 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-modified zinc oxide, a highly active vis-photocatalyst: the nature and reactivity of photoactive centres
Jonathan Z.
Bloh†
*,
Ralf
Dillert‡
and
Detlef W.
Bahnemann
Institut für Technische Chemie, Leibniz Universität Hannover, Callinstr. 3, 30167 Hannover, Germany. E-mail: bahnemann@iftc.uni-hannover.de
First published on 30th January 2014
Abstract
We recently reported a highly active photocatalyst, ruthenium-modified zinc oxide, which was found to be able to utilise the red part of the visible light spectrum for photocatalytic reactions [Bloh et al., Environ. Sci. Pollut. Res., 2012, 19, 3688–3695]. However, the origin and mechanism of the observed activity as well as the nature of the photoactive centres are still unknown. Herein, we expand on that by reporting a series of experiments specifically designed to unravel the mechanism of the visible light induced photocatalytic reactions. The absolute potentials of the valence and the conduction band edge are identified by the combined use of electrochemical impedance and UV-vis diffuse reflectance spectroscopy. The conduction band electron and the valence band hole activity are assessed through a novel approach tracing their signature oxidative species, i.e., hydrogen peroxide and hydroxyl radicals, respectively. Oxygen reduction currents are measured at different potentials to investigate the role of molecular oxygen as an electron scavenger as well as the underlying reduction pathways. Additionally, the photocatalytic activity of the samples is verified using another (ISO standard) degradation test, the gas-phase oxidation of nitric oxide. The experimental results reveal that the employed synthetic route yields a unique mixture of ruthenium(VI)-doped zinc oxide and ruthenium(VI) oxide particles with both forms of the ruthenium playing their own independent role in the enhancement of the photocatalytic activity. The ruthenium ions acting as dopants enable a better charge separation as well as the absorption of red light resulting in the direct promotion of electrons from the Ru(VI)-species to the conduction band. Both, the conduction band electrons and the thus formed Ru(VII) subsequently participate in the degradation of the pollutant molecules. The ruthenium dioxide particles, on the other hand, act as catalysts increasing the efficiency of the reaction by improving the oxygen reduction properties of the material.
1 Introduction
Since the discovery of water-splitting on illuminated titanium dioxide in the early 1970's, semiconductor photocatalysis has attracted ever increasing interest, owing to its ability to create reactive electron–hole pairs and strongly oxidising species upon illumination with light of sufficient energy.1–5 Consequently, there have been several initiatives to harness this effect in a variety of applications such as self-cleaning and self-sterilising surfaces, disinfection and deodorisation of indoor air, removal of air pollutants and waste water remediation.6–17 Photocatalysts are also used for the photo-electrochemical splitting of water as alternatives to photovoltaic systems in a hydrogen economy and for artificial photosynthesis.18–21 In particular, photocatalysis is currently considered for the large scale application in concrete and other construction materials in order to reduce ambient pollutant levels such as nitrogen oxides (NOx).22–24 This application has already made it out of the lab-scale and there are currently several real world pilot projects assessing the impact of photocatalytic materials such as paving stones and noise barriers on the air pollutant concentrations.25–27The major limitation at this point is that most of these applications are designed for the use of sunlight or artificial indoor lighting. Those light sources, however, mostly contain visible light with insufficient energy to excite the large band gap semiconductors commonly used for photocatalysis. Both, titanium dioxide and zinc oxide, the two most popular photocatalysts, require ultraviolet light for bandgap excitation. However, even under ideal conditions with the sun at the zenith near the equator (AM1.0), the ultraviolet part only constitutes about 5% of the sunlight.27 Ultraviolet light is also strongly scattered and absorbed in the atmosphere, so when the sun is at a lower angle and the light has to travel a longer path through the atmosphere to reach the Earth's surface, the ultraviolet portion is considerably lowered. This is the case in more extreme latitudes, in winter, or at any time of the day other than solar noon. Consequently, the UV portion usually constitutes significantly less than 5% of the solar energy available to initiate any photocatalytic process. In extreme cases, such as winter in northern Europe, only as little as 0.1% of the sunlight is ultraviolet light, even at solar noon.27
Applications designed for the use under such low UV conditions should therefore employ a photocatalyst capable of utilising part of the visible spectrum of the sunlight as well. Since none of the traditionally used photocatalysts are capable of doing so, a tremendous effort has been devoted to the development of vis-photocatalysts leading to the discovery of several such materials, mostly based on doped or metal modified titanium dioxide.28–30 However, even in the more successful cases such as nitrogen-, carbon- or sulphur-doped titanium dioxide, the extension of the absorption edge is usually limited to the blue part of the visible spectrum, leaving the majority of the visible light energy untapped.31–36 Also, while photocatalytically active under illumination with visible light, these materials often exhibit a considerably lower photocatalytic activity under UV illumination, possibly due to the introduction of crystallographic defects. Therefore, the remaining challenge is to extend the absorption edge even further and to utilise even the low energy portion of the visible light while not lowering the overall photocatalytic activity in the process.
Unfortunately, in most cases, knowledge of the underlying mechanism of the visible light induced photoreaction is limited to an observed smaller bandgap or an additional absorption edge. Changes in the actual positions of the band edges, the reaction mechanism itself and the nature of the photoactive centres are often unknown or poorly understood. This knowledge, however, is imperative for the development of next generation visible-light active photocatalysts and to precisely engineer photocatalysts with specific properties.
A major part of the early research in the field of photocatalysis was focused on zinc oxide,37–40 however, there was a steady decline of interest in this material afterwards. Instead, most of the investigations were then diverted to titanium dioxide, owing to its advantageous physical and chemical properties.41–44 However, due to the fact that zinc oxide has not been investigated as thoroughly as titanium dioxide, it may yet possess a lot of untapped potential.
One example for this is ruthenium-modified zinc oxide, which was just recently discovered as a promising photocatalyst. We previously reported that this catalyst exhibits exceptional photocatalytic activity under both, UV and visible light irradiation.45 This activity is especially remarkable since the reaction appears to be driven by red and even near-infrared light, which is the least energetic part of the visible light spectrum. The origin and mechanism of the observed activity as well as the nature of the photoactive centres, however, are still unknown.
With the present contribution we attempt to fill this knowledge gap by reporting a series of experiments specifically designed to unravel the mechanism of the visible light induced photocatalytic reaction. First, the photocatalytic activity of the samples is verified using another degradation test, i.e., the gas-phase oxidation of nitric oxide, the results of which are of critical importance for the application in air depollution. Then, in order to gain insight into the nature and reactivity of photo-generated charge carriers, the absolute positions of the valence and the conduction band edge are identified by the combined use of electrochemical impedance and UV-vis diffuse reflectance spectroscopy. Additionally, the conduction band electron and the valence band hole activity are observed by tracing their signature oxidative species, i.e., hydrogen peroxide and hydroxyl radicals, respectively. Finally, the role of molecular oxygen as an electron scavenger and its reduction pathways are investigated by measuring oxygen reduction currents at different potentials.
2 Experimental section
Synthesis
Ruthenium-modified zinc oxide was prepared using a facile, previously reported, sol–gel method.45 In a typical preparation, 60 mmol zinc acetate dihydrate (99.5%, Carl Roth GmbH, Germany) was suspended in 200 mL of ethanol (99.8%, Carl Roth GmbH, Germany). To stabilise the suspension, 20 mL of a complexing solution consisting of 3 mol L−1 diethanolamine (99%, Sigma Aldrich, USA) and 3 mol L−1 water in ethanol was added dropwise to the suspension, resulting in a clear solution. This solution was also used directly for the electrode preparation. For the powder synthesis, however, it was aged for 24 h under ambient conditions and the resulting gel was subsequently transferred into a furnace for calcination. The calcination was conducted under air with the applied temperature program being as follows: heating up to 100 °C at a rate of 2 K min−1, maintaining at 100 °C for 60 min, heating up to 500 °C with 2 K min−1, maintaining at 500 °C for 300 min and subsequently cooling to room temperature with 2 K min−1. Finally, the obtained powders were ground in an agate mortar. For the introduction of the ruthenium, a fraction of the zinc acetate dihydrate, corresponding to 0.1, 0.3 and 1.0 at% was substituted by ruthenium(III) acetylacetonate (97%, Sigma-Aldrich, USA).Physico-chemical characterisation
X-ray diffraction (XRD) spectra were collected on a STADI P diffractometer (STOE, Germany) with a positional sensitive detector using monochromatic CuKα radiation. Average volume weighted grain sizes and crystallite strain were determined from Williamson–Hall plots of the peak broadening using a shape factor of 0.9.The BET surface area of the samples was determined using a FlowSorb II 2300 (Micromeritics, USA) with a 30% nitrogen–70% helium gas mixture as the adsorbate.
UV-vis diffuse reflectance spectra were collected on a Cary 4000 (Varian) UV-vis spectrophotometer fitted with an integrating sphere. Polytetrafluoroethylene was used as reference and the wavelength range studied was 200 nm to 800 nm. The resulting reflectance spectra were transformed into apparent absorption spectra using the Kubelka–Munk function:46
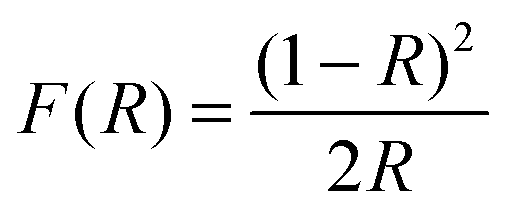 | (1) |
Nitric oxide (NO) oxidation
Measurements of the photonic efficiency for the oxidation of nitric oxide were carried out on the basis of the ISO standard 22197-1.49 Samples of the zinc oxide powders were pressed into a 5 × 10 cm2 acrylic glass moulding with a resulting uniform and planar powder surface with an area of 3.926 × 10−3 m2. These mouldings were placed in an acrylic glass reactor with a laminar volumetric flow rate of 5.114 × 10−5 m3 s−1 with a nitric oxide concentration of 1 ppm. The concentrations of NO, NO2 and total NOx in the outlet gas flow were monitored using an APNA-360 Ambient Monitor (Horiba, Japan). Each sample was measured in the dark until an equilibrium concentration was reached and afterwards under illumination until an equilibrium concentration was reached. For the irradiation with ultraviolet light, a Cleo Compact (Philips, Netherlands) light source in combination with a LC-HU02 bandpass filter (Laser Components GmbH, Germany) was employed. The resulting photon flux was determined using ferrioxalate actinometry50,51 and was found to be 1.121 × 10−5 mol s−1 m2. For experiments under visible light irradiation, a 500 W halogen lamp with a LC-Y420 bandpass filter (Laser Components GmbH, Germany) and a resulting photon flux of 8.609 × 10−5 mol s−1 m2 was used instead. Finally, the photonic efficiency ξ, which depicts the ratio of degraded molecules to impinging photons, was calculated according to eqn (2), where cd is the concentration under dark conditions, ci the concentration under illumination,![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) the volumetric flow rate, p the pressure, A the irradiated area, R the gas constant, T the absolute temperature and Φ the photon flux impinging the photocatalyst surface as determined by actinometry. The photonic efficiency was determined separately for NO, NO2 and total NOx.
the volumetric flow rate, p the pressure, A the irradiated area, R the gas constant, T the absolute temperature and Φ the photon flux impinging the photocatalyst surface as determined by actinometry. The photonic efficiency was determined separately for NO, NO2 and total NOx.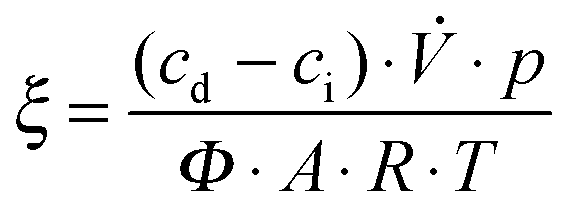 | (2) |
Electrochemical characterisation
Electrodes of indium doped tin oxide (ITO) glass coated with ruthenium-modified zinc oxide were prepared for the electrochemical characterisation. Freshly cleaned (through subsequent rinsing with 2-propanol and deionised water followed by drying in a dry nitrogen stream) 1 × 2 cm2 pieces of ITO were dip-coated at half their area (1 × 1 cm2) in the solution that was used for the powder synthesis, prior to ageing. The dip-coating procedure consisted of 20 dips with a drawing speed of 2 mm s−1 and subsequent calcination at the same temperature program as the powder synthesis. The whole procedure was repeated four times to yield 351 ± 66 nm thick layers of the modified zinc oxide, as estimated by gravimetric analysis. Finally, a copper wire was glued to the uncoated part of the glass piece with a conducting epoxy resin and the whole electrode except the coated area was insulated with silicone.All electrochemical measurements were performed using a IviumStat potentiostat (Ivium Technologies, Netherlands) under dark conditions in a three-electrode setup, using the sample-coated ITO-glass as a working electrode, a platinum coil as a counter electrode and an Ag/AgCl (3 mol L−1 KCl) reference electrode. A 0.1 mol L−1 KCl solution, brought to pH 10 with KOH, was used as the electrolyte. This pH was chosen since zinc oxide exhibits its highest chemical and electrochemical stability at pH 9–11.52,53 All potential values presented herein are corrected for the reference electrode and pH and consequently given as vs. RHE (reversible hydrogen electrode).
Impedance spectra were collected in the potential range of +0.8 to −0.2 V vs. RHE with a step width of 50 mV. Each spectrum was recorded at a fixed potential in the frequency range of 1 × 10−1 Hz to 1 × 105 Hz with 10 steps per decade and an amplitude of 20 mV. The spectra were subsequently fitted to an equivalent circuit to derive the values for the space charge layer capacitance (Csc) in dependence of the applied potential. In this case, a double randles circuit (used to represent a simple electrode/electrolyte interface) was used as an equivalent circuit to represent the electrolyte/zinc oxide interface and the interface between zinc oxide and the ITO glass, cf.Fig. 1.54 Instead of ordinary capacitance, constant phase elements (CPE) were used to account for the unideal behaviour of the uneven surface.55 The exponential parameter of the CPE was found to be between 0.9 and 1.0, indicating almost ideal capacitance behaviour. Consequently, the space charge layer capacitance was assumed to be equal to the pseudo-capacitance of the CPE of the electrolyte/zinc oxide interface.55,56
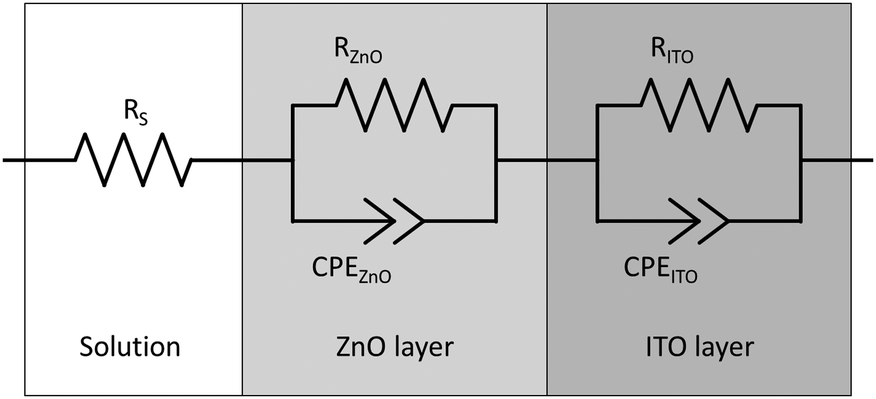 | ||
| Fig. 1 The double randles equivalent circuit used to analyse the impedance spectra, consisting of a solution resistance and a charge transfer resistance and a constant phase element for the electrolyte/zinc oxide and the zinc oxide/ITO interface, respectively. | ||
A Mott–Schottky plot was then constructed for each sample by plotting the inversed square of the space charge layer capacitance Csc normalised for the contact area against the potential at which it was derived. The extrapolation of the linear trend in the Mott–Schottky plot to the x-axis (potential) led to the quantity 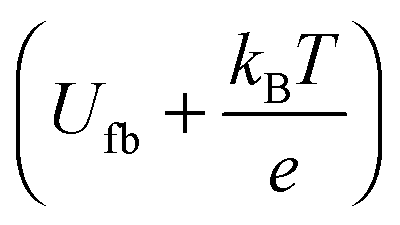 (see eqn (3)) from which the flat band potential (Ufb, also representing the Fermi level since there is no band bending at flat band conditions) for a given semiconductor oxide could be calculated.57–59
(see eqn (3)) from which the flat band potential (Ufb, also representing the Fermi level since there is no band bending at flat band conditions) for a given semiconductor oxide could be calculated.57–59
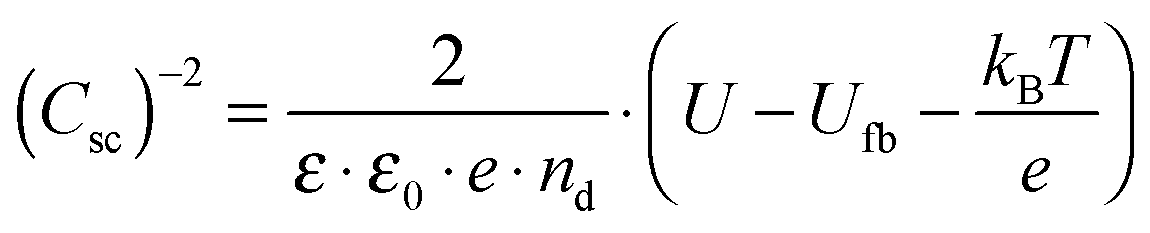 | (3) |
Additionally, the donor density nd was calculated from the slope of the linear part of this plot using a value of 8.15 for the dielectric constant of zinc oxide.60–62 As the donor density was found to be higher (≥1 × 1019 cm−3) than the effective density of states in the conduction band (5.824 × 1018 cm−3 assuming a value of 0.38 me for the electron effective mass63) in all cases, the energy difference between the Fermi level and the conduction band edge (Ucb) is very small 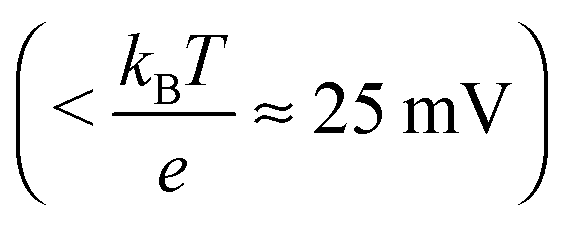 and both values can be assumed to be approximately identical (Ucb ≈ Ufb).
and both values can be assumed to be approximately identical (Ucb ≈ Ufb).
Oxygen reduction currents were obtained by performing a potentiodynamic scan both in a nitrogen and an oxygen-saturated solution in a potential range of +0.8 V to −0.2 V vs. RHE with a step width of 10 mV and a scan rate of 1 mV s−1. The difference of both scans constitutes the oxygen reduction current. Additionally, chronoamperometric measurements with a duration of 1 h were done to ensure the observed currents were stable and not transient adsorption phenomena.
Quantification of oxidative species
For the determination of the amount of photocatalytically formed hydroxyl radicals, a previously described fluorescence based method was adopted.64,65 For the analysis, 10 mmol L−1 of coumarin has been added to a suspension of 10 μg mL−1 of the powder sample in 0.1 mol L−1 potassium chloride, brought to pH 10 with potassium hydroxide. This suspension was then illuminated for a set length of time, either by UV or by visible light (illumination setup identical as that used for the NO degradation test). Hydroxyl radicals formed during the illumination react with the coumarin, forming fluorescent 7-hydroxycoumarin. Subsequently, the fluorescence of the product (λex = 332 nm, λem = 450 nm) was measured using a Spectrofluorophotometer RF-540 (Shimadzu, Japan). Finally, the photonic efficiency of the reaction was calculated as the ratio of the rate of the formed product to the photon flux.Hydrogen peroxide evolution was measured in a similar manner, using a suspension of 10 μg mL−1 of the powdered sample in 0.1 mol L−1 potassium chloride, brought to pH 10 with potassium hydroxide also containing 2 mmol L−1 potassium acetate as a hole scavenger.40,65,66 This suspension was then irradiated for a set amount of time and subsequently filtered through a 0.45 μm PVDF-filter (CHROMAFIL(R) Xtra PVDF-45/25, Macherey-Nagel, Germany). A 2 mL aliquot of this filtrate was then added to 100 μL of the development reagent (160 mg L−1 freshly recrystallised p-hydroxyphenylacetic acid and 40 U L−1 horseradish peroxidase in 0.1 Tris-buffer pH 8.8). This reagent will catalyse the dimerisation of p-hydroxyphenylacetic acid to a fluorescent product, i.e., 5,5′-dicarboxymethyl-2,2′-dihydroxybiphenyl (λex = 315 nm, λem = 410 nm) which was subsequently analysed using the spectrofluorophotometer followed by the transformation of the hydrogen peroxide evolution rate into photonic efficiency values.
3 Results
Three samples of ruthenium-modified zinc oxide and, for comparison, a pure zinc oxide sample were synthesised using the facile, sol–gel based method described above. The ruthenium-content of the samples was 0.1, 0.3 and 1.0 at%. The materials were confirmed as being zinc oxide using X-ray diffraction analysis, cf.Fig. 2. A control sample constituting of 100 at% ruthenium prepared under the same synthetic conditions yielded a mixture of metallic ruthenium and ruthenium dioxide. The diffractograms of the modified zinc oxides also revealed a small quantity of ruthenium dioxide present, but no metallic ruthenium was detected. The primary particle size as determined by Williamson–Hall plots was 56 nm for the unmodified zinc oxide and 44 nm to 50 nm for the ruthenium modified ones, indicating a reduction in size of 10% to 20% by the presence of ruthenium. Inhomogeneous strains were not detected in any of the samples with the strain parameter always being lower than 0.4%. The zinc oxide lattice parameters for the pure zinc oxide agree well with previously reported values.63 They were relatively unchanged upon addition of ruthenium, only at the highest concentration, i.e. 1.0 at%, there was a slight reduction of 0.06% in both lattice parameters (see Table 1 for the complete set of data).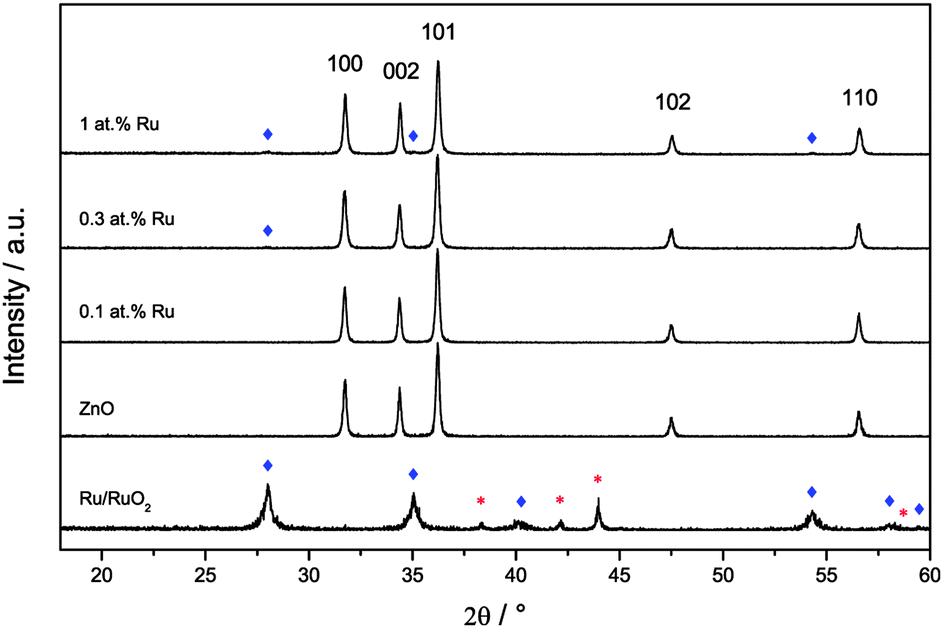 | ||
| Fig. 2 X-ray diffractograms of the synthesised samples and the pure ruthenium control. Reflexes belonging to hexagonal zinc oxide are marked by their respective faces, ruthenium dioxide by blue diamonds and metallic ruthenium by red asterisks. | ||
| Sample | Size/nm | Strain/% | a/Å | c/Å | BET/m2 g−1 |
|---|---|---|---|---|---|
| Pure ZnO | 55.7 ± 3.7 | 0.03 ± 0.01 | 3.255 | 5.216 | 5.26 |
| ZnO + 0.1 at% Ru | 49.5 ± 10.0 | 0.02 ± 0.01 | 3.256 | 5.217 | 6.54 |
| ZnO + 0.3 at% Ru | 44.4 ± 2.9 | 0.01 ± 0.01 | 3.256 | 5.217 | 6.67 |
| ZnO + 1.0 at% Ru | 48.5 ± 8.1 | 0.02 ± 0.03 | 3.253 | 5.213 | 8.20 |
Since photocatalysis is a surface phenomenon, the surface area should play a pivotal role in the activity of the catalysts. The surface area values of the samples, measured by the BET-method, are given in Table 1. While the surface area of the pure zinc oxide sample is rather small (5.26 m2 g−1), it gradually increases with higher ruthenium content, reaching 8.20 m2 g−1 at 1.0 at% Ru, an increase of 56%. It should be noted that these surface area values are smaller by a factor of 2.7 to 3.6 than expected from the particle sizes calculated from the XRD data, indicating that the particles might be considerably agglomerated.
Optical spectroscopy was used to measure the light absorption capabilities and the optical band gaps of the samples. The obtained diffuse reflectance spectra were transformed using the Kubelka–Munk function and then used to construct Tauc plots for the band gap determination. Pure zinc oxide showed an optical band gap of 3.27 ± 0.01 eV, which is in excellent agreement with other reports.67–69 This band gap was unchanged by the ruthenium modification, measuring the exact same value for all samples.
The visible light absorption of the samples, however, is changed drastically upon ruthenium addition, cf.Fig. 3. While pure zinc oxide appears as a pristine perfect white powder, modification with ruthenium turned the samples dark green, more pronounced at a higher concentration. Ruthenium-modified zinc oxides apparently exhibit a pronounced optical absorption of red light, as seen in their absorption spectra. While there is also some absorption over the whole visible spectrum, the main spectral feature that is apparent upon ruthenium addition is a broad absorption peak centred at 800 nm. Since the optical band gap of the samples was unchanged, it is likely that this absorption feature will be responsible for any visible light response of the materials.
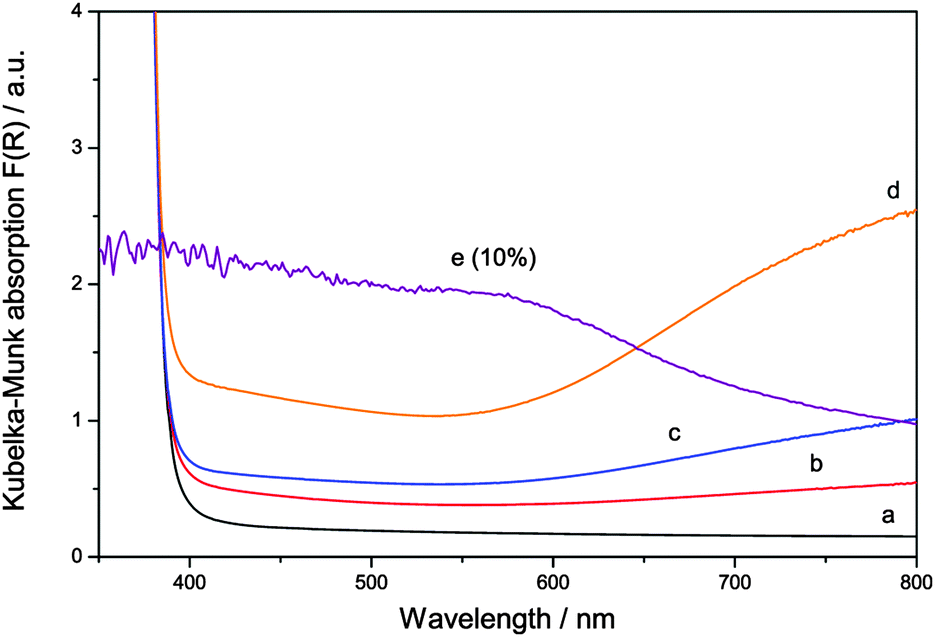 | ||
| Fig. 3 The optical absorption of pure zinc oxide (a, black) and of ruthenium modified zinc oxide with 0.1 (b, red), 0.3 (c, blue) and 1.0 at% (d, orange) ruthenium content as well as the Ru/RuO2 reference sample (e, purple, resized to 10% of its value) given as Kubelka–Munk transformed apparent absorption as calculated from the diffuse reflectance spectra. | ||
The photocatalytic activity of the samples has previously been reported45 for the gas-phase degradation of acetaldehyde, according to ISO 22197-2.70 The measurements showed that for both ultraviolet and visible light irradiation, the sample containing 0.1 at% ruthenium exhibited the highest activity, surpassing the pure zinc oxide sample by a factor of 3.6 and 168, respectively.45 In addition to that, the activity measurements are supplemented in the present report by gas-phase nitric oxide oxidation measurements, according to ISO 22197-1.49 Nitric oxide (NO) is a major air pollutant compound and can be photocatalytically oxidised to nitrate in a three-step reaction, eqn (4).71 Each step represents the transfer of a single electron and can be realised by the reaction with either a valence band hole, hydroxyl radicals or reactive oxygen species derived from reduced molecular oxygen, i.e. superoxide or hydrogen peroxide. It should be noted that both intermediate products, nitrous acid and nitrogen dioxide, are toxic compounds, more so than nitric oxide itself. As a consequence, formation and release of these intermediates should be minimised for an efficient detoxification of the air.
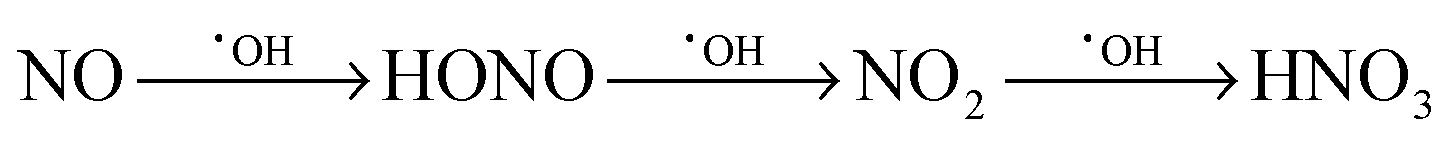 | (4) |
To achieve a good comparability with other reports and to put values into perspective, two commercially available photocatalysts were also measured as standards. One is the well known Aeroxide P25 (Evonik Degussa, Germany), a titanium dioxide based UV photocatalyst with both rutile and anatase phases. The other standard is PP10 (Toho, Japan), a visible light photocatalyst based on sulphur-doped anatase.
The results of the activity measurements under ultraviolet illumination are depicted in Fig. 4. In terms of nitric oxide removal, the titanium dioxide standards easily outperform all zinc oxide catalysts, with photonic efficiencies of 0.76% and 1.97% for P25 and PP10, respectively. Pure zinc oxide achieves a value of 0.45%, while the ruthenium-modified samples show even smaller activities of 0.28 to 0.35%.
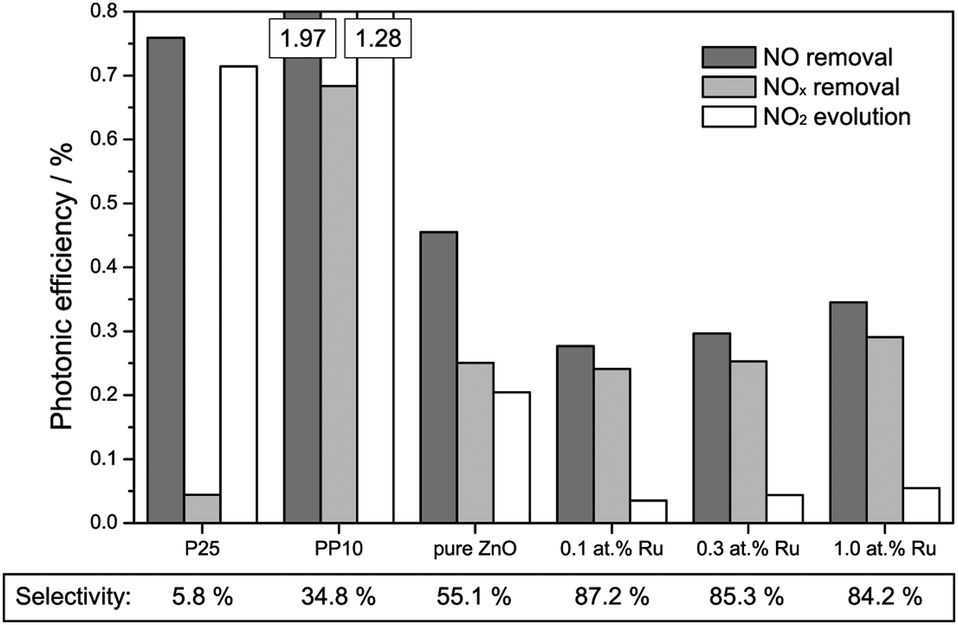 | ||
| Fig. 4 Photonic efficiencies for the removal of NO (dark grey bars), NOx (light grey bars) and for the evolution of NO2 (white bars) under illumination with ultraviolet light. Where the scale exceeds the boundaries exact numbers are given on the top of the bar. Additionally, the selectivity of the NO conversion to nitrate is given at the bottom of the figure. | ||
However, for a correct evaluation of the activity, only the complete oxidation of nitric oxide to nitrate, i.e., the removal of NOx, should be considered. An incomplete oxidation, producing significant amounts of (more toxic) nitrogen dioxide, may actually have detrimental effects on the air quality, and should at the very least not be counted as an improvement.
The total NOx removal rates of the titanium dioxide standards are in fact significantly lower than their NO removal rates, with photonic efficiencies of 0.04% and 0.68% for P25 and PP10, respectively. This is a consequence of their low selectivity for the total NO oxidation towards nitrate of 5.8% and 34.8%, respectively. In fact, in both cases the majority of the converted nitric oxide was detected as nitrogen dioxide in the outlet gas stream. The zinc oxide powders on the other hand display a significantly higher selectivity for the complete NO oxidation. While pure zinc oxide shows a selectivity of 55.1%, selectivity for the ruthenium-modified samples ranges from 84.2% to 87.2%. This results in NOx removal photonic efficiencies of 0.25% for the pure zinc oxide remaining almost unchanged for the ones with ruthenium modification (0.24% to 0.29%).
More importantly, the nitric oxide oxidation was also studied under visible light irradiation, the results of the measurements are displayed in Fig. 5. When only the removal of nitric oxide is considered, the PP10 standard catalyst powder shows the highest activity, achieving a photonic efficiency of 0.269%. Both, P25 and pure zinc oxide exhibit less activity as expected of pure UV-active photocatalysts, showing efficiencies of 0.063% and 0.007%, respectively. The fact that they display any activity at all is probably due to residual UV intensity “leaking” through the filter and, in the case of P25, the fact that rutile can absorb part of the blue spectrum. The ruthenium-modified zinc oxides all exhibit significantly enhanced visible light activity compared to the pure zinc oxide sample. Their respective photonic efficiencies, ranging from 0.043% to 0.086% are (in the case of the sample with 0.1 at% ruthenium content) up to 11.9 times higher than that of pure zinc oxide.
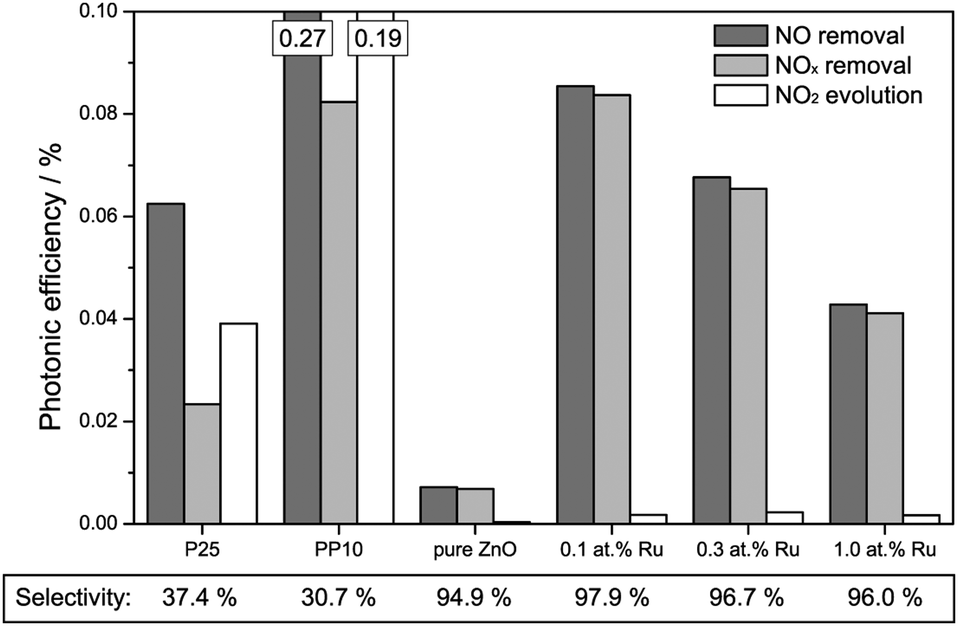 | ||
| Fig. 5 Photonic efficiencies for the removal of NO (dark grey bars), NOx (light grey bars) and for the evolution of NO2 (white bars) under illumination with visible light. Where the scale exceeds the boundaries exact numbers are given at the top of the bar. Additionally, the selectivity of the total NO conversion to nitrate is given at the bottom of the figure. | ||
This behaviour is not significantly altered when the total NOx removal is considered since all zinc oxide samples feature at least 94.9% selectivity towards nitrate formation, i.e., they only form traces of NO2 during the reaction. The titanium dioxide based standards, however, show similarly low selectivity as seen under ultraviolet illumination: 37.4% and 30.7% for P25 and PP10, respectively. As a consequence of the vastly different selectivity, it is the 0.1 at% Ru sample which displays the highest total NOx removal rate with a photonic efficiency of 0.084% rather than the PP10 sample even though the latter has a more than three times higher NO removal rate.
The following experiments were carried out to further elucidate the mechanism of the visible light induced photoactivity. One of the most characteristic properties of a semiconductor is the absolute position of its band edges, i.e., as it governs which reactions are possible after photoexcitation. Since the position and consistence of the bands can change upon doping with foreign elements, so can the possible reactions. Therefore, the positions of the band edges need to be determined to elucidate the mechanism of the photocatalytic reactions of a doped semiconductor. As a first step, the respective conduction band edges were measured using electrochemical impedance spectroscopy and Mott–Schottky plots. As shown in Fig. 6, the Mott–Schottky plots feature a linear regime which could be extrapolated to the x-axis to calculate the respective conduction band edge. Pure zinc oxide yielded a conduction band edge of −0.31 V vs. RHE, well in agreement with previously reported values.69 The modification with ruthenium gradually shifted the conduction band edge to more positive values, varying from −0.24 V to −0.14 V vs. RHE.
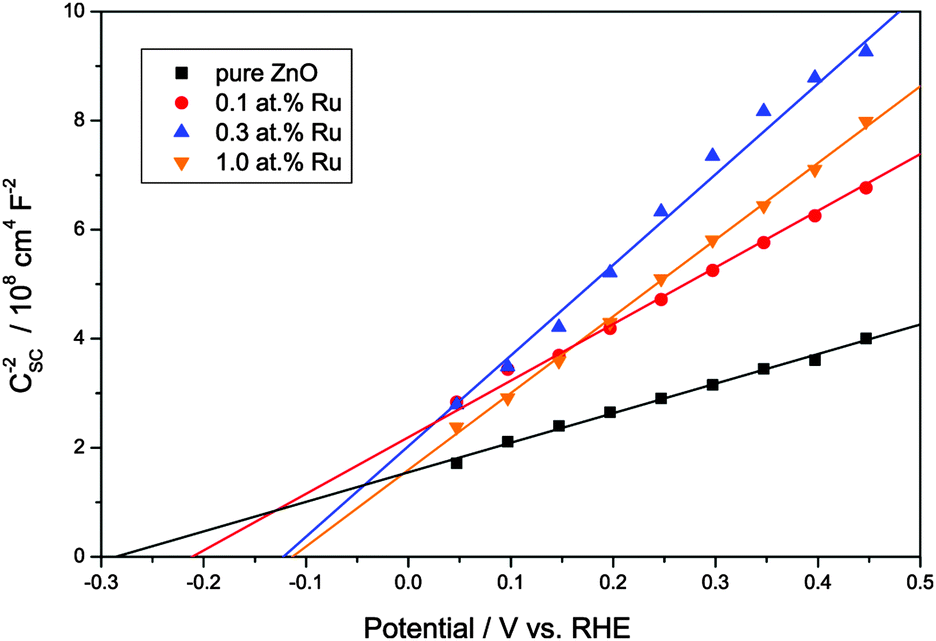 | ||
Fig. 6 Mott–Schottky plots of the synthesised zinc oxide powders in the linear regime. The intercept of the linear extrapolation with the x-axis (potential) marks the flatband potential as 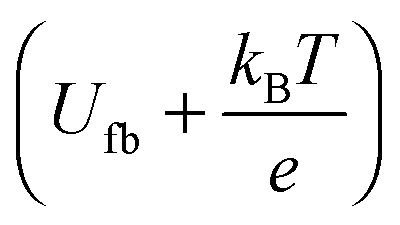 . . | ||
The valence band position is accordingly calculated by adding the value of the band gap, obtained from optical spectroscopy, to the conduction band potential, see Fig. 7. While not exactly known at this point, the mid-band gap state responsible for the visible light absorption is approximately positioned at 1.64 ± 0.09 eV above the valence band, depending on whether the excitation occurs from the valence band to the mid gap state or from the latter to the conduction band. The difference between both cases is relatively small since the visible light excitation energy is about half the value of the band gap, hence either case yields a similar result.
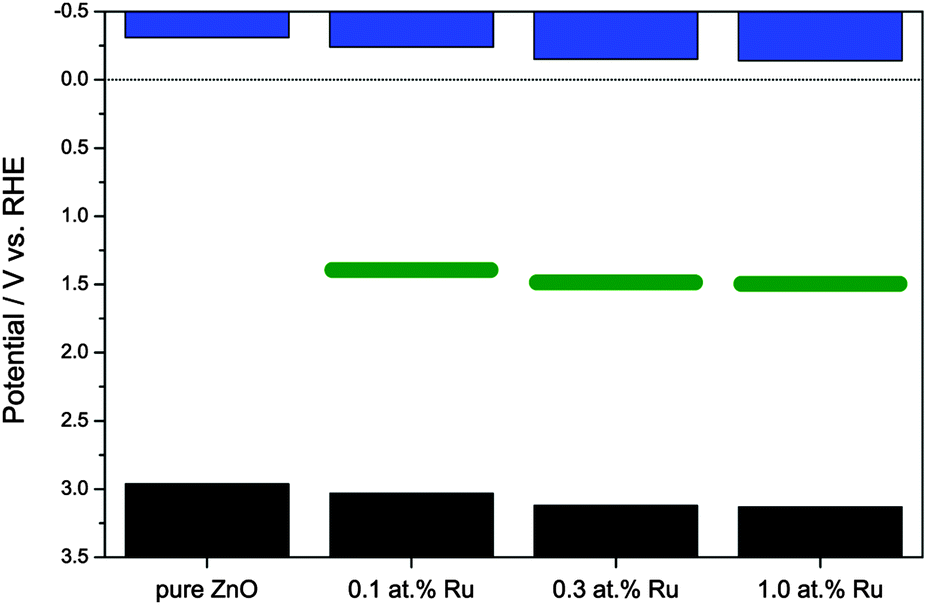 | ||
| Fig. 7 Band structure of the synthesised zinc oxide powders. Displayed are, from top to bottom, the conduction band (blue bars), the approximate position of the mid-band gap energy level (green bars), and the valence band (black bars). | ||
The Mott–Schottky plots also allow the determination of the donor density from the slope of the linear regime. This was found to be 3.14 × 1019 cm−3 for the pure ZnO powder and to be slightly lower for the ruthenium-modified samples with 1.65 × 1019, 1.13 × 1019 and 1.35 × 1019 cm−3 for 0.1, 0.3 and 1.0 at% ruthenium content, respectively. This lower donor density might be a consequence of a lower number of oxygen vacancies in the zinc oxide, which are the primary source of free conduction band electrons in zinc oxide.
Since in the majority of applications, molecular oxygen is the only available electron acceptor, its reduction mechanism plays a pivotal role for most photocatalytic reactions. There are several possible reaction routes, depending on how many electrons are transferred in the reaction:
| O2 + H+ + e− → HO2˙ E0 = −0.05 VRHE (ref. 72) | (5) |
| O2 + 2H+ + 2e− → H2O2 E0 = +0.70 VRHE (ref. 73) | (6) |
| O2 + 4H+ + 4e− → 2H2O E0 = +1.23 VRHE (ref. 73) | (7) |
In unmodified semiconductors, mainly the one-electron reduction is observed since there is no efficient mechanism to store electrons and they are therefore usually only locally available as single electrons. For this reason, materials with a conduction band edge more positive than −0.046 V vs. RHE (WO3, Fe2O3) are unable to reduce molecular oxygen in an efficient way and are thus photocatalytically inactive unless supplied with an alternative electron acceptor. By modifying these materials with suitable co-catalysts, multi-electron processes can be enabled and this limitation can thus be overcome. A good example for this approach is the preparation of photocatalytically active tungsten oxide loaded with platinum.74,75
In order to study whether the modified zinc oxides are also able to promote the multi-electron reduction of oxygen, the oxygen reduction currents were measured at different potentials, cf.Fig. 8. As a positive control, a pure zinc oxide loaded with 0.1 wt% platinum was used, prepared by a method described elsewhere.65,76 Platinum is well known to act as an electron sink and to thus enable the multi-electron oxygen reduction when loaded onto a photocatalyst.
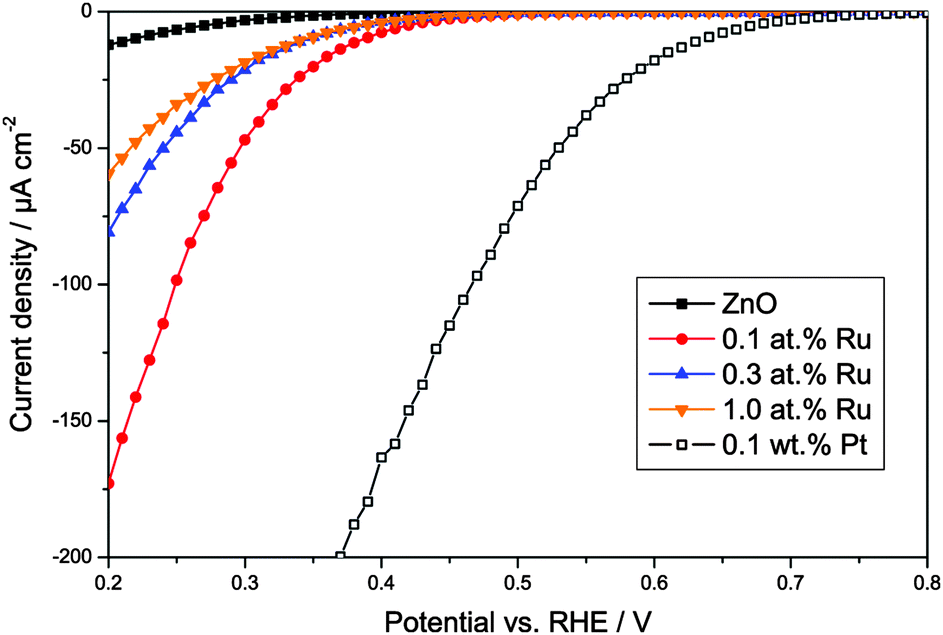 | ||
| Fig. 8 Oxygen reduction currents versus applied potential for pure zinc oxide, the synthesised ruthenium-modified zinc oxides, and, as a positive control, zinc oxide loaded with 0.1 wt% platinum. | ||
The unmodified zinc oxide shows a negligible oxygen reduction current at positive potentials, indicating that no multi-electron oxygen reduction is happening. This behaviour was expected for this ordinary unmodified photocatalyst. Once modified with platinum though, a significant reduction current is observed up to a potential of about +0.7 V vs. RHE, indicating that at least a two-electron reduction is happening, the potential for which is +0.695 V vs. RHE. As shown in Fig. 8, the ruthenium-modification enables reduction currents at higher potentials as well. While the onset of the reduction current and the magnitude are not as high as with platinum, they are significantly enhanced when compared to ordinary zinc oxide. The reduction current onset is observed at a potential of about +0.45 V vs. RHE, clearly indicating a two-electron electron reduction. The highest current was observed with the sample modified with 0.1 at% ruthenium, surpassing that of the other two ruthenium-modified samples by a factor of two to three and that of pure zinc oxide by a factor of 14. These results suggest that the ruthenium-modification enables the two-electron oxygen reduction in a similar but less efficient way as the platinum-loading.
In order to assess the role of conduction band electrons and valence band holes in the mechanism of the visible light induced photocatalytic reaction, the nature of the photogenerated oxidative species was studied in detail. Conduction band electrons primarily react with molecular oxygen to eventually form hydrogen peroxide, either directly in a two-electron reaction (eqn (8)) or with superoxide as the intermediate (eqn (9)–(11)).
| O2 + 2H+ + 2ecb− → H2O2 E0 = +0.70 VRHE (ref. 73) | (8) |
| O2 + H+ + ecb− → HO2˙ E0 = −0.05 VRHE (ref. 72) | (9) |
| HO2˙ + H+ + ecb− → H2O2 E0 = +1.47 VRHE (ref. 72) | (10) |
| 2HO2˙ → H2O2 + O2 | (11) |
The amount of hydrogen peroxide formed through the symproportionation of hydroxyl radicals formed by valence band holes should be negligible, especially since a hole or hydroxyl radical scavenger (acetate) was used in the respective experiments.77 Therefore, any detected hydrogen peroxide should be a very specific indicator for a conduction band activity.
Fig. 9 shows the rate of the hydrogen peroxide formation under illumination with ultraviolet or visible light, expressed as photonic efficiency. When the photocatalysts are illuminated with ultraviolet light, the amount of detected hydrogen peroxide is relatively constant, measuring a photonic efficiency of 0.33% for the pure zinc oxide and from 0.24% to 0.36% for the ruthenium modified ones.
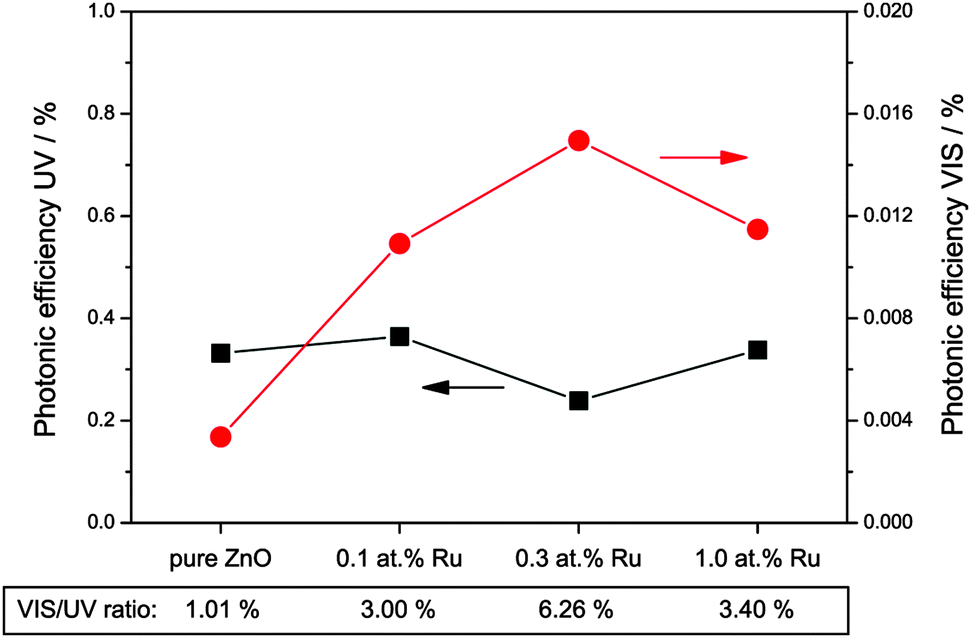 | ||
| Fig. 9 Photonic efficiencies of hydrogen peroxide formation in the presence of a hole scavenger and under illumination with ultraviolet (black squares, left scale) or visible (red dots, right scale) light. Additionally, the ratio of visible to ultraviolet efficiency is given at the bottom of the figure. | ||
However, when the samples are exposed to visible light only, they exhibit a significantly different photocatalytic behaviour. Under these conditions, pure zinc oxide exhibits a photonic efficiency for hydrogen peroxide formation of 0.0036%, i.e., about one percent of the rate observed under ultraviolet conditions. The modification with ruthenium increases the amount of hydrogen peroxide formed under visible light irradiation, with the photonic efficiencies measured being 0.0109%, 0.0150% and 0.0115% for 0.1, 0.3 and 1.0 at% ruthenium, respectively. This corresponds to a visible to ultraviolet ratio of 3% to 6%, up to six times higher than for pure zinc oxide. While the visible light activity of the ruthenium-free sample may be attributed to residual UV irradiation in the experiment, this “leakage activity” should then be a fixed percentage of the UV activity. Consequently, the rate observed for the ruthenium modified samples represents hydrogen peroxide formation due to “real” visible light activity, since the visible to UV ratio exceeds the one percent “leakage activity” by a factor of three to six.
The involvement of valence band holes in the mechanism was investigated in a similar manner. The signature oxidative species of valence band holes are generally considered to be hydroxyl radicals, which are predominantly formed by the reaction of the holes with water, eqn (12). The presence of hydroxyl radicals should therefore indicate an involvement of valence band holes in the overall reaction mechanism.
| H2O + hvb+ → ˙OH + H+ E0 = +2.72 VRHE (ref. 72) | (12) |
The formation of hydroxyl radicals was determined indirectly by a fluorometric assay based on the reaction of coumarin with hydroxyl radicals. This reaction forms a single, fluorescent product (7-hydroxycoumarin) and while not necessary reflecting the total number of hydroxyl radicals, it should be proportional to it. The results of this assay are displayed in Fig. 10. The photonic efficiencies observed are about three orders of magnitude lower than for the hydrogen peroxide measurements, i.e., an efficiency of only 4.5 × 10−6 was found for the pure zinc oxide under ultraviolet illumination. The introduction of ruthenium to the zinc oxide results in an increase in the photonic efficiency to 5.7 × 10−6 to 7.5 × 10−6, with the highest value being obtained for the sample with 0.1 at% ruthenium.
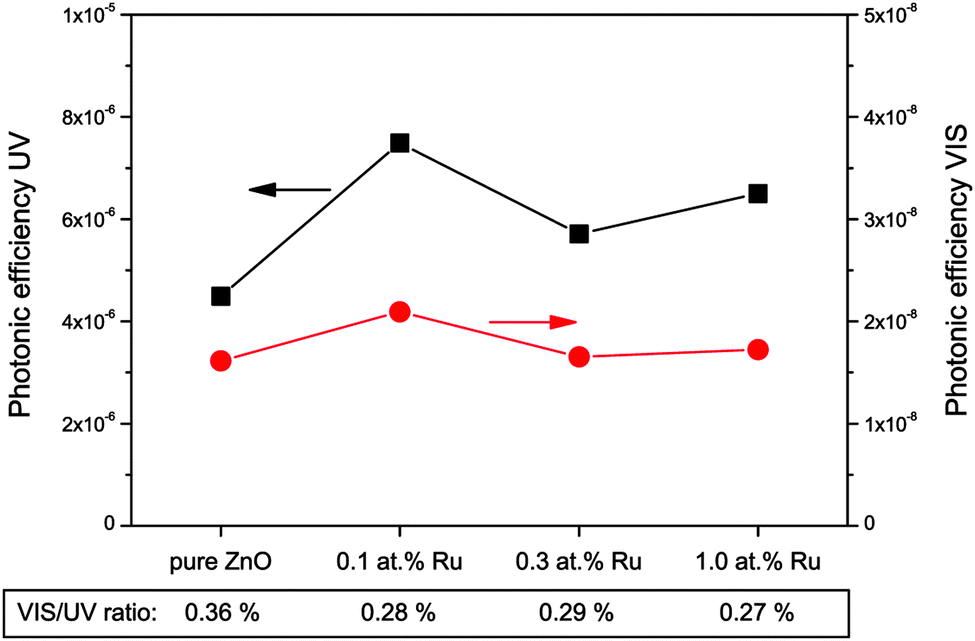 | ||
| Fig. 10 Photonic efficiencies of coumarin to 7-hydroxycoumarin conversion as an indicator for hydroxyl radical formation under illumination with ultraviolet (black squares, left scale) or visible (red dots, right scale) light. Additionally, the ratio of visible to ultraviolet efficiency is given at the bottom of the figure. | ||
If visible light is used instead, the situation is very similar. The observed photonic efficiencies are smaller by a factor of about 300, starting from 1.6 × 10−8 for pure zinc oxide and reaching values from 1.7 × 10−8 to 2.1 × 10−8 for the ruthenium modified powders. This also results in similar visible to ultraviolet ratios of 0.36% for the unmodified and 0.27% to 0.29% for the ruthenium containing samples. This suggests that the observed reaction under visible light is probably the result of residual ultraviolet light and not related to the mechanism of the visible light induced photocatalytic reaction.
4 Discussion
Photocatalytically highly active materials were obtained with the addition of ruthenium to zinc oxide. These ruthenium-modified zinc oxides exhibit increased photocatalytic activities upon illumination with ultraviolet light as compared to pristine zinc oxide. They are also able to utilise visible light with their visible light activity surpassing even the best reference material used (Toho PP10). The visible light activity of the samples was observed for both acetaldehyde and nitric oxide gas phase degradation measurements. The highest activities were recorded using zinc oxide with a ruthenium content of 0.1 at%, both for ultraviolet- and visible light illumination.These high photocatalytic activities are especially remarkable since the materials exhibit a rather low surface area as compared to the titanium dioxide standards used. As photocatalysis is a surface phenomenon, it is usually assumed that a high surface area contributes to a high activity.78 However, the prepared ruthenium-modified zinc oxide samples only show a specific BET surface area of only up to 8.2 m2 g−1 while the titanium dioxide standards show 50 m2 g−1 to over 300 m2 g−1. This indicates that the degradation tests used for the measurements are not operated in an adsorption-governed regime and are therefore ideal for measuring the actual photocatalytic activity of the materials rather than their adsorption capabilities.
The modification with ruthenium does not have any significant effect on the morphology of the zinc oxide particles. The particle size of the ruthenium-modified samples is slightly decreased, ranging from 44 nm to 48 nm as compared to 56 nm for pristine zinc oxide prepared by the same method. Correspondingly, the surface area of the former is a little larger, showing values of 6.54 m2 g−1 to 8.20 m2 g−1 in contrast to the 5.26 m2 g−1 for unmodified zinc oxide. This can be explained by the decreased particle size and the thus larger geometric surface assuming a roughly spherical particle shape. Another explanation is the presence of small amounts of RuO2-particles (as evidenced by XRD) also contributing to the overall surface area. These small changes in the morphology are, however, unlikely to be the cause for the dramatic increase in photocatalytic activity upon ruthenium addition.
The increased photocatalytic activity under illumination with ultraviolet light can instead be explained by an improved charge separation through the introduction of electron- and/or hole-traps as proposed earlier.45,79–81 Ruthenium ions occupying zinc positions in the zinc oxide lattice enable the immobilisation of charge carriers and thus a better charge separation. This leads to a decrease in the recombination rate and therefore an increase in the photocatalytic activity.
Another reason for the increased photocatalytic activity could also be the improved oxygen reduction properties of the ruthenium-modified samples. Since for pristine zinc oxide, the conduction band potential is just barely above the one-electron-reduction potential of oxygen (−0.31 and −0.05 V vs. RHE, respectively), this is a potentially rate-limiting factor. Although the modification with ruthenium lowers the conduction band even further to −0.24 V to −0.14 V vs. RHE, the oxygen-reduction current measurements suggest that these materials can also reduce molecular oxygen via a two-electron transfer process, utilising a much lower potential at +0.70 V vs. RHE, resulting in a much higher actual potential difference. Additionally, the observed shift of the valence band potential to more positive values could have a beneficial effect on the activity. The difference in potentials between valence band and hydroxyl radical formation (+2.98 and +2.73 V vs. RHE, respectively) is again very small for pure zinc oxide. The ruthenium modified samples exhibit a higher difference in potential here as well. In fact, the experimental results show a slightly increased level of hydroxyl radical formation for the ruthenium modified samples. This may, however, also be a result of the overall increased activity due to one of the aforementioned reasons.
For the discussion of the mechanism of the observed photocatalytic activity under visible light irradiation, it is very important to know in which form the added ruthenium ions are actually present in the samples. In principle, the ruthenium ions can either exist as a completely separate phase, adsorbed at the surface of the zinc oxide particles or they could be incorporated into the zinc oxide lattice.
While there was evidence for a small quantity of ruthenium dioxide in the XRD patterns, this does not account for the strong deep green colouration of the material, as ruthenium dioxide is black. This colouration might be the result of defects or dopants in the crystal lattice of the zinc oxide. However, the optical properties of native defects such as interstitial zinc or oxygen or vacancies of zinc or oxygen are well studied and none of them match the absorption feature observed here.82,83 This leaves the incorporation of ruthenium ions into the zinc oxide lattice, i.e., an actual doping of ZnO with Ru as a possible cause, either arising from interstitial or substitutional doping. Interstitial ruthenium is known to increase the lattice parameters of the host zinc oxide lattice.84 Such an increase was, however, not observed in the samples. On the contrary, they even exhibited a decrease in the lattice parameters, making the presence of interstitial ruthenium highly unlikely. In substitutional doping, ruthenium cations occupy zinc cation positions in the zinc oxide lattice and they will consequently be positioned in a tetrahedral coordination.67 However, this coordination is only known for ruthenium with higher valencies. The observed small decrease in the volume of the unit cell is another sign for a higher valency of the ruthenium, as the ionic radius decreases with increasing oxidation state. Additionally, the colour of the material has to be considered. Ru(III) and Ru(IV) species usually display a red colour that originates from the electronic transition in the octagonal ligand field.85 There are, however, several reports evincing that Ru(VI) species in tetrahedral coordination show a deep green colour.86,87
In tetrahedral coordination, the Ru(VI)-ion exhibits three characteristic absorption bands.88 Two of the bands, at 300 and 350 nm, respectively, are assigned to a ligand-to-metal charge transfer. Additionally, there is a band at about 800 nm which is caused by a 3A2 → 3T2 transition. This represents an electronic transition between the d-orbitals, the energetic levels of which are split by the tetrahedral ligand field.88,89
This behaviour is an good agreement with the experimentally determined absorption spectra of the ruthenium-modified ZnO powders. The two absorption bands at 300 and 350 nm are probably masked by the band gap of zinc oxide and cannot be separately identified. However, the very characteristic absorption band of tetrahedrally coordinated Ru(VI) at 800 is clearly observed in the ruthenium-modified zinc oxide samples (cf.Fig. 3).
The ionic radius of Ru(VI) in tetrahedral coordination, 36 pm,86,87 is significantly lower than that of Zn(II), 60 pm,90 which it is replacing in the zinc oxide lattice. This explains the observation of a slightly smaller unit cell for the ruthenium-modified samples.
Due to these considerations, it is assumed that in addition to separate ruthenium dioxide particles, part of the ruthenium ions are incorporated (doped) into the zinc oxide lattice as Ru(VI) in tetrahedral coordination, replacing a zinc ion (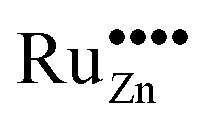 in the Kröger–Vink notation commonly used for crystallographic defects). Since Ru(VI) has a higher valency than Zn(II), the additional charge has to be compensated. This can be done either by ionic, eqn (13), or by electronic, eqn (14), compensation:91
in the Kröger–Vink notation commonly used for crystallographic defects). Since Ru(VI) has a higher valency than Zn(II), the additional charge has to be compensated. This can be done either by ionic, eqn (13), or by electronic, eqn (14), compensation:91
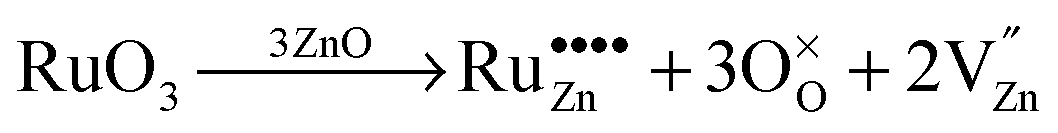 | (13) |
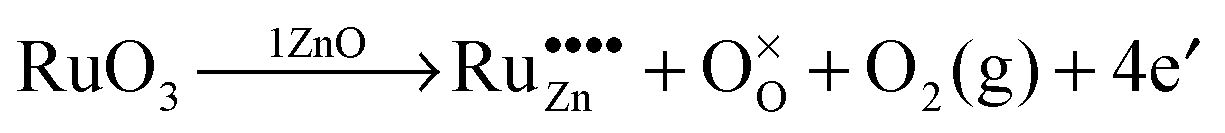 | (14) |
Ionic compensation demands that the excess charge be compensated by other crystal defects, with the most probable ones in this case being zinc vacancies (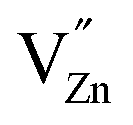 ).68 Two zinc vacancies are necessary to compensate the charge of a single Ru(VI) dopant. Another possibility is the electronic compensation. In this mechanism, the excess charge is compensated by additional free electrons, four electrons per ruthenium ion in this case. This has the additional effect of increasing the donor density of the material. Such an increase, however, was not observed in the ruthenium-modified materials. Therefore, it is assumed that the charge is balanced by ionic compensation in these materials.
).68 Two zinc vacancies are necessary to compensate the charge of a single Ru(VI) dopant. Another possibility is the electronic compensation. In this mechanism, the excess charge is compensated by additional free electrons, four electrons per ruthenium ion in this case. This has the additional effect of increasing the donor density of the material. Such an increase, however, was not observed in the ruthenium-modified materials. Therefore, it is assumed that the charge is balanced by ionic compensation in these materials.
Judging from the change in visible light absorption at 800 nm, the ruthenium doping ratio seems to be roughly proportional to the amount of ruthenium added during the synthesis. However, due to the presence of other ruthenium species the real doping ratio is assumed to be significantly lower. A previously proposed model for transition metal doped photocatalysts suggests an optimal doping ratio of about 0.005 at% for zinc oxide particles of this size, which is 5% of the amount of ruthenium added to the optimal sample here (0.1 at%).80 This might be a good indication of the approximate amount of ruthenium ions that are incorporated into the zinc oxide as dopants.
The presence of the ruthenium dioxide particles may also play an important role in the increased photocatalytic activity. Ruthenium dioxide is known for being a very good catalyst for both, the oxidation of water to molecular oxygen as well as for the corresponding reverse reductive reaction.92–95 RuO2/Ti electrodes, for example, show an oxygen reduction current even at a potential of +0.73 V vs. RHE.96 This reaction involves either a two- or a four-electron transfer, depending on the potential.94 Therefore, it is proposed here that the improved oxygen reduction properties of the materials are the result of the ruthenium dioxide particles present acting as electron transfer catalysts.
The prerequisite for visible light driven photocatalysis is the absorption of light with the respective wavelengths. This is the case for the ruthenium-modified samples as they exhibit a dark green colour due to the absorption of red light. As the dominant absorption feature of these materials in the visible region is the one at 800 nm, this will most likely be the cause of the observed activity. This absorption edge is caused by an electron transfer in the ligand field of the tetragonal coordinated Ru(VI) ions. In principle, the energy absorbed could promote an electron from the valence band of the semiconductor to the doped ruthenium-species, thus reducing it in the process, eqn (15). The other possibility is the promotion of an electron from the doped ruthenium-species to the conduction band of the semiconductor, i.e., the oxidation of the dopant, eqn (16).
As shown by the qualitative and quantitative determination of oxidative species (see Fig. 9 and 10), illumination with visible light causes the formation of hydrogen peroxide but not of hydroxyl radicals. This is a strong indication for the involvement of conduction band electrons in the process, since they are responsible for the formation of hydrogen peroxide. Valence band holes, however, do not seem to be involved in this mechanism, as they would create hydroxyl radicals, which were not observed.
Based on these considerations we propose the following mechanism to explain the visible light driven photocatalytic activity of the ruthenium modified zinc oxides, illustrated in Fig. 11. First, photons with a wavelength of approximately 800 nm (1.55 eV) are absorbed by the material. This causes an electron associated with a Ru(VI) species in the zinc oxide lattice to be excited into the conduction band of the semiconductor, leaving behind an oxidised Ru(VII) species (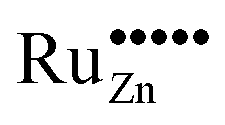 ), eqn (16). The conduction band electron subsequently reduces adsorbed molecular oxygen, forming superoxide or hydrogen peroxide (eqn (17) and (18)). The oxidised ruthenium species may in turn react with electron donors (D) to regenerate itself, eqn (19). According to the data analysis presented here, the redox potential of this Ru(VI)/Ru(VII) redox pair lies at +1.31, +1.40, and +1.41 V vs. RHE for 0.1, 0.3, and 1.0 at% ruthenium content, respectively. This is in very good agreement with known values for this redox pair, placing it at +1.35 to +1.45 V vs. RHE.97,98 This redox potential is still high enough to oxidise most pollutants, e.g., nitric oxide with +0.98 V vs. RHE.73
), eqn (16). The conduction band electron subsequently reduces adsorbed molecular oxygen, forming superoxide or hydrogen peroxide (eqn (17) and (18)). The oxidised ruthenium species may in turn react with electron donors (D) to regenerate itself, eqn (19). According to the data analysis presented here, the redox potential of this Ru(VI)/Ru(VII) redox pair lies at +1.31, +1.40, and +1.41 V vs. RHE for 0.1, 0.3, and 1.0 at% ruthenium content, respectively. This is in very good agreement with known values for this redox pair, placing it at +1.35 to +1.45 V vs. RHE.97,98 This redox potential is still high enough to oxidise most pollutants, e.g., nitric oxide with +0.98 V vs. RHE.73
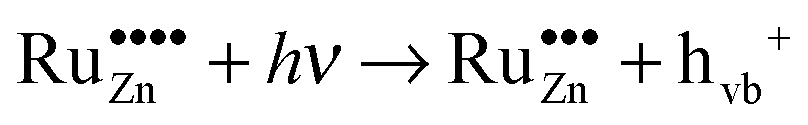 | (15) |
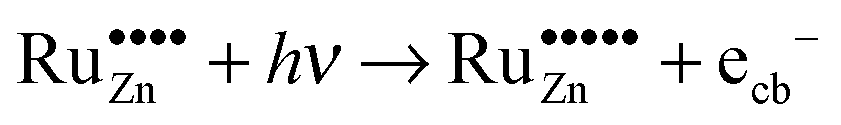 | (16) |
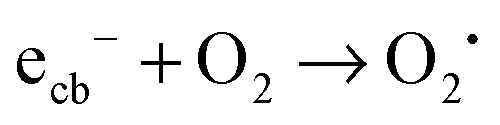 | (17) |
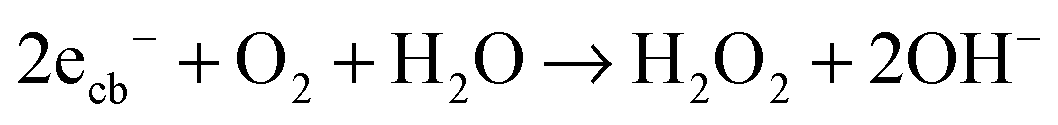 | (18) |
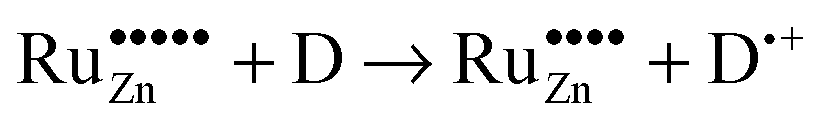 | (19) |
In contrast to ordinary zinc oxide, the reduction of molecular oxygen by conduction band electrons may not only be a one-electron transfer, eqn (17), but also a two-electron transfer, eqn (18). This is evidenced by the oxygen-reduction current measurements showing reduction currents for the ruthenium modified samples at potentials that are not possible with merely a one-electron reduction. This multi-electron transfer is likely to be catalysed by the ruthenium dioxide particles present in the samples. The reduced oxygen species, superoxide and hydrogen peroxide, are strong oxidising agents themselves and will subsequently partake in the oxidation of adsorbed pollutant molecules.
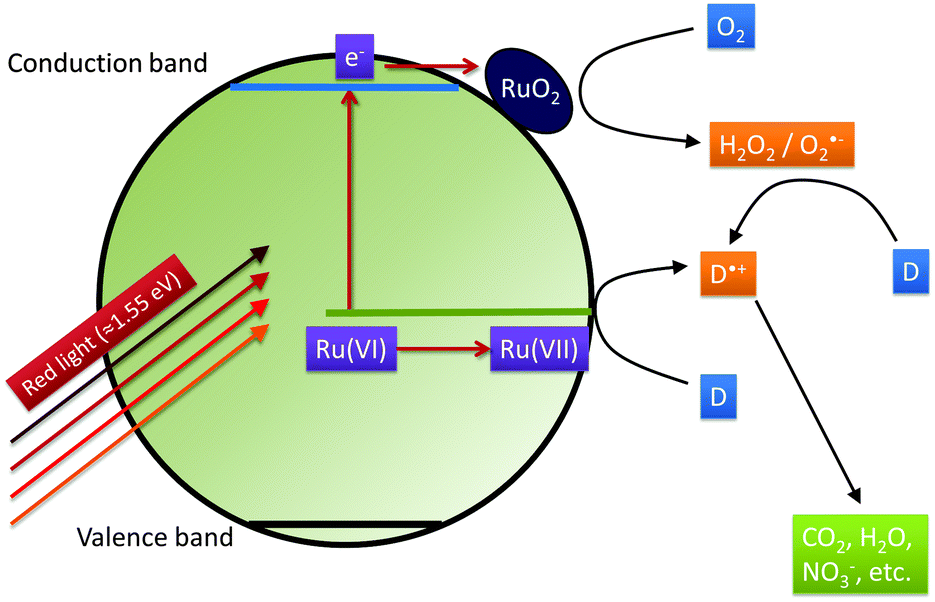 | ||
| Fig. 11 The proposed mechanism explaining the photocatalytic activity of the Ruthenium-modified zinc oxides under illumination with visible light. Excitation by red light (≈800 nm = 1.55 eV) creates conduction band electrons and an oxidised Ru(VII) species. The former react with molecular oxygen to form reactive oxygen species, catalysed by RuO2-particles and the latter directly oxidise donor molecules (D), i.e., the pollutants such as NO or acetaldehyde, and eventually mineralise them. | ||
5 Conclusions
Ruthenium-modified zinc oxide, a visible light photocatalyst already reported previously, was synthesised with three different ruthenium concentrations. The visible light activity that was already observed for the gas-phase degradation of acetaldehyde was confirmed by a second (ISO standard) test method, i.e., by the oxidation of nitric oxide in the gas phase. In addition to confirming the previously observed visible light activity, which surpassed even the best titanium dioxide standards, this test also revealed a vastly increased selectivity towards nitrate formation, suppressing the formation of the toxic nitrogen dioxide. The ruthenium-modified zinc oxide is therefore a considerably better photocatalyst for nitric oxide oxidation than the commercial titanium dioxide standards used here, since both the photocatalytic activity and the selectivity towards nitrate formation are significantly higher.Even though the material and its visible light response were reported before, the nature and reaction of the responsible photoactive centres remained a mystery. Through further investigations, this mystery was now elucidated using a series of experiments specifically designed to do so. It was discovered hence that this synthetic route likely yields a unique mixture of Ru(VI)-doped zinc oxide and ruthenium dioxide particles. Both, the ruthenium ions acting as dopants of the ZnO particles and the ruthenium dioxide particles play their own independent role in the enhancement of the photocatalytic activity. The doped ruthenium ions enable a better charge separation as they can trap charge carriers and reduce their recombination rate. Additionally, they allow the absorption of low energy light from the red part of the visible light spectrum which can then be utilised in photocatalytic reactions. The absorption of red light leads to the promotion of electrons from the Ru(VI)-species, which is in turn oxidised, to the conduction band. Both, the conduction band electrons and the oxidised Ru(VII) participate in the degradation of the pollutant molecules, either directly or indirectly via activated oxygen species. The ruthenium dioxide particles, on the other hand, improve the oxygen reduction properties of the material, which is often a bottleneck for photocatalytic reactions, thereby increasing the efficiency of the reaction. Even though the oxidation state of the doped ruthenium ions could not be directly experimentally confirmed, the change in unit cell volume, the optical absorption properties and the redox potential of the species all confirm this theory.
Tracing of signature oxidative species (i.e., hydroxyl radicals and hydrogen peroxide) has been used here to unravel the valence and conduction band involvement in the mechanism of a visible light induced photocatalytic reaction. To the best of our knowledge, this is the first time that such an approach has been used for this purpose. We are convinced that this approach has proven to be a valuable tool to elucidate the photocatalytic reaction mechanism in our case. A similar approach could most certainly also be adapted to validate the proposed reaction mechanisms of various other visible light photocatalysts.
Acknowledgements
Financial support from the German Federal Ministry of Education and Research (BMBF Project “HelioClean”, grant no. 03X0069F) is gratefully acknowledged. R. D. thanks the Leibniz University Hannover and Prof. Dr D. Bahnemann for granting him a leave of absence.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Fujishima, X. Zhang and D. Tryk, Int. J. Hydrogen Energy, 2007, 32, 2664–2672 CrossRef CAS.
- J. M. Herrmann, Top. Catal., 2005, 34, 49–65 CrossRef CAS.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005, 44, 8269–8285 CrossRef CAS.
- O. Carp, C. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS.
- D. Bahnemann, Sol. Energy, 2004, 77, 445–459 CrossRef CAS.
- O. Alfano, D. Bahnemann, A. Cassano, R. Dillert and R. Goslich, Catal. Today, 2000, 58, 199–230 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- U. Gaya and A. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- J. Herrmann, Catal. Today, 1999, 53, 115–129 CrossRef CAS.
- S. Malato, P. Fernández-Ibáñez, M. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- S. Noimark, C. W. Dunnill and I. P. Parkin, Adv. Drug Delivery Rev., 2013, 65, 570–580 CrossRef CAS PubMed.
- I. Oller, S. Malato and J. a. Sánchez-Pérez, Sci. Total Environ., 2011, 409, 4141–4166 CrossRef CAS PubMed.
- I. P. Parkin and R. G. Palgrave, J. Mater. Chem., 2005, 15, 1689 RSC.
- J. Peral, X. Domènech and D. F. Ollis, J. Chem. Technol. Biotechnol., 1997, 70, 117–140 CrossRef CAS.
- S. Pasternak and Y. Paz, ChemPhysChem, 2013, 14, 2059–2070 CrossRef CAS PubMed.
- J. Zhao and X. Yang, Build. Environ., 2003, 38, 645–654 CrossRef.
- K. Nakata, T. Ochiai, T. Murakami and A. Fujishima, Electrochim. Acta, 2012, 84, 103–111 CrossRef CAS.
- M. Ni, M. K. Leung, D. Y. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- J. Lasek, Y.-H. Yu and J. C. Wu, J. Photochem. Photobiol., C, 2013, 14, 29–52 CrossRef CAS.
- G. Hüsken, M. Hunger and H. Brouwers, Build. Environ., 2009, 44, 2463–2474 CrossRef.
- A. Folli, C. Pade, T. B. k. Hansen, T. De Marco and D. E. Macphee, Cem. Concr. Res., 2012, 42, 539–548 CrossRef CAS.
- M. Ballari and H. Brouwers, J. Hazard. Mater., 2013, 254–255, 406–414 CrossRef CAS PubMed.
- G. L. Guerrini, Constr. Build. Mater., 2012, 27, 165–175 CrossRef.
- A. Folli, J. Z. Bloh, M. Strøm, T. Pilegaard Madsen, T. Henriksen and D. E. Macphee, J. Phys. Chem. Lett., 2014, 5 CAS.
- S. Rehman, R. Ullah, A. M. Butt and N. D. Gohar, J. Hazard. Mater., 2009, 170, 560–569 CrossRef CAS PubMed.
- A. Kudo, H. Kato and I. Tsuji, Chem. Lett., 2004, 33, 1534–1539 CrossRef CAS.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505–516 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- C. Di Valentin and G. Pacchioni, Catal. Today, 2013, 206, 12–18 CrossRef CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed.
- N. Todorova, T. Vaimakis, D. Petrakis, S. Hishita, N. Boukos, T. Giannakopoulou, M. Giannouri, S. Antiohos, D. Papageorgiou, E. Chaniotakis and C. Trapalis, Catal. Today, 2013, 209, 41–46 CrossRef CAS.
- C. Di Valentin, G. Pacchioni and A. Selloni, Chem. Mater., 2005, 17, 6656–6665 CrossRef CAS.
- V. Çelik and E. Mete, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 205112 CrossRef.
- W. Doerffler and K. Hauffe, J. Catal., 1964, 3, 156–170 CrossRef CAS.
- W. Doerffler and K. Hauffe, J. Catal., 1964, 3, 171–178 CrossRef CAS.
- D. W. Bahnemann, C. Kormann and M. R. Hoffmann, J. Phys. Chem., 1987, 91, 3789–3798 CrossRef CAS.
- C. Kormann, D. W. Bahnemann and M. R. Hoffmann, Environ. Sci. Technol., 1988, 22, 798–806 CrossRef CAS PubMed.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- A. Fujishima and X. Zhang, C. R. Chim., 2006, 9, 750–760 CrossRef CAS.
- J. Z. Bloh, R. Dillert and D. W. Bahnemann, Environ. Sci. Pollut. Res., 2012, 19, 3688–3695 CrossRef CAS PubMed.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef CAS.
- International Organization for Standardization, ISO 22197-1: Fine ceramics (advanced ceramics, advanced technical ceramics) - Test method for air-purification performance of semiconducting photocatalytic materials - Part 1: Removal of nitric oxide, International organization for standardization technical report, 2007.
- C. A. Parker, Proc. R. Soc. A, 1953, 220, 104–116 CrossRef.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. A, 1956, 235, 518–536 CrossRef CAS.
- E. M. P. Steinmiller and K.-S. Choi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20633–20636 CrossRef CAS PubMed.
- S. J. Limmer, E. A. Kulp and J. A. Switzer, Langmuir, 2006, 22, 10535–10539 CrossRef CAS PubMed.
- J. E. B. Randles, Discuss. Faraday Soc., 1947, 1, 11 RSC.
- S. P. Harrington and T. M. Devine, J. Electrochem. Soc., 2008, 155, C381 CrossRef CAS.
- S. P. Harrington and T. M. Devine, J. Electrochem. Soc., 2009, 156, C154 CrossRef CAS.
- N. F. Mott, Proc. R. Soc. A, 1939, 171, 27–38 CrossRef.
- W. Schottky, Z. Phys., 1939, 113, 367–414 CrossRef CAS.
- W. Schottky, Z. Phys., 1942, 118, 539–592 CrossRef CAS.
- T. Alexander, T. Bukowski, D. Uhlmann, G. Teowee, K. McCarthy, J. Dawley and B. Zelinski, ISAF '96. Proceedings of the Tenth IEEE International Symposium on Applications of Ferroelectrics, 1996, 2, 585–588.
- H. Frederikse, Handbook of Chemistry & Physics, CRC Press, 91th edn, 2010 Search PubMed.
- K. F. Young and H. P. R. Frederikse, J. Phys. Chem. Ref. Data, 1973, 2, 313 CrossRef.
- L. Berger, Handbook of Chemistry & Physics, CRC Press, 91th edn, 2010 Search PubMed.
- K.-i. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- C. Baumanis, J. Z. Bloh, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2011, 115, 25442–25450 CAS.
- G. G. Guilbault, P. J. Brignac and M. Juneau, Anal. Chem., 1968, 40, 1256–1263 CrossRef CAS PubMed.
- C. Klingshirn, ChemPhysChem, 2007, 8, 782–803 CrossRef CAS PubMed.
- A. Janotti and C. G. Van de Walle, Rep. Prog. Phys., 2009, 72, 126501 CrossRef.
- Y. Xu and M. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- International Organization for Standardization, ISO 22197-2: Fine ceramics (advanced ceramics, advanced technical ceramics) – Test method for air-purification performance of semiconducting photocatalytic materials – Part 2: Removal of acetaldehyde, International organization for standardization technical report, 2011.
- R. Dillert, J. Stötzner, A. Engel and D. W. Bahnemann, J. Hazard. Mater., 2012, 211–212, 240–246 CrossRef CAS PubMed.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637–1755 CrossRef CAS.
- P. Vanýsek, Handbook of Chemistry & Physics, CRC Press, 91th edn, 2010 Search PubMed.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780–7781 CrossRef CAS PubMed.
- U. A. Joshi, J. R. Darwent, H. H. P. Yiu and M. J. Rosseinsky, J. Chem. Technol. Biotechnol., 2011, 86, 1018–1023 CrossRef CAS.
- P.-A. Brugger, P. Cuendet and M. Grätzel, J. Am. Chem. Soc., 1981, 103, 2923–2927 CrossRef CAS.
- A. J. Hoffman, E. R. Carraway and M. R. Hoffmann, Environ. Sci. Technol., 1994, 28, 776–785 CrossRef CAS PubMed.
- A. C. Dodd, A. J. McKinley, M. Saunders and T. Tsuzuki, J. Nanopart. Res., 2006, 8, 43–51 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- J. Z. Bloh, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2012, 116, 25558–25562 CAS.
- J. Z. Bloh, R. Dillert and D. W. Bahnemann, ChemCatChem, 2012, 5, 774–778 CrossRef.
- Z. Lin-Li, G. Chang-Xin and H. Jun-Tao, Chin. Phys., 2005, 14, 586–591 CrossRef.
- A. Janotti and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165202 CrossRef.
- L. Mančić, S. Grgurić-Šipka, V. Djinović, Z. Marinković, T. Sabo and O. Milošević, Mater. Sci. Forum, 2005, 494, 149–154 CrossRef.
- S. C. Navale, V. Ravi and I. Mulla, Sens. Actuators, B, 2009, 139, 466–470 CrossRef CAS.
- D. Fischer and R. Hoppe, Z. Anorg. Allg. Chem., 1992, 617, 37–44 CrossRef CAS.
- D. Fischer and R. Hoppe, Z. Anorg. Allg. Chem., 1990, 691, 87–94 CrossRef.
- T. C. Brunold and H. U. Gu, Inorg. Chem., 1997, 36, 2084–2091 CrossRef CAS PubMed.
- A. Rauk, T. Ziegler and D. E. Ellis, Theor. Chim. Acta, 1974, 34, 49–59 CrossRef CAS.
- W. Haynes, Handbook of Chemistry & Physics, CRC Press, 91th edn, 2010 Search PubMed.
- R. van de Krol, in Photoelectrochemical Hydrogen Production, ed. R. van de Krol and M. Grätzel, Springer US, Boston, MA, 2012, vol. 102 Search PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- C. Chang and T. Wen, J. Appl. Electrochem., 1997, 27, 355–363 CrossRef CAS.
- D. Duonghong, E. Borgarello and M. Graetzel, J. Am. Chem. Soc., 1981, 103, 4685–4690 CrossRef CAS.
- Y. Takasu, K. Oohori, N. Yoshinaga and W. Sugimoto, Catal. Today, 2009, 146, 248–252 CrossRef CAS.
- L. D. Burke and D. P. Whelan, J. Electroanal. Chem. Interfacial Electrochem., 1979, 103, 179–187 CrossRef CAS.
- L. D. Burke and J. Healy, J. Electroanal. Chem. Interfacial Electrochem., 1981, 124, 327–332 CrossRef CAS.
Footnotes |
| † Present address: Department of Chemistry, University of Aberdeen, Meston Walk, AB24 3UE Aberdeen, UK. E-mail: jonathanbloh@gmx.net. |
| ‡ Present address: Department of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo 102-8554, Japan. |
| This journal is © the Owner Societies 2014 |