-hydrogen atoms (C60O90 stoichiometry) and up to 780 valence electrons.
The located minima and transition states have been confirmed employing harmonic frequency analysis, and the connections between transition states and minima have been mapped using intrinsic reaction coordinate (IRC) calculations. The calculated IR spectra show B3LYP/6-31G* harmonic frequencies scaled by a factor of 0.96. All of the energies presented below have been zero-point corrected.
Results
Primary-ozonide formation
The mechanism of POZ formation on C60 has been explored in detail in prior work,22,25 and we provide only a brief review here for the sake of completeness. There are two symmetry-inequivalent types of C–C bonds in C60: those between hexagons, and those between a pentagon and a hexagon. For both sites, the addition of ozone to generate the POZ is highly exothermic, but addition to a 6,6 site is thermodynamically more favorable than to a 6,5 site. The B3LYP/6-31G* zero-point corrected energies are −44.0 and −28.4 kcal mol−1, respectively for the 6,6 and 6,5 additions. More importantly, there is also a significant difference in the barriers. Thus, while the zero-point-corrected barrier for 6,6 addition is −2.2 kcal mol−1, that for 6,5 addition is 4.3 kcal mol−1. The sizeable difference in the barrier translates into a thermal rate constant at room temperature that is orders of magnitude larger for addition to 6,6 than to 6,5, and it suggests the exclusive formation of the 6,6 POZ under the experimental conditions of Davis et al.27 The fact that the barrier for the 6,6 reaction is negative implies a transition state that is below the asymptote of the reagents and follows the formation of a pre-transition-state complex formed by long-range van der Waals forces, as has been described before.25 The energy of the van der Waals minimum is −2.6 kcal mol−1 at the B3LYP/6-31G* level. We note that the barrier calculated in this work is in stark contrast to the one previously calculated with the lower-level AM1 semiempirical Hamiltonian (+20 kcal mol−1),22 suggesting inaccuracy in the semiempirical Hamiltonian for the transition state of this reaction.Fig. 1 shows the IR spectrum measured during exposure of a C60 film to 100 L (1 L = 1 × 10−6 Torr s) of pure ozone (red trace). This spectrum differs from that of C60 primarily in the absence of bands at 1428 and 1128 cm−1 and the presence of intense absorption at 977 cm−1. In Fig. 1a and b, the experimental spectrum is compared with the calculated ones for the 6,6 and 6,5 POZ, respectively. In addition to the intense band at 977 cm−1 in the experiment, two bands of decreasing intensity appear at slightly lower frequencies. The 6,6 calculated spectrum agrees well with the experiment and replicates the peak structure seen around 977 cm−1, with the calculated peaks being consistently shifted by about 20 cm−1 toward lower frequencies from the experiment. In contrast, the calculated 6,5 POZ (Fig. 1b) does not agree as well with the experiment around 977 cm−1, but more noticeably, it exhibits an intense peak at ∼1400 cm−1, which is absent in the measurements. The agreement between the 6,6 POZ calculated spectrum and the lack of agreement between the 6,5 POZ calculated spectrum and experiment demonstrate that 6,6 POZ formation is exclusive under thermal conditions. Fig. 1c shows the calculated IR spectrum of the C60O epoxide proposed as the end product of gas-phase C60 ozonolysis in the literature.22 Clearly, the spectrum recorded in the laboratory under short-time ozone exposure to C60 does not support the formation of this product.
 | ||
| Fig. 1 Calculated infrared spectra of (a) 6,6 C60 POZ, (b) 6,5 C60 POZ, and (c) C60O epoxide. The red trace corresponds to the experiments of ref. 27 after 100 L of ozone exposure to a C60 film. The inset in panel (c) shows the calculated IR spectrum of C60O in the 1000–2000 cm−1 region. The spectra are normalized to highest peak height. | ||
To calibrate the accuracy of the B3LYP/6-31G* combination for the title reaction, we have computed the reaction energies and barrier using higher-level methods as described above. The hybrid QM:QM CCSD(T)/6-311G*:B3LYP/6-31G* calculations provide a reaction energy for the 6,6 POZ formation of −46.2 kcal mol−1, which compares well with the B3LYP/6-31G* value (−44.0 kcal mol−1). In addition, the B3LYP/6-31G* barrier (−2.2 kcal mol−1) is also a good representation of the more accurate hybrid result (−0.5 kcal mol−1). These comparisons lend confidence to the use of B3LYP/6-31G* calculations for the rest of this paper.
Because of the large amount of ozone with which the C60 film was dosed in the experiment when the spectrum of Fig. 1 was obtained (100 monolayers), and because B3LYP/6-31G* predicts that the addition of ozone to C60 is barrierless, a question emerges as to whether POZ formation on more than one 6,6 bond of the C60 cage can lead to IR spectra that also agree with experiment. Fig. 2 shows a Schlegel representation of all eight possible double 6,6 POZ isomers that can be formed on C60. The reaction energies for the di-POZ formation (−45.9, −42.1, −40.5, −43.9, −43.4, −42.5, −42.7, and −42.3 kcal mol−1 for isomers A–H, respectively) are very similar to that of the single POZ (−44.0 kcal mol−1), which suggests that the thermodynamics of formation of a second POZ on a C60 cage that already possesses one POZ is not significantly affected by the first POZ. Moreover, calculation of the barriers for the formation of the di-POZs A–H (−0.9, −1.3, −1.1, −2.2, −1.8, −1.6, −1.7, and −1.3 kcal mol−1, respectively) indicates that the addition of a second POZ to a C60O3 POZ continues to be kinetically favored. This result is especially poignant for the di-POZ A, as in this isomer both primary ozonides reside in nearest-neighbor 6,6 sites. However, as the calculations show, the presence of two POZs in the same hexagon does not significantly affect the energetics of POZ formation.
 | ||
| Fig. 2 Schlegel diagram of C60 showing the eight possible symmetry-inequivalent 6,6 di-POZ isomers. The 6,6 bond with the black shade represents the location of the first 6,6 POZ. | ||
Fig. 3 shows the calculated IR spectra of some selected di-POZ isomers compared to the spectrum of the single 6,6 POZ (Fig. 3a). The IR trace of the di-POZ species in which both ozonides are nearest neighbors (isomer A, Fig. 3b) is significantly different from that of the single POZ. Therefore, it seems unlikely that the di-POZ isomer A forms in the low-exposure regime of the study of Davis et al.27 For other isomers, the differences between the IR spectra of the di-POZ species and the primary ozonide are small enough that we cannot determine unequivocally whether the experimental IR spectrum corresponds to a single POZ, to a species that contains two POZs, or to a combination in which some of the C60 molecules in the film contain a single POZ and some others possess two. For instance, even though the IR spectra of di-POZ isomers B and C exhibit more bands in the 977 cm−1 region than the single POZ, these bands have a smaller intensity and are sufficiently close to other, more intense peaks to be enveloped by the broader experimental bands. IR spectra for the rest of the di-POZ isomers not shown in Fig. 3 are similar to those of isomers B, C, and H.
 | ||
| Fig. 3 Theoretical IR spectra of selected 6,6 di-POZ C60 isomers compared to the IR spectrum of the 6,6 POZ (a). | ||
Since the calculations of reaction energetics for di-POZ isomers and the data in Fig. 3 show that the possibility exists that more than one ozonide could be formed on the C60 cage, we have performed a study of POZ formation on all available 6,6 bonds of C60. Thus, starting from the POZ, we have calculated the reaction energy of successive addition of O3 to the remaining 29 6,6 bonds of C60 to form poly-POZ products. Because the number of potential isomers for the tri-, tetra-, etc., adducts quickly becomes intractably large, we have calculated the reaction energy along two exemplary ozonation routes. Fig. 4 shows the order of formation of primary ozonides along these two routes. Pathway A considers that ozonation occurs in 6,6 sites that are furthest from other 6,6 bonds that already contain a primary ozonide. Along this route, one can generate POZs on 6,6 bonds that are not in the same hexagon up to the 7th addition. Similarly, addition of the 17th ozone molecule and beyond necessitates the placement of three ozonides on the same hexagon. Pathway B considers ozonation that grows from an initial 6,6 bond toward 6,6 bonds that are in the vicinity of the original POZ and then disseminate throughout the entire cage. The plot shown in Fig. 4 exhibits the zero-point-corrected reaction energy for the C60O3(n−1) + O3 → C60O3n reactions, where n, the number of final primary ozonides that the cage contains after the reaction, serves as the abscissa.
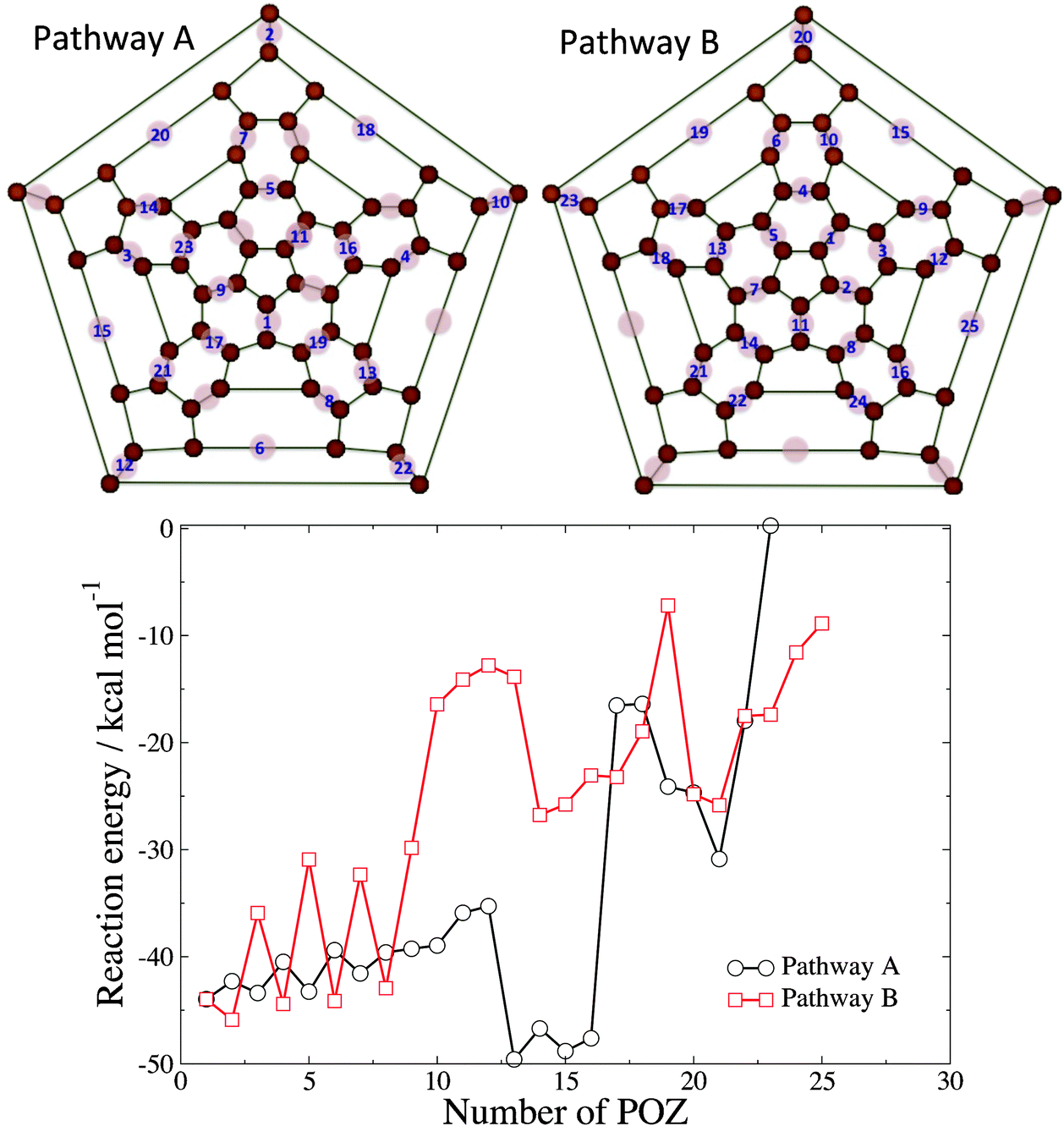 | ||
| Fig. 4 (top) Ozonation pathways. The numbers on 6,6 bonds indicate the order in which primary ozonides are formed on the C60 cage for both pathways. (bottom) Zero-point corrected energies for the C60O3(n−1) + O3 → C60O3n (POZ) reactions. | ||
As expected, successive ozonation generally leads to increasingly less exothermic processes due to, first, growing repulsion between neighboring POZs, and second, growing cage strain as C atoms undergo sp2 to sp3 rehybridization upon addition. Pathway A leads to products that are generally more thermodynamically stable than along pathway B, as the former pathway avoids as much as possible the initial formation of nearest-neighbor ozonides and therefore reduces inter-POZ repulsion and local strain. There are several points of interest along pathway A. First, there is a noticeable increase in the reaction energy for the formation of the 13th ozonide. We attribute the increased exothermicity in this reaction to the release of strain in the vicinity of the bond that undergoes POZ formation. As ozonation proceeds up to the 12th addition, strain is generated along the bond that undergoes the 13th cycloaddition. Fig. S2 (ESI†) shows the relevant region of the molecule for reagents and products, where the twist around the CC bond that forms part of the 13th POZ can be appreciated. The change in hybridization of the C atoms undergoing addition serves to release some of this strain, which leads to an increased exothermicity. Second, there is a sharp decrease in the reaction exothermicity from the 16th to the 17th ozonide. As mentioned above and shown in the Schlegel representation of Fig. 4, the 17th POZ formation in pathway A is the first that involves the ozonation of all three 6,6 bonds in a hexagon. Both the repulsions between the oxygen atoms in the proximal POZs and the strain generated in the C-atom hybridization change are likely contributors to the destabilization of this molecule.
Pathway B represents an ozonation route in which primary ozonides are formed in close proximity to each other. This route is intended to capture a reaction pathway in which an O3 molecule reacts upon collision adjacent to an existing POZ (i.e. no thermalization and subsequent diffusion occurs). Clustering primary ozonides in the same region of the C60 results in less exothermic reactions than pathway A on average, owing to inter-POZ repulsion and the strain generated along the same region of the cage. However, at large loadings (for 17 POZs and above), pathway A and B provide reaction energies that are similar, as at those loadings, repulsion or strain cannot be easily avoided along either route. At large loadings, the reaction energies become less and less negative, suggesting that full ozonation is not favored. In fact, while we tried to optimize the structures of C60O3n with n up to 30, the larger species (n > 25) did not converge to stable minima.
To further estimate the likelihood of large ozone loading in a single C60 cage, we have complemented the analysis of thermodynamic stability shown in Fig. 4 by calculating barriers of POZ formation for a few selected ozonation steps. Along pathway B, the barriers for the 1st, 2nd, and 3rd ozonations are −2.2, −0.9, and +3.5 kcal mol−1. These barriers indicate that ozonation of the third 6,6 bond of a hexagon is considerably slower at room temperature than addition to a 6,6 bond in a hexagon that is pristine or has only one POZ. Regarding route A, the barrier for the 17th ozonation, which is the first one that completes a hexagon, is as large as 33.9 kcal mol−1, strongly suggesting that high loadings are unlikely. To sample intermediate loadings, we have calculated the barrier for addition of the 7th ozone molecule along pathways A and B. These barriers (2.8 and 3.3 kcal mol−1 for pathways A and B, respectively) are significantly smaller than those found at larger loadings and are of the same size as the one for ozonation of ethylene.11 This analysis of the barriers indicates that full ozonation of C60 is highly unlikely under thermal conditions. The formation of a few ozonides is, however, highly exothermic and occurs through small barriers, suggesting the possibility that C60 might accommodate reactions with several ozone molecules under high-flux conditions. Fig. S3 (ESI†) shows the structures of sample poly-POZ species. The IR spectra for these sample poly-POZ species show results similar to those described above for the di-POZ species, in which the main features of the experimental spectrum are borne out by the calculations, but the presence of various POZ on the cage gives rise to additional low-intensity peaks in close proximity to the more intense bands.
Decomposition of the primary ozonide
A critical simplification in the examination of the formation of multiple primary ozonides on C60 presented above is that the POZ does not decompose prior to addition of subsequent ozone molecules. However, the POZ is known to be short-lived at room temperature, and consequently, an unequivocal characterization of this intermediate outside the solution phase was not accomplished until 2012.27 We now investigate potential decomposition pathways of a single POZ in an attempt to provide a more complete description of this reaction.As mentioned above, the experiments of Davis et al.27 detected the POZ after 100 L exposure. IR spectra recorded at larger exposures and longer reaction times showed the disappearance of the POZ peak around 977 cm−1 with simultaneous growth of a broad band at 1700–1900 cm−1 (see Fig. 1 of ref. 27). Prior gas-phase ozonolysis studies of C60 have considered the decomposition of the POZ into an epoxide and O2 as the only reaction pathway,22 and analogous results have been presented for C70.23,26 However, as Fig. 1c shows, epoxide formation is inconsistent with experimental IR data at short times. Epoxide formation at longer times is also inconsistent with experiment, as the spectrum in the inset of Fig. 1c does not show any peaks in the 1700–1900 cm−1 region. In search of reaction products that are consistent with the experiment, we have characterized additional stationary points on the POZ decomposition potential energy surface beyond the primary ozonide.
The full reaction pathway, including the POZ formation for completeness, is presented in Fig. 5, and shows the formation of products that are far more stable than C60O and singlet O2. Structures of the minima and transition states along the reaction pathway are shown in Fig. 6, energetic data are given in Table 1, and geometric data are presented in S4 (ESI†). The first step in the decomposition of the POZ (labeled A in Fig. 5) is the concerted breakage of the C–C and an O–O bond. Detailed structures of the relevant sections of the molecule are shown in Fig. 6, where the atoms of the bonds undergoing bond breakage are labeled as 1C–2C and 1O′–3O′. Following breakage of these bonds, the 1C–2C distance increases from 1.61 Å in the POZ to 2.67 Å in the Criegee intermediate B, indicating an opening of the fullerene cage. Rotation of the carbonyl oxide (2C2O′3O′) moiety also occurs after the transition state. The electronic structure of species B indicates that this carbonyl oxide is better described as a biradical than a zwitterionic species. That is, the O–O and C–O bond lengths are nearly identical (reflecting similar bond orders), which is counter to the characteristics of carbonyl oxide zwitterions.31 In addition, the bond rotation seen on the pathway from the A → B transition state to the B species in Fig. 6 is consistent with the absence of a double C–O bond in these structures, which would be expected if they possessed zwitterionic character.
 | ||
| Fig. 5 B3LYP/6-31G* stationary points on the reaction pathway for addition of one ozone molecule to C60 fullerene. | ||
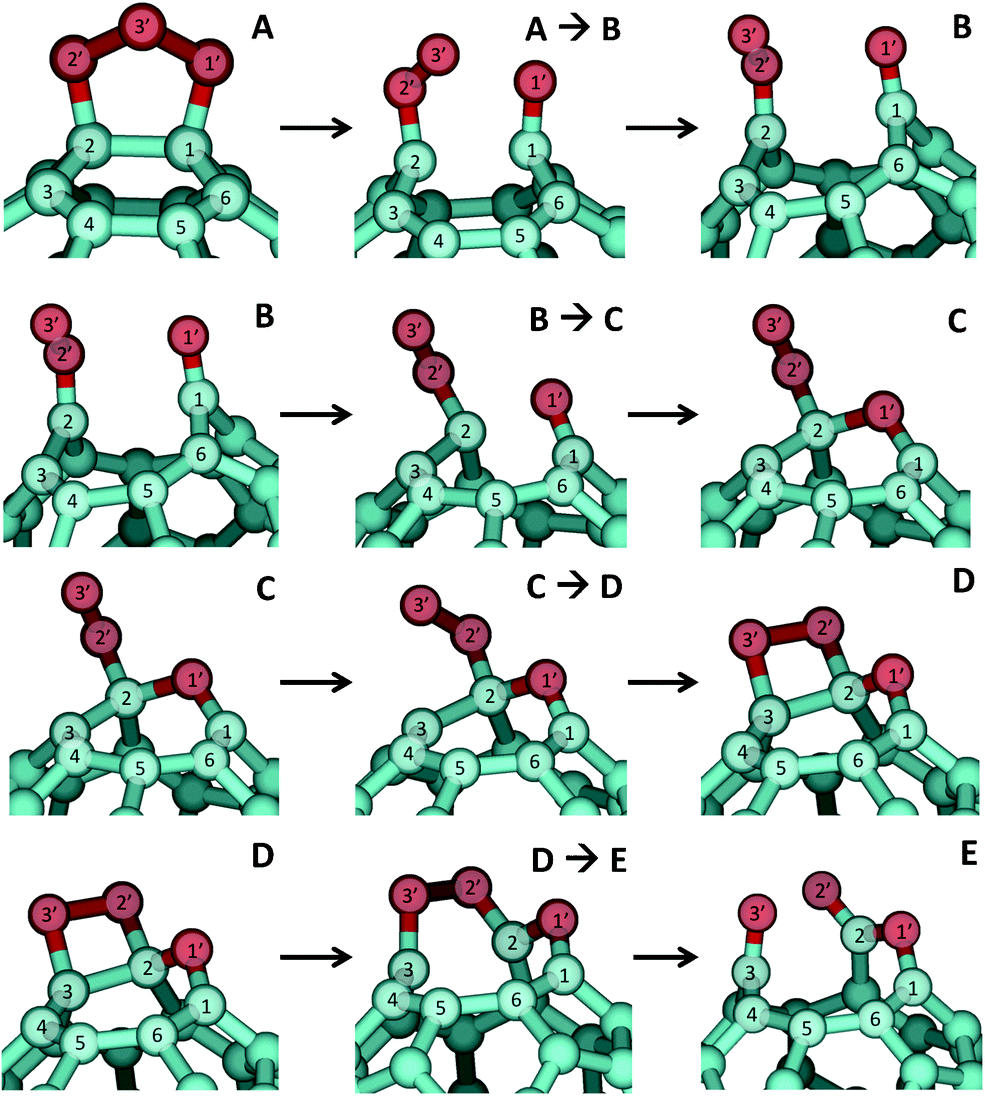 | ||
| Fig. 6 Selected regions of optimum structures of minima A–E and transition states between them in the lowest-energy reaction pathway profile of Fig. 5. | ||
| Intermediate | Energy | Energy (TS) |
|---|---|---|
| a Energies correspond to zero-point-corrected B3LYP/6-31G* calculations. | ||
| C60 + O3 | ||
| A | −44.0 | −2.2 |
| B | −29.6 | −22.0 |
| C | −23.7 | −20.1 |
| D | −48.7 | −23.4 |
| E | −120.3 | −27.9 |
| F + O2 | −21.1 | −14.2 |
| C60O3(E) + O3 | ||
| G | −50.3 | −3.6 |
| H | −68.8 | −35.7 |
| J | −184.7 | +7.7 |
The Criegee intermediate (B) rearranges to form a bond between 1O′ and 2C that results in a seven-membered oxepin ring (C). Note that the 2C carbon atom of the carbonyl oxide moiety rehybridizes from sp2 to sp3 at the transition state. Subsequent to oxepin ring formation, the O–O moiety bound to 2C aligns with a C–C bond to form a dioxetane ring (D). This step occurs through a very small barrier (0.3 kcal mol−1) and is the first exothermic step following POZ formation. The C–D step also represents the key difference between the pathway for epoxide + O2 formation described in prior work22 and the new mechanism that we present here. Contrary to the low barrier and exothermic formation of the dioxetane species D, a much larger barrier of 9.4 kcal mol−1 must be surmounted from the oxepin species C to produce C60O epoxide and O2, which are about 30 kcal mol−1 less thermodynamically stable than product D. Under thermal conditions, the rate of the reaction that yields the epoxide will be about 106 smaller than the rate of dioxetane-species formation, thereby explaining the complete absence of a spectral signature for the epoxide product in the experiments of Davis et al.27
The final step in the decomposition pathway leads from dioxetane intermediate (D) to ketoester product (E). Animation of the imaginary frequency of the intervening transition state shows the breakage of the bond between 2C and 3C. Therefore, the scission of the 2O′–3O′ bond that accompanies this step occurs after the transition state, and this has been corroborated by IRC calculations. The high exothermicity of this last step (−71.7 kcal mol−1) results from the further opening of the fullerene cage and from the formation of very stable carbonyl and ester moieties at the seam of the opened cage. A noticeable feature of the potential energy surface is that once minimum D has been formed, the reaction to form product E has an energy barrier 4.5 kcal mol−1 lower than that of the back-reaction that returns intermediate D into C (towards epoxide product formation). Therefore, under thermal conditions, the forward reaction toward low-energy products will be considerably faster.
Harmonic frequency calculations of the stationary points of the low-energy reaction pathway shown in Fig. 5 can be used to provide further insight into the experimental results. As mentioned above, Davis et al.27 performed RAIRS analysis during the reaction between ozone and C60 fullerene under ultra-high vacuum conditions. Concomitant with the disappearance of RAIRS peaks characteristic of the POZ is the formation of an intense, broad infrared band characteristic of CO stretching in the 1750–1875 cm−1 region.32 This suggests the formation of carbonyl groups following the degradation of the primary ozonide. Of the B–E minima examined in this work, only those with carbonyl moieties yield theoretical vibrational modes with frequencies greater than 1650 cm−1: minimum B exhibits a CO stretching mode at 1731 cm−1, and product E shows similar stretching modes at 1710 cm−1 (ketone) and 1811 cm−1 (ester). The fact that the low-energy product E possesses vibrations that absorb IR wavelengths in the range measured by Davis et al. at long reaction times provides credence to the feasibility of the ozonolysis reaction pathway mapped in this paper.
Reactions with a second ozone molecule
Two additional pieces of experimental information are available from the work of Davis et al. First, at very long exposures (5000 L and beyond), in addition to the carbonyl fingerprints in the IR spectrum, a lower-intensity yet noticeable band appears at about 2200 cm−1. None of the calculated species described so far in this paper can account for that peak. Second, the rate at which ozone reacts with the C60 film becomes smaller at high exposures, indicating a substantial change in the C60 film that leads to reaction retardation. In addition, earlier solution experiments have described the appearance of an “ozopolymer” species at high ozone exposures.19 Taken together, these qualitative experimental results might suggest the degradation of the C60 cage to yield a stable, heavily oxidized substrate that does not react readily with additional ozone. The products of the ozonolysis reaction just described lead to an open-cage structure with an oxidized seam, with many available sites for subsequent reaction with additional ozone molecules. Since our calculations of multiple additions of ozone molecules to a single cage indicate that several ozone molecules can add through low barriers, an open question remains about the mechanism of further reactions that lead to further cage degradation.In the remainder of this paper, we describe a first attempt at characterizing those reactions by investigating the effect of the addition of a second molecule to the end product of the ozonolysis of C60 by one ozone molecule. Contrary to our assumption in the calculations presented in Fig. 4, we assume that the reaction of ozone to C60 leading to the ketoester product E is rapid and that a second ozone molecule does not add to the cage before product E is formed.
From the discussion above, it is clear that there is a large number of 6,6 sites on product E to which a second ozone molecule can add. Because we are interested in pathways that lead to a further opening of the cage, we present here calculations of a pathway that considers the addition of a second ozone molecule to a 6,6 bond belonging to the seam of the open cage. Obviously, this is just one the many possible pathways for the reaction of a second ozone molecule with an oxidized C60 cage. The full potential energy profile of the reaction is shown in Fig. 7, the key regions of the structures of the stationary points are in Fig. 8, and corresponding energetic data are given in Table 1.
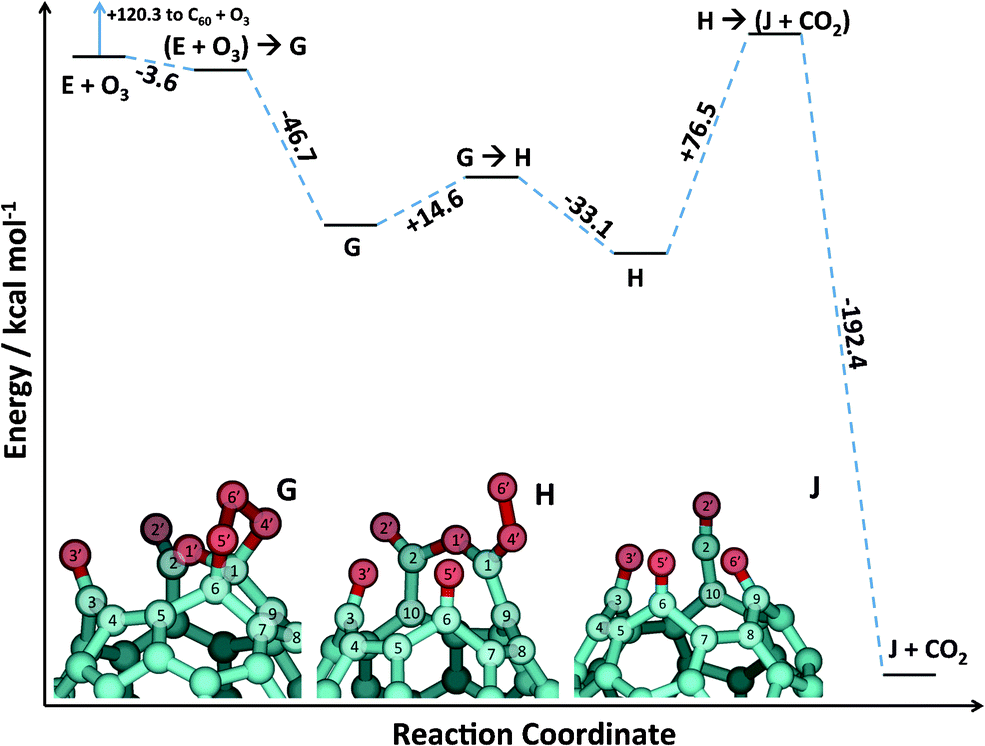 | ||
| Fig. 7 B3LYP/6-31G* stationary points on the reaction pathway for addition of a second ozone molecule to product E in Fig. 5 and 6. | ||
 | ||
| Fig. 8 Selected regions of optimum structures of minima E–J and transition states between them in the reaction pathway for addition of a second ozone molecule to product E in Fig. 7. | ||
In the specific pathway we follow here, the reaction starts with the addition of a second ozone molecule across the 1C6C bond adjacent to the 1C–1O′–2C moiety of product E. Similar to the initial formation of the C60 POZ, this reaction has a transition state below the reagents' asymptote. The addition of a second ozone molecule to product E leads to a POZ formation (G). Both the mechanism and the reaction energy (−50.3 kcal mol−1) are analogous to the ones described above for POZ formation on a pristine C60 cage.
Following the formation of the POZ species G, a concerted exothermic dissociation of the POZ, followed by the rotation of the 6O′–4O′ moiety of the resulting carbonyl oxide, yields species H. Since the original CC bond to which the second ozone molecule has added belonged to the seam of the open cage, this POZ dissociation leads to a further opening of the fullerene, evidenced by the increase in distance between 1C and 6C from 1.60 Å in minimum G to 2.05 Å at the transition state, and ultimately to 3.07 Å in minimum H.
From minimum H, a final complex step leads to product J with concomitant evolution of gaseous CO2. The step starts with a concerted breakage of the bond between 1C and 9C and the formation of a bond between 5O′ and 9C leading to the transition state (Fig. 8). This portion of the step leads to the formation of a furan ring at the transition state. After the transition state, several changes in bonding occur, which are shown in the IRC plot that connects this transition state to products J + CO2 (Fig. 9). (The energy values reported in Fig. 9 have not been ZPE-corrected.) The IRC calculation shows that following the transition state, the 1C4O′6O′ moiety rotates to allow for subsequent bond formation between 6O′ and 9C. Next, the bond is broken between oxygen atoms 4O′ and 6O′, followed by bond breakage between atoms 5O′ and 9C of the furan ring, and π-bond formation between 5O′ and 6C. The final portion of the step is the scission of the 1O′–2C bond that ultimately leads to gaseous CO2 accompanied by formation of a ketene moiety at the seam of the cage. To verify that none of these changes result in stable minima prior to products J + CO2, each structure depicted in Fig. 9 was subjected to geometry optimization, and each optimization yielded products J + CO2. Therefore, the mechanism shown in Fig. 9 is indeed continuously downhill.
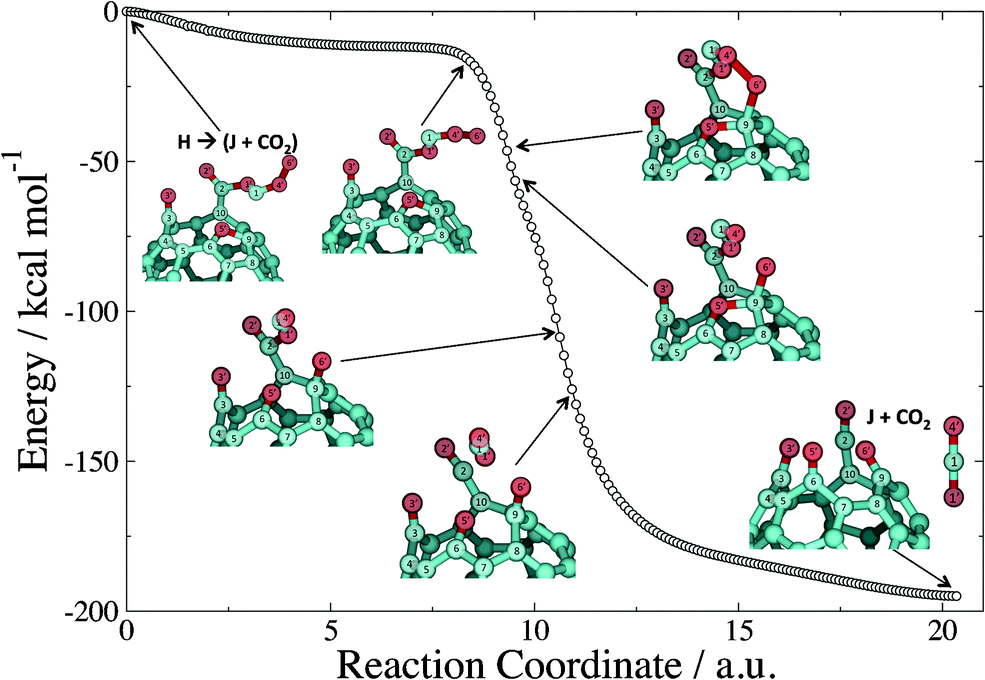 | ||
| Fig. 9 B3LYP/6-31G forward IRC calculation connecting the transition state of the H → J + CO2 step of Fig. 7 and 8 with products. | ||
While the formation of CO2 and stable carbonyl and ketene products at the seam of the cage leads to a tremendously exothermic last step in the mechanism (−115.9 kcal mol−1), the barrier for this step is quite large (76.5 kcal mol−1). In fact, this step is clearly the rate-limiting one, as its barrier is 7.7 kcal mol−1 above the E + O3 reagents' asymptote. The large barrier from H to J + CO2 might make it possible for other processes, including isomerization and addition of other ozone molecules, to occur, but these processes have not been explored in detail in this work. It is therefore unclear whether, under room-temperature conditions, mass loss of the cage occurs at any appreciable rate. While these calculations might trigger further experiments aimed at identifying cage degradation, we can probe the plausibility of product J by comparing calculated and experimental IR signals. As was the case for addition of the first ozone, very few theoretical vibrational modes with frequencies above 1650 cm−1 were found for the E → J + CO2 mechanism, and all of them are carbonyl stretching modes. Furthermore, the asymmetric ketene stretching mode of product J was the only one amongst all the minima with a frequency calculated above 2000 cm−1. This mode, with a theoretical frequency of 2106 cm−1, is in the region of the low-intensity but broad experimental peak around 2200 cm−1 obtained at high levels of ozone exposure. While we cannot unequivocally attribute product J to the experimental spectrum, the agreement between the calculated frequency for this stretch and the experimentally measured IR feature strongly suggests the formation of a ketene moiety. Although the mechanism for ketene formation in the experiment may differ from the one suggested here, this work can serve as a blueprint for future calculations that will explore cage-opening pathways that are possible when a second ozone molecule reacts with a CC bond different from the one considered here.
Concluding remarks
Fullerenes are currently being produced by the metric ton, yet little is known about the fate of these carbonaceous materials in the environment, where they are processed by atmospheric oxidizers. In contrast to the wealth of ozonation studies of C60 in solution, detailed gas-phase or gas-surface studies are more sparse, and until now have focused on characterizing the first intermediate in the reaction pathway, the primary ozonide, and the C60O epoxide + O2 products. Drawing motivation from very recent gas-surface UHV experiments, we have carried out an extensive investigation of the C60 + O3 reaction that transcends the formation of the primary ozonide and the C60O + O2 products using electronic structure methods. We have identified a decomposition pathway of the primary ozonide that is of substantially lower energy than the C60O + O2 products previously reported. This decomposition entails permanent breakage of the C–C bonds in the C60 cage that ultimately results in an open-cage structure. The seam of the open cage contains stable ketone and ester moieties, whose IR absorption is consistent with experiments. The new final product of the reaction of ozone with C60 is ∼100 kcal mol−1 more stable than the C60O + O2 products, which explains the lack of evidence for the latter products in the measured IR spectra of recent experiments. While studies in which the cage of fullerenes can be opened via solution chemistry abound,33 the work presented here serves to fill the gap for this type of studies beyond the solution phase.We have also learned that while ozonation of all 6,6 bonds in C60 is unlikely even under high-flux conditions, addition of a few ozone molecules to a single cage is exothermic and occurs over low barriers. With this in mind, we have explored one of the many possible reactions between the ketoester product of the C60 + O3 reaction and a second ozone molecule that adds to a CC double bond at the seam of the open cage. The dissociation of the primary ozonide of this addition ultimately leads to a further opening of the cage, mass loss due to CO2 evolution, and stable ketone and ketene products at the seam that are consistent with IR measurements. Solution studies of extensive ozonation have found evidence of an ozopolymer product whose formation has been postulated to entail the formation of covalent bonds between adjacent C60 cages. However, those studies have not been able to determine with certainty whether the fullerenes present an open-cage structure. While understanding the formation of bridges between C60 cages during ozonolysis is outside the scope of this paper, the calculations that we show here present pathways for low-energy open-cage structures that might intervene in the solution ozonation studies.
The ultimate proof of C60 cage opening due to ozonolysis will be provided by future experimental gas-surface studies. Nevertheless, the possibility that ozone can degrade a C60 cage stimulates investigation of the potential release of a range of materials that endohedral fullerenes are known to encapsulate, such as heavy metals.34 The release of an endohedral fullerene payload upon ozonolysis will be the subject of future theoretical studies.
Acknowledgements
The support of the National Science Foundation CHE-0547543 and CHE-0948293 is gratefully acknowledged. The authors also acknowledge Advanced Research Computing at Virginia Tech for providing computational resources and technical support that have contributed to the results reported within this paper. URL: http://www.arc.vt.edu.References
- M. B. Rubin, Helv. Chim. Acta, 2003, 86, 930 CrossRef CAS.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259 RSC.
- B. E. Krisyuk and A. V. Maiorov, Russ. J. Phys. Chem. B, 2011, 5, 790 CrossRef CAS.
- O. B. Gadzhiev, S. K. Ignatov, B. E. Krisyuk, A. V. Maiorov, S. Gangopadhyay and A. E. Masunov, J. Phys. Chem. A, 2012, 116, 10420 CrossRef CAS PubMed.
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745 CrossRef.
- E. J. Feltham, M. J. Almond, G. Marston, V. P. Ly and K. S. Wiltshire, Spectrochim. Acta, Part A, 2000, 56, 2605 CrossRef CAS.
- B. E. Coleman and B. S. Ault, J. Phys. Chem. A, 2010, 114, 12667 CrossRef CAS PubMed.
- Y.-T. Su, Y.-H. Huang, H. A. Witek and Y.-P. Lee, Science, 2013, 340, 174 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177 CrossRef CAS PubMed.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Chem. Phys., 2013, 138, 244307 CrossRef PubMed.
- J. M. Anglada, R. Crehuet and J. M. Bofill, Chem.–Eur. J., 1999, 5, 1809 CrossRef CAS.
- D. Johnson and G. Martson, Chem. Soc. Rev., 2008, 37, 699 RSC.
- N. M. Donahue, G. T. Drozd, S. A. Epstein, A. A. Presto and J. H. Kroll, Phys. Chem. Chem. Phys., 2011, 13, 10848 RSC.
- R. Atkinson, J. Phys. Chem. Ref. Data, 1997, 26, 215 CrossRef CAS PubMed.
- M. F. A. Hendrickx and C. Vinckier, J. Phys. Chem. A, 2003, 107, 7574 CrossRef CAS.
- S. N. Chu, S. Sands, M. R. Tomasik, P. S. Lee and V. F. McNeill, J. Am. Chem. Soc., 2010, 132, 15968 CrossRef CAS PubMed.
- F. Cataldo, Carbon, 2002, 40, 1457 CrossRef CAS.
- F. Cataldo, Fullerenes, Nanotubes, Carbon Nanostruct., 2003, 11, 1 CrossRef CAS PubMed.
- F. Cataldo and D. Heymann, Polym. Degrad. Stab., 2000, 70, 237 CrossRef CAS.
- J. P. Deng, C. Y. Mou and C. C. Han, Fullerenes Sci. Technol., 1997, 5, 1033 CrossRef CAS.
- D. Heymann and R. B. Weisman, C. R. Chim., 2006, 9, 1107 CrossRef CAS PubMed.
- Z. Shang, Y. Pan, Z. Cai, X. Zhao and A. Tang, J. Phys. Chem. A, 2000, 104, 1915 CrossRef CAS.
- Y.-S. Huang and G.-W. Wang, THEOCHEM, 2008, 860, 24 CrossRef CAS PubMed.
- D. S. Sabirov and R. G. Bulgakov, Comput. Theor. Chem., 2011, 963, 185 CrossRef CAS PubMed.
- D. S. Sabirov, S. L. Khursan and R. G. Bulgakov, J. Mol. Graphics Modell., 2008, 27, 124 CrossRef CAS PubMed.
- A. Bil, Z. Latajka and C. A. Morrison, J. Phys. Chem. A, 2009, 113, 9891 CrossRef CAS PubMed.
- E. D. Davis, A. Wagner, M. McEntee, M. Kaur, D. Troya and J. R. Morris, J. Phys. Chem. Lett., 2012, 3, 3193 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennuci and G. A. Petersson, et al., Gaussian 09, Revision A.1, 2009 Search PubMed.
- D. S. Sabirov, S. L. Khursan and R. G. Bulgakov, Russ. Chem. Bull., 2008, 57, 2520 CrossRef CAS.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, THEOCHEM, 1999, 462, 1 CrossRef.
- D. Cremer, J. Gauss, E. Kraka, J. F. Stanton and R. J. Bartlett, Chem. Phys. Lett., 1993, 209, 547 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, in The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, ed. D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, Academic Press, San Diego, 1991, p. 73 Search PubMed.
- G. C. Vougioukalakis, M. M. Roubelakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 39, 817 RSC.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp55212h |
| This journal is © the Owner Societies 2014 |