 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards a structure-based exciton Hamiltonian for the CP29 antenna of photosystem II†
Frank
Müh
*,
Dominik
Lindorfer
,
Marcel
Schmidt am Busch
and
Thomas
Renger
Institute for Theoretical Physics, Johannes Kepler University Linz, Altenberger Str. 69, 4040 Linz, Austria. E-mail: frank.mueh@jku.at
First published on 17th February 2014
Abstract
The exciton Hamiltonian pertaining to the first excited states of chlorophyll (Chl) a and b pigments in the minor light-harvesting complex CP29 of plant photosystem II is determined based on the recent crystal structure at 2.8 Å resolution applying a combined quantum chemical/electrostatic approach as used earlier for the major light-harvesting complex LHCII. Two electrostatic methods for the calculation of the local transition energies (site energies), referred to as the Poisson–Boltzmann/quantum chemical (PBQC) and charge density coupling (CDC) method, which differ in the way the polarizable environment of the pigments is described, are compared and found to yield comparable results, when tested against fits of measured optical spectra (linear absorption, linear dichroism, circular dichroism, and fluorescence). The crystal structure shows a Chl a/b ratio of 2.25, whereas a ratio between 2.25 and 3.0 can be estimated from the simulation of experimental spectra. Thus, it is possible that up to one Chl b is lost in CP29 samples. The lowest site energy is found to be located at Chl a604 close to neoxanthin. This assignment is confirmed by the simulation of wild-type-minus-mutant difference spectra of reconstituted CP29, where a tyrosine residue next to Chl a604 is modified in the mutant. Nonetheless, the terminal emitter domain (TED), i.e. the pigments contributing mostly to the lowest exciton state, is found at the Chl a611–a612–a615 trimer due to strong excitonic coupling between these pigments, with the largest contributions from Chls a611 and a612. A major difference between CP29 and LHCII is that Chl a610 is not the energy sink in CP29, which is presumably to a large extent due to the replacement of a lysine residue with alanine close to the TED.
1 Introduction
Life on earth is powered by the sun owing to the action of photosynthetic organisms which developed the ability to convert solar energy efficiently into chemical energy. Most of the biomass is produced by oxygenic photosynthesis, where water is the ultimate electron source and dioxygen is released into the atmosphere.1 In higher plants, the relevant pigment–protein complexes (PPCs) involved in solar energy conversion are embedded in the thylakoid membrane.2,3 Here, photosystem I (PSI), the cytochrome b6f complex and photosystem II (PSII) operate in series with the light-induced charge separation performed by the two photosystems. At one end of the chain is PSII, the site of oxidative water cleavage, and at the other end is PSI, from where the electrons are delivered to enzymes synthesizing nicotinamide adenine dinucleotide phosphate (NADPH). In general, photosynthetic PPCs can be divided into antenna proteins or light-harvesting complexes (LHCs) and reaction centers (RCs). The former absorb light and transfer the excitation energy to the latter, where it is used to separate charges.4 Both PSI5,6 and PSII7 contain a core antenna at a fixed ratio of antenna to RC pigments. Besides, there are outer antenna proteins that further increase the effective absorption cross section of the RC and allow for the regulation of excitation energy transfer (EET).8–11Plant PSII is largely organized in supercomplexes comprising besides the PSII core complex (PSIIcc) the major light-harvesting complex LHCII and the minor complexes Lhcb4–6 (CP29, CP26, and CP24, respectively).12 In order to understand the EET in such a supercomplex, it is necessary to know the excited state energy levels of the involved pigments, in particular, the chlorophylls. Although the optical spectra of PPCs contain information about these energy levels, it is usually impossible to determine these unambiguously. Rather, information from structural biology has to be taken into account. However, linking structure and optical spectra of PPCs is a difficult task. Nonetheless, progress has been made in recent years by combining quantum chemistry of pigments in vacuo with classical electrostatic computations.13,14 A prerequisite for an application of these methods is that a crystal structure of the PPC is available, which according to our experience, should have a resolution better than about 3 Å. This is currently the case for PSIIcc of cyanobacteria15–18 as well as LHCII19–21 and CP2922 of higher plants. In the present work, we focus on CP29 and a comparison of CP29 with LHCII, on which such calculations have been performed earlier.23–25
According to the crystallographic work by Pan et al.,22 the apoprotein of CP29 follows the general fold expected for a membrane protein of the Lhc family (Fig. 1). The amino acid sequence was traced from Gly 88 to Leu 243, but the extended N-terminus of CP29 is missing. This is due to partial degradation in the process of crystallization as well as due to its inherent flexibility.22 The latter is also evident from spin-labeling studies.26 Based on the electron density map, 13 chlorophyll (Chl) binding sites were assigned in CP29. Of these, nine are occupied with Chl a and four with Chl b according to PDB file 3PL9 as indicated in Fig. 1 and Table 1. This assignment results in a Chl a/b ratio of Ra/b = 2.25. The Chl numbering follows that of analogous Chls in LHCII according to Liu et al.20 (Table 1). Note that there is no counterpart of Chl a615 in LHCII, while Chls b601 and b605 are not found in CP29. Furthermore, the analogs of Chls a609 and b614 in CP29 are Chl b and Chl a, respectively, in LHCII. From the crystallographic side, there remains an uncertainty as to whether the site 610 in CP29 is occupied with Chl a or b. Assigning it to Chl b results in Ra/b = 1.6, whereas assuming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixed occupancy yields Ra/b = 1.89. The latter value is close to the experimentally determined ratio of Ra/b = 1.8 determined from redissolved crystals, while the original preparations used by Pan et al. for crystallization showed Ra/b = 2.3.22 This has to be compared to ratios varying between 2.3 and 3.4 reported in the literature for various optical samples.27–32 In addition, the crystal structure of CP29 contains three different carotenoid pigments, lutein (Lut), violaxanthin (Vio) and neoxanthin (Neo), as well as one glyceraldehyde 3-phosphate (G3P) that is sandwiched by the two Chls a611 and a615. G3P, also known as triose phosphate, is an intermediate in the Calvin–Benson cycle,33 and its assignment in the crystal structure is tentative.22
:1 mixed occupancy yields Ra/b = 1.89. The latter value is close to the experimentally determined ratio of Ra/b = 1.8 determined from redissolved crystals, while the original preparations used by Pan et al. for crystallization showed Ra/b = 2.3.22 This has to be compared to ratios varying between 2.3 and 3.4 reported in the literature for various optical samples.27–32 In addition, the crystal structure of CP29 contains three different carotenoid pigments, lutein (Lut), violaxanthin (Vio) and neoxanthin (Neo), as well as one glyceraldehyde 3-phosphate (G3P) that is sandwiched by the two Chls a611 and a615. G3P, also known as triose phosphate, is an intermediate in the Calvin–Benson cycle,33 and its assignment in the crystal structure is tentative.22
 | ||
| Fig. 1 (A) Arrangement of π-skeletons of Chl a (green) and Chl b (blue) in CP29 relative to the protein backbone (cyan), viewed along the membrane plane. Helices are shown as columns. The Chl numbering is according to Pan et al.22 The figure is made using VMD.71 (B) Schematic representation of the location of Chl a (green) and Chl b (blue) pigments with their axial ligands (black arrows) relative to the major helices (cyan) of CP29. The Chl numbering is according to Pan et al.,22 while two numbering schemes are given for the major helices. Axial ligands are labeled as are amino acid residues in the beginning or end of a helix (italics). Chls without explicitly shown axial ligands are ligated by G3P (a611, a615) or water (a604, b606, b607, b608). N and C label the amino- and carboxy-terminus, respectively, where the dashed blue line indicates that the N-terminus is missing up to Gly 88 in the crystal structure. | ||
| m | CP29 | LHCII | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Chl #a | Chl type | E m/cm−1 (PBQC)b,c | E m/cm−1 (CDC)b,d | E m/cm−1 (fit)b | Chl #e | Chl type | E m/cm−1 (PBQC)c,f | E m/cm−1 (fit)f | |
a PDB 3PL9, Pan et al.22
b
E
(a)0 = 14900 cm−1 (671 nm), E(b)0 = 15674 cm−1 (638 nm).
c
![[small epsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0de.gif) p = 1.8, εmem = 2.0, εsolv = 5.0, Chl charge sets from HF-CIS.
d
ε
eff = 2.0, Chl charge sets from HF-CIS.
e PDB 1RWT, Liu et al.20
f
E
(a)0 = 14900 cm−1 (671 nm), E(b)0 = 15385 cm−1 (650 nm); data from ref. 25.
p = 1.8, εmem = 2.0, εsolv = 5.0, Chl charge sets from HF-CIS.
d
ε
eff = 2.0, Chl charge sets from HF-CIS.
e PDB 1RWT, Liu et al.20
f
E
(a)0 = 14900 cm−1 (671 nm), E(b)0 = 15385 cm−1 (650 nm); data from ref. 25.
|
|||||||||
| 1 | — | — | — | — | — | 601 | b | 15319 |
15405 |
| 2 | 602 | a | 14777 |
14828 |
14980 |
602 | a | 14930 |
14940 |
| 3 | 603 | a | 14885 |
14973 |
14900 |
603 | a | 14871 |
14850 |
| 4 | 604 | a | 14827 |
14816 |
14810 |
604 | a | 14868 |
14820 |
| 5 | — | — | — | — | — | 605 | b | 15497 |
15465 |
| 6 | 606 | b | 15626 |
15699 |
15684 |
606 | b | 15365 |
15385 |
| 7 | 607 | b | 15649 |
15671 |
15439 |
607 | b | 15347 |
15225 |
| 8 | 608 | b | 15439 |
15464 |
15439 |
608 | b | 15186 |
15215 |
| 9 | 609 | a | 14935 |
14964 |
14980 |
609 | b | 15423 |
15475 |
| 10 | 610 | a | 14897 |
14984 |
14920 |
610 | a | 14786 |
14790 |
| 11 | 611 | a | 14756 |
14839 |
14850 |
611 | a | 14930 |
14950 |
| 12 | 612 | a | 14898 |
14971 |
14900 |
612 | a | 14924 |
14940 |
| 13 | 613 | a | 14847 |
14891 |
14880 |
613 | a | 14858 |
14840 |
| 14 | 614 | b | 15682 |
15735 |
15674 |
614 | a | 14904 |
14940 |
| 15 | 615 | a | 14919 |
14922 |
14940 |
— | — | — | — |
Theoretical modeling is pivotal to establishing structure–function relationships of PPCs as exemplified through a recent study of PSII supercomplexes34 (see also the references therein). The central quantity in these models is the exciton Hamiltonian that, when represented as a matrix in the basis of localized first excited states of the PPC, contains the excitonic couplings between QY (S0 → S1) transitions of the Chls in the off-diagonal and the site energies (local QY transition energies in the absence of excitonic couplings) in the diagonal. Herein, we describe our efforts to determine the exciton Hamiltonian of CP29 based on the crystal structure by Pan et al.22 The work is organized as follows: first, we describe theory and computational methods. Then, we present the results of our computation of excitonic couplings and site energies. For the latter, we compare two different electrostatic approaches referred to as the Poisson–Boltzmann/quantum chemical (PBQC) method applied earlier to the Fenna–Matthews–Olson (FMO) protein,35,36 LHCII,23 and the CP43 core antenna of PSII,37 and the charge density coupling (CDC) method applied previously to FMO36,38 and cyanobacterial PSI.39 We note that a direct comparison of the two methods under matchable conditions (i.e., with the PPC having the same protonation pattern) has not been performed before. The calculated site energies are evaluated by comparison with experimental absorbance, linear dichroism (LD), circular dichroism (CD), and fluorescence spectra of native CP29 as well as wild-type-minus-mutant difference (WMD) spectra of reconstituted CP29. Further, we determine the exciton levels of CP29 and the contributions of pigments to the exciton states, identify the location of the lowest exciton state referred to as “terminal emitter domain” (TED) and compare with LHCII. In the discussion, we address the question of pigment composition in conjunction with the possibility to map out the exciton Hamiltonian by combining the quantum chemical/electrostatic approach with site-directed mutagenesis, the degree of conservation of the TED in CP29 and LHCII40 and implications of our results for the orientation of CP29 within the supercomplex.
2 Theory and computational methods
2.1 Protein structure and electrostatic modeling
The calculations are based on the crystal structure of CP29 from spinach (Spinacia oleracea) at 2.8 Å resolution by Pan et al.22 (PDB entry 3PL9). The pseudo-N-terminus at Gly 88, which is the first modeled residue in the amino acid sequence, is capped with an acetyl group. Besides the protein and the cofactors introduced above, six water molecules are modeled explicitly, because they are involved in the axial ligation of Chls. Specifically, H2O molecules are axial ligands to Chls a604, b606, b607, and b608. In the case of Chl b607, the axial H2O forms a hydrogen bond with the 7-formyl group of Chl b606. The axial ligand of Chl b606 forms hydrogen bonds with the side chain carboxyl group of Glu 151 and with another water molecule that in turn is hydrogen bonded to the backbone carbonyl group of Leu 147. In the case of Chl b608, the axial ligand forms a hydrogen bond with another water molecule that in turn is hydrogen bonded to the 133-methylester carbonyl group of Chl b608 and to the side chain carboxyl group of Glu 159.In the electrostatic calculations, protein and cofactors are modeled as a set of atomic partial charges embedded in a dielectric medium with the positions of heavy atoms taken from the crystal structure and those of hydrogen atoms inferred from molecular modeling with CHARMM.41,42 Atomic partial charges of the protein are taken from the CHARMM22 force field,43 while those of cofactors are the same as used in our earlier work on LHCII.23 To model the dielectric properties of the thylakoid membrane (or that of a detergent belt in the vicinity of the protein) in the framework of Poisson–Boltzmann calculations (see below), we divide space into four regions with three different (static) dielectric constants as described earlier.13,23
2.2 Protonation states
The calculation of protonation states of titratable groups in CP29 follows the same procedures as described earlier for LHCII.13,23 The experimental spectra that we are simulating here were measured on buffered samples containing 10–20 mM 4-(2-hydroxymethyl)-1-piperazineethanesulfonic acid (HEPES) at pH 7.5 and either glycerol or sucrose as a glass-forming agent.27,40,44 Cooling causes a pH increase of 0.01 pH K−1 for HEPES.45 At about 210 K, the protonation equilibria are frozen in,46 so that the proton activity in cryo-samples below the glass transition temperature corresponds to pH ≈ 8.4. The Monte-Carlo titration of protonation states47 was carried out at T = 210 K for cryo-samples. A suitable set of dielectric constants for the Poisson–Boltzmann calculations with TAPBS48 under these conditions is εp = 4.0 for the protein, εmem = 2.0 for the membrane slab, and εsolv = 80 for the outer medium.23,392.3 Site energies
The site energies (local transition energies) of the S0 → S1 transition of Chl a and Chl b pigments in CP29 were calculated with two different methods. In the PBQC method, the calculations are performed as described earlier for LHCII23 using TAPBS48 and the different static dielectric constantsp = 1.8 for the protein, εmem = 2.0 for the membrane slab, and εsolv = 5.0 for the outer medium in cryo-samples below the glass transition temperature. The dielectric constants used for the site energy calculations differ from those used for the calculation of protonation states (see above). The reasons are (i) that the medium is in a different state (below the glass transition temperature, hence a change of εsolv) and (ii) that the protein dielectric constant p is chosen in such a way as to optimize simulated optical spectra and, therefore, does not solely represent the static polarizability of the protein, as discussed in detail earlier.13
The site energy of Chl(a/b) in site m of CP29 is given as
| Em = E(a/b)0 + ΔEm | (1) |
Here, E(a)0 and E(b)0 are two free parameters that are determined from a comparison of simulated and measured spectra and
| ΔEm = ΔGsp,m(S1) − ΔGsp,m(S0) | (2) |
In the CDC method13,36,38,39
 | (3) |
p in PBQC and εeff in CDC account for the dielectric properties of the medium and also serve to compensate for possible deficiencies of the quantum-chemically calculated atomic partial charges of the pigments.
2.4 Excitonic couplings
The excitonic couplings between QY transitions of Chls in CP29 are calculated on the basis of the Chl coordinates from the crystal structure22 by using the Poisson-TrEsp method introduced earlier.36,39 In this method, the transition density of the S0 → S1 transition of each Chl is represented by atomic partial charges (transition charges) q(m)I(0,1) as described.13,14,39,53,54 These charges are based on quantum chemical calculations using either TD-DFT with the B3LYP XC-functional or HF-CIS and are rescaled according to![[q with combining tilde]](https://www.rsc.org/images/entities/i_char_0071_0303.gif) (m)I(0,1) = χq(m)I(0,1) (m)I(0,1) = χq(m)I(0,1) | (4) |
The correction factor χ is chosen such that the dipole strength obtained from the first moment of the transition charges matches the experimental value for vacuum. Since the latter cannot be measured directly for Chls, we use values for the dipole strengths of 21.0 D2 for Chl a and 14.7 D2 for Chl b as determined in an empty cavity analysis by Knox and Spring55 from absorbance data in different solvents. The dielectric environment of the Chls influencing the excitonic couplings is modeled as shown elsewhere for PSI,39,54 where the Chls are represented by vacuum cavities in a homogeneous medium with optical dielectric constant εopt = n2 and refractive index n. A value of εopt = 2.0 is used, which is in the range estimated51 on the basis of the integrated dipole strength of protein-bound and solvent-extracted Chl a.56 To obtain the excitonic coupling Vmn between pigments m and n, the Poisson equation is solved numerically using MEAD57 with the charge distribution given by the rescaled transition charges as
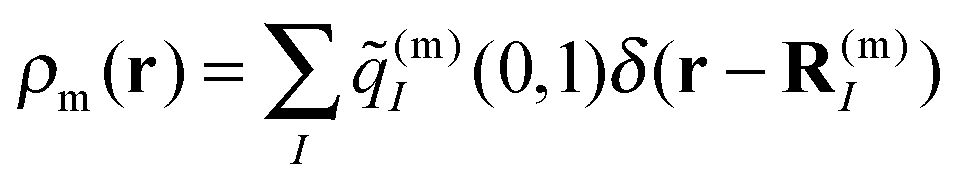 | (5) |
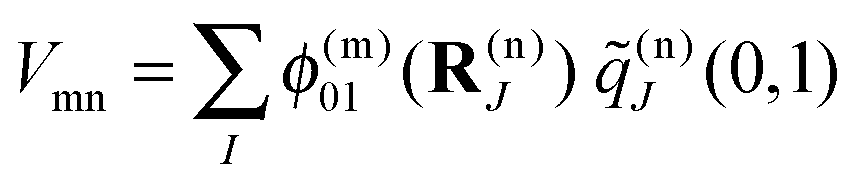 | (6) |
2.5 Linear optical spectra
The calculation of linear absorbance (OD, optical density), LD, CD, and fluorescence spectra is based on the dynamical theory of optical spectra detailed elsewhere58,59 and is performed as previously described for refined simulations of LHCII.25 Detailed formulae are given in the ESI.†3 Results
3.1 Protonation states
As explained and discussed earlier,13,23 we take the protonation pattern of the protein established at 210 K as representative for all temperatures below 210 K. Based on the temperature coefficient of the buffer used in the experimental work (see Section 2.2), we can estimate the pH representing the proton activity below 210 K to be ∼8.4. The combined Poisson–Boltzmann/Monte Carlo calculations revealed that only two titratable groups in CP29 have apparent pKa values in this range (8.3 for Glu 131 and 8.4 for Glu 165) so that their protonation states are not well defined. In addition, we found Glu 128, Glu 151, and His 235 to be protonated throughout, and thus in their non-standard protonation state, as well as an apparent pKa value of 9.0 for the C-terminus. To test the influence of those groups with an uncertain protonation state, we performed site energy calculations (PBQC method) with three different protonation patterns (P0: Glu 131, Glu 165, and C-terminus deprotonated; P1: Glu 131 and Glu 165 protonated and C-terminus deprotonated; P2: Glu 131, Glu 165, and C-terminus protonated; all other titratable groups unchanged including Glu 128, Glu 151, and His 235 in their non-standard states). In this way, the influence of the protonation state of the C-terminus on the site energies was found to be negligible. Site energy differences between P0 and P1 were found to be below 11 cm−1, and thus insignificant, except for Chl b608, where the difference was between 35 and 46 cm−1 depending on the quantum chemical method underlying the atomic partial charges. This situation is similar to that in LHCII,23 where only the site energy of Chl b608 was found to be sensitive to protonation state changes in the relevant pH range (site energy shift of 25 cm−1). We note that Glu 131 and Glu 165 are located on different sides of the membrane (see Fig. 1B) and thus will have different effects on specific site energies. Therefore, the above results also imply that the effects of changing their protonation states independently are small. In the following, we base all computations on the protonation pattern P0, keeping in mind that the site energy of Chl b608 has a somewhat larger uncertainty.3.2 Excitonic couplings
In Table 2 are compiled the excitonic couplings Vmn between the QY transitions of Chls in CP29 calculated with the Poisson-TrEsp method in combination with TD-DFT/B3LYP (for comparison, values obtained with HF-CIS instead of TD-DFT/B3LYP are listed in the ESI,† Table S5). The calculations are based on the assignment of Chl-types to sites as in the crystal structure (Table 1). The largest excitonic couplings (in the order of 70–80 cm−1) are found for the Chl pairs a604–b606, a603–a609, a611–a612, and a611–a615. Thus, a611, a612, and a615 form a strongly coupled trimer, while a603–a609 form a dimer and a604–b606 a Chl a–Chl b heterodimer. The latter is homologous to the a604–b606 pair in LHCII. In contrast, a615 is missing in LHCII. However, the strong coupling between a611 and a612 is also found there.23,60,61 Sites 603 and 609 are also strongly coupled in LHCII, but there, 609 is a Chl b.| m | n | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |
| 2 | 14 | 6 | 5 | 6 | −7 | −26 | −4 | 0 | 7 | −1 | 0 | 34 |
| 3 | — | 0 | −3 | 5 | 4 | 81 | 6 | −1 | 1 | 2 | −4 | −2 |
| 4 | — | 67 | 24 | −4 | −5 | −1 | −3 | 2 | 2 | −2 | −3 | |
| 6 | — | 20 | −4 | 2 | −1 | −2 | 2 | 1 | −1 | −2 | ||
| 7 | — | −3 | −6 | 1 | −2 | 2 | 2 | −1 | −2 | |||
| 8 | — | 27 | 38 | 5 | −1 | −2 | 1 | 5 | ||||
| 9 | — | −1 | 4 | −1 | −3 | 1 | 6 | |||||
| 10 | — | −31 | 14 | 5 | 0 | −9 | ||||||
| 11 | — | 73 | −3 | 1 | 70 | |||||||
| 12 | — | 1 | 0 | −1 | ||||||||
| 13 | — | −9 | −4 | |||||||||
| 14 | — | 0 | ||||||||||
Another excitonic heterodimer with a coupling of about 40 cm−1 is formed by Chls b608 and a610 in both CP29 and LHCII. The moderately strong coupling of about 35 cm−1 between a602 and a615 is unique to CP29, while a similarly strong coupling exists in LHCII between a602 and b601. The next category of excitonic couplings with absolute values between 24 and 31 cm−1 forms a remarkably conserved pattern in the two PPCs concerning the coupled sites and the sign of the coupling, i.e., such couplings are found between sites 602 and 609, 604 and 607, 608 and 609 as well as 610 and 611. The only difference between CP29 and LHCII in this respect is the different type of Chl in site 609. The conserved pattern of excitonic couplings reflects the structural homology of the two PPCs. Another group with a conserved pattern is found for couplings in the range 15–20 cm−1, namely between Chls a602 and a603, b606 and b607 as well as a610 and a612. All other excitonic couplings have magnitudes below 10 cm−1 in CP29. In contrast, there is a homodimer with a moderately strong coupling of –36 cm−1 in LHCII (a613–a614), which is absent in CP29, because 614 is a Chl b and the coupling is weaker. Further, there is a coupling of 20 cm−1 between Chls b601 and a611 in LHCII, which does not exist in CP29, because pigment 601 is absent.
3.3 Calculated site energies
In Fig. 2 are shown the site energy shifts ΔEm obtained with different quantum chemical methods (where cHF indicates the different amount of exact exchange with cHF = 1 representing HF-CIS) and two different types of electrostatic computations. Again, the calculations are based on the assignment of Chl-types to sites as in the crystal structure (Table 1). The site energies Em resulting for HF-CIS are listed in Table 1. Values of ΔEm for all methods can be found in ESI,† Table S6. Similar to the observation made earlier with LHCII,23 the value of cHF, influencing the shape of the difference in electrostatic potential between the first excited and the ground state of the Chls, has some influence on the calculated site energy shift depending on the site. The strongest influence of this kind is found for Chls a611 and a615. As shown below, the best agreement with experimental data is obtained for atomic partial charges calculated with HF-CIS. Therefore, we concentrate on data pertaining to this quantum chemical method in the following.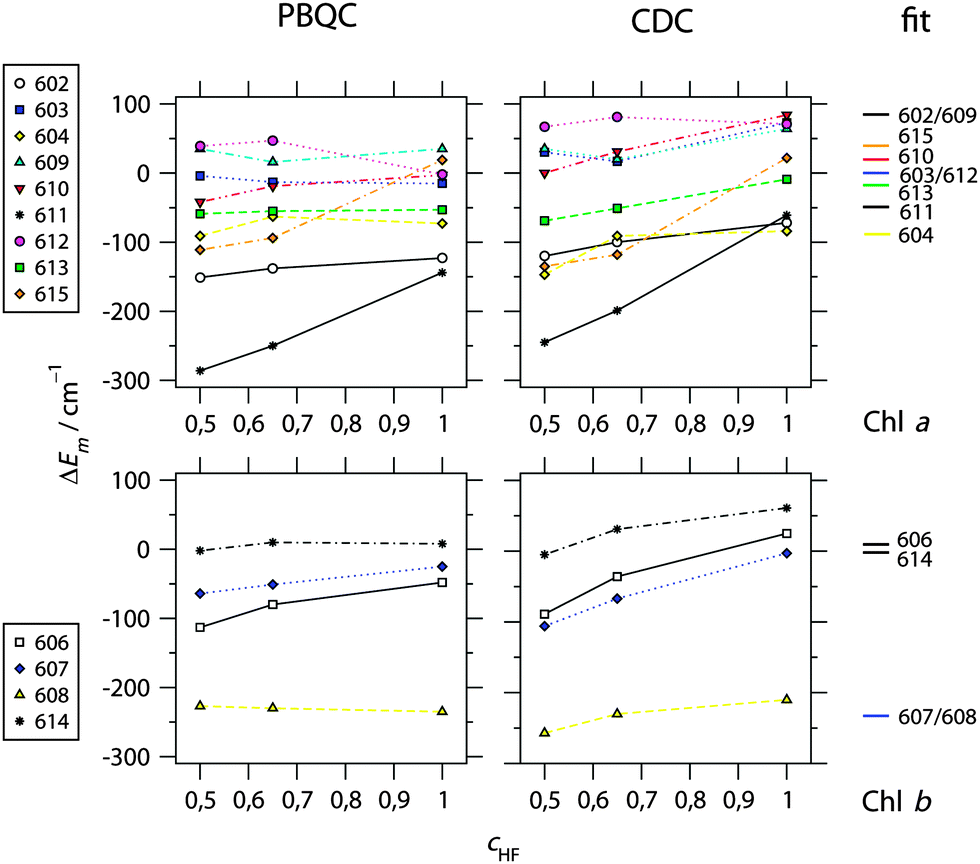 | ||
| Fig. 2 Calculated site energy shifts ΔEm of Chl a (top) and Chl b (bottom) bound to CP29 calculated on the basis of the 2.8 Å resolution structure (PDB 3PL922) using either the PBQC method (p = 1.8, εmem = 2.0, εsolv = 5.0) or the CDC method (εeff = 2.0) as a function of the amount of exact exchange (cHF) used in the quantum chemical calculation of atomic partial charges. For comparison, site energy shifts obtained from a refinement fit of linear optical spectra (see Fig. 3 and 4) are shown at the right side. Chl numbering is according to Pan et al.22 Absolute site energies for cHF = 1.0 and the fit are listed in Table 1. | ||
For Chl a, the PBQC data allow for a rough classification of sites into I: strongly redshifted (ΔEm < −100 cm−1 for a602 and a611), II: moderately redshifted (−100 cm−1 < ΔEm < −50 cm−1 for a604 and a613), and III: essentially unshifted (−50 cm−1 < ΔEm < +50 cm−1 for a603, a609, a610, a612, and a615). The most significant differences compared to LHCII23 as regards conserved Chls are that Chl a610 is not strongly redshifted and Chls a611 and a602 are redshifted. However, the redshift of the latter in CP29 is probably an artifact as will be discussed below. Based on the CDC data, a similar classification is obtained, but all categories are upshifted by about 50 cm−1, i.e., I: redshifted, II: unshifted, and III: blueshifted. Also, two Chls change the category: Chl a615 belongs to category II instead of III, and Chl a604 to I instead of II. In fact, Chl a604 has the lowest site energy when calculated with CDC and HF-CIS (Table 1). It should be noted that an overall offset of site energy shifts ΔEm between PBQC and CDC is to be expected, since the reference values E(a)0 in eqn (1) refer to different situations in the two methods: PBQC determines site energy shifts with respect to a hypothetical Chl in aqueous solution, whereas CDC does the same with respect to vacuum. Furthermore, the offset depends on the dielectric constants. Therefore, only the order of site energy shifts is important. Then, given a certain error limit, all what matters here is that Chl a615 and a604 belong to different categories in the two methods. Note that, nonetheless, the same value of E(a)0 for both methods is used in Table 1 for the calculation of Em, since this turns out to be suitable based on a comparison with experimental spectra (see below).
For Chl b, the same classification arises from both PBQC and CDC, namely one Chl b (b608) is strongly redshifted, while all others (b606, b607, and b614) are essentially unshifted (Fig. 1 and Table 1; ESI,† Table S6). This situation is similar to LHCII, where also Chl b608 is calculated to be strongly redshifted. However, as shown below, two strongly redshifted Chl b are required to adequately model the experimental spectra of CP29.
3.4 Calculated spectra and fitted site energies
To optimize the simulated linear spectra, the site energies were varied by hand around the directly calculated values. The resulting spectra are compared to experimental spectra in Fig. 3 and 4 (blue solid versus black dashed lines), while the fitted site energies are listed in Table 1 and also indicated in Fig. 2. The correlation between calculated and fitted site energies for the different quantum chemical and electrostatic methods is plotted in ESI,† Fig. S9. As can be seen from Fig. 3, the set of fitted site energies together with the calculated excitonic couplings (Table 2) provides an adequate description of the absorption and fluorescence spectra at different temperatures below 210 K. We note that the same inhomogeneous width of 130 cm−1 was used for all sites for simplicity and to keep the number of adjustable parameters as small as possible. Since, in fact, the width can be site-dependent, some of the residual differences between experimental and simulated line shapes may result from the neglect of this dependence. Apart from that, the agreement is good enough to draw two main conclusions: (i) the match of simulated and measured absorption spectra indicates that the pigment assignment in the crystal structure, particularly the resulting Chl a/b ratio, is reasonable. However, there remain uncertainties that we address below. (ii) The match of simulated and measured fluorescence spectra indicates that the effective modes used to complete the optical line shape (see ESI†) are suitable for CP29, although they have been inferred from a simulation of fluorescence spectra of LHCII.24 Thus, these modes seem to be transferable between PPCs and suitable for the simulation of optical Chl a line shapes in general. However, we cannot decide faithfully whether they are also suitable for Chl b, as assumed here for simplicity. | ||
| Fig. 3 Linear absorption and fluorescence spectra of CP29 at different temperatures simulated by using the fitted site energies (Table 1) and the excitonic couplings from the Poisson-TrEsp method (Table 2) compared to the experimental data from Pascal et al.27 Blue curves are simulations based on the Chl inventory of the crystal structure, whereas green curves (largely hidden underneath the blue curves) refer to simulations, where Chl b614 was assigned a zero oscillator strength. Parameters: σ = 130 cm−1, Vc = 20 cm−1, S0 = 0.5. | ||
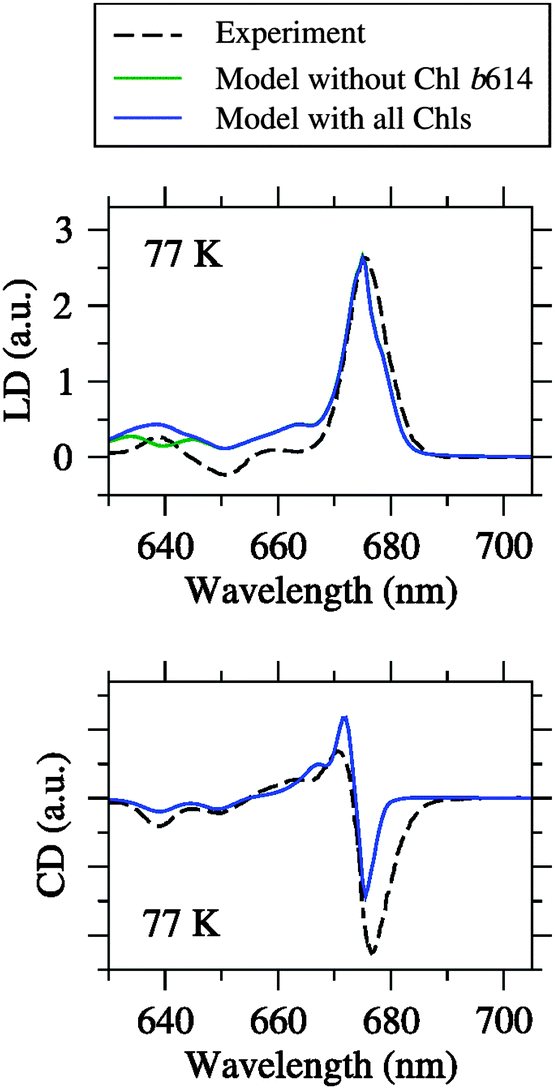 | ||
| Fig. 4 Linear dichroism (LD) and circular dichroism (CD) spectra at different temperatures simulated by using the fitted site energies (Table 1) and the excitonic couplings from the Poisson-TrEsp method (Table 2) compared to the experimental data from Pascal et al.27 Blue curves are simulations based on the Chl inventory of the crystal structure, whereas green curves (largely hidden underneath the blue curves) refer to simulations, where Chl b614 was assigned a zero oscillator strength. Parameters: σ = 130 cm−1, Vc = 20 cm−1, S0 = 0.5. | ||
It can be seen from Fig. 4 that the agreement between simulation and experiment is somewhat less good for LD and CD, especially for CD. The latter finding conforms to our earlier experience with LHCII.23,25 In the case of LD, the overall appearance of the spectrum is well reproduced. In particular, the spectral intensity is weak in the Chl b region, and there is one major positive peak in the Chl a region. However, the LD is somewhat too positive in the Chl b region. One possible error source is that the pigment orientations are not exactly the same in the LD samples as in the crystal structure. This possibility has been demonstrated recently in an analysis of LD spectra of LHCII.25 Another error source could be the Chl a/b ratio treated below. In the case of CD, there is a reasonable agreement between simulation and experiment in the Chl b region in fair contrast to LHCII.25 The Chl a region is less satisfactory, but the best compromise achieved so far after extensive fitting. One major problem is the non-conservative nature of the experimental CD spectrum, indicating the limits of a model that considers only excitonic CD effects between QY transitions.
A comparison of calculated and fitted site energies (see correlation plots in ESI,† Fig. S9) shows at first glance no good correlation between the data sets. However, a closer look reveals that in the case of Chl a, in fact, most data points are close to the diagonal. The best agreement is achieved with the charge sets based on HF-CIS. This is reflected not only in the lowest standard deviations between the two data sets of 90 cm−1 for PBQC and 85 cm−1 for CDC (for all Chl types, see ESI,† Table S7), but also in the lowest number of significant outliers, which is two (Chl a602 and Chl a611) for PBQC and only one (Chl a602) for CDC (upper part of ESI,† Fig. S9). The blueshifted site energy for Chl a611 obtained with CDC compared to PBQC is in better agreement with experiment. For both methods, Chl a602 remains a significant outlier. The reason is that the site energy of Chl a602 had to be upshifted significantly in the fit compared to the direct calculations (152–203 cm−1, Table 1) in order to achieve the aforementioned compromise concerning the CD spectra. Without this upshift, we were unable to create any reasonable resemblance between simulated and measured CD spectra. For all other Chl a, the site energies calculated directly with the HF-CIS method are within 45 cm−1 of the fitted values for PBQC and within 73 cm−1 for CDC (see Table 1). We consider this as a good correlation. Note that the fits are based upon the calculated site energies. So these results indicate that the least changes in site energies have to be made in order to achieve good agreement with experiment, when starting from the HF-CIS data. In this respect, CDC performs slightly better than PBQC due to its prediction of a less redshifted Chl a611. In the case of Chl b, the major outlier is Chl b607, the site energy of which had to be significantly downshifted in the fit compared to the direct calculations (210–232 cm−1, Table 1) in order to achieve the cancelation of LD signals (similar to LHCII23) and also to obtain a reasonable CD spectrum in the Chl b region. At present, we have no explanation for the outliers, but for Chl a602, a possible reason is discussed below.
In Fig. 5, the differences in site energy shifts obtained from fits to experimental spectra between conserved sites in CP29 and LHCII are shown. Taking the standard deviation between calculated and fitted site energies of CP29 (90 cm−1) as a rough error estimate, most of the differences are smaller than this limit, reflecting again the structural homology of the two PPCs. However, two site energies differ substantially: Chl a610 is significantly blueshifted in CP29 compared to LHCII (130 cm−1, Table 1), while Chl a611 is redshifted (100 cm−1, Table 1).
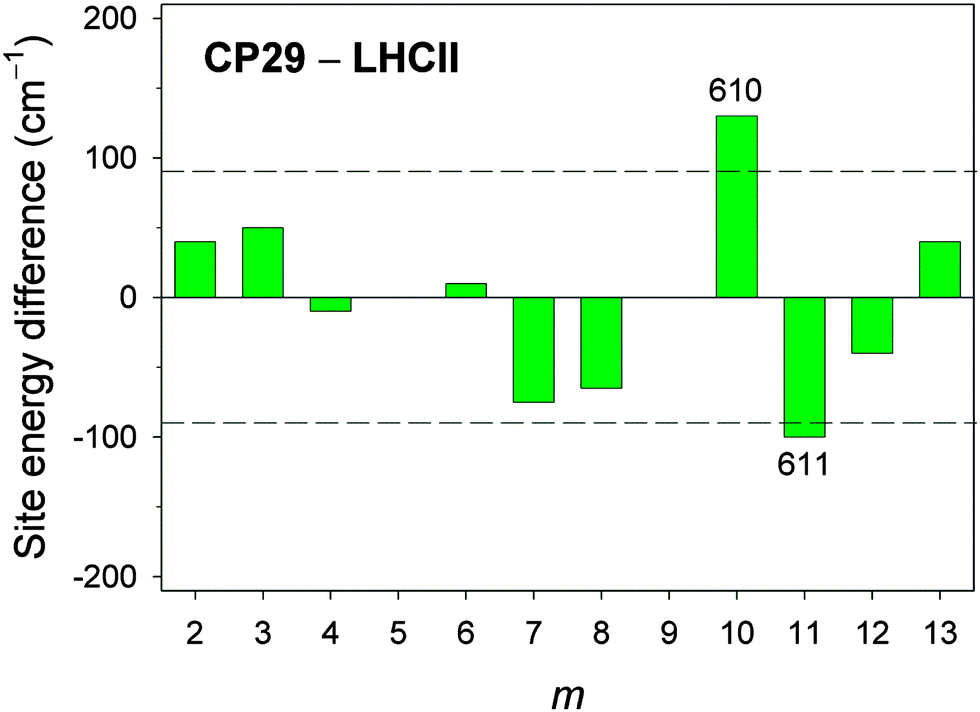 | ||
| Fig. 5 Site energy differences between CP29 and LHCII for Chl sites m that are conserved with regard to pigment type and position within the PPC (i.e., m = 2–4, 6–8, 10–13). The dashed lines indicate the estimated error of ∼90 cm−1 for calculated site energies based on the standard deviation between calculated (cHF = 1.0) and fitted site energies for CP29 (see ESI,† Table S7). | ||
As described in the Introduction, the Chl a/b ratio of the optical samples is usually higher (2.3–3.0) than that resulting from the crystal structure (2.25). Indeed, the simulated absorption and LD spectra appear to have a too high intensity in the Chl b region compared to the Chl a region. In particular, there is too much intensity around 640 nm in the absorption spectra (Fig. 3, left). To estimate possible uncertainties in the Chl a/b ratio, we performed additional simulations (green curves in Fig. 3 and 4), where a zero oscillator strength was assigned to Chl b614, which in our formalism is equivalent to depletion of this pigment. We note that Chl b614 is located at the C-terminus of CP29 (see Fig. 1) and is strongly solvent-exposed so that it might be lost during the biochemical preparation of CP29. Depleting Chl b614 results in Ra/b = 3.0. The absorption spectra simulated with this Chl a/b ratio provide a somewhat better description of the experiment in the Chl b region, but there is no improvement of the LD spectrum. No changes are observed for fluorescence and CD. The latter effect is due to the fact that Chl b614 forms a single-pigment exciton domain, i.e., is weakly coupled to all other pigments. A further readjustment of site energies could be done to further improve the agreement between simulation and experiment, especially for LD, but we shall not further pursue this goal in the present paper. The main point is that a reasonable description of the experimental data is still possible with Ra/b increased up to a value of 3.0 (taking into account uncertainties in site energies and line widths). Loss of Chl b614 would be a possible explanation, and a partial loss could well explain the value of Ra/b = 2.85 (as well as the Chl/carotenoid ratio of ∼4) determined experimentally for these particular samples.27 However, the entire replacement of one Chl b with a Chl a would result in a Chl a/b ratio that is too high and more difficult to reconcile with the experimental spectra. Also, a replacement of Chl a610 with a Chl b as suggested on the basis of uncertainties in the crystallographic assignment22 would be in conflict with the intensity ratios of Chl b to Chl a of the experimental spectra.
3.5 Spectra of mutant CP29
Neoxanthin (Neo) is bound to both CP29 and LHCII in a conserved motif.20–22 Part of the Neo binding site is a tyrosine residue forming a hydrogen bond with the 3′-hydroxyl group of Neo. This tyrosine is Y112 in LHCII and Y135 in CP29. The binding motif is adjacent to Chl a604. In LHCII, it was found by structure-based computations23 that the hydrogen bond network of the two hydroxyl groups of Y112 and Neo can exist in two different orientations (see Figure 10 in ref. 23). In the more stable conformation, the 3′-hydroxyl group of Neo points towards the tetrapyrrole macrocycle of Chl a604 and accepts a hydrogen bond from Y112. In this conformation, the side chain of Y112 has a specific effect on the site energy of Chl a604, contributing a blueshift of 76 cm−1, whereas Neo has no significant effect on this site energy. Turning both hydroxyl groups into the less stable conformation, so that the 3′-hydroxyl group of Neo points away from the macrocycle and donates a hydrogen bond to Y112, causes a significant redshift of the site energy of Chl a604 by 212 cm−1. Because of the conserved binding motif, we expect the same to happen in CP29. Our modeling of hydrogen atoms revealed the same stable orientation of hydroxyl groups, i.e., Neo accepts a hydrogen bond from Y135. To test the influence of Y135 on the site energy of Chl a604, we constructed a mutant in silico, where tyrosine is replaced with phenylalanine, i.e., YF(135). This was done by replacing the hydroxyl group of Y135 with a hydrogen atom and reoptimizing the hydrogen positions with CHARMM. This mutation has a very specific effect on the site energy of Chl a604, namely, it causes a redshift by 76 cm−1 (based on PBQC and the HF-CIS charge sets), while leaving all other site energies unchanged in complete agreement with the results obtained for LHCII.The corresponding YF mutant in CP29 has been constructed earlier by Caffarri et al.,44 and the recombinant and reconstituted PPCs analyzed with optical absorption spectroscopy. The WMD spectrum indeed exhibits one clear feature in the QY region that is in accordance with a redshift of a single Chl a in the mutant. In Fig. 6 is shown a comparison of the experimental WMD spectrum of Caffarri et al.44 with our simulation. In the simulation of the mutant, we used the fitted site energies for wild type listed in Table 1, but with site 604 redshifted by 76 cm−1. The good match of experimental and simulated WMD spectra confirms the assigned site energies to Chl a604 in both wild type and mutant, and thus provides evidence that Chl a604 has the most redshifted site energy in CP29. However, this does not necessarily imply that Chl a604 is the energy sink, as shown below.
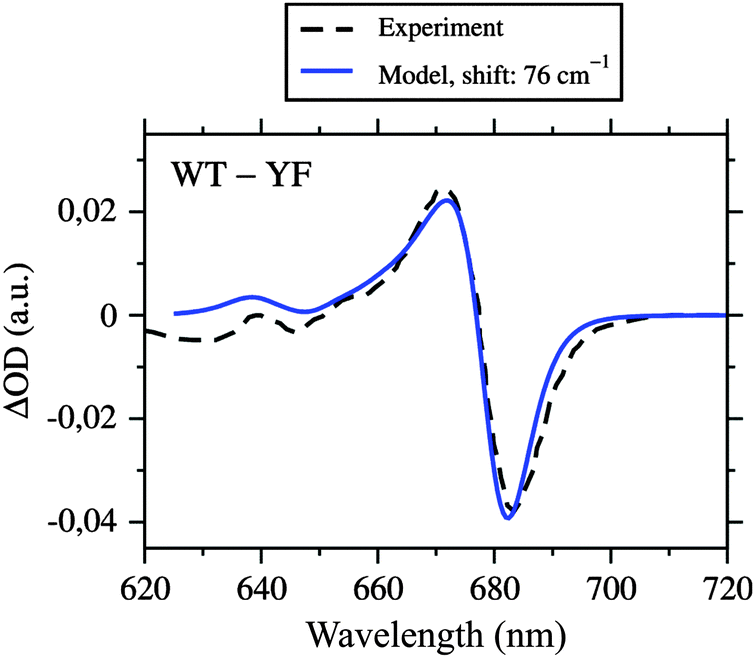 | ||
| Fig. 6 Absorbance difference (ΔOD) spectrum of wild type CP29 and the YF mutant, where tyrosine 135 close to Chl a604 is replaced with phenylalanine, simulated by using the fitted site energies (Table 1), the excitonic couplings from the Poisson-TrEsp method (Table 2), and a redshift of E4 by 76 cm−1 (calculated with PBQC and HF-CIS charges) compared to the experimental data from Caffarri et al.44 Parameters: σ = 130 cm−1, Vc = 20 cm−1, S0 = 0.5. | ||
3.6 Different contributions to site energy shifts
The CDC method is suitable for a detailed analysis of the contributions of different parts of the PPC to individual site energy shifts.36,38,39 As an example, we show in Table 3 the contribution of selected groups to the site energy of Chl a611. For comparison, we recalculated the contributions of homologous groups in LHCII with the same method. In accordance with our earlier data,23 the largest individual contributions to the site energy of Chl a611 in LHCII originate from the phospholipid (PG) and Lys 182, largely compensating each other. This situation is conserved in CP29 with G3P and Lys 199 taking over the roles of PG and Lys 182, respectively. However, the redshift induced by G3P is rather strong and only partly compensated by the blueshift due to Lys 199. This contributes to the more strongly redshifted site energy of Chl a611 in CP29 compared to LHCII (Table 1). Since the assignment of G3P in the crystal structure is tentative,22 it follows that the structure-based site energy of Chl a611 is uncertain.| Group | CP29 | Group | LHCII | |||||
|---|---|---|---|---|---|---|---|---|
| ΔE11 | ΔE11 | |||||||
| Complete residue | Side chain | Complete residuea | ||||||
| BHHLYP | B65LYP | HF-CIS | BHHLYP | B65LYP | HF-CIS | HF-CIS | ||
| a Average of the three equivalent sites in the LHCII trimer. | ||||||||
| G3P | −531 | −411 | −433 | — | — | — | PG | −123 |
| Lut 620 | 1 | 0 | 2 | — | — | — | Lut 620 | 2 |
| Arg 101 | 13 | −4 | 23 | 13 | −5 | 23 | Arg 70 | 22 |
| Met 104 | 0 | 0 | 0 | 0 | 1 | 0 | Met 73 | 0 |
| Glu 159 | −9 | 1 | −15 | −9 | 1 | −15 | Glu 139 | −15 |
| Arg 162 | 10 | −1 | 16 | 10 | −1 | 16 | Arg 142 | 16 |
| Asp 179 | −10 | 6 | −16 | −10 | −1 | −16 | Asp 162 | −16 |
| Ile 192 | −2 | 0 | −3 | 0 | 0 | 0 | Glu 175 | −43 |
| Gln 194 | 0 | 0 | −2 | −1 | −1 | −1 | Lys 177 | 18 |
| Ala 196 | 3 | 4 | 1 | 1 | 1 | 0 | Lys 179 | 46 |
| Glu 197 | −12 | 11 | −28 | −14 | 8 | −28 | Glu 180 | −28 |
| Lys 199 | 312 | 252 | 331 | 319 | 261 | 331 | Lys 182 | 273 |
| His 200 | 1 | 0 | 2 | 1 | 0 | 2 | Asn 183 | −5 |
Another interesting residue is Ala 196 in CP29, which is homologous to Lys 179 in LHCII. These residues are located next to Chls a610 and a611 (ESI,† Fig. S10). Lys 179 in LHCII contributes 46 cm−1 to the site energy E11. However, more significant is its influence on E10, where it contributes −90 cm−1. A mutation AK(196) in CP29 can be expected to cause a similar redshift and, thus, is a means to determine the site energy of Chl a610. Most likely, the replacement of Lys with Ala at this position is responsible for at least part of the blueshift of Chl a610 and the redshift of Chl a611 so that, in contrast to LHCII, Chl a610 is not the pigment with the lowest site energy in CP29.
We also studied the influence of residues 88 to 101 on the site energies of the nearby Chls a602 and a603 (see Fig. 1). It turns out that this part of the protein causes a somewhat stronger redshift of E2 than the homologous part in LHCII (while the effect on E3 is very similar; see ESI,† Tables S8 and S9). However, the missing N-terminus in CP29 is a problem. An overlay of the structures of CP29 and LHCII (ESI,† Fig. S11) suggests that the N-terminus of CP29, when folding in a structure analogous to LHCII, would come close to Chl a602 and thus could have a major influence on the site energy E2. On the other hand, the high inherent flexibility of the N-terminus22,26 seems to imply that the structure is different, so that the influence of the N-terminus remains obscure. It cannot be excluded that the N-terminus contributes to a blueshift of E2, which then would provide an explanation for the discrepancy between calculated and fitted site energy (Table 1). The overlay of the two structures of CP29 and LHCII suggests that Chl a615 could be strongly influenced by the N-terminus as well (ESI,† Fig. S11). Thus, the site energy of this pigment likewise is afflicted with some uncertainty.
3.7 Exciton states and pigment contributions
In Fig. 7 are shown the exciton state pigment distribution functions62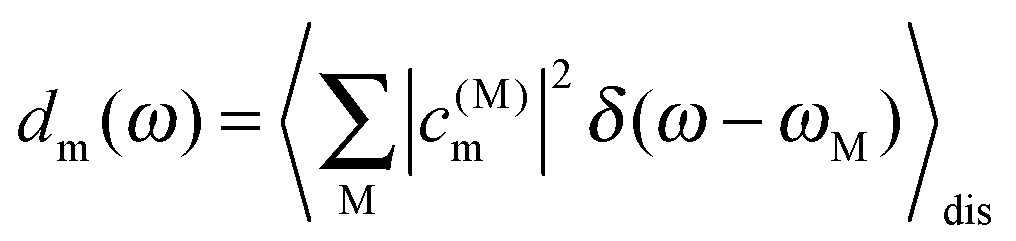 | (7) |
 | ||
| Fig. 7 Exciton state pigment distribution functions62dm (eqn (7)) of the 13 different Chl sites m in CP29 at 77 K based on the fitted site energies (Table 1) and the excitonic couplings from the Poisson-TrEsp method (Table 2). (A) Stromal layer without the terminal emitter domain; (B) terminal emitter domain; (C) lumenal layer. Parameters: σ = 130 cm−1, Vc = 20 cm−1, S0 = 0.5. | ||
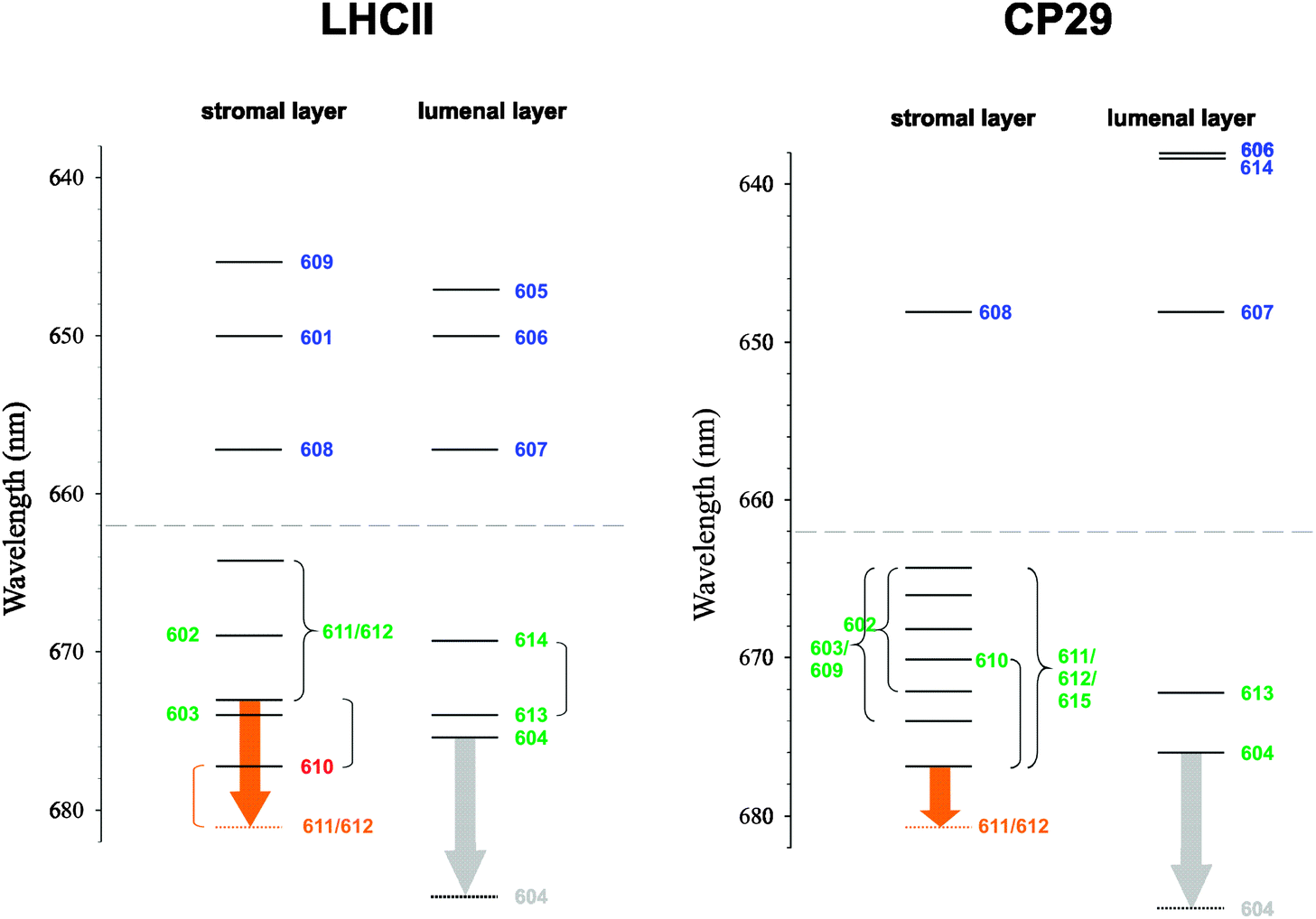 | ||
| Fig. 8 Exciton energy level scheme for the stromal and lumenal layer of pigments in trimeric LHCII, based on the exciton energies εM from ref. 25, and in CP29, based on the values of εM from this work (Table 4). Dominant pigment contributions are indicated by numbers following the numbering scheme of ref. 20 and 22. Curly brackets connect exciton states that are significantly delocalized between pigments, whereas round brackets connect exciton states that are delocalized, but the contribution of the indicated pigment is mainly to one of the exciton states. The orange arrows represent a possible temperature-dependent shift of an exciton state involving Chl a612.40,68 The gray arrows represent the hypothetical redshift of the site energy of Chl a604 due to a conformational change of neighboring hydroxyl groups. The dashed lines indicate the borderline between Chl a and Chl b region. | ||
| Chl type | M | CP29 | LHCIIa | ||
|---|---|---|---|---|---|
| ε M | Major contributing pigments | ε M | Major contributing pigments | ||
| a From ref. 25, valid at low temperatures. Chl b region tentative based on the present results. | |||||
| a | 1 | 14770 |
611, 612, 615 | 14770 |
610 |
| a | 2 | 14800 |
604 | 14810 |
604 |
| a | 3 | 14840 |
603, 609 | 14830 |
613, 614 |
| a | 4 | 14880 |
613 | 14840 |
603 |
| a | 5 | 14890 |
602, 603, 610, 611, 612, 615 | 14860 |
610, 611, 612 |
| a | 6 | 14930 |
602, 603, 609, 610, 611, 612, 615 | 14950 |
613, 614 |
| a | 7 | 14980 |
602, 603, 609, 610, 611, 612 | 14950 |
602 |
| a | 8 | 15020 |
602, 603, 609, 611, 612, 615 | 15060 |
611, 612 |
| a | 9 | 15060 |
602, 603, 609, 615 | — | — |
| b | 10 | 15430 |
607 | 15210 |
608 |
| b | 11 | 15440 |
608 | 15210 |
607 |
| b | 12 | 15670 |
614 | 15390 |
601 |
| b | 13 | 15690 |
606 | 15390 |
606 |
| b | 14 | — | — | 15460 |
605 |
| b | 15 | — | — | 15500 |
609 |
Despite the occurrence of significant excitonic couplings between Chl a and Chl b pigments, their site energies are too different to allow for a substantial exciton delocalization between pigments of different type. Therefore, it is possible to ascribe to each exciton state in CP29 either Chl a or Chl b character (Table 4). Due to the choice of the cutoff parameter Vc = 20 cm−1 (see ESI†), the network of coupled Chls decomposes into four domains: Domain 1 encompasses all Chls of the stromal layer (a602, a603, b608, a609, a610, a611, a612, and a615), i.e., any two pigments of this domain are connected either directly or via a chain of intermediate pigments with excitonic couplings ≥20 cm−1 (Table 2). Within this domain, Chl b608 dominates the highest exciton level because of the aforementioned site energy differences (Fig. 7A). All other Chls in the stromal layer are Chl a and contribute more or less to all of the remaining seven exciton states. The lowest exciton state is dominated by Chls a611, a612 and a615, so that these pigments form the “terminal emitter domain” (TED) with the highest contribution from a611 and a612 (Fig. 7B). At least at cryogenic temperatures, the situation is different in LHCII, where the lowest exciton state is dominated by Chl a610.23,25,60
In both CP29 and LHCII, there are no couplings larger than 20 cm−1 between the two layers of pigments. Domains 2–4 in CP29 all comprise Chls of the lumenal layer with a604, b606, and b607 forming one domain as well as Chls a613 and b614 forming single-pigment domains. Because of the site energy differences between Chl a and Chl b, all exciton states in the lumenal layer are ultimately localized (Fig. 7C). Although Chl a604 contributes to the emission of CP29, it does not contribute to the lowest exciton state (based on the values of εM) despite having the lowest site energy. The reason is the large exciton splitting between pigments a611, a612, and a615, causing the most redshifted exciton state to be located in the stromal layer.
We note that in order to obtain good fits of the Chl b region of the spectra, we had to use different values of E(b)0 for CP29 and LHCII. This is clearly an inconsistency that has to be removed, but the solution to this problem is beyond the scope of the present work. Therefore, the apparent offset of Chl b energy levels between CP29 and LHCII is an artifact, and the corresponding absolute transition energies for LHCII are tentative (see further discussion below).
4 Discussion
We performed structure-based simulations using a combination of quantum chemistry and classical electrostatics to obtain an exciton Hamiltonian for the CP29 antenna of PSII that, in conjunction with a dynamical theory of optical spectra, was used to model linear absorption, LD, CD, fluorescence and WMD spectra of isolated CP29. These simulations reveal on the one hand the high degree of homology between CP29 and LHCII as regards conserved pigment sites, and on the other hand, point to a specific difference between the two PPCs, namely that besides differences in pigment composition of other sites, Chl a610 is more blueshifted and Chl a611 more redshifted in CP29 compared to LHCII. This has consequences for the electronic structure of the so-called “TED”, i.e., the set of pigments contributing mostly to the lowest exciton state (see below).For the first time, we made here a direct comparison of the two electrostatic methods PBQC and CDC (explained in recent reviews13,14) on the same PPC under matchable conditions, i.e., based on the same protonation pattern. As regards the quantum chemical methods underlying the atomic partial charges, it turns out that HF-CIS performs best among the three methods tested. This result is in agreement with recent LHCII data.25 Then, a comparison of calculated and fitted site energies shows both electrostatic methods to be similarly accurate. Actually, CDC performs better in that it predicts a less redshifted site energy for Chl a611, which is in closer agreement with the fit (ESI,† Fig. S9).
Concerning the atomic partial charges, we have presently no explanation for the good performance of the partial charges derived from HF-CIS. This result is entirely empirical. In particular, it does not allow us to conclude that HF-CIS provides the most realistic description of the changes in the electronic wave function of a Chl upon light excitation. Our findings merely indicate that HF-CIS is the most suitable among the methods tested so far to produce parameters (partial charges) for an effective description of the electrostatic pigment–protein coupling within the approximations used. These approximations have been discussed earlier in detail.13,23 Here, we would like to stress one aspect related to the difference between PBQC and CDC, namely the distinction between first- and second-order effects. Based on quantum mechanical perturbation theory, the leading first-order term in the pigment–protein coupling is the Coulomb interaction between permanent charge distributions of a Chl and its protein environment. The charge distributions are represented by atomic partial charges, and these are the same in CDC and PBQC. In both methods, the difference in charge distribution between S1 and S0 states of the Chl is the main reason for the site energy shift.
Nonetheless, there are second-order contributions, which can be divided into inductive effects (i.e., polarization of the pigment's electron cloud by the permanent charge distribution of the protein and vice versa) and dispersive interactions (i.e., mutual polarization of the electron clouds of Chl and protein also known as van der Waals interactions). The latter are neglected in our approach based on the assumption that the differences in dispersive interactions between sites are small. The inductive effects are approximated by allowing the medium, in which the atomic partial charges reside, to be polarizable. In CDC, a single effective dielectric constant is assigned to the whole medium. This merely allows for a screening of the Coulomb interaction between the atomic partial charges. However, there are contributions to the site energy shifts due to the difference in inductive couplings between the excited and the ground state charge density of the pigments. In CDC, these contributions are only accounted for implicitly and averaged by adjusting the reference energy E0. In contrast, the inductive site energy shifts are calculated explicitly in the PBQC method. By assigning different dielectric constants to protein, membrane and solvent environment and by solving the Poisson–Boltzmann equation, it is possible to consider the influence of inductive effects on site energy differences in a way that is impossible by merely adjusting E0. Since it turns out that CDC and PBQC are of similar accuracy when tested against fits, we can conclude that the inclusion of inductive site energy shifts as done in the PBQC method does not result in a significant improvement of the calculated site energies, and the simpler CDC method is actually sufficient at the present level of theory. Most likely, there is a certain amount of error compensation at work, that is, the rather small differences in inductive site energy shifts are over-compensated by other effects such as the differences in dispersive interactions, which are neglected so far in both CDC and PBQC. We note that it is possible with the PBQC method to consider the non-equilibrium nature of inductive shifts that arise from the different time scales of electronic and nuclear motion. However, the non-equilibrium corrections were found to be small.13
Also, a word of caution is in order: The evaluation of methods performed in the present paper suffers from two major problems: (i) the missing N-terminus of CP29 can be expected to cause errors of at least some site energies for which the electrostatic methods cannot be made responsible, and (ii) the fitted site energies are still sub-optimal due to residual deviations between simulated and fitted spectra, in particular, the CD spectrum. In addition, we are presently upgrading the quantum chemical methods for the calculation of atomic partial charges of the pigment states. Therefore, it may become necessary to re-evaluate the electrostatic methods in future work based on improved benchmark data.
The deviation between simulated and experimental CD spectra is a persistent problem. Given our experience with LHCII,23,25 we were rather surprised to find such a good match between theory and experiment in the Chl b region of CP29. However, different values of the reference transition energy E(b)0 are obtained for CP29 and LHCII (difference: 289 cm−1) and a significant offset of Chl b energy levels between the two PPCs results (Fig. 8). Since the calculated site energy shifts of conserved Chl b sites do not differ by such an amount (Fig. 5), there is no structure-based rationale for this offset, so that it is an artifact. The existence of high-lying Chl b excited states in CP29 is apparent from experimental data. Femtosecond pump–probe,63,64 nonphotochemical hole-burning31,32,65 and 2D electronic spectroscopy66 all show the presence of essentially two Chl b pools absorbing around 640 and 650 nm. Our assignment of Chl b states in CP29 is basically in agreement with these findings. As a consequence, the value of E(b)0 chosen for CP29 is very likely correct, and the Chl b energy levels of LHCII rather than those of CP29 will have to be revised in the future. Such refinements should take into account the non-conservativity of the CD spectra.
Similar to LHCII, the Chl a region of the spectra can be regarded as the sum of the spectra of one large stromal domain and two small lumenal domains. In contrast to LHCII, there is no lumenal Chl a homodimer in CP29, because site 614 is occupied by a Chl b. Accordingly, there is one absorption due to Chl a613 peaking at 672 nm (Fig. 7C). The Chl a604 binding motif is conserved between CP29 and LHCII. The excellent agreement between simulated and measured WMD spectra of the YF(135) mutant of CP29 is direct evidence for a low-lying absorption of Chl a604, having its maximum at 676 nm (Fig. 7C). This assignment can be considered the most stringent result of the present paper, as it refers to a direct experimental verification of one particular calculated site energy. Note that fitted and directly calculated site energies are essentially the same for Chl a604 (Table 1). Effects resulting from the strong excitonic coupling between Chls a604 and b606 (Table 2) are weak because of the large site energy difference, but nonetheless visible in both the simulated and the experimental WMD spectra (Fig. 6). Additional changes in the experimental spectrum in this spectral region could be due to a perturbation of the Chl b606 binding pocket as a result of the loss of Neo found in the mutant.44 These findings suggest that the assignment of site energy E6 is correct with Chl b606 having a maximum absorption at 638 nm (Fig. 7C and 8) and thus being the most blueshifted pigment in CP29.
According to our model, the loss of Neo observed experimentally in the YF mutant44 cannot be made responsible for the spectral change, as this would require a different orientation of the hydroxyl groups of Neo and Y135 in the wild type. In the more stable conformation predicted by modeling with CHARMM, the direct influence of Neo on the site energy E4 is negligible. The relevant shift is due to the hydroxyl group of the tyrosine. Caffarri et al.44 also constructed homologous YF mutants in LHCII and CP26. In both cases, a difference peak similar to the one of CP29 is found at the same wavelength, but is partly overlain by other spectral shifts of unknown origin. Thus, the result is not as clear as in the case of CP29, but nonetheless, we take this as evidence that Chl a604 in LHCII has an absorption around 675 nm in compliance with our earlier assignment.25 This assignment implies that the long-lived excited state of LHCII in the 660 nm region, referred to as “bottleneck” state and hitherto ascribed to Chl a604,60 has to be explained differently.‡ We can also anticipate that a similar binding motif for Neo and Chl a604 with a similar site energy E4 exists in CP26.
The case of Chl a604 demonstrates that it is possible in principle to determine site energies with some confidence by combining site-directed mutagenesis, optical difference spectroscopy and structure-based quantum chemical/electrostatic computations. Prerequisites for such a determination are a suitable mutagenesis system (including the possibility to reconstitute the PPC in a native-like state as is necessary for plant antenna proteins), reliable structural information and single amino acid residues that can be mutated without causing major structural changes and that have a specific effect on one or only a few site energies. Information about the latter can be obtained from the computations in a first step as demonstrated here for CP29. As an example, Ala 196 is of high interest as its replacement with, e.g., lysine is expected to have a strong and relatively specific effect on Chl a610. Also, the question of mixed occupancy of the 610 site could be tackled in this way. A Lys at position 196 would affect the absorption of either Chl type and ideally should cause shifts in different wavelength regions, if there is a heterogeneity of this type. Clearly, this approach is limited by possible side effects of mutations such as long-distance structural changes affecting many pigment sites or pigment loss. However, even these problems could principally be circumvented by combining the site energy calculations with molecular dynamics simulations, albeit at a higher computational cost.
While it is well possible that mixed binding sites exist in reconstituted CP29, the crystal structure22 argues against the existence of such sites in native CP29 with the possible exception of site 610. Our results corroborate this argument, as there is no need for the assumption of mixed binding sites for a reasonable simulation of the optical spectra of native CP29. Nonetheless, on the basis of the stationary spectra alone, mixed sites cannot be strictly excluded. For example, the replacement of Chl a610 with a Chl b in only a subset of PPCs in a sample and the concomitant replacement of another Chl b with a Chl a in a way that the Chl a/b ratio remains between 2.3 and 3.0 is conceivable. Note, however, that this would be in conflict with the interpretation of the crystallographic data.
Salverda et al.67 argued in favor of mixed binding sites in native CP29 based on three pulse echo peak shift (3PEPS) measurements. According to their interpretation, the data indicate the presence of slow Chl b → Chl b EET processes besides fast Chl b → Chl a EET. The slow EET processes should not be observable, however. With the assumption made by Salverda et al.67 that two Chl b per CP29 are present, at least one of them should be coupled strongly enough to Chl a to allow for the fast Chl b → Chl a EET. Then the population of the excited state of the acceptor in the slow Chl b → Chl b EET should always be too small to allow for detection of the latter process. If such a process is observed, though, it implies that in a fraction of CP29 complexes in the sample, the fast Chl b → Chl a EET is abolished. This is interpreted as a replacement of some Chl a with Chl b, i.e., the existence of mixed binding sites. Based on the recent crystal structure22 and our simulations, we have to start with the assumption of three to four Chl b per CP29. Given the computed excitonic couplings (Table 2), only Chl b614 is weakly coupled to all other Chls, whereas the remaining Chl b should be able to deliver excitation energy quickly to Chl a. Thus, the argument of Salverda et al.67 concerning the observability of the slow Chl b → Chl b EET still holds. However, in our view, this fact is no strict proof of the existence of mixed binding sites: (i) the observed process is not necessarily Chl b → Chl b EET, but could be something else, e.g., a structural relaxation of the protein matrix that leads to a shift of the site energy of a Chl b. (ii) If a slow Chl b → Chl b EET is present and the fast Chl b → Chl a EET is abolished in a fraction of PPCs, the latter is not necessarily due to a replacement of Chl a with Chl b, but could be due to static disorder leading to a significant slow down of Chl b → Chl a EET in some PPCs. We think that at present these possibilities cannot be excluded, and further work including the structure-based simulation of non-linear spectroscopic experiments is required to clarify the issue.
It has been argued on the basis of mutagenesis experiments and optical difference spectroscopy that the TED, which is also a possible site of nonphotochemical quenching (see below), is essentially conserved in LHCII and CP29 (and also in CP26),40 meaning in particular that Chl a612 is involved. On the basis of our computations, we come to a similar conclusion as regards the location of the TED, but there are differences between CP29 and LHCII that deserve further comments. Two major differences between the PPCs are (i) the extra Chl a615 in CP29, being as strongly coupled to Chl a611 as Chl a612, and (ii) the site energy blueshift of Chl a610 and redshift of Chl a611 in CP29 compared to LHCII. The extra Chl a615 in CP29 forms an unusual H-type aggregate with Chl a611 in a way that the two Chls sandwich their common axial ligand, the phosphate group of the putative G3P. At first glance, this seems to indicate the presence of a low exciton state with no or a small oscillator strength as typical for H-aggregates. In fact, there is no such dark state due to the coupling between Chls a611 and a612, leading to a strongly allowed lowest exciton state that is dominated by these two Chls (Fig. 7B). We note that this dominance is a consequence of the relatively high site energy of Chl a615. In a recent analysis of hole-burning data, Feng et al.32 independently assigned the same pigments to the TED, but assigned a higher site energy to Chl a611 (15010 instead of 14850 cm−1). As a consequence, the lowest exciton state in their model has a slightly smaller contribution from Chl a611 than ours, but still the TED is dominated by Chls a611–a612.
Thus, according to the modeling, Chl a612 contributes to the lowest exciton state in CP29, but not in LHCII.23,25 In order to properly assess the meaning of this difference, we have to take into account the temperature dependence of WMD spectra. Mutants have been constructed in LHCII, CP29, CP26, and CP24, in which the (putative) axial ligand to Chl a612 is replaced with a non-ligating residue.40,68 In all cases, at least one pigment is lost, which is interpreted as the removal of Chl a612. Provided this loss causes no major structural changes, then the WMD spectrum should yield information about the exciton state (or states) to which Chl a612 contributes significantly. In the case of LHCII, this interpretation is supported by the fact that the WMD spectrum can be properly modeled by simply assigning a zero oscillator strength to Chl a612 in the simulation of the mutant spectrum, while leaving all other parameters unchanged to those of the wild type (see Figure 8B in ref. 23). The WMD spectra of LHCII, CP29, and CP26 all show a very similar positive peak (in CP24, the situation is different).40 The maxima of the WMD spectra at 77 K are at 678 nm for LHCII,40,68 at 676 nm for CP29,40 and at 677 nm for CP26.40 However, at room temperature, they are redshifted to 681 nm in all of the three antenna complexes.40,68,69 This is interpreted as a structural change of the PPCs involving the Chl a612 binding site in a way that an exciton state, to which this pigment contributes significantly, is redshifted (orange arrows in Fig. 8). The origin of the shift is unknown, but a change in the excitonic coupling between Chls a611 and a612 is a possibility. The important detail as regards the comparison of CP29 and LHCII is the different site energy of Chl a610. Assuming this site energy to be temperature-independent, a shift of the lowest exciton state in CP29 due to a change of Chl a612 would have no major impact on the character of this state, as the contribution of the blueshifted Chl a610 is small anyway. However, the situation is different in LHCII: here, Chl a610 is redshifted and dominates the lowest exciton state at 77 K. A redshift of the Chl a612 state would then allow this state to outcompete Chl a610 as an energy sink and to take over at higher temperatures. As a consequence, the TED is dominated by Chls a611 and a612 at physiological temperatures in both CP29 and LHCII. It is in this sense that the TED is conserved between the two PPCs (and likely also in CP26).
The assignment of the TED has consequences for the structural modeling of the C2S2M2 supercomplex.12 This complex consists of two copies of each of the PSII core complex (C), a strongly bound LHCII trimer (S), and a moderately bound LHCII trimer (M). In addition, the complex contains two copies of each of CP24, CP26, and CP29. Caffarri et al.12 constructed a model of this supercomplex based on their 12 Å resolution projection map from single particle electron microscopy of C2S2M2, the 2.9 Å resolution structure of cyanobacterial PSIIcc,16 and the 2.72 Å resolution structure of spinach LHCII.20 The minor antenna complexes were modeled based on their homology to monomeric LHCII, including CP29, whose high-resolution structure was not yet available at that time. In this model, CP29 is located between LHCII-M and CP24 on the one hand and CP47 of PSIIcc on the other hand, so that it can serve to transfer excitation energy from the former to the latter. The orientation is such that Chl a603 faces CP47, while the Chl a611–a612 pair points in the opposite direction. Based on our computations and the premise that the TED should face the complex to which excitation energy is transferred, as the lowest exciton states indicate the preferred direction of energy flow, we come to the conclusion that CP29 should be rotated such that the Chl a611–a612 pair comes closest to the energy accepting pigments of CP47. In this conclusion, we agree with Feng et al.32 However, this proposal is made with the proviso that the location of the energy sink is not affected by interactions between PPCs within the supercomplex.
There has been a debate concerning the preferable site for nonphotochemical quenching in the antenna system of PSII as a means of EET regulation,9 and the minor complexes CP29, CP26, and CP24 were implicated to play a particularly important role.70 It is clearly reasonable to look for quenching sites among the energy sinks, as they attract the excitons. However, there are two problems: (i) the energy sinks need not be the same in a quenching and a non-quenching conformation of a PPC. However, a recent analysis of LHCII conformations seems to indicate that they are the same.25 (ii) The TED is a rather shallow energy sink. For example, the energy difference between the lowest exciton state of the TED in CP29 and the state dominated by Chl a604 is just 30 cm−1 (see Fig. 8 and Table 4), which is far below the thermal energy at physiological temperatures. Thus, from an energetic point of view, the TED is not necessarily the quenching site, but the excitation energy may easily go to Chl a604 and be quenched there. Since both the TED (see above) and the Chl a604 site are conserved among CP29 and LHCII, our data provide no rationale for preferring the minor complexes to LHCII as quenching sites.
5 Summary and conclusions
The combined quantum chemical/electrostatic approach used earlier to determine the exciton Hamiltonian of Chl a and b S1 states in LHCII23 was extended here to CP29 based on recent crystal structure data.22 Two electrostatic methods for the calculation of site energies, PBQC and CDC, that differ in the treatment of polarization effects13,14 were compared and tested against site energies obtained from a fit of linear absorption, LD, CD, and fluorescence spectra of isolated CP29. Both methods give similar results, but CDC performs slightly better. The computations provide a structure-based rationale for the optical spectra, implying that there is no serious conflict between the assignment of Chl types to sites in the crystal structure and the spectroscopic data, although the loss of up to one Chl b is possible. At present, we cannot completely rule out the possibility of mixed binding sites. This issue requires further simulations and analyses which are beyond the scope of the present paper. However, the problem of assigning site energies and Chl types to sites can be tackled by a combination of site-directed mutagenesis, optical difference spectroscopy and structure-based calculations. As an example, we demonstrated the principle feasibility of this approach by simulating the effect of replacing tyrosine 135 close to Chl a604 with phenylalanine, yielding a good match of computed and measured WMD spectra and thus confirming that Chl a604 has a redshifted site energy. The TED, however, is located at the Chl a611–a612 pair in keeping with earlier proposals.40 This assignment implies that the TED is essentially conserved between CP29 and LHCII despite the extra Chl a615 being strongly coupled to Chl a611 in CP29, the significant site energy differences of Chls a610 and a611 between the two PPCs and provided the temperature dependence of exciton states involving Chl a612 is taken into account. Uncertainties remain because of the missing N-terminus in the structure of CP29 and difficulties with an appropriate simulation of the CD spectra. In particular, the Chl b energy levels of LHCII will have to be reanalyzed in future work to obtain a consistent theoretical description of the two antenna complexes. Of prime importance is to gain a better understanding of the non-conservativity of the CD spectra. Such an understanding is necessary to further optimize the exciton Hamiltonian of the PSII antenna system for a faithful simulation of excitation energy flow.Acknowledgements
Financial support by the Austrian Science Fund (FWF: P 24774-N27) is gratefully acknowledged.References
- G. Renger, Primary Processes of Photosynthesis, Principles and Apparatus, Royal Society Chemistry Publishing, Cambridge, U.K., 2008 Search PubMed.
- R. Nevo, D. Charuvi, O. Tsabari and Z. Reich, Plant J., 2012, 70, 157–176 CrossRef CAS PubMed.
- R. Kouril, J. P. Dekker and E. J. Boekema, Biochim. Biophys. Acta, 2012, 1817, 2–12 CrossRef CAS PubMed.
- F. Müh, C. Glöckner, J. Hellmich and A. Zouni, Biochim. Biophys. Acta, 2012, 1817, 44–65 CrossRef PubMed.
- A. Amunts and N. Nelson, Plant Physiol. Biochem., 2008, 46, 228–237 CrossRef CAS PubMed.
- A. Amunts and N. Nelson, Structure, 2009, 17, 637–650 CrossRef CAS PubMed.
- F. Müh, T. Renger and A. Zouni, Plant Physiol. Biochem., 2008, 46, 238–264 CrossRef PubMed.
- R. Croce and H. van Amerongen, J. Photochem. Photobiol., B, 2011, 104, 142–153 CrossRef CAS PubMed.
- A. V. Ruban, M. P. Johnson and C. D. P. Duffy, Biochim. Biophys. Acta, 2012, 1817, 167–181 CrossRef CAS PubMed.
- M. Ballottari, J. Girardon, L. Dall'osto and R. Bassi, Biochim. Biophys. Acta, 2012, 1817, 143–157 CrossRef CAS PubMed.
- J. D. Rochaix, Antioxid. Redox Signaling, 2013, 18, 2184–2201 CrossRef CAS PubMed.
- S. Caffarri, R. Kouril, S. Kereiche, E. J. Boekema and R. Croce, EMBO J., 2009, 28, 3052–3063 CrossRef CAS PubMed.
- T. Renger and F. Müh, Phys. Chem. Chem. Phys., 2013, 15, 3348–3371 RSC.
- T. Renger, M. E. Madjet, M. Schmidt am Busch, J. Adolphs and F. Müh, Photosynth. Res., 2013, 116, 367–388 CrossRef CAS PubMed.
- A. Guskov, A. Gabdulkhakov, M. Broser, C. Glöckner, J. Hellmich, J. Kern, J. Frank, F. Müh, W. Saenger and A. Zouni, ChemPhysChem, 2010, 11, 1160–1171 CrossRef CAS PubMed.
- A. Guskov, J. Kern, A. Gabdulkhakov, M. Broser, A. Zouni and W. Saenger, Nat. Struct. Mol. Biol., 2009, 16, 334–342 CAS.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- F. H. M. Koua, Y. Umena, K. Kawakami and J. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3889–3894 CrossRef CAS PubMed.
- T. Barros and W. Kühlbrandt, Biochim. Biophys. Acta, 2009, 1787, 753–772 CrossRef CAS PubMed.
- Z. F. Liu, H. C. Yan, K. B. Wang, T. Y. Kuang, J. P. Zhang, L. L. Gui, X. M. An and W. R. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed.
- J. Standfuss, A. C. Terwisscha van Scheltinga, M. Lamborghini and W. Kühlbrandt, EMBO J., 2005, 24, 919–928 CrossRef CAS PubMed.
- X. W. Pan, M. Li, T. Wan, L. F. Wang, C. J. Jia, Z. Q. Hou, X. L. Zhao, J. P. Zhang and W. R. Chang, Nat. Struct. Mol. Biol., 2011, 18, 309–315 CAS.
- F. Müh, M. E. A. Madjet and T. Renger, J. Phys. Chem. B, 2010, 114, 13517–13535 CrossRef PubMed.
- T. Renger, M. E. Madjet, A. Knorr and F. Müh, J. Plant Physiol., 2011, 168, 1497–1509 CrossRef CAS PubMed.
- F. Müh and T. Renger, Biochim. Biophys. Acta, 2012, 1817, 1446–1460 CrossRef PubMed.
- A. A. Kavalenka, R. B. Spruijt, C. J. A. M. Wolfs, J. Strancar, R. Croce, M. A. Hemminga and H. van Amerongen, Biophys. J., 2009, 96, 3620–3628 CrossRef CAS PubMed.
- A. Pascal, C. Gradinaru, U. Wacker, E. Peterman, F. Calkoen, K. D. Irrgang, P. Horton, G. Renger, R. van Grondelle, B. Robert and H. van Amerongen, Eur. J. Biochem., 1999, 262, 817–823 CrossRef CAS.
- A. V. Ruban, P. J. Lee, M. Wentworth, A. J. Young and P. Horton, J. Biol. Chem., 1999, 274, 10458–10465 CrossRef CAS PubMed.
- S. K. Das and H. A. Frank, Biochemistry, 2002, 41, 13087–13095 CrossRef CAS PubMed.
- M. Rätsep, J. Pieper, K. D. Irrgang and A. Freiberg, J. Phys. Chem. B, 2008, 112, 110–118 CrossRef PubMed.
- X. M. Feng, X. W. Pan, M. Li, J. Pieper, W. R. Chang and R. Jankowiak, J. Phys. Chem. B, 2013, 117, 6585–6592 CrossRef CAS PubMed.
- X. M. Feng, A. Kell, J. Pieper and R. Jankowiak, J. Phys. Chem. B, 2013, 117, 6593–6602 CrossRef CAS PubMed.
- M. Stitt, Curr. Opin. Biotechnol., 2013, 24, 229–238 CrossRef CAS PubMed.
- D. I. Bennett, K. Amarnath and G. R. Fleming, J. Am. Chem. Soc., 2013, 135, 9164–9173 CrossRef CAS PubMed.
- F. Müh, M. E. Madjet, J. Adolphs, A. Abdurahman, B. Rabenstein, H. Ishikita, E. W. Knapp and T. Renger, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16862–16867 CrossRef PubMed.
- J. Adolphs, F. Müh, M. E. Madjet and T. Renger, Photosynth. Res., 2008, 95, 197–209 CrossRef CAS PubMed.
- F. Müh, M. E. Madjet and T. Renger, Photosynth. Res., 2012, 111, 87–101 CrossRef PubMed.
- M. Schmidt am Busch, F. Müh, M. E. Madjet and T. Renger, J. Phys. Chem. Lett., 2011, 2, 93–98 CrossRef CAS.
- J. Adolphs, F. Müh, M. E. Madjet, M. Schmidt am Busch and T. Renger, J. Am. Chem. Soc., 2010, 132, 3331–3343 CrossRef CAS PubMed.
- M. Mozzo, F. Passarini, R. Bassi, H. van Amerongen and R. Croce, Biochim. Biophys. Acta, 2008, 1777, 1263–1267 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS.
- S. Caffarri, F. Passarini, R. Bassi and R. Croce, FEBS Lett., 2007, 581, 4704–4710 CrossRef CAS PubMed.
- S. C. Baicu and M. J. Taylor, Cryobiology, 2002, 45, 33–48 CrossRef CAS.
- H. Schulze, O. Ristau and C. Jung, Biochim. Biophys. Acta, 1994, 1183, 491–498 CrossRef CAS.
- B. Rabenstein and E. W. Knapp, Biophys. J., 2001, 80, 1141–1150 CrossRef CAS.
- G. Kieseritzky and E. W. Knapp, Proteins, 2008, 71, 1335–1348 CrossRef CAS PubMed.
- J. Kong, C. A. White, A. I. Krylov, D. Sherrill, R. D. Adamson, T. R. Furlani, M. S. Lee, A. M. Lee, S. R. Gwaltney, T. R. Adams, C. Ochsenfeld, A. T. B. Gilbert, G. S. Kedziora, V. A. Rassolov, D. R. Maurice, N. Nair, Y. H. Shao, N. A. Besley, P. E. Maslen, J. P. Dombroski, H. Daschel, W. M. Zhang, P. P. Korambath, J. Baker, E. F. C. Byrd, T. Van Voorhis, M. Oumi, S. Hirata, C. P. Hsu, N. Ishikawa, J. Florian, A. Warshel, B. G. Johnson, P. M. W. Gill, M. Head-Gordon and J. A. Pople, J. Comput. Chem., 2000, 21, 1532–1548 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- T. Renger, M. E. Madjet, F. Müh, I. Trostmann, F. J. Schmitt, C. Theiss, H. Paulsen, H. J. Eichler, A. Knorr and G. Renger, J. Phys. Chem. B, 2009, 113, 9948–9957 CrossRef CAS PubMed.
- M. E. Madjet, F. Müh and T. Renger, J. Phys. Chem. B, 2009, 113, 12603–12614 CrossRef PubMed.
- M. E. Madjet, A. Abdurahman and T. Renger, J. Phys. Chem. B, 2006, 110, 17268–17281 CrossRef CAS PubMed.
- T. Renger and F. Müh, Photosynth. Res., 2012, 111, 47–52 CrossRef CAS PubMed.
- R. S. Knox and B. Q. Spring, Photochem. Photobiol., 2003, 77, 497–501 CrossRef CAS.
- F. Müh and A. Zouni, Biochim. Biophys. Acta, 2005, 1708, 219–228 CrossRef PubMed.
- D. Bashford, Front. Biosci., 2004, 9, 1082–1099 CrossRef CAS PubMed.
- T. Renger and R. A. Marcus, J. Chem. Phys., 2002, 116, 9997–10019 CrossRef CAS PubMed.
- G. Raszewski and T. Renger, J. Am. Chem. Soc., 2008, 130, 4431–4446 CrossRef CAS PubMed.
- V. I. Novoderezhkin, M. A. Palacios, H. van Amerongen and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 10493–10504 CrossRef CAS PubMed.
- J. S. Frähmcke and P. J. Walla, Chem. Phys. Lett., 2006, 430, 397–403 CrossRef PubMed.
- G. Raszewski, W. Saenger and T. Renger, Biophys. J., 2005, 88, 986–998 CrossRef CAS PubMed.
- C. C. Gradinaru, A. A. Pascal, F. van Mourik, B. Robert, P. Horton, R. van Grondelle and H. van Amerongen, Biochemistry, 1998, 37, 1143–1149 CrossRef CAS PubMed.
- R. Croce, M. G. Müller, R. Bassi and A. R. Holzwarth, Biophys. J., 2003, 84, 2508–2516 CrossRef CAS.
- J. Pieper, K. D. Irrgang, M. Rätsep, J. Voigt, G. Renger and G. J. Small, Photochem. Photobiol., 2000, 71, 574–581 CrossRef CAS.
- N. S. Ginsberg, J. A. Davis, M. Ballottari, Y. C. Cheng, R. Bassi and G. R. Fleming, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3848–3853 CrossRef CAS PubMed.
- J. M. Salverda, M. Vengris, B. P. Krueger, G. D. Scholes, A. R. Czarnoleski, V. Novoderezhkin, H. van Amerongen and R. van Grondelle, Biophys. J., 2003, 84, 450–465 CrossRef CAS.
- H. Rogl, R. Schödel, H. Lokstein, W. Kühlbrandt and A. Schubert, Biochemistry, 2002, 41, 2281–2287 CrossRef CAS PubMed.
- R. Remelli, C. Varotto, D. Sandona, R. Croce and R. Bassi, J. Biol. Chem., 1999, 274, 33510–33521 CrossRef CAS PubMed.
- T. K. Ahn, T. J. Avenson, M. Ballottari, Y. C. Cheng, K. K. Niyogi, R. Bassi and G. R. Fleming, Science, 2008, 320, 794–797 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed formulae for the calculation of linear optical spectra, excitonic couplings calculated with Poisson-TrEsp/HF-CIS (Table S5), site energy shifts calculated with different quantum chemical and electrostatic methods (Table S6), standard deviations between calculated and fitted site energies (Table S7), contributions of selected amino acid residues to site energy shifts of Chl a602 (Table S8) and a603 (Table S9), correlation between calculated and fitted site energies (Fig. S9), location of Lys 179 in LHCII and Ala 196 in CP29 relative to Chls a610 and a611 (Fig. S10), overlay of the peptide backbones of CP29 and LHCII and positions of Chls a602 and a615 relative to the backbone (Fig. S11). See DOI: 10.1039/c3cp55166k |
| ‡ More precisely, the long-lived bleaching in transient spectra cannot be due to the 0–0 transition of Chl a604. Other possibilities are a 0–1 transition of Chl a604 or of any other Chl a that belongs to an exciton domain involved in slow Chl a → Chl a EET. |
| This journal is © the Owner Societies 2014 |