 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-range proton-coupled electron transfer in phenol–Ru(2,2′-bipyrazine)32+ dyads†
Catherine
Bronner
and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, Spitalstrasse 51, CH-4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
First published on 13th January 2014
Abstract
Two dyads in which either 4-cyanophenol or un-substituted phenol is connected via a p-xylene spacer to a Ru(bpz)32+ (bpz = 2,2′-bipyrazine) complex were synthesized and investigated. Selective photo-excitation of Ru(bpz)32+ at 532 nm in a CH3CN–H2O mixture leads to the formation of 4-cyanophenolate or phenolate along with Ru(bpz)32+ in its electronic ground state. This apparent photoacid behavior can be understood on the basis of a reaction sequence comprised of an initial photoinduced proton-coupled electron transfer (PCET) during which 4-cyanophenol or phenol is oxidized and deprotonated, followed by a thermal electron transfer event in the course of which 4-cyanophenoxyl or phenoxyl is reduced by Ru(bpz)3+ to 4-cyanophenolate or phenolate. Conceptually, this reaction sequence is identical to a sequence of photoinduced charge-separation and thermal charge-recombination events as observed previously for many electron transfer dyads, with the important difference that the initial photoinduced electron transfer process is proton-coupled. The dyad containing 4-cyanophenol reacts via concerted-proton electron transfer (CPET) whereas the dyad containing un-substituted phenol appears to react predominantly via a stepwise PCET mechanism. Long-range PCET is a key reaction in photosystem II. Understanding the factors that govern the kinetics of long-range PCET is desirable in the broader context of light-to-energy conversion by means of proton–electron separation across natural or artificial membranes.
Introduction
Many important redox reactions including water oxidation and carbon dioxide reduction must be coupled to acid/base chemistry in order to proceed efficiently.1 Thus, there is significant interest in understanding proton-coupled electron transfer (PCET) reactions at the most fundamental level both in artificial and biological systems.2–8 Phenol molecules have played a particularly prominent role in mechanistic PCET studies because their redox and acid/base chemistry are strongly interrelated.9–12 Much research focused on bimolecular PCET in which phenols form hydrogen-bonded encounter complexes with reaction partners that can act as electron and/or proton acceptors.13–19 In addition, there has been significant work on dyads in which phenols are connected covalently to an electron-accepting moiety and where the solvent (or added base) acts as a proton acceptor.20–27 The importance of the distance between electron/proton donating and accepting sites in covalent PCET dyads has received increasing attention recently.28–34 Long-range electron transfer which is not coupled to proton transfer is of course rather well understood.35,36Our prior studies demonstrated that bimolecular PCET between various phenols and photoexcited Ru(bpz)32+ (bpz = 2,2′-bipyrazine) occurs via a concerted proton–electron transfer (CPET) mechanism which takes place when the reactants form weak hydrogen-bonded encounter adducts in solution (Scheme 1a).37,38 In the case of 4-cyanophenol (R = CN) the CPET process was associated with an H/D kinetic isotope effect (KIE) of 10.2 ± 0.6, while for phenol (R = H) we found KIE = 3.4 ± 0.2.37 In this paper we report on the two new covalent dyads shown in Scheme 1b in which 4-cyanophenol or phenol is connected covalently to Ru(bpz)32+via a p-xylene spacer. We aimed to explore how driving-force changes (brought about by the change from 4-cyanophenol to un-substituted phenol) affect the PCET mechanisms and rates in the two different settings shown in Scheme 1. This is interesting because in both settings the same reactants are involved, but in one case the PCET reaction resembles hydrogen atom transfer (Scheme 1a) whereas in the other case a multi-site PCET is operative (Scheme 1b). In other words, the electron and proton transfer directions are the same in Scheme 1a but different in Scheme 1b.
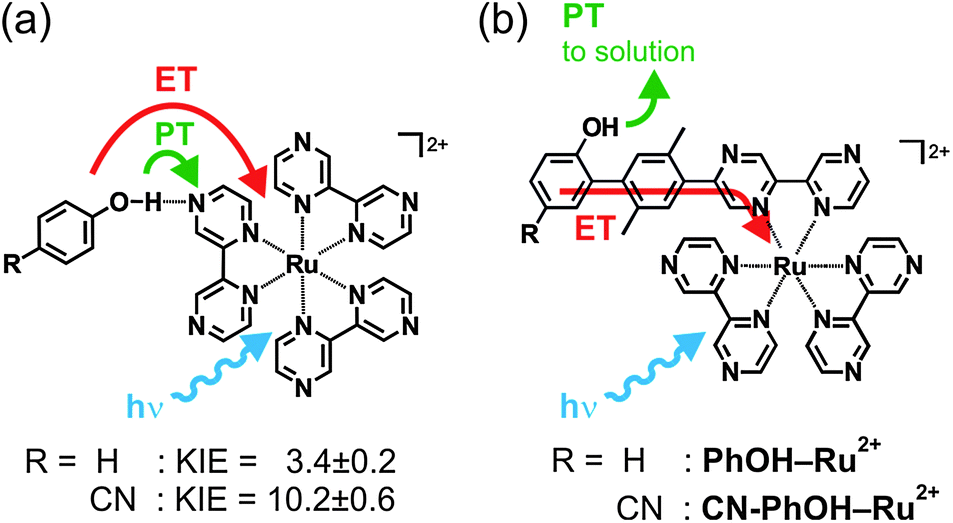 | ||
| Scheme 1 (a) Phenol–Ru(bpz)32+ reaction pairs which were previously investigated in the context of hydrogen-atom transfer (HAT)-like PCET.37 (b) Dyads investigated in this work. ET = electron transfer, PT = proton transfer, KIE = H/D kinetic isotope effect. | ||
Our PCET study involving photoexcited Ru(bpz)32+ as a key reactant in the two dyads is possible because this particular complex is a substantially stronger photooxidant than the more commonly used Ru(2,2′-bipyridine)32+ complex.
Results and discussion
Syntheses and structural aspects
The more common 2,2′-bipyridine ligand has very frequently been used in electron or energy transfer dyads39,40 but for 2,2′-bipyrazine (bpz) this is not the case, and the covalent attachment of rigid rod-like bridges (including phenol units) to bpz had to be newly developed (Scheme 2). Commercially available 2-amino-pyrazine (1) was reacted with N-bromosuccinimide to afford 2-amino-5-bromo-pyrazine (2).41 Treatment of molecule 2 with HI, I2, and NaNO2 gave 2-bromo-5-iodo-pyrazine (3)42 which was reacted with 4-trimethylsilyl-2,5-dimethylphenylboronic acid (4)43 to afford the Suzuki coupling product 5. Subsequent Stille coupling to tri-(n-butyl)stannylpyrazine (6)44 led to the bipyrazine ligand with one attached p-xylene unit (7). Deprotection of the TMS group with Br2 gave molecule 8 which was reacted with boronic acid derivatives of phenol and 4-cyanophenol. In the case of the simple phenol commercial 2-hydroxyphenylboronic acid (9) could be used directly, whereas in the case of 4-cyanophenol an appropriate boronic acid coupling partner (10) with the phenolic group protected by a methoxymethyl ether had to be prepared. The final ligands (11, 12) were reacted with Ru(bpz)2Cl2. Detailed synthetic protocols including product characterization data are given in the ESI.†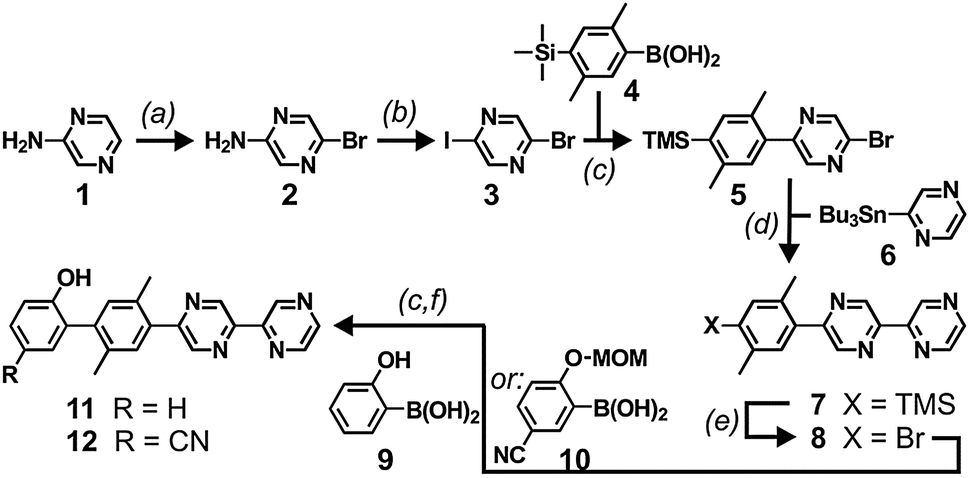 | ||
| Scheme 2 Synthesis of the ligands for the two dyads. (a) NBS, CH2Cl2; (b) aq. HI, I2, NaNO2; (c) Pd(PPh3)4, Na2CO3, EtOH, toluene; (d) Pd(PPh3)4, m-xylene; (e) Br2, NaOAc, THF; (f) HCl, dioxane. See ESI† for details. | ||
The result of an X-ray structure analysis of 11 is shown in Fig. 1. This ligand crystallizes in the monoclinic space group P21 with a single molecule in the asymmetric unit. Molecule 11 arranges in sheets along the crystallographic a- and c-axes with hydrogen bonds between the phenol and a nitrogen atom of bpz (the closest intermolecular O1–N2 distance is 2.798(2) Å) but there is no evidence for π–π interactions. The two pyrazine units of a given bpz ligand are oriented in a parallel fashion, and they are nearly coplanar with a torsion angle (N3–C5–C4–C3) of only 12.5(2)° between them. The dihedral angle between the xylene unit and the neighboring pyrazine (C6–C7–C9–C14) is 37.0(2)° and the dihedral angle between the phenol and the xylene (C13–C12–C17–C18) is 72.5(2)°, in line with the equilibrium torsion angles expected for oligo-p-xylenes.45,46
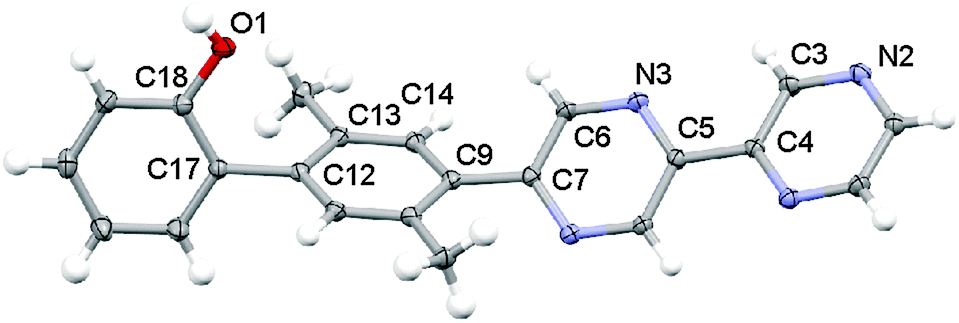 | ||
| Fig. 1 Crystallographic structure of ligand 11. Anisotropic displacement parameters are drawn at the 50% probability level. | ||
Photochemistry of the dyads in pure CH3CN and in CH3CN–H2O mixtures
In pure (aerated) CH3CN photoexcitation of the ruthenium unit of the two dyads simply produces 3MLCT luminescence but there is no photochemistry. The luminescence spectra and the excited-state lifetimes (τ = 474 ns for CN–PhOH–Ru2+; τ = 503 ns for PhOH–Ru2+) are virtually identical to those of the Ru(bpz)32+ reference complex (τ = 520 ns) under the same conditions (Fig. S1a and b, ESI†). The transient absorption spectra of the dyads resemble those of the reference complex (Fig. S2, ESI†). Obviously, under these conditions electron transfer (ET) from 4-cyanophenol and phenol to *Ru(bpz)32+ is highly inefficient. This makes sense because 4-cyanophenol and phenol are oxidized at potentials above 1.2 V vs. Fc+/Fc in CH3CN47 whereas *Ru(bpz)32+ is reduced at ca. 1.0 V vs. Fc+/Fc48 hence simple ET is endergonic by ≥0.2 eV in both dyads (the standard oxidation potentials of 4-cyanophenol and phenol in CH3CN are 1.40 V and 1.25 V vs. Fc+/Fc, respectively.).47In CH3CN–H2O mixtures the 3MLCT luminescence of the dyads is quenched, and the excited-state lifetimes shorten to 148 ns for CN–PhOH–Ru2+ and ∼20 ns for PhOH–Ru2+ while that of the reference complex is 772 ns (Fig. S3, ESI†). The black traces in Fig. 2a and b are transient absorption spectra obtained for ∼10−5 M solutions of CN–PhOH–Ru2+ and PhOH–Ru2+ in CH3CN–H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) for CN–PhOH–Ru2+ in Fig. 2a and 1:1 (v:v) for PhOH–Ru2+ in Fig. 2b). Selective Ru(bpz)32+ excitation occurred at 532 nm with laser pulses of ∼10 ns duration. In the case of CN–PhOH–Ru2+ detection took place with a time delay of 2 μs and subsequent time-averaging over 200 ns whereas in the case of PhOH–Ru2+ no time delay was necessary because the photoproducts form more rapidly in this dyad (see below). For CN–PhOH–Ru2+ one observes bleaching at ∼375 nm and absorption bands at ∼420 and ∼440 nm (black trace in Fig. 2a). For PhOH–Ru2+ one detects the same three bands but at slightly longer wavelengths (black trace in Fig. 2b).
:1 (v:v) for CN–PhOH–Ru2+ in Fig. 2a and 1:1 (v:v) for PhOH–Ru2+ in Fig. 2b). Selective Ru(bpz)32+ excitation occurred at 532 nm with laser pulses of ∼10 ns duration. In the case of CN–PhOH–Ru2+ detection took place with a time delay of 2 μs and subsequent time-averaging over 200 ns whereas in the case of PhOH–Ru2+ no time delay was necessary because the photoproducts form more rapidly in this dyad (see below). For CN–PhOH–Ru2+ one observes bleaching at ∼375 nm and absorption bands at ∼420 and ∼440 nm (black trace in Fig. 2a). For PhOH–Ru2+ one detects the same three bands but at slightly longer wavelengths (black trace in Fig. 2b).
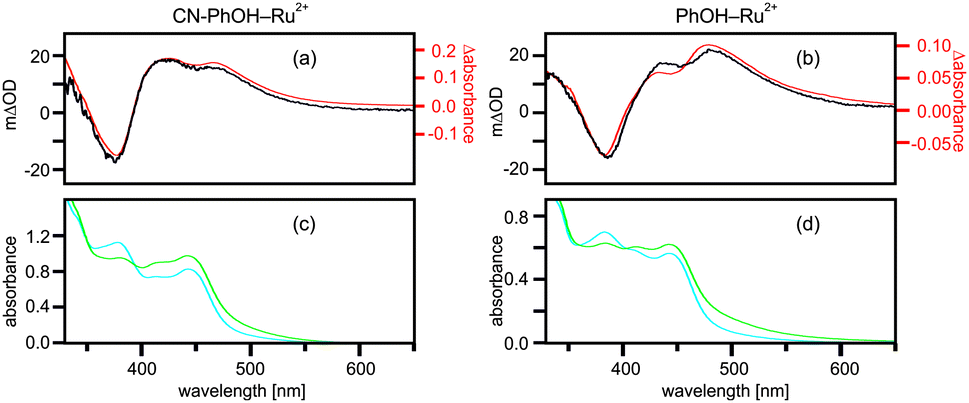 | ||
| Fig. 2 (a, b) Black traces: transient absorption spectra of the CN–PhOH–Ru2+ (a) and PhOH–Ru2+ (b) dyads in CH3CN–H2O recorded by time-averaging over 200 ns after excitation at 532 nm (time delay of 2 μs in (a), no time delay in (b)). The red traces in (a, b) were obtained by subtracting the blue traces from the green traces in (c, d). (c, d) UV-Vis spectra of CN–PhOH–Ru2+ (c) and PhOH–Ru2+ (d) in CH3CN–H2O at pH 7 (blue traces) and at pH 11 (green traces). | ||
Identification of the photoproducts formed in CN–PhOH–Ru2+ and PhOH–Ru2+ is straightforward when considering the UV-Vis spectra of the dyads shown in Fig. 2c and d (blue traces) and the spectra of their deprotonated forms (green traces). (Deprotonation of the phenolic units occurred by addition of excess NaOH to the CH3CN–H2O mixtures.) When subtracting the blue traces from the green traces in Fig. 2c and d one obtains the difference spectra shown as red traces in Fig. 2a and b. The agreement between these derived difference spectra and the experimental transient absorption spectra (black traces in Fig. 2a and b) is nearly perfect, indicating that the photoproducts in the dyads are the phenolates and Ru(bpz)32+ in the electronic ground state. It is as if the two dyads merely acted as photoacids.
Photochemical reaction pathways in CH3CN–H2O
The apparent photoacid behavior of the two dyads shown in Scheme 1b is unlikely to be the result of direct phenol deprotonation after excitation of the Ru(bpz)32+ complex. Phenol excitation at 532 nm is highly inefficient, and it is not obvious why the phenols should become significantly more acidic when the metal complex is excited, particularly in view of the fact that the two moieties are kept apart by a p-xylene spacer.If direct phenol deprotonation after selective ruthenium excitation is impossible, then how else can the photochemical formation of 4-cyanophenolate and phenolate in combination with Ru(bpz)32+ in its electronic ground state be explained? The reaction sequence illustrated by Scheme 3 provides a plausible explanation. Selective excitation of the metal complex at 532 nm triggers electron transfer (ET) from CN–PhOH or PhOH to Ru(bpz)32+ coupled to release of the phenolic proton (PT) to water (step 1 in Scheme 3). Whether this PCET process occurs in a concerted or consecutive (stepwise) fashion is a separate question that we address later. The key point here is that phenol oxidation is coupled to release of the phenolic proton (by whichever mechanism), as is commonly observed when phenols are oxidized.9–27 This PCET process produces H3O+, neutral 4-cyanophenoxyl or phenoxyl radicals, and Ru(bpz)3+ (step 2 in Scheme 3). Thermal electron transfer in the reverse direction from Ru(bpz)3+ to 4-cyanophenoxyl or phenoxyl can then produce the spectroscopically observed photoproducts comprised of 4-cyanophenolate or phenolate in addition to Ru(bpz)32+ (step 3 in Scheme 3). In principle, this reaction sequence is completely analogous to the sequence of photoinduced charge-separation followed by thermal charge-recombination events previously observed in many electron transfer dyads. The only difference in our dyads is that the initial photoinduced electron transfer step is proton-coupled. Thermal back electron transfer is rapid because the electron transfer distance is short and because the process is highly exergonic; thermal re-protonation of 4-cyanophenolate or phenolate is comparatively slow because this is a bimolecular process (step 4 in Scheme 3).
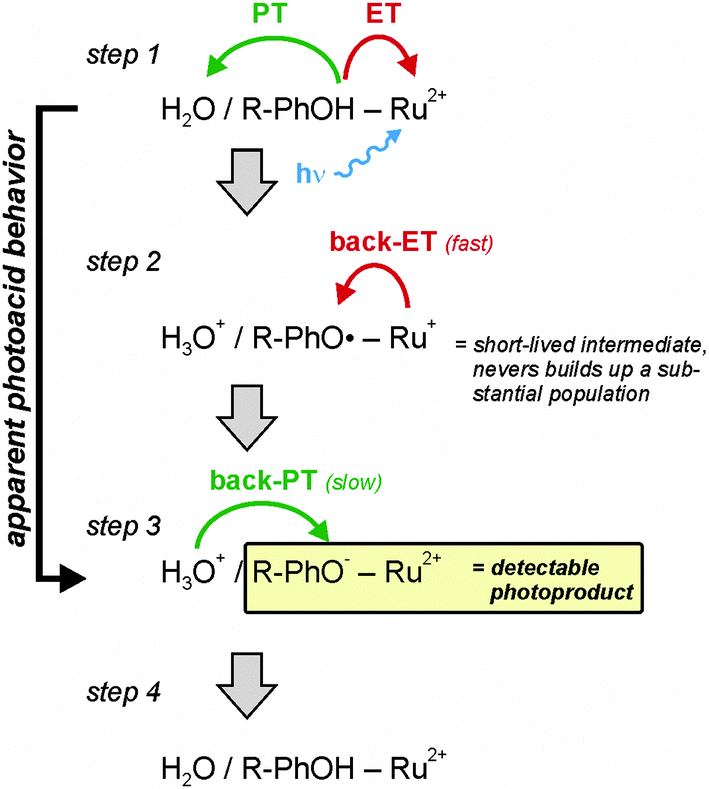 | ||
| Scheme 3 Reaction sequence explaining the apparent photoacid behavior of the two dyads shown in Scheme 1b. | ||
It is tempting to make free energy estimates for the individual reaction steps shown in Scheme 3 based on electrochemical potentials and acidity constants. However, for two reasons we refrain from implementing this idea: (i) the pKa values of the 4-cyanophenoxyl and phenoxyl radical cations (CN–PhOH+/PhOH+) would be of key importance for this purpose but these acidity constants are not known for CH3CN–H2O mixtures, and they are experimentally very tricky to determine. (ii) Phenol oxidation in CH3CN–H2O mixtures is irreversible (either due to loss of the phenolic proton or fast dimerization of the phenoxyl radicals) hence accurate determination of standard potentials is not possible under these conditions.
The reaction sequence in Scheme 3 provides the only viable explanation for the occurrence of 4-cyanophenolate or phenolate following selective Ru(bpz)32+ excitation at 532 nm. There is no spectroscopic evidence for 4-cyanophenoxyl or phenoxyl radicals in transient absorption spectra (in particular the 4-cyanophenoxyl radical has a rather diagnostic spectral signature that we and others have detected many times before).37,49,50 From this we conclude that the initial PCET event (step 1 in Scheme 3) is rate-determining while the thermal back-ET process (step 2 in Scheme 3) is rapid. In this scenario a significant population of 4-cyanophenoxyl or phenoxyl radical intermediates never builds up.
The key question then is whether the initial rate-determining PCET step is a concerted or a stepwise reaction. As noted above, the phenols do not become more acidic upon excitation of the Ru(bpz)32+ complex hence direct photoacid behavior is excluded. However, in our CH3CN–H2O solutions the phenols are in equilibrium with their phenolate forms (eqn (1)).
| R–PhOH–Ru2+ + H2O ⇆ R–PhO−–Ru2+ + H3O+ | (1) |
In principle it is conceivable that phenol deprotonation first has to occur via this chemical equilibrium before photoexcited Ru(bpz)32+ oxidizes the phenolate. In other words, there could be a rate-determining phenol deprotonation step followed by a rapid (intramolecular and highly exergonic) ET event (which would then continuously induce a shift in chemical equilibrium from phenol to phenolate). In this scenario the overall reaction rate cannot be faster than the rate of phenol deprotonation (k−H+). 4-Cyanophenol has pKa ≈ 8 in H2O.12 It follows that Ka = (k−H+/k+H+) = 10−8 M, where k−H+ and k+H+ are the rate constants of 4-cyanophenol deprotonation and 4-cyanophenolate protonation, respectively. Even under the assumption that protonation of 4-cyanophenolate occurs at a diffusion-controlled rate of k+H+ = 1011 M−1 s−1, the rate of 4-cyanophenolate deprotonation will be limited to k−H+ = 103 s−1. For deprotonation of un-substituted phenol (pKa ≈ 10 in H2O)12 an upper limit of k−H+ = 101 s−1 can be estimated. These estimated maximal rate constants of 4-cyanophenol and phenol deprotonation cannot be reconciled with the nanosecond kinetics reported in the next section. Consequently, a PT–ET mechanism based on the chemical equilibrium in eqn (1) is unlikely.
We are thus left with the mechanistic possibilities of (i) a rate-determining CPET (concerted proton–electron transfer) or (ii) a rate-determining ET process leading to phenol oxidation followed by subsequent release of the phenolic proton (before back-ET produces the observable phenolate and Ru(bpz)32+ photoproducts). This mechanistic issue will be addressed further in the following section.
Reaction kinetics
Fig. 3a shows the temporal evolution of the transient absorption signals at 380 nm (black trace), 420 nm (blue trace), and 475 nm (green trace) after excitation of the CN–PhOH–Ru2+ dyad in aerated 4:1 CH3CN–H2O at 532 nm. An average time constant of 139 ns is extracted from kinetic analysis of the three single-exponential transients, in line with the luminescence lifetime of 148 ns reported above. Fig. 3b shows the same set of data for the deuterated analog (CN–PhOD–Ru2+ in 4:1 CH3CN–D2O) yielding an average time constant of 285 ns. Thus, an H/D kinetic isotope effect (KIE) of 2.0 ± 0.2 is associated with the formation of the photoproducts, indicating that the rate-determining step involves proton motion. From this we conclude that the initial PCET reaction in Scheme 3 (step 1) is indeed a concerted proton–electron transfer (CPET) event; as noted above, a PT–ET sequence is impossible after selective Ru(bpz)32+ excitation at 532 nm.
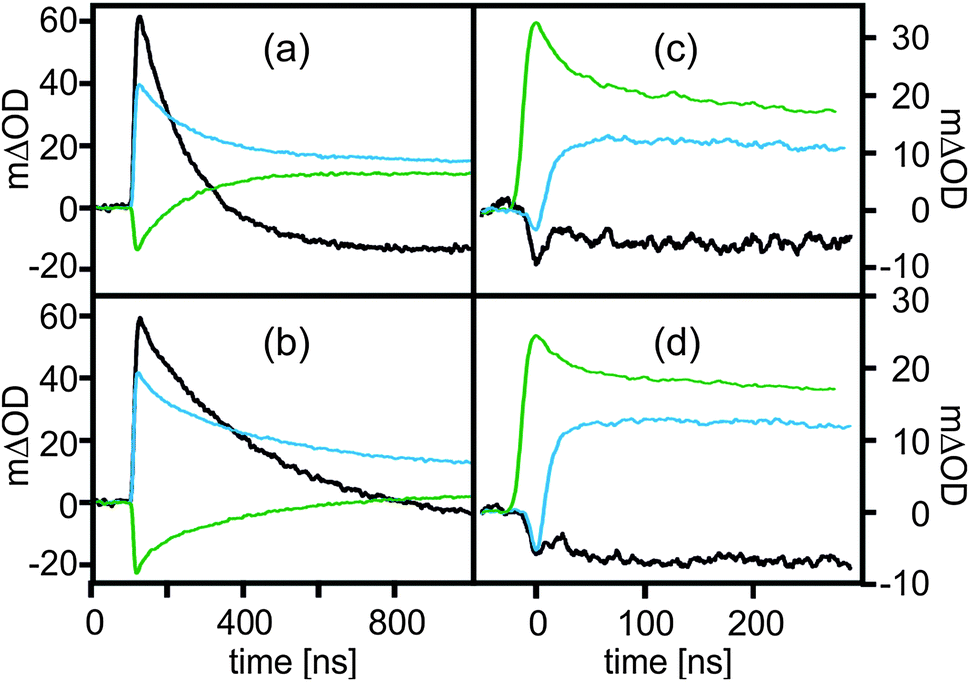 | ||
| Fig. 3 (a) Temporal evolution of the transient absorption signals at 380 nm (black), 420 nm (blue), and 475 nm (green) after 532 nm excitation of CN–PhOH–Ru2+ in 4:1 (v:v) CH3CN–H2O. (b) The same experiments for CN–PhOD–Ru2+in 4:1 (v:v) CH3CN–D2O. (c) The same experiments for PhOH–Ru2+ in 1:1 (v:v) CH3CN–H2O. (d) The same experiments for PhOD–Ru2+ in 1:1 (v:v) CH3CN–D2O. The excitation pulse width was ∼10 ns in all cases. | ||
Analogous kinetic experiments were performed with the PhOH–Ru2+ dyad in 1:1 (v:v) CH3CN–H2O (Fig. 3c) and with its deuterated analog in 1:1 CH3CN–D2O (Fig. 3d). The reaction kinetics are markedly faster in this case. The time constants of product formation are 17 ns (PhOH–Ru2+) and 18 ns (PhOD–Ru2+). Evidently, there is no significant H/D KIE in this case, and thus a distinction between concerted and stepwise PCET mechanisms is not possible on the basis of the kinetic data. We note that the absence of an H/D KIE is not an argument against CPET because several other examples are known in which concerted proton–electron transfers were unambiguously identified as rate-determining reaction steps, yet no significant KIEs could be detected.30
One piece of evidence points towards CPET as a rate-determining reaction step in PhOH–Ru2+: in anhydrous CH3CN no photochemistry occurs (see above). Water must be present in order for photochemistry to happen, and the likely role of H2O is that of a proton acceptor. If an ET–PT sequence with a rate-determining ET step was operative for PhOH–Ru2+ in CH3CN–H2O why would the rate-determining ET step not occur in pure CH3CN also where there is no proton acceptor present? Thus one could argue that phenol oxidation in PhOH–Ru2+ is only possible when occurring in concert with deprotonation. On the other hand we note that the change from pure CH3CN to CH3CN–H2O entails a change in phenol oxidation potentials that might render ET more favorable, i.e., even a simple photoinduced ET step could possibly become operative only upon solvent change. As noted above, phenol oxidation in CH3CN–H2O is irreversible, making the determination of accurate standard potentials impossible and hence we refrain from detailed thermodynamic considerations which are bound to ultimately provide no unambiguous answer either. On a qualitative level it seems obvious that one-electron oxidation of phenol proceeds at less positive potentials than oxidation of 4-cyanophenol hence an ET–PT mechanism is inherently more likely for PhOH–Ru2+ than for CN–PhOH–Ru2+.
We conclude that in PhOH–Ru2+ the rate-determining photochemical reaction step is most likely the ET step of an ET–PT sequence, but a CPET mechanism cannot be ruled out completely. In a scenario in which both dyads react via a rate-determining CPET step, the difference in reaction rates (139 ns vs. 17 ns) could easily be explained by the difference in O–H bond dissociation free energies between 4-cyanophenol (92.6 kcal mol−1 in DMSO) and phenol (88.3 kcal mol−1 in DMSO).12
Summary and conclusions
PCET between 4-cyanophenol or phenol and photoexcited Ru(bpz)32+ has been explored in two different settings, namely in bimolecular reactions resembling hydrogen atom transfer (Scheme 1a) and in multi-site PCET reactions with covalent dyads (Scheme 1b). The bimolecular reactions shown in Scheme 1a are clear-cut cases of concerted PCET processes. The new dyads shown in Scheme 1b exhibit apparent photoacid behavior which can only be explained by a sequence of photoinduced PCET followed by a thermal electron transfer event in the reverse direction (Scheme 3). An H/D kinetic isotope effect of 2.0 ± 0.2 for CN–PhOH–Ru2+ indicates that the rate-determining step in this dyad is a concerted proton–electron release at 4-cyanophenol. This KIE is markedly lower than that of the bimolecular reaction between 4-cyanophenol and Ru(bpz)32+ (10.2 ± 0.6), but of course the proton acceptors in the two different scenarios are not the same (H2O versus a bpz ligand). For PhOH–Ru2+ no significant H/D KIE is detected hence the initial PCET reaction is most likely comprised of a rate-determining electron release followed by subsequent deprotonation of a phenoxyl radical cation before thermal back electron transfer from Ru(bpz)3+ produces the observable phenolate photoproduct. The photochemical behavior of the PhOH–Ru2+ dyad thus appears to be in contrast to the bimolecular reaction between phenol and photoexcited Ru(bpz)32+ (Scheme 1a) which involves concerted proton–electron release at the phenol.The photochemical reaction sequence occurring in our dyads is conceptually identical to a sequence of photoinduced charge-separation and thermal charge-recombination events as previously reported for many electron transfer dyads. The key characteristic of the dyads shown in Scheme 1b is that the initial photoinduced charge-separation event is proton-coupled, involving either concerted proton–electron transfer (CN–PhOH–Ru2+) or a sequence of ET and PT events (PhOH–Ru2+).
Acknowledgements
This work was supported by the Swiss National Science Foundation (grant number 200021_146231/1). Pierre Dechambenoit (Université de Bordeaux) is thanked for acquiring the X-ray diffraction data.Notes and references
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163 CrossRef CAS.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363 CrossRef CAS PubMed.
- S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Acc. Chem. Res., 2009, 42, 1881 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Chem. Rev., 2010, 110, 7024 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2010, 43, 1019 CrossRef CAS PubMed.
- L. Hammarström and S. Styring, Energy Environ. Sci., 2011, 4, 2379 Search PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016 CrossRef CAS PubMed.
- I. J. Rhile and J. M. Mayer, J. Am. Chem. Soc., 2004, 126, 12718 CrossRef CAS PubMed.
- I. J. Rhile, T. F. Markle, H. Nagao, A. G. DiPasquale, O. P. Lam, M. A. Lockwood, K. Rotter and J. M. Mayer, J. Am. Chem. Soc., 2006, 128, 6075 CrossRef CAS PubMed.
- T. F. Markle, I. J. Rhile, A. G. DiPasquale and J. M. Mayer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8185 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961 CrossRef CAS PubMed.
- L. Biczok, N. Gupta and H. Linschitz, J. Am. Chem. Soc., 1997, 119, 12601 CrossRef CAS.
- J. L. Cape, M. K. Bowman and D. M. Kramer, J. Am. Chem. Soc., 2005, 127, 4208 CrossRef CAS PubMed.
- J. J. Concepcion, M. K. Brennaman, J. R. Deyton, N. V. Lebedeva, M. D. E. Forbes, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2007, 129, 6968 CrossRef CAS PubMed.
- J. Bonin, C. Costentin, C. Louault, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2011, 133, 6668 CrossRef CAS PubMed.
- J. Bonin, C. Costentin, M. Robert and J. M. Savéant, Org. Biomol. Chem., 2011, 9, 4064 CAS.
- C. Costentin, M. Robert and J. M. Savéant, Phys. Chem. Chem. Phys., 2010, 12, 11179 RSC.
- N. V. Lebedeva, R. D. Schmidt, J. J. Concepcion, M. K. Brennaman, I. N. Stanton, M. J. Therien, T. J. Meyer and M. D. E. Forbes, J. Phys. Chem. A, 2011, 115, 3346 CrossRef CAS PubMed.
- A. Magnuson, H. Berglund, P. Korall, L. Hammarström, B. Åkermark, S. Styring and L. C. Sun, J. Am. Chem. Soc., 1997, 119, 10720 CrossRef CAS.
- O. Johansson, H. Wolpher, M. Borgström, L. Hammarström, J. Bergquist, L. C. Sun and B. Åkermark, Chem. Commun., 2004, 194 RSC.
- T. Lachaud, A. Quaranta, Y. Pellegrin, P. Dorlet, M. F. Charlot, S. Un, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2005, 44, 1536 CrossRef PubMed.
- T. Irebo, S. Y. Reece, M. Sjödin, D. G. Nocera and L. Hammarström, J. Am. Chem. Soc., 2007, 129, 15462 CrossRef CAS PubMed.
- G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466 CrossRef CAS PubMed.
- T. Irebo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2008, 130, 9194 CrossRef CAS PubMed.
- A. A. Pizano, J. L. Yang and D. G. Nocera, Chem. Sci., 2012, 3, 2457 RSC.
- T. Irebo, M.-T. Zhang, T. F. Markle, A. M. Scott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 16247 CrossRef CAS PubMed.
- V. W. Manner, A. G. DiPasquale and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 7210 CrossRef CAS PubMed.
- V. W. Manner and J. M. Mayer, J. Am. Chem. Soc., 2009, 131, 9874 CrossRef CAS PubMed.
- T. F. Markle, I. J. Rhile and J. M. Mayer, J. Am. Chem. Soc., 2011, 133, 17341 CrossRef CAS PubMed.
- M.-T. Zhang, T. Irebo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2011, 133, 13224 CrossRef CAS PubMed.
- C. Costentin, M. Robert, J. M. Savéant and C. Tard, Phys. Chem. Chem. Phys., 2011, 13, 5353 RSC.
- J. J. Warren, A. R. Menzeleev, J. S. Kretchmer, T. F. Miller, H. B. Gray and J. M. Mayer, J. Phys. Chem. Lett., 2013, 4, 519 CrossRef CAS PubMed.
- M. Kuss-Petermann, H. Wolf, D. Stalke and O. S. Wenger, J. Am. Chem. Soc., 2012, 134, 12844 CrossRef CAS PubMed.
- B. Albinsson, M. P. Eng, K. Pettersson and M. U. Winters, Phys. Chem. Chem. Phys., 2007, 9, 5847 RSC.
- C. Shih, A. K. Museth, M. Abrahamsson, A. M. Blanco-Rodriguez, A. J. Di Bilio, J. Sudhamsu, B. R. Crane, K. L. Ronayne, M. Towrie, A. Vlcek, J. H. Richards, J. R. Winkler and H. B. Gray, Science, 2008, 320, 1760 CrossRef CAS PubMed.
- C. Bronner and O. S. Wenger, J. Phys. Chem. Lett., 2012, 3, 70 CrossRef CAS.
- O. S. Wenger, Acc. Chem. Res., 2013, 46, 1517 CrossRef CAS PubMed.
- V. Balzani, Electron transfer in chemistry, VCH Wiley, Weinheim, 2001, vol. 3 Search PubMed.
- O. S. Wenger, Coord. Chem. Rev., 2009, 253, 1439 CrossRef CAS PubMed.
- D. A. Debie, A. Ostrowicz, G. Geurtsen and H. C. Vanderplas, Tetrahedron, 1988, 44, 2977 CAS.
- N. Dales and Z. Zhang, US Pat., WO 2008/0245390 A2, 2008 Search PubMed.
- D. Hanss and O. S. Wenger, Inorg. Chem., 2009, 48, 671 CrossRef CAS PubMed.
- M. Darabantu, L. Boully, A. Turck and N. Ple, Tetrahedron, 2005, 61, 2897 CrossRef CAS PubMed.
- D. Hanss, M. E. Walther and O. S. Wenger, Coord. Chem. Rev., 2010, 254, 2584 CrossRef CAS PubMed.
- E. Lörtscher, M. Elbing, M. Tschudy, C. von Hänisch, H. B. Weber, M. Mayor and H. Riel, ChemPhysChem, 2008, 9, 2252 CrossRef PubMed.
- M. Yamaji, J. Oshima and M. Hidaka, Chem. Phys. Lett., 2009, 475, 235 CrossRef CAS PubMed.
- P. A. Anderson, R. F. Anderson, M. Furue, P. C. Junk, F. R. Keene, B. T. Patterson and B. D. Yeomans, Inorg. Chem., 2000, 39, 2721 CrossRef CAS.
- C. Bronner and O. S. Wenger, Inorg. Chem., 2012, 51, 8275 CrossRef CAS PubMed.
- P. K. Das, M. V. Encinas and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 4154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols and product characterization data, additional luminescence and transient absorption data, and crystallographic data for ligand 11 (CIF). CCDC 951403. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cp55071k |
| This journal is © the Owner Societies 2014 |