 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium coordination in protic ionic liquids†
S.
Menne
,
T.
Vogl
and
A.
Balducci
*
Westfälische Wilhelms-Universität, Institut für Physikalische Chemie-MEET, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: andrea.balducci@uni-muenster.de
First published on 27th January 2014
Abstract
The lithium ion–ion interactions in protic ionic liquids can be very different compared to those in aprotic ionic liquids. In this study we show that, for equal lithium ion concentration, the lithium coordination number in protic ionic liquids is lower than that in aprotic ones. This lower coordination makes lithium ions more “free” to move in protic ionic liquids and it might have an important consequence in the lithium mobility.
1. Introduction
Ionic liquids (ILs) are presently regarded as one of the most interesting chemical compounds.1,2 ILs display ionic character, negligible vapor pressure, high chemical and thermal stability. These properties make ILs very attractive for use in catalysis, as a solvent in synthetic processes and as a heat carrier in solar thermal energy generators.3–5 Moreover, because of their high ionic conductivity and large electrochemical stability, ILs are presently considered as promising electrolyte candidates for fuel cells, lithium batteries and electrochemical double-layer capacitors.6–12 As ILs display negligible vapor pressure, their introduction in these electrochemical storage devices would be extremely beneficial for the safety of these systems, which presently use flammable and volatile organic solvents.13–15 To date, however, the introduction of ILs in commercial devices has been hindered by the lower performance and higher costs of IL-based systems compared to that of systems containing organic solvents. In lithium-ion batteries (LIBs), for example, the higher viscosity of ILs compared to that of an organic-based electrolyte might dramatically reduce the lithium mobility and consequently the electrochemical performance of these devices, particularly when high current densities are applied. In order to overcome this limitation, and increase the lithium mobility in IL-based electrolytes, several strategies have been proposed in the last few years, e.g. modification of the cation side-chain and the use of mixtures of ILs and organic solvents.16,17Recently, we proposed the use of protic ionic liquids (PILs) as electrolytes for LIBs. We showed that the use of PILs makes possible the realization of LIBs displaying promising performance also at high current densities at room temperature.18 Taking into account these results, PILs can be regarded as an interesting class of IL-based electrolytes for LIBs. Additionally, since they are typically cheaper than AILs, their use could be helpful to overcome the cost limitation related to the use of the IL-based electrolytes. Nevertheless, in order to understand the advantages and the limitations of these electrolytes a deeper investigation of their chemical–physical properties is needed. Particularly, the lithium mobility in PIL-based electrolytes, which has never been studied, should be carefully investigated as it is a key factor for the development of advanced electrolytes for high performance LIBs.
Herein, we report for the first time an investigation on the lithium coordination in PIL-based electrolytes. Two PILs, pyrrolidinium bis(trifluoromethanesulfonyl)imide (PYRHHTFSI) and N-butylpyrrolidinium bis(trifluoromethanesulfonyl)imide (PYRH4TFSI), were synthesized and used in combination with the lithium salt lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) for the realization of electrolytes suitable for LIBs. With the aim to compare the behavior of PIL-based electrolytes with that of “conventional” aprotic ionic liquid (AIL) based electrolytes, electrolytes containing N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (PYR14TFSI) and LiTFSI were also considered. Initially, the conductivity and the viscosity at 30 °C of the IL-based electrolytes were determined. Afterward, the lithium coordination as well as the influence of the salt concentration on the lithium coordination of these IL-based electrolytes were investigated via Raman spectroscopy.
2. Experimental
Pyrrolidine (Aldrich >99%) and 1-butylpyrrolidine (Aldrich >98%) were distilled directly before use. HCl (37%), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), N-methylpyrrolidine (>97%) 1-iodobutane (>99%), and ethyl acetate (ACS grade, >99.5%) were used as received. The pyrrolidinium-based ILs, PYRHHTFSI and PYRH4TFSI, were synthesized following a procedure similar to that described elsewhere.11 PYR14TFSI was synthesized following the procedure described in ref. 16. At the end of the synthesis the obtained ILs were dried under vacuum at 60 °C. The water content of the ILs was measured using coulometric Karl-Fisher titration, and was found to be less than 10 ppm.Binary mixtures containing PYR14TFSI, PYRH4TFSI and PYRHHTFSI with a molar ratios of LiTFSI between 0.03 and 0.22 were prepared. All binary solutions displayed a water content less than 10 ppm.
The conductivity and the viscosity of the prepared solutions were determined as reported in ref. 17 and 18.
The Raman spectra were recorded using a RAM II FT-Raman module of a Bruker Vertex70 FT-IR spectrometer with a laser wavelength of 1064 nm. The collected spectra are the average of 512 scans at an optical resolution of 2 cm−1. The samples were sealed in glass ampules under vacuum and measured at room temperature. The resultant spectrum was fit with the multipeak fitting package in IGOR PRO 6.22A using a Voigt function with a fixed Lorentzian![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Gaussian ratio.
:Gaussian ratio.
3. Results and discussion
Table 1 presents the conductivity and the viscosity at 30 °C of several electrolytes containing PYR14TFSI, PYRH4TFSI and PYRHHTFSI, with LiTFSI. In these binary mixtures the molar ratio of LiTFSI was between 0.03 and 0.22 (the values of conductivity and viscosity of other binary mixtures are reported in the ESI†). It is important to notice that the presence of water might have dramatic effects on the physical–chemical properties of ILs.1,2 As indicated above, all ILs and binary solutions investigated in this work display comparable water content (less than 10 ppm) and therefore their properties can be safely compared.As shown in the table, when dry PILs are considered, a marked difference in terms of ionic conductivity between PILs and AILs cannot be observed.18 For example, the solution containing PYRHHTFSI, PYRH4TFSI and PYR14TFSI and having a molar ratio of LiTFSI equal to 0.03 displayed a conductivity of 3.6, 3.5 and 3.3 mS cm−1, respectively. For all binary mixtures an increase of the molar ratio of LiTFSI led to a decrease of conductivity. As shown, the viscosity of all binary solutions was also rather comparable. Nevertheless, it is interesting to note that the solution containing PYRHHTFSI displayed higher viscosity compared to the one containing PYRH4TFSI and PYR14TFSI. Taking into account the conductivities of the electrolytes, this difference appears somehow surprising. This behavior might have originated due to the different sizes of the cations of the ILs as well as their different interaction with the TFSI anion. The PYRHH+ cation is smaller than the other cations. At the same time, as it is less shielded, the cation PYRHH+ is probably subjected to a stronger interaction with the anion TFSI− compared to the cation PYRH4+ and PYR14+, and this interaction might explain the higher viscosity of the electrolyte containing PYRHHTFSI compared to that containing PYRH4TFSI and PYR14TFSI. Nevertheless, in order to understand such behavior further investigations (in progress) appear necessary. For all binary solutions an increase of the molar ratio of LiTFSI leads to an increase of the viscosity.
In ionic liquids containing the TFSI anion, the peak located at wavenumbers around 742 cm−1 in the Raman spectra is typically considered one of the most suitable to investigate the TFSI− environment.19,20 Such a peak is associated to contraction/expansion of the whole TFSI− anion as well as to the presence of a transoid (C2) and cisoid (C1) conformation in the liquid state19,21,22 and this distinct mode is very sensitive to the interaction of the TFSI− anion with the surrounding ions, e.g. lithium.
In the past years, the term “free” was commonly used to describe (lithium) non-coordinating TFSI− anions. This term was used because it is assumed that no interactions, except the urge for electroneutrality, between cations and TFSI− anions are present in ILs. A comparison of the peak wavenumbers available in the literature assigned to contraction/expansion modes of “free” TFSI− anions of several aprotic ILs indicates that the type of cation as well as the length and chemical composition of its side chains do not have a strong influence on the peak wavenumbers, which is for ILs at around 742 cm−1 (see Table in S2, ESI†).23–28 It is worth mentioning, however, that in the literature almost no studies have reported investigation on the “free” TFSI− in protic ionic liquids. To the best of our knowledge only Martinelli et al.22 have reported a study about the cis-/trans-isomerism of the TFSI− anion in protic as well as aprotic ionic liquids in which they showed a comparable conformer ratio in both types of ILs.
Fig. 1 compares the peak of the “free” TFSI− as observed in the pure ILs PYR14TFSI, PYRH4TFSI and PYRHHTFSI. As shown, the peak wavenumber of PYR14TFSI was found at around 742 cm−1. This value is in accordance with those reported in the literature. Interestingly, the peak wavenumbers of PYRH4TFSI and PYRHHTFSI shifted to higher values of 744 cm−1 and 745 cm−1, respectively. Most likely, such a shift was caused by the withdrawal of electron density from the TFSI− anion, which could be generated by the less shielded positive charge (compared to AILs) of the PIL-cations and/or by the presence of hydrogen bonds. Considering these results, it appears reasonable to suppose that the presence of a proton in the cation of ILs might have an impact on the ion–ion interactions of ILs and it can generate differences in terms of the TFSI− environment between PILs and AILs. Taking into account this point, in the following we will use the term “lithium non-coordinating TFSI− anions” (TFSInon−) instead of the commonly used “free”.
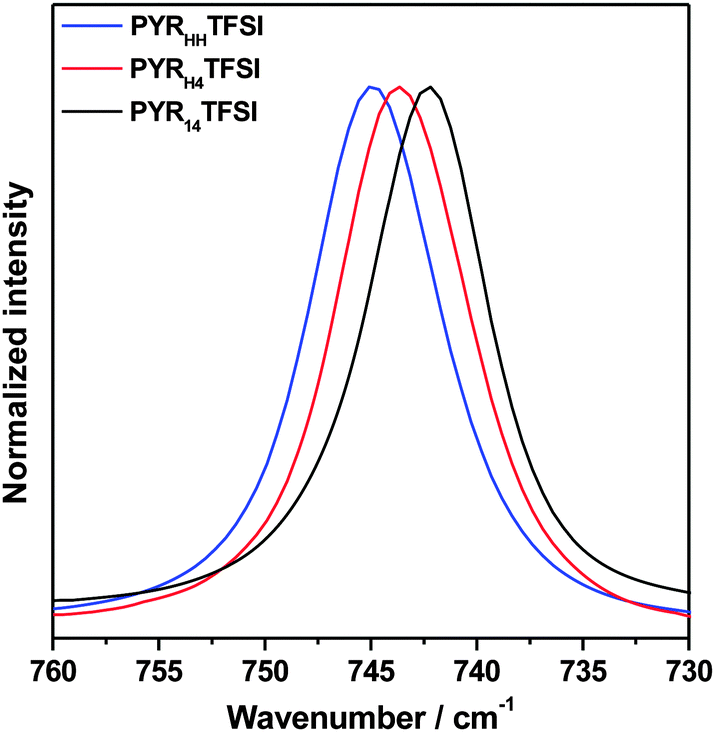 | ||
| Fig. 1 Raman mode of the TFSI− anions in the ionic liquids PYR14TFSI, PYRH4TFSI and PYRHHTFSI. | ||
When lithium ions are introduced in TFSI-based ILs, the TFSI− anions coordinating with lithium (TFSIcoor−) generate an additional signal, which is shifted to higher wavenumbers in the Raman spectra. The comparison of the area of this additional signal with that of the peak at around 742 cm−1 can be used to determine the lithium coordination of these binary systems.23,24Fig. 2 shows the peak fittings for TFSInon− and TFSIcoor− in electrolytes containing PYR14TFSI, PYRH4TFSI and PYRHHTFSI having a molar ratio of 0.22 LiTFSI. As shown, in the case of the aprotic PYR14TFSI, the additional signal for the Li coordinating TFSI− anions can be clearly seen in the shoulder at around 748 cm−1 (Fig. 2A). Such a shoulder was already reported for electrolytes containing PYR14TFSI.23,24 In the case of the two PIL-based electrolytes, due to the shift of the TFSInon peak to higher wavenumbers (see Fig. 1), such a distinct shoulder cannot be observed. Instead, a broadening and asymmetric deformation of the peak (to higher wavenumbers) was observed for both electrolytes (Fig. 2B and C). Despite this difference, for all investigated electrolytes it was possible to fit the areas assigned to coordinating and non-coordinating TFSI anions and consequently to determine the TFSIcoor ratio in the ILs.
 | ||
| Fig. 2 Raman mode of lithium non-coordinating TFSI− anions and lithium coordinating TFSI− anions in binary solutions containing LiTFSI and PYR14TFSI (A), LiTFSI and PYRH4TFSI (B) and LiTFSI and PYRHHTFSI (C). In all solutions the molar ratio of LiTFSI was equal to 0.22. | ||
Fig. 3A shows the averaged ratios of TFSIcoorversus the molar fraction of LiTFSI in the ILs. In all electrolytes the amount of coordinating TFSI anions increases with increasing amounts of LiTFSI. However, it is important to notice that in the case of low molar fractions of LiTFSI (0.03) the TFSI− anion coordinated to lithium in PYR14TFSI is one order of magnitude higher than that coordinated in the protic PYRH4TFSI and PYRHHTFSI. With increasing molar fraction of LiTFSI the relative amount of TFSIcoor− increases linearly for all electrolytes. Here, PYR14TFSI shows the stiffest slope, followed by PYRH4TFSI and finally by PYRHHTFSI.
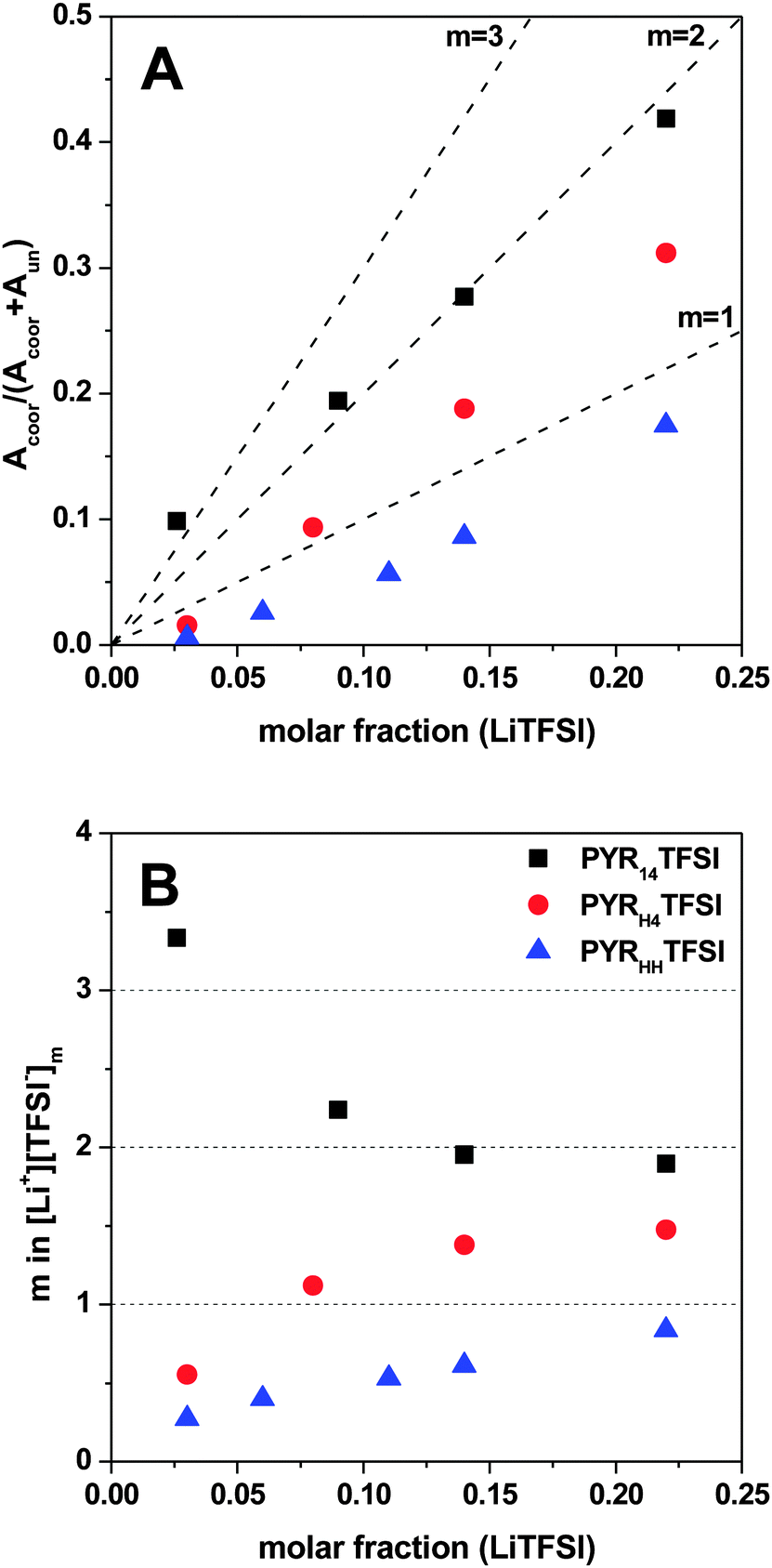 | ||
| Fig. 3 Ratio of lithium coordinated TFSI− anions versus molar fraction of LiTFSI (A) and the lithium coordination number (m) versus molar fraction of LiTFSI (B) in PYR14TFSI, PYRH4TFSI and PYRHHTFSI. The dashed lines represent the lithium coordination number m as a guide to the eye. | ||
A better understanding of the ion–ion interplay in these salt mixtures can be achieved by plotting the Li+ coordination number, m, versus the molar fraction of LiTFSI. Fig. 3B shows that the trend observed for PYR14TFSI is completely different with respect to the one observed for PYRH4TFSI and PYRHHTFSI. As a matter of fact, in the case of PYR14TFSI the lithium coordination number decreases when the molar fraction of LiTFSI increases. In contrast, when the molar fraction of LiTFSI increases, the lithium coordination number for the protic ionic liquids PYRH4TFSI and PYRHHTFSI increases.
4. Conclusions
The results of this investigation show that the lithium ion coordination in PIL-based electrolytes can be extremely different compared to that in AIL-based electrolytes. This difference can be mainly attributed to the different ion–ion interactions occurring in these two types of ILs. In AILs, due to sterical shielding of the IL cations, the interactions between the cation and TFSI anions are very weak.22–24 In PILs, on the other hand, interactions between the ions of ILs take place due to the less shielded (positive) charge of the cation. When a lithium salt (in the case of this work, LiTFSI) is introduced into these ILs, this different ion–ion interaction has an important consequence in the lithium coordination. In the case of AILs, the TFSI− ions preferentially coordinate to the lithium ions and, due to thermodynamics, when the concentration of the lithium inside the electrolytes increases such coordination decreases. In the case of PILs, due to the presence of interactions between the cation of the PIL and the anion, the TFSI− ions are coordinated by both, the lithium ions and the cation of the IL. Therefore, in this type of ILs a sort of “competition” for the coordination of the TFSI− anion occurs between these two positively charged ions. Here, when the lithium concentration is increased, the coordination number of lithium increases, too. It is important to notice that as a consequence of the “competition” between cation and lithium ions, in the case of equal lithium ion concentration, the coordination number of lithium in PIL-based electrolytes is always lower than that of AIL-based electrolytes. This lower coordination makes lithium ions more “free” to move in PIL-based electrolytes than in AIL-based electrolytes and it might have an important consequence in the lithium mobility. As mentioned above, the low lithium mobility in IL-based electrolytes is one of the main drawbacks holding the introduction of these safe electrolytes in LIBs. Taking into account the results of this study, although the conductivity and viscosity of AILs and (dry) PILs appear comparable, the use of ILs containing low-shielded cations, like PILs, might be regarded as a promising strategy to improve the lithium mobility of IL-based electrolytes for LIBs.Acknowledgements
The authors wish to thank the University of Münster, the Ministry of Innovation, Science and Research of North Rhine-Westphalia (MIWF) within the project “Superkondensatoren und Lithium-Ionen-Hybrid-Superkondensatoren auf der Basis ionischer Flüssigkeiten” for the financial support.Notes and references
- C. A. Angell, N. Byrne and J.-P. Belieres, Acc. Chem. Res., 2007, 40, 1228–1236 CrossRef CAS PubMed.
- P. Wasserscheid, Handbook of Green Chemistry, Volume 6: Ionic Liquids Handbook of Green Chemistry, 2010 Search PubMed.
- X. Chen, R. Liu, Y. Xu and G. Zou, Tetrahedron, 2012, 68, 4813–4819 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2008, 37, 1709–1726 RSC.
- M. E. V. Valkenburg, R. L. Vaughn, M. Williams and J. S. Wilkes, Thermochim. Acta, 2005, 425, 181–188 CrossRef.
- F. Endres, MRS Bull., 2013, 38, 567–571 CrossRef CAS.
- L. J. Hardwick, M. Holzapfel, A. Wokaun and P. Novák, J. Raman Spectrosc., 2007, 38, 110–112 CrossRef CAS.
- C. Largeot, P. L. Taberna, Y. Gogotsi and P. Simon, Electrochem. Solid-State Lett., 2011, 14, A174–A176 CrossRef CAS.
- M. Lazzari, M. Mastragostino, A. G. Pandolfo, V. Ruiz and F. Soavi, J. Electrochem. Soc., 2011, 158, A22–A25 CrossRef CAS.
- B. H. R. Suryanto, C. A. Gunawan, X. Lu and C. Zhao, Electrochim. Acta, 2012, 81, 98–105 CrossRef CAS.
- L. Timperman, P. Skowron, A. Boisset, H. Galiano, D. Lemordant, E. Frackowiak, F. Beguin and M. Anouti, Phys. Chem. Chem. Phys., 2012, 14, 8199–8207 RSC.
- T. Yasuda and M. Watanabe, MRS Bull., 2013, 38, 560–566 CrossRef CAS.
- A. Guerfi, M. Dontigny, P. Charest, M. Petitclerc, M. Lagacé, A. Vijh and K. Zaghib, J. Power Sources, 2010, 195, 845–852 CrossRef CAS.
- Y. Kai, A. Jin Jing and C. Shi, J. Therm. Anal. Calorim., 2010, 99, 515–521 CrossRef.
- H. Nakagawa, J. Power Sources, 2007, 174, 1021 CrossRef CAS.
- M. Kunze, E. Paillard, S. Jeong, G. B. Appetecchi, M. Schönhoff, M. Winter and S. Passerini, J. Phys. Chem. C, 2011, 115, 19431–19436 CAS.
- S. Menne, R. S. Kühnel and A. Balducci, Electrochim. Acta, 2013, 90, 641–648 CrossRef CAS.
- S. Menne, J. Pires, M. Anouti and A. Balducci, Electrochem. Commun., 2013, 31, 39–41 CrossRef CAS.
- D. Brouillette, D. E. Irish, N. J. Taylor, G. Perron, M. Odziemkowski and J. E. Desnoyers, Phys. Chem. Chem. Phys., 2002, 4, 6063–6071 RSC.
- I. Rey, P. Johansson, J. Lindgren, J. C. Lassègues, J. Grondin and L. Servant, J. Phys. Chem. A, 1998, 102, 3249–3258 CrossRef CAS.
- M. Herstedt, M. Smirnov, P. Johansson, M. Chami, J. Grondin, L. Servant and J. C. Lassègues, J. Raman Spectrosc., 2005, 36, 762–770 CrossRef CAS.
- A. Martinelli, A. Matic, P. Johansson, P. Jacobsson, L. Börjesson, A. Fernicola, S. Panero, B. Scrosati and H. Ohno, J. Raman Spectrosc., 2011, 42, 522–528 CrossRef CAS.
- J.-C. Lassègues, J. Grondin, C. Aupetit and P. Johansson, J. Phys. Chem. A, 2008, 113, 305–314 CrossRef PubMed.
- M. Kunze, S. Kunze, E. Jeong, M. Paillard, M. Schönhoff, S. Winter and M. Passerini, Adv. Energy Mater., 2011, 1, 274–281 CrossRef CAS.
- G. B. Appetecchi, S. Scaccia, C. Tizzani, F. Alessandrini and S. Passerini, J. Electrochem. Soc., 2006, 153, A1685–A1691 CrossRef CAS.
- S. Duluard, J. Grondin, J.-L. Bruneel, I. Pianet, A. Grélard, G. Campet, M.-H. Delville and J.-C. Lassègues, J. Raman Spectrosc., 2008, 39, 627–632 CrossRef CAS.
- J.-C. Lassegues, J. Grondin and D. Talaga, Phys. Chem. Chem. Phys., 2006, 8, 5629–5632 RSC.
- M. Castriota, T. Caruso, R. G. Agostino, E. Cazzanelli, W. A. Henderson and S. Passerini, J. Phys. Chem. A, 2004, 109, 92–96 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp55183k |
| This journal is © the Owner Societies 2014 |