 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A water-soluble tin(IV) porphyrin as a bioinspired photosensitiser for light-driven proton-reduction†
Anne-Marie
Manke
a,
Karen
Geisel
b,
Anne
Fetzer
a and
Philipp
Kurz
*a
aInstitute for Inorganic and Analytical Chemistry, Albert-Ludwigs-University Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: philipp.kurz@ac.uni-freiburg.de; Tel: +49-7611-203-6127
bInstitute for Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52056 Aachen, Germany
First published on 31st January 2014
Abstract
The water-soluble tin(IV) porphyrin dichlorido-5,10,15,20-tetrakis(p-carboxyphenyl)-porphyrinato-tin(IV) (SnTPPC, 1) was synthesised as a mimic of biological chlorophyll photosensitisers. In natural photosynthesis, chlorophyll pigments start the multi-electron transfer processes resulting in water-oxidation and NADP+-reduction. The photochemical properties of compound 1 were characterised by measuring absorption and fluorescence spectra. Electrochemical measurements in water revealed well-suited redox potentials of 1 for both proton-reduction to H2 as well as water-oxidation to O2. The tin(IV) porphyrin was then used as a photosensitiser in model systems for light-induced proton-reduction in aqueous solution, where an optimization of the experimental conditions was carried out to achieve reaction rates comparable to those found for [Ru(bipy)3]2+, a standard dye in artificial photosynthesis. By employing UV/Vis-spectroelectrochemistry, we found that the porphyrin ligand of 1 is redox non-innocent in water. A complex set of reduction reactions of the porphyrin macrocycle occurs during photocatalytic experiments involving the ligand's chlorin form as a key intermediate. On the basis of these results, a potential reaction sequence for light-driven H2-formation is formulated, where the reductive quenching of 1 forms the initial reaction step and reduced forms of 1 serve as hydride transfer agents to the H2 evolution catalyst. The spectroscopic, electrochemical and catalytic properties of SnTPPC make this compound class an attractive, affordable and easily accessible choice for photosensitisers in artificial photosynthetic systems. Finally, the detected complicated redox reactions of the porphyrin ring in water offer a possible explanation of why the chlorophylls of P680 or P700 are carefully wrapped in a water-free part of the PSII and PSI proteins.
Introduction
Many designs for solar energy conversion are based on light-driven water-splitting.1,2 The first step of any such system is light absorption followed by charge separation. The most important process for solar energy conversion on earth is oxygenic photosynthesis as carried out by green plants and algae. Chlorophyll molecules are key players during this process, as they act both as light absorbing molecules and electron transfer agents. In this way, the biological system generates the strongly oxidising potentials needed for water-oxidation in Photosystem II (PSII) and the strong reducing agent for NADP+-reduction in PSI via light absorption by chlorophyll dyes.3,4Chlorophylls are substituted porphyrin-type ligands coordinating a redox-inactive Mg2+-ion. X-ray crystallography revealed that the chlorophyll molecules of the primary donors of the photosynthetic machinery are arranged in ensembles of four chlorophylls, the pigments P6805 of PSII and P7006 of PSI. Both are perfectly positioned within the protein environments for efficient electron transfer after light excitation.
In their electronic ground states, both P680 and P700 are poor electron donors, but in their excited states electron transfer from the chlorophylls to the primary acceptors within PSI and PSII takes place with a quantum efficiency of nearly 100%. After light-induced oxidation (oxidative quenching), the oxidised forms of the pigments, P680˙+ and P700˙+, are then available as ground state oxidation agents within the respective electron transfer chains to drive the oxidation of water (in PSII) or plastocyanin (in PSI), respectively.
| 2H2O + 4hν → O2 + 4H+ + 4e− | (1) |
| 2H+ + 2e− + 2hν → H2 | (2) |
We work on the development and understanding of biomimetic systems for light-driven water-oxidation (“PSII mimics”, eqn (1)) and proton-reduction (“PSI mimics”, eqn (2)). This field has seen vast developments in recent years. In addition to the fundamental interest in the chemistry of the photosystems, research groups all over the world try to find strategies for an efficient and affordable conversion of solar energy into “solar fuels”.1–3,7 Suitable light absorbing molecules are central components of any such devices. Most systems reported so far which are able to photocatalyse reaction (1) or (2) use either ruthenium polypyridyl complexes, polyaromatic organic species or semiconductor systems as photosensitisers. There are many examples of all three approaches in the literature.4
In the study presented here, we “rediscover” a more biomimetic approach for the development of photosensitisers for reactions (1) or (2). Inspired by the structures of the biological chlorophyll dyes, we present a detailed investigation of the porphyrin complex dichlorido-5,10,15,20-tetrakis(p-carboxyphenyl)-porphyrinato-tin(IV) (1, Scheme 1) concerning its suitability to act as a chlorophyll mimic. Water-soluble tin(IV) porphyrins have already been used for the photocatalysis of both reactions (1) and (2) in studies which were published 10 or 30 years ago, respectively.8,9 However, details concerning their syntheses are sketchy, the redox reactions involved in the photocatalytic process remain unclear and these dyes have not been studied in this context since. The last point is hard to understand because tin(IV) porphyrins are interesting candidates for bioinorganic model chemistry and artificial photosynthetic systems for a number of reasons. They are:
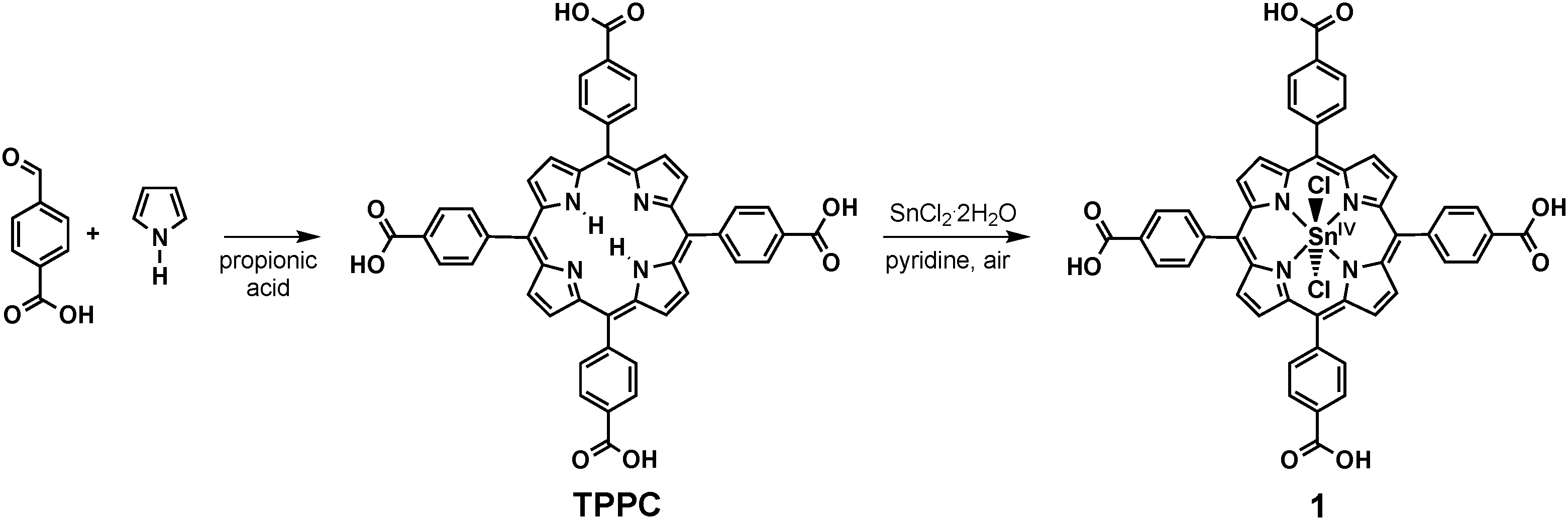 | ||
| Scheme 1 Two-step synthesis of dichlorido-5,10,15,20-tetrakis(p-carboxyphenyl)-porphyrinato-tin(IV) (1). | ||
• water-soluble, and therefore present in homogenous solution with the substrates of both reactions (1) and (2).
• easily accessible via a two-step synthesis (see below).
• built around a central main group ion (Sn4+) with a redox-inert electron configuration ([Kr]4d105s0) similar to the Mg2+ center of chlorophylls ([Ne]3s0).
• known to have suitable redox potentials for water-splitting.10–13
• very stable, as the large, highly-charged Sn4+ ion fits perfectly into the porphyrin ring system.10,12
Results and discussion
Syntheses and purification
The synthesis of symmetrically substituted meso-tetraphenyl-porphyrins (TPP) has been known for decades since the work of Adler et al.14 Thus several methods exist to obtain water-soluble porphyrins by introducing polar substituents to the phenyl rings of the tetraphenylporphyrin backbone.10,12 Many meso-tetraphenyl-porphyrins and some of their metal-complexes are even commercially available. However, their syntheses and purification methods are rarely or only partially described in the often rather dated literature and this is also the case for tin(IV) porphyrins. As purity and an affordable and easy synthesis are major requirements for a photosensitiser in an artificial photosynthetic system, we thus had to re-investigate the preparation routes for these complexes first.To achieve solubility in water, we started from reported procedures for the preparation of the well-known sulphonato8,13 and carboxylato15,16 derivatives of tetraphenylporphyrin and their corresponding tin(IV) complexes. None of the published syntheses yielded truly satisfactory results and thus had to be modified. After an extensive study, we found that the synthesis and purification of the carboxyphenyl-porphyrin complex of tin(IV) (compound 1) were far easier and yielded much better results. Aside from impurities caused by inorganic salts like carbonates and sulphates, an additional reason for the difficult purification of the sulphonato derivative appeared to be that the reported preparation methods for the tetrakis(p-sulfophenyl)porphyrin (TPPS) ligand also yield mono-, di-, and trisulfonated porphyrins,17 which are very hard to separate from the desired ligand bearing four –SO3−-groups. Interestingly, all so far published reports on the syntheses of tin(IV) TPPS complexes lack detailed analytical data, leaving the question of purity of these products unclear.8,13,18
After some optimisation, we found that an analytically pure product of the carboxylato complex 1 could be obtained following a manageable two-step procedure. First, the water-soluble porphyrin 5,10,15,20-tetrakis(p-carboxyphenyl)-porphyrine (TPPC) could be prepared following the established synthetic route of Adler and co-workers by condensing pyrrole and a benzaldehyde, in this case 4-formylbenzoic acid (Scheme 1).14,16,19 The complexation of the Sn4+ cation could then be achieved in the reaction of this ligand with tin(II) chloride in the presence of pyridine acting as a base and air as a mild oxidant.12,20
We carried out a comprehensive characterisation of complex 1 concerning its purity and its spectroscopic and electrochemical properties. NMR spectra (1H, 13C, 119Sn) showed that the TPPC ligand and also the tin(IV) porphyrin complex were obtained in pure form and (especially important here, see below) with no indication for otherwise commonly found chlorin impurities21 (see ESI,† Fig. S1a and b). In addition to the NMR data, IR spectroscopy and elemental analyses (CHN and Cl−) confirmed that our preparation of 1 contains no noteworthy inorganic impurities (see experimental section).
For a solution of 1 in dimethyl sulfoxide, the chemical shift of −590 ppm found for 119Sn by NMR shows that there are two axial chlorido ligands coordinated to the tin centre of the porphyrin complex.12,22 If complex 1 is dissolved in D2O/2 M NaOD, three signals could be found in the 119Sn-NMR spectrum (Fig. 1). The complex with two axial chlorido ligands can still be observed at δ = −590 ppm in aqueous solution, but there are two more signals visible in the NMR spectrum.
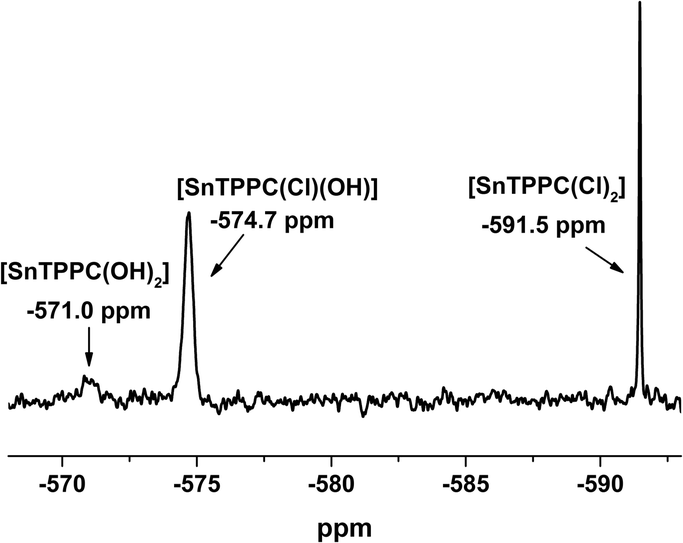 | ||
| Fig. 1 119Sn-NMR spectrum of 1 in D2O/2M NaOD (identical spectra were observed for freshly prepared solutions and a sample kept for 3 days at room temperature). | ||
For the complex with two axial hydroxido ligands [SnTPPC(OH)2], a shift of the 119Sn-NMR signal to about −570 ppm would be expected,12,22 but the signal with this chemical shift is very small (Fig. 1), indicating that no full exchange of the two axial Cl− ligands for OH− occurs. An additional signal can be observed at −574.7 ppm. We assume that this is the mixed complex with one chlorido and one hydroxido ligand coordinated to the tin(IV) metal centre, which would show that partial exchange of the axial ligands occurs in aqueous solution. After leaving the sample at room temperature for extended time periods, no further exchange of the axial ligands can be observed in the 119Sn-NMR-spectra. The spectrum remained unchanged after 3 days, which leads to the conclusion that no complete exchange of the axial ligands from chlorido to hydroxido happens in aqueous solution even at high pH.
Spectroscopic characterisation and zero–zero energy (E00)
For pH-values above pH 7, clear solutions of 1 in water can be prepared, which are intense purple in colour and show the expected UV/Vis absorption bands for metal porphyrins23 (Fig. 2): an intense Soret band at 421 nm and Q-bands at 517 (Q0–2), 557 (Q0–1) and 596 nm (Q0–0). Steady state fluorescence spectra of 1 in water exhibit two emission bands at 600 nm and 654 nm (Fig. 2), which are due to fluorescence out of the singlet Q-states and can be observed for excitation into both the Soret or the Q0–2 absorption bands.24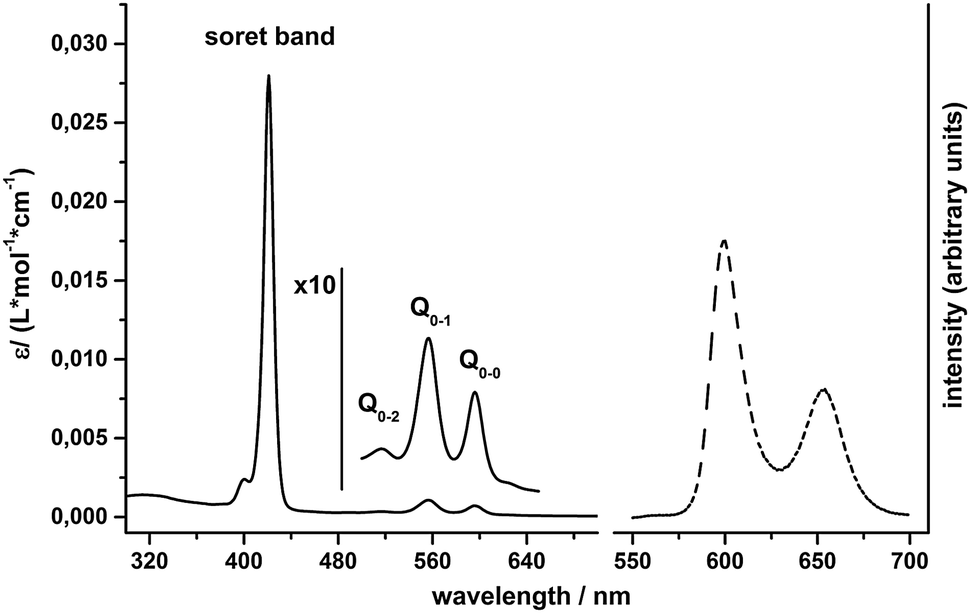 | ||
| Fig. 2 Left: absorption spectrum (solid line) of 1 in water, measured at pH = 7 (0.1 M phosphate buffer). Right: fluorescence spectrum (dashed line) of 1 in water (pH = 7, 0.1 M phosphate buffer, [1] = 1 μM, excitation wavelength: 420 nm). | ||
The zero–zero energy E00 is the energy difference between the molecule's ground state and the vibrational ground state of the electronic excited state of the compound (THEXI, the THermally Equilibrated eXcIted state).25,26 An estimate of this energy difference is needed to calculate the redox potentials of the excited state of a compound from its ground state redox potentials using the Rehm–Weller equation (see below).27 Different methods exist to derive E00 from absorption and fluorescence spectra.26,28 First, we calculated E00 using the medium wavelength values between corresponding absorbance and fluorescence peaks for both the singlet Q0–1 and the Q0–0 states. Additionally, we used Gaussian fits of corresponding absorption and emission bands and derived E00 from the intercept of the normalized curves (see ESI,† Fig. S2 and Table S1). The E00 values obtained from both methods are similar and yield average excited state energies of 1.99 eV for E00(Q0–0) and 2.14 eV for E00(Q0–1).
Light-driven water-splitting reactions in a homogenous system are diffusion controlled. The lifetimes of the singlet THEXI state of water-soluble metalloporphyrins of ZnII, PdII or SnIV are only about 1 ns long, so one also has to consider the possibility of photochemistry involving the much longer-lived triplet states with lifetimes of about ∼1 ms.24,29 This is especially important in the case of tin(IV) porphyrins like 1, where the efficiency of S → T intersystem crossing is known to be rather high.15 As we unfortunately did not have the possibility to record phosphorescence spectra to determine E00 of the triplet state by spectroscopic methods, we used the singlet–triplet energy difference found by Harriman et al.29 for [SnIVCl2TPPS] (0.48 eV) to calculate the triplet E00 for 1 from our data. The approximate E00 value thus determined from the Q0–1 data is 1.66 eV, very similar to the 1.65 eV reported for [SnIVCl2TPPS] in water.29
The possibility of aggregation is always an issue for solutions of porphyrins.30,31 We measured absorption spectra for different concentrations of 1 in neutral phosphate buffer and found that the Q0–1 absorbance at 557 nm follows the Beer–Lambert law for the concentration range from 10−6 M to 10−3 M (see ESI,† Fig. S3). This indicates that the tin(IV) porphyrin molecules form no aggregates at least up to concentrations of 1 mM and thus the later presented data on spectroscopic, but also redox and catalytic behaviour represent the properties of isolated molecules of 1 in solution.
Redox properties of the ground and photoexcited states
The redox properties of the ground state of 1 in water were determined by cyclic voltammetry (CV) and differential pulse voltammetry (DPV). In order to record electrochemical signals for a maximum potential range in water, a hanging-mercury-drop-electrode was used for reductive and a glassy carbon electrode for oxidative scans. In this way we could benefit from the high overpotentials of these two electrode materials for proton-reduction (Hg) and water-oxidation (C), respectively, and thus achieved a measurement range of −1.8 V < E < +1.8 V in aqueous solution (all redox potentials here and throughout the text are given vs. the normal hydrogen potential, NHE).Within this potential window, the CV of compound 1 shows two irreversible reduction waves at −0.73 V and −0.95 V (Fig. 3). The DPV of the same solution of compound 1 shows two reduction events at the same potentials (see ESI,† Fig. S4). For oxidizing potentials, an irreversible process was found with a peak potential of +1.42 V. In the reductive CV, a re-oxidation wave can be observed at −0.42 V (see ESI,† Fig. S5), but this is apparently not the re-oxidation of the product of the reduction events. Instead, a comparison of a re-oxidation DPV of 1 with that of a buffered solution of Sn2+ showed that the re-oxidation peak most likely originates from tin amalgam formation during the reductive part of the measurements, as both the scans for 1 and for Sn2+ showed a large re-oxidation event at −0.4 V (see ESI,† Fig. S6). Thus, at potentials suitable for tin(IV) reduction, a fraction of the tin from the tin(IV) porphyrin apparently deposits as tin amalgam at the mercury drop and is then later re-oxidized at −0.42 V. The fact that we could not observe this peak during measurements of the free ligand and also not during measurements with a glassy carbon electrode supports this interpretation (see ESI,† Fig. S7).
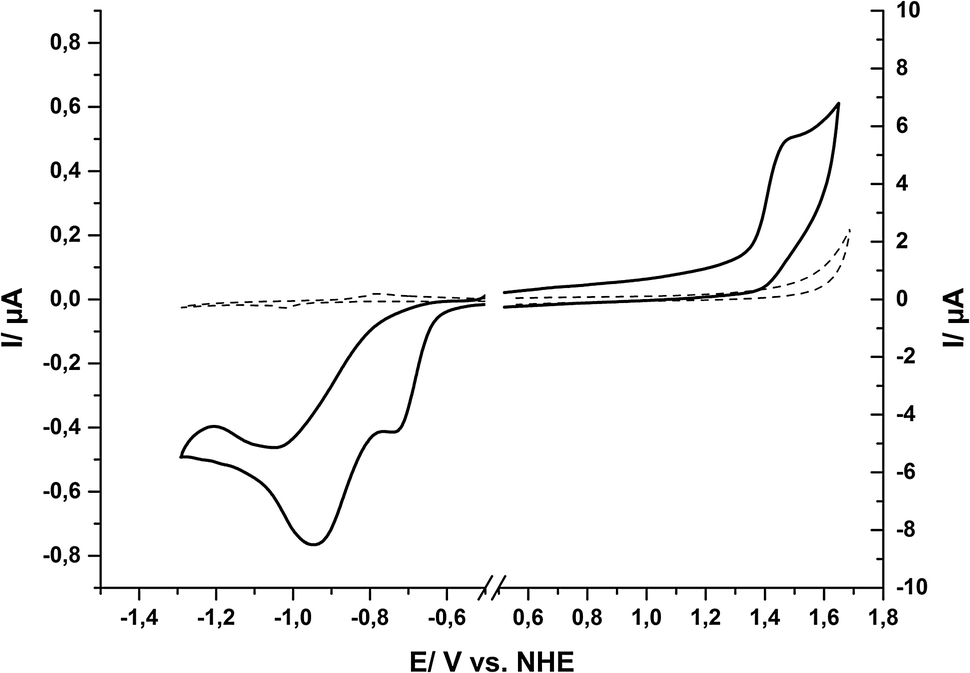 | ||
| Fig. 3 Cyclic voltammograms for oxidative and reductive scans of 1 in aqueous phosphate buffer ([1] = 0.5 mM, total phosphate concentration 0.5 M, pH = 7). The dashed line shows the electrolyte background and the sweep rate was 100 mV s−1 in all cases. The different current ranges occur because the used electrodes differed in surface areas and shapes: spherical (∼0.4 mm2) in the case of the mercury-drop-electrode vs. planar (3.1 mm2) in the case of the glassy carbon electrode. | ||
Using the Rehm–Weller equation,27,32 we calculated redox potentials for the singlet THEXI state of 1 in water from the electrochemical data and the smaller E00 value (1.99 eV) derived from spectroscopy (see above). Table 1 shows an overview of the values obtained for 1 in this way in comparison to the redox properties reported for [Ru(bpy)3]2+ (Ru, the currently most commonly used dye in artificial photosynthetic systems), the related water soluble zinc(II) porphyrin ZnTPPC and P680/P700 (the primary donors of PSII and PSI).
| SnTPPC (1) | [Ru(bipy)3]2+a | ZnTPPCb | P680c | P700d | |||
|---|---|---|---|---|---|---|---|
| Singlet | Triplet | Singlet | Triplet | ||||
| a From ref. 32. b From ref. 24. c From ref. 32 and 33. d From ref. 34. e In eV. f In V vs. NHE. g Not known. | |||||||
| E 00 | 1.99 | 1.61 | 2.12 | 2.03 | 1.60 | ∼1.8 | ∼1.8 |
| E red | −0.73 | −1.35 | −1.25 | ||||
| E red*f | 1.26 | 0.88 | 0.77 | 0.78 | 0.35 | ||
| E ox | 1.42 | 1.29 | 0.80 | ∼1.25 | ∼0.40 | ||
| E ox*f | −0.57 | −0.19 | −0.83 | −1.23 | −0.80 | ∼−0.55 | ∼−1.40 |
An analysis of the data in Table 1 shows that 1 has indeed very promising redox properties both for oxidative and reductive photochemistry in the context of artificial photosynthesis, because both the redox potentials necessary for reactions (1) and (2) are easily reached by the oxidised and reduced forms of the dye, respectively. In comparison to other compounds for light-driven electron transfer, the singlet excited state 1* is a stronger oxidant but a weaker reductant than Ru*. It is also a stronger oxidant but a much weaker reductant than the excited state of ZnTPPC, thus following the previously established observation that metalloporphyrins of SnIV are especially electron poor.11,12 For reductive chemistry, the single-electron reduced species Ru− and [ZnTPPC]˙− are on the other hand much stronger electron donors than 1 in their reduced form. Concerning the potential model character of 1 for the natural photosynthetic pigment, SnTPPC's oxidation potentials in ground and singlet excited states are very suited to mimic those of P680 in Photosystem II, while they are much higher than those found for P700 of PSI.
In photocatalytic test systems, sacrificial quencher molecules are often used to start the reaction sequence by irreversible electron transfer reactions between the excited state of the photosensitisers and the quencher molecules in their ground states. The redox potentials of typical quenchers, like the electron donors triethanolamine (TEOA) and ethylendiamine-tetraacetic acid (EDTA) or the electron acceptor methyl viologen (MV2+), were determined in the same aqueous electrolyte system as used before for the electrochemistry of 1. For the oxidations of TEOA and EDTA single, irreversible oxidation waves can be observed in the CVs (see ESI,† Fig. S8). The respective oxidation potentials were determined by DPV to be 1.07 V for TEOA and 1.17 V for EDTA, respectively (see ESI,† Fig. S8). The CV of the electron acceptor MV2+ shows two partially reversible reduction waves, positioned at −0.43 V and −0.74 V according to the analysis of the DPV (see ESI,† Fig. S8).
As a summary of the redox properties of interest, the oxidation and reduction potentials of quenchers and the tin(IV) porphyrin 1 in its ground and excited states are shown in Scheme 2. One can see that the singlet excited state (S*) of compound 1 (Ered*S = 1.26 V) can be reductively quenched by both TEOA and EDTA while the triplet excited state (T*) of 1 is not oxidising enough for efficient oxidation of TEOA or EDTA (Ered*T = 0.88 V). The singlet excited state of 1 (Eox*S = −0.57 V) can also react with the electron acceptors MV2+ and [CoIII(NH3)5Cl]2+ (Co3+), while the triplet excited state (Eox*T = −0.19 V) can be oxidised only by Co3+. On the basis of these findings, all four quencher molecules could theoretically be used for light-driven electron-transfer reactions with SnTPPC as a photosensitiser. However, in certain cases this is only true if the more energetic singlet state is indeed accessible for redox chemistry despite its short lifetime.
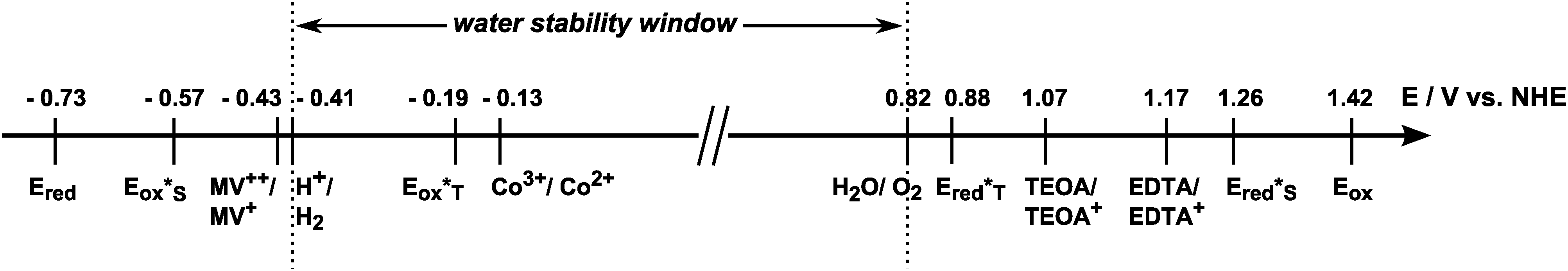 | ||
| Scheme 2 Ground and excited state redox potentials of 1 in water at pH = 7 in relation to those of the redox couples H+/H2 and H2O/O2, the oxidative quenchers [CoIII(NH3)5Cl]2+ (Co3+) and methylviologen (MV2+) and the reductive quenchers triethanolamine (TEOA) and ethylendiamine-tetraacetic acid (EDTA). | ||
Photocatalytic proton-reduction
After establishing a feasible synthesis route and the basic redox parameters for 1 in aqueous medium, we next used the metalloporphyrin as a photosensitiser for light-driven proton-reduction in systems inspired by the work of Fuhrhop and Krüger (“System 1”) or Kalyanasundaram (“System 2”), respectively.9,35If a mixture containing SnTPPC as a photosensitiser (PS), platinum dioxide (PtO2) as a heterogeneous proton-reduction catalyst (Catred) and triethanolamine (TEOA) or ethylenediamine-tetraacetic acid (EDTA) as sacrificial electron donors (D) is illuminated with visible light (λ > 400 nm), a continuous evolution of H2 can be detected by headspace GC for at least 1 h in neutral phosphate buffer. Such a reaction mixture is similar to the system investigated by Fuhrhop and Krüger in 1982, who used a water-soluble, β-substituted tin(IV) porphyrin as a photosensitiser, EDTA as an electron donor and colloidal platinum as a catalyst (System 1).9
The electron flow in such systems starts with the oxidation of the sacrificial electron donor and ends by the use of the reduction equivalents for H2 formation at Catred (Scheme 3). During the suggested reaction sequence for this process, the excited state of the photosensitiser is reductively quenched by the electron donor, which results in the formation of the single-electron reduced form of PS, which might undergo further reaction steps in the following (“PSred” in Scheme 3; for details on its nature, see below). The electrons are then transferred to the catalyst, where the reduction of protons to hydrogen takes place.
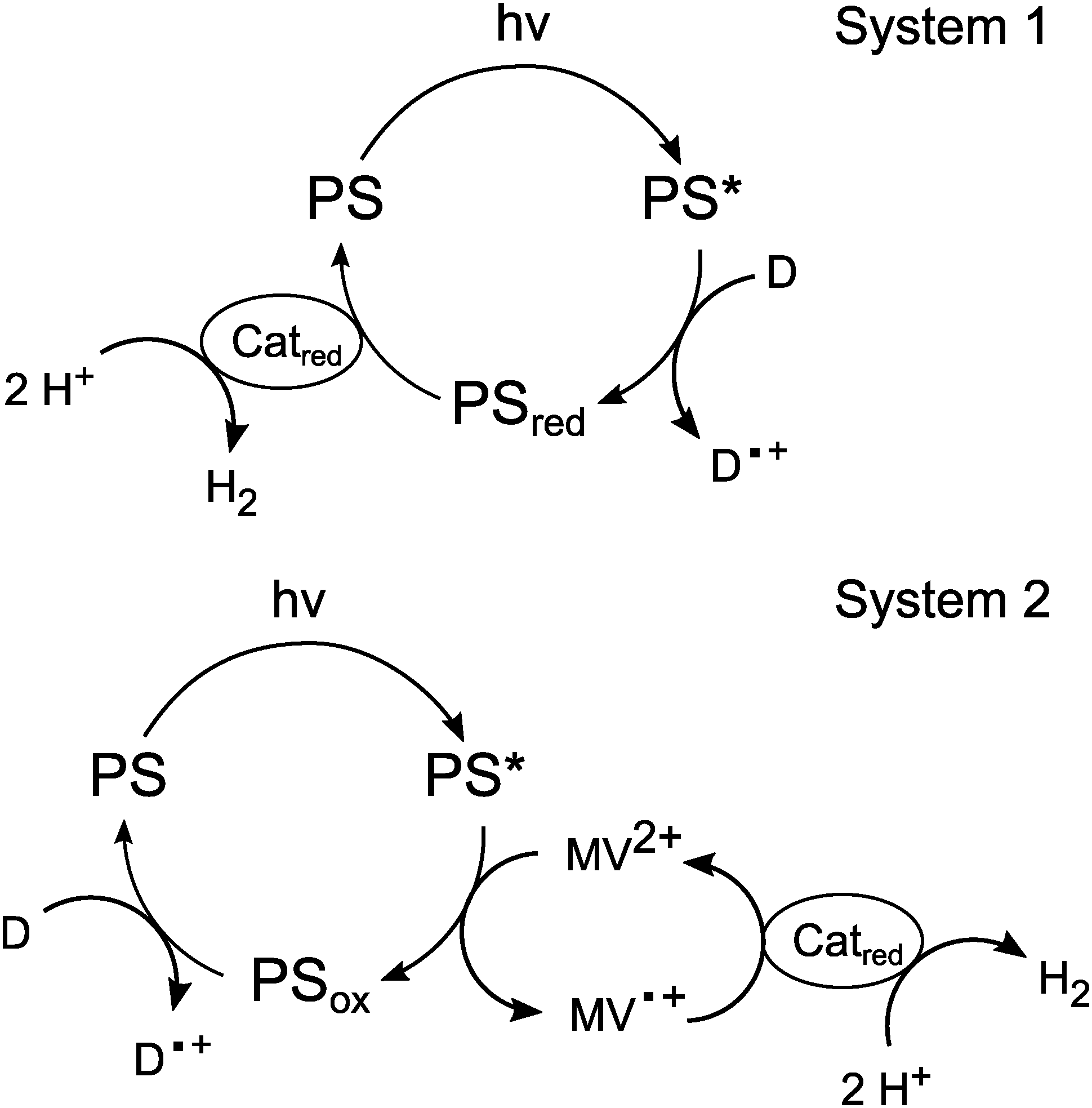 | ||
| Scheme 3 Suggested reaction sequences for light-driven proton-reduction in Systems 1 and 2 with photosensitisers (PS), electron donors (D) and proton-reduction catalysts (Catred). | ||
In the system developed by Kalyanansundaram (System 2), the electron transfer agent methyl viologen (MV2+) is also present in the reaction mixture.35 Here, [Ru(bpy)3]2+ serves as the photosensitiser, TEOA as an electron donor and PtO2 powder as a catalyst. This system is at first very similar to System 1, but the key difference is the quenching mechanism of the excited state of the photosensitiser. In System 1, PS* is reductively quenched by D while in System 2 the excited state is oxidatively quenched by the acceptor MV2+. The electron donor (TEOA) is added to regenerate the original redox state of the photosensitiser in a dark reaction.
In the following we first present an optimization of the reaction conditions for System 1 using SnTPPC as a photosensitiser, secondly the use of 1 as a PS for System 2 and a comparison of its performance with the established photosensitiser [Ru(bpy)3]2+ and finally details concerning the reaction sequence in System 1.
Optimization of the light-driven proton-reduction System 1
In order to optimize the reaction conditions for this system, we investigated the dependence of the reaction rate on the buffer system and pH of the aqueous reaction medium, the type and concentration of the quencher and also the concentrations of catalyst and dye. A summary of the catalytic experiments can be found in Table 2.| Optimization parameter | [SnTPPC]/mM | [Quencher]/mM | [PtO2]/mg L−1 | [MV2+]a/mg mL−1 | pHb (5 mL) | Quenching mechanism of 1* | H2 evolution/μmol h−1 | |
|---|---|---|---|---|---|---|---|---|
| Reductive (System 1) | Oxidative (System 2) | |||||||
| a Methyl viologen was purchased as MVCl2·xH2O. b The pH was controlled by use of an aqueous phosphate buffer (0.1 M). c For pH 4, an acetic acid buffer (0.1 M) was used. | ||||||||
| pH | 0.5 | TEOA/50 | 100 | — | 7 | ✓ | 1.42 | |
| 0.5 | TEOA/50 | 100 | — | 5.5 | ✓ | 0.84 | ||
| 0.5 | TEOA/50 | 100 | — | 4c | ✓ | — | ||
| Quencher | 0.5 | TEOA/50 | 100 | — | 7 | ✓ | 1.42 | |
| 0.5 | EDTA/50 | 100 | — | 7 | ✓ | 0.42 | ||
| [SnTPPC]/mM | 0 | TEOA/50 | 100 | — | 7 | ✓ | — | |
| 0.1 | 0.33 | |||||||
| 0.6 | 1.46 | |||||||
| 1 | 2.06 | |||||||
| 1.5 | 1.84 | |||||||
| 2.5 | 1.05 | |||||||
| 6.6 | 0 | |||||||
| [TEOA]/M | 0.5 | 0 | 100 | — | 7 | ✓ | — | |
| 25 | 0.51 | |||||||
| 50 | 1.42 | |||||||
| 75 | 1.76 | |||||||
| 100 | 1.22 | |||||||
| [PtO2]/mg mL−1 | 0.5 | TEOA/50 | 0 | — | 7 | ✓ | — | |
| 22 | 1.04 | |||||||
| 100 | 1.42 | |||||||
| 233 | 1.14 | |||||||
| 340 | 0.68 | |||||||
| Reaction system | 0.5 | TEOA, 50 | 100 | — | 7 | ✓ | 1.42 | |
| 0.5 | TEOA, 50 | 100 | 1 | 7 | ✓ | 0.11 | ||
| Optimized system | 1 | TEOA, 60 | 22 | — | 7 | ✓ | 4.8 | |
In a typical experiment, 5 mL of argon-flushed buffer solution was prepared in a 20 mL septum-capped vial in the dark. The solution, typically containing 0.5 mM photosensitiser, 100 mg L−1 platinum dioxide and 50 mM quencher was irradiated with a slide projector while stirring. The amount of hydrogen in the vial's headspace was measured by gas chromatography after one hour of irradiation, disregarding the amount of hydrogen dissolved in the solution. This approach is feasible, because the solubility of hydrogen in water is very low.36
To prove that such a system produces H2 only if all components are present in the reaction mixture and if the mixture is irradiated with visible light, we performed a series of negative tests. Four experiments were made, each of it omitting one of the necessary components of System 1: SnTPPC, PtO2, TEOA or irradiation. In none of the experiments hydrogen evolution could be detected if any of the four components is absent. This points out that proton-reduction takes place only light-induced and that all reaction partners are indeed needed to achieve hydrogen evolution in System 1.
In a typical experiment utilising compound 1 as a photosensitiser (e.g. the first experiment in Table 2), we observed 1.42 μmol of evolved hydrogen after one hour of irradiation. An extension of the irradiation time to three hours increased the amount of evolved hydrogen only to 1.88 μmol, indicating that the catalytic process slowly stops due to unproductive side reactions (see below).
By variation of the reaction parameters, we firstly found a strong dependence of the amount of evolved H2 on the pH of the solution and the buffer system (Table 2, optimization parameter pH). In a system containing 0.5 mM SnTPPC, 100 mg L−1 PtO2 and 0.05 M TEOA, we observed 1.42 μmol evolved hydrogen after one hour of irradiation at neutral pH in 0.1 M phosphate buffer. Under otherwise identical conditions, only 0.84 μmol H2 per h could be detected for the more acidic solution at pH 5.5, although Krüger and Fuhrhop reported pH 5 as the best pH condition for hydrogen evolution in their study.9 If the pH of the reaction medium is reduced further to pH 4 (0.1 M acetate buffer), hydrogen formation ceases.
Next the irreversible electron donor TEOA was replaced by EDTA (Table 2, optimization parameter quencher). Using otherwise identical reaction conditions as before at pH 7, only 0.42 μmol H2 were detected after 1 h and thus only about one third of the amount that was found when TEOA acted as electron donor.
Furthermore, we find that the hydrogen evolution rate is higher if the tin(IV) porphyrin 1 is used in reaction System 1 than in System 2, as we found a marked decrease in the amount of evolved hydrogen to 0.11 μmol h−1 if MV2+ is added to the reaction mixture (Table 2, optimization parameter reaction system).
Having identified a preferable pH range (pH 7) and a quencher molecule (TEOA) for light-driven proton reduction reactions using SnTPPC, we next performed measurements to optimize the concentrations of the individual components of System 1.
To visualize the results, hydrogen evolution rates as functions of the concentrations of TEOA, SnTPPC and PtO2 are presented in Fig. 4. At the top of Fig. 4 the rate dependence on the quencher concentration is shown for solutions containing 0.5 mM SnTPPC, 100 mg L−1 PtO2 and a variable concentration of TEOA in phosphate buffer (pH 7). At first, the amount of evolved hydrogen increases with increasing quencher concentrations to reach a maximum for [TEOA] = 75 mM. For an even higher quencher concentration of 100 mM, the hydrogen evolution decreases markedly.
 | ||
| Fig. 4 Hydrogen formation after 1 h of visible light irradiation as function of reactant concentrations using reaction System 1: variations in the amount of TEOA (top), SnTPPC (middle) and PtO2 (bottom) in the reaction mixture (total volume 5 mL) are shown. | ||
The relationship between the photosensitiser concentration [SnTPPC] and the hydrogen evolution rate is shown in the middle of Fig. 4. For this series, solutions contained different concentrations of SnTPPC, 100 mg L−1 PtO2 and 0.05 M TEOA in phosphate buffer (pH 7). The hydrogen evolution rate reached a pronounced maximum at a value of [SnTPPC] = 1 mM, after which the reaction rate decreased gradually until no catalysis is observed any more for a porphyrin concentration of 6.5 mM.
Finally, the third graph of Fig. 4 illustrates the dependence of the reaction rate on the amount of PtO2 catalyst present in the system. The sample solutions contained different quantities of PtO2, 0.5 mM SnTPPC and 0.05 M TEOA in phosphate buffer (pH 7). Again, we were able to clearly identify an optimum range for catalysis, in this case a concentration of 100 mg L−1 platinum oxide.
Considering the combined data of Table 2 and the trends shown in the graphs of Fig. 4, one realises that there seem to exist rather pronounced “optimal conditions” for the varied concentrations [H+] (pH), [TEOA], [SnTPPC] and [PtO2]. In all four cases, very low or very high concentrations never result in saturation, but rather an inhibition behaviour concerning the catalytic rate.
First and foremost, we find that the pH of the reaction mixture plays an important role in light-driven proton-reduction. The pH value should be low, because proton-reduction according to eqn (2) is more efficient at high [H+]. However, at too low pH values the quenching reaction with the amines TEOA and EDTA is not possible anymore, as their free electron pairs of the amine functions (the electron source in the molecule) are protonated. These opposing trends could explain the observation that pH 7 was found to be the best pH value for proton-reduction in System 1.
Next, the catalyst to photosensitiser ratio seems to play an important role in the catalytic activity of the system. Hydrogen evolution becomes more efficient when the relative concentration of photosensitiser to catalyst is high. Proton-reduction to H2 is a multi-electron process and additionally a certain degree of “reductive charging” of the PtO2 particles seems necessary before H2 formation occurs. Thus higher amounts of the photosensitiser are needed. Aside from that, the concentration of the photosensitiser should not be increased too much, as very high concentrations of 1 decrease and even stop hydrogen evolution (Fig. 4, middle). This may be caused by aggregation of the porphyrin at higher concentrations, as porphyrins are well-known to form stacked assemblies in solution (H- and J-aggregates).30,31 As we found that the absorption of 1 does not follow the Beer–Lambert law any more for concentrations above 1 mM (see ESI,† Fig. S9), this seems reasonable.
The concentration of the electron donor is generally chosen to be much higher than the concentrations of the other compounds. This is meant to lead to more efficient quenching of the excited state. Dynamic quenching is a diffusion controlled process and therefore one would expect that the reaction rate should increase continually for increasing quencher concentrations. In contrast, we also observed a concentration optimum for [TEOA] in our system (Fig. 4, top). As TEOA can also act as a base (pKB 6.2), higher [TEOA] increase the pH of the solution when the system reaches the capacity of the phosphate buffer. The resulting lower [H+] again disfavours proton-reduction (see above).
The comparison of the two studied donor molecules showed that TEOA is better suited for the investigated system than EDTA. TEOA is a slightly stronger reductant (Eox = 1.07 V) than EDTA (Eox = 1.17 V) (Scheme 2), which may be one reason for the more efficient reductive quenching of 1*. Another reason could be that there are more free amine units available for TEOA at pH 7 than for EDTA (pKB(EDTA4−) 3.7).
After the described optimization rounds for the catalytic system SnTPPC/TEOA/PtO2, we continued our study using the developed optimized system for light-driven hydrogen evolution consisting of 1 mM tin(IV) porphyrin 1, 20 mg L−1 PtO2 and 60 mM TEOA in phosphate buffer (pH 7). Through the optimization procedure, we were able to increase the reaction rate to an amount of 4.8 μmol of evolved hydrogen after one hour of irradiation. During this time continuous H2 evolution can be observed (Fig. 5). In agreement with our previous arguments, the time-course of H2 evolution shows a lower rate during the first 20 min (build-up of reduced photosensitiser and catalyst). After about 50 min of irradiation H2 production slows down again, which is probably caused by the various side reactions of the reduced tin porphyrin (see below). In contrast to 1, no hydrogen evolution can be detected in such a system if the commonly used [Ru(bpy)3]2+ serves as a photosensitiser, most probably because the excited state of the ruthenium complex Ru* cannot be reductively quenched by TEOA (see Scheme 4).35
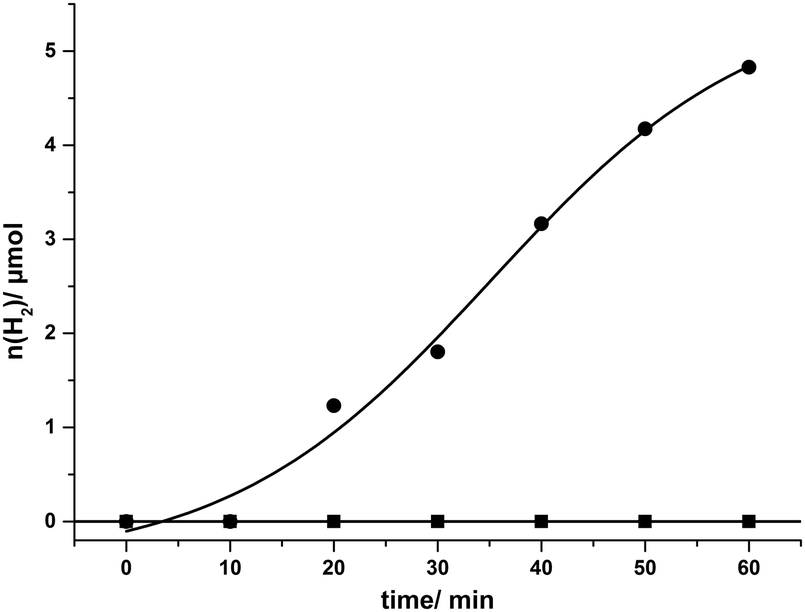 | ||
| Fig. 5 Time course of photoinduced hydrogen evolution under visible light irradiation with tin(IV) porphyrin 1 (●) and [Ru(bpy)3]2+ (■) as photosensitisers in System 1. Conditions: 1 mM dye, 20 mg L−1 PtO2 and 60 mM TEOA in phosphate buffer (pH 7). | ||
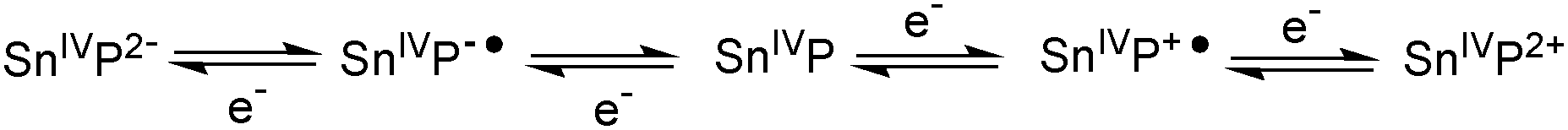 | ||
| Scheme 4 Ligand-centred redox processes reported for tin(IV) porphyrins in organic solvents.10,11 P denotes the aromatic part of the porphyrin ring system. | ||
Krüger and Fuhrhop detected hydrogen evolution rates of 18 mol(H2)/mol(PS) after one hour of irradiation. In a typical experiment containing 5 × 10−5 M photosensitiser, EDTA as quencher and 2 × 10−5 M colloidal platinum as catalyst in 1 L solution at pH 5, they detected about 900 μmol of hydrogen after an irradiation time of one hour.9 We thus find similar amounts of hydrogen per solution volume here (970 μmol(H2) h−1 L−1), but our hydrogen evolution rate per photosensitiser (0.96 mol(H2)/mol(1)) is much lower as we used a higher photosensitiser concentration in our experiments.
Comparison with the established photosensitiser [Ru(bpy)3]2+
As mentioned above, Kalyanasundaram detected hydrogen evolution for an alternative reaction mixture containing the photosensitiser [Ru(bpy)3]2+ in combination with the acceptor methyl viologen (MV2+) acting as an oxidative quencher for Ru* (Scheme 3, System 2).35 Proton reduction takes place at the catalyst after electron transfer from the reduced relay (MV˙+) to the catalyst and the oxidised form of the photosensitiser (Ru+) reacts with the electron donor in a “dark reaction” back to its initial state.35Illuminating a mixture of 0.5 mM [Ru(bpy)3]2+, 1 mg mL−1 methyl viologen, 100 mg mL−1 PtO2 and 0.05 M electron donor in phosphate buffer (pH 7), we were able to reproduce the good photocatalytic performance of the Kalyanasundaram system with [Ru(bpy)3]2+ and found a hydrogen evolution rate of 2.21 μmol(H2) h−1 (0.88 mol(H2)/mol(Ru)) with EDTA as an electron donor. For TEOA as an electron donor, only 0.16 μmol(H2) h−1 (0.06 mol(H2)/mol(Ru)) could be detected. Kalyanasundaram found 9 mol(H2)/mol(Ru) during typical experiments (4 × 10−5 M [Ru(bpy)3]2+, 10−2 M MV2+, 0.05 M TEOA and PtO2 as catalyst in 100 mL solution at pH 7).35 We thus again detected similar amounts of hydrogen per hour and volume here (440 μmol(H2) h−1 L−1) as in the original report (360 μmol(H2) h−1 L−1).
However, when we exchanged the photosensitiser for the tin(IV) porphyrin 1, reaction rates were very low. A blank experiment for Ru as a photosensitiser without MV2+ present (0.5 mM [Ru(bpy)3]2+, 100 mg mL−1 PtO2 and 0.05 M electron donor (TEOA, EDTA) in phosphate buffer (pH 7)), confirmed that the addition of methyl viologen to the reaction mixture is necessary to observe any amount of hydrogen using Ru.
To summarize the observations of the photocatalysis experiments, Fig. 6 shows a comparison of hydrogen evolution rates for the use of either [Ru(bpy)3]2+ or SnTPPC as photosensitisers in both Systems 1 and 2. Additionally, the dependence of the reaction rates on the electron donor is illustrated.
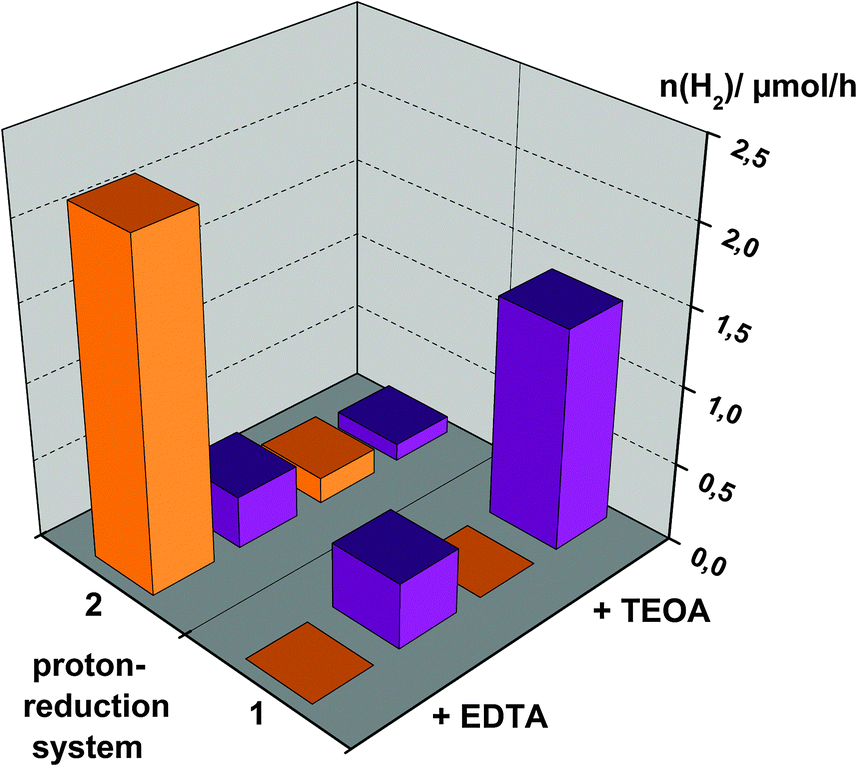 | ||
| Fig. 6 Comparison of hydrogen evolution rates in Systems 1 and 2 depending on the used photosensitiser: [Ru(bpy)3]2+ (orange) or SnTPPC (purple). All of the sample solutions contained a 0.5 mM photosensitiser, 100 mg mL−1 PtO2 and 0.05 M electron donor in phosphate buffer (pH 7). The electron donor was either EDTA (two rows at the front/left) or TEOA (two rows at the back/right). | ||
In general, hydrogen evolution in System 2 is much more efficient if the ruthenium complex [Ru(bpy)3]2+ is used as a dye for electron transfer (Fig. 6). Considering redox potentials, it is possible for both photosensitisers to start the electron transfer chain as the excited states of both the ruthenium complex (Ru*) and the singlet excited state of the tin(IV) porphyrin (1*) have suitable potentials to be oxidatively quenched by MV2+ (Scheme 2). On the other hand, reductive quenching of Ru* is not possible with EDTA or TEOA according to Scheme 2. As clearly visible in Fig. 6, oxidative quenching of 1* by MV2+ is apparently not a very productive route to obtain H2, but rather negatively competes with the reductive route, so using the tin(IV) porphyrin in System 2 leads to virtually no hydrogen production.
Fig. 6 also shows that more hydrogen is evolved using EDTA as an electron donor in System 2 in comparison to TEOA. This may be affected by more efficient electron transfer from EDTA to the oxidised form of the ruthenium complex Ru+ than with TEOA as an electron donor. Taking into account that TEOA (Eox = 1.07 V) is a stronger reductant than EDTA (Eox = 1.17 V), we can give no reasonable explanation for this observation.
In summary, we were able to show that light-driven hydrogen evolution is possible using the tin(IV) porphyrin 1 as a photosensitiser in a simple system in neutral aqueous solution. The fastest formation of hydrogen could be detected for SnTPPC in an optimized form of System 1, where reaction rates comparable to the established photosensitiser [Ru(bpy)3]2+ in System 2 could be reached (Fig. 6).
Reaction sequence
For aqueous solutions, it is well-documented that follow-on reactions of the metalloporphyrin can occur after oxidation or reduction events.9,29,37 For example, two of the single-electron-oxidised or -reduced porphyrin species can disproportionate leading to doubly reduced or doubly oxidised species, which react with water in substitution reactions of the porphyrin heterocycle. In such cases, the addition of protons to the doubly reduced macrocycle results in the formation of chlorin species as the products of +2e−/+2H+- events (see Scheme 5), while the addition of hydroxide to the doubly oxidised form yields the −2e−/+2OH−- product isoporphyrin. For tin(IV) porphyrins it is known that the first process can easily occur as photochemical conversions to the chlorin form have already been observed in non-aqueous systems if only trace amounts of reducing agents were present.21,38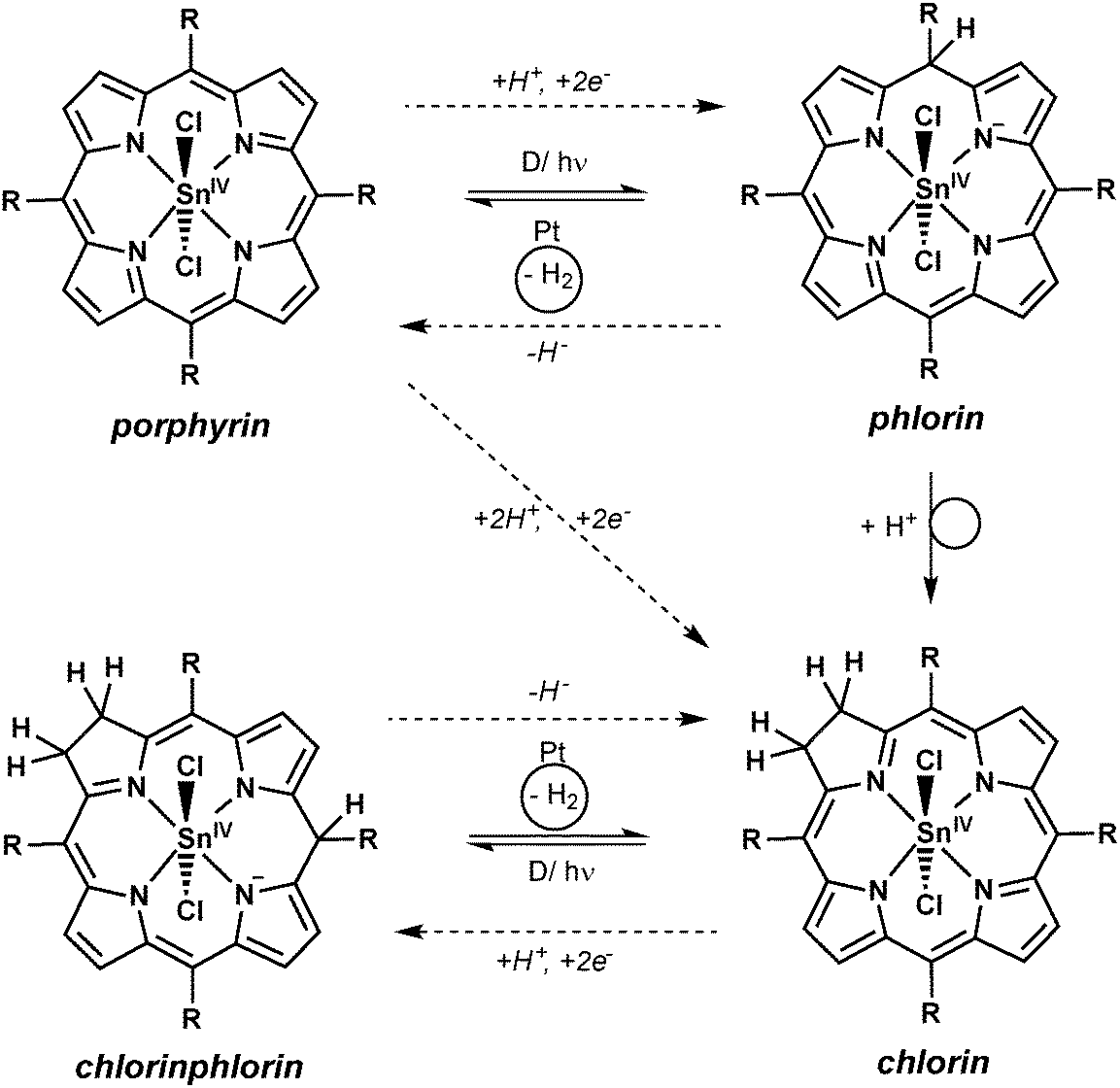 | ||
| Scheme 5 Possible reactions of the macrocycle of the tin(IV) porphyrin 1 during light-driven proton-reductions in water. Reductive quenching by an electron donor D is most likely the first step of a proton-coupled reaction sequence leading to the tin(IV) chlorin species. Phlorin and chlorinphlorin intermediates might act as hydride-transfer agents to the platinum catalyst where the evolution of hydrogen occurs. | ||
In all light-driven proton-reduction experiments studied here, a colour change of the solutions from purple to green was observed during the reaction if SnTPPC is used as a photosensitiser. This colour change was more or less pronounced for different experiments, but occurred in all cases and was found to be irreversible even if the product mixture was exposed to air.
In Fig. 7 the absorption spectra of a “System 1 mixture” (0.5 mM SnTPPC, 100 mg L−1 PtO2 and 0.05 M TEOA in neutral phosphate buffer) after 0, 30, 90 and 180 minutes of visible light irradiation are shown. The formation of a new Q-band at 625 nm can be observed, while the Soret-band maximum at 421 nm remains unchanged. The other Q-bands at 556 and 596 nm slightly decrease in intensity during irradiation. The observation of an additional Q-band at 625 nm has been identified as characteristic of chlorin formation before,9,39 and we thus conclude that a reduction of 1 to the corresponding chlorin also occurs here during the photoreactions of System 1. If the reaction mixture of System 1 is irradiated without the platinum dioxide catalyst, the same colour and spectral changes of the reaction mixture can be detected (see ESI,† Fig. S10), but – as the catalyst is missing – no evolution of H2 occurs.
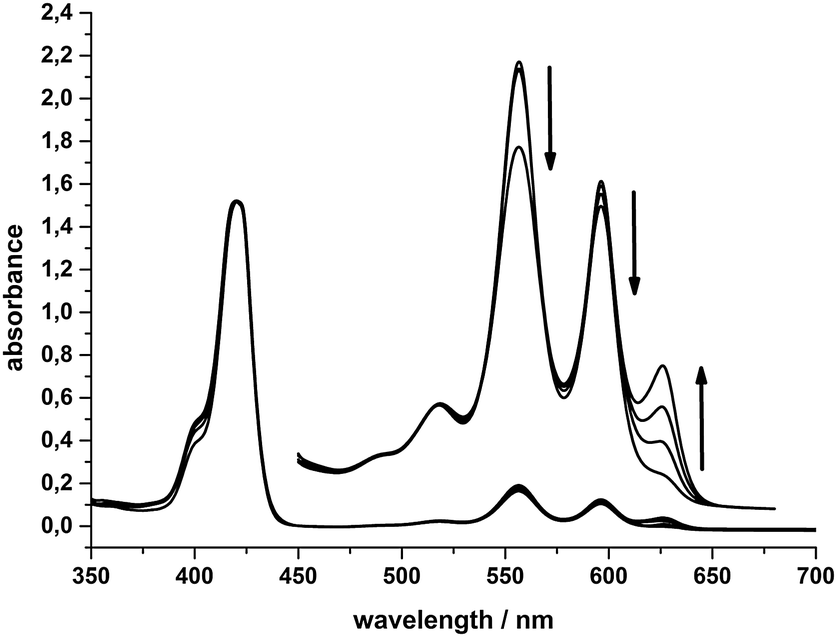 | ||
| Fig. 7 Absorption spectra of 10 μM SnTPPC (Q-bands 100 μM) in System 1 after irradiation for 0, 30, 90 and 180 minutes. | ||
The timescale of the photochemical chlorin formation does not behave proportionally to the timescale of light-induced hydrogen evolution in System 1. The absorption at 625 nm due to chlorin formation increases linearly (∼0.16 a.u. h−1) during an irradiation time of three hours. In contrast to that, hydrogen evolution takes place at a rate of 1.42 μmol(H2) h−1 during the first hour of irradiation and then slows down markedly in the next two hours, where we found a rate of 0.23 μmol(H2) h−1 (see above). Apart from a possible deactivation of the photosensitiser and/or the catalyst, the competition between chlorin formation and hydrogen evolution might be a reason for that. Another reason could be that the catalytic route involving the tin(IV) chlorin is less efficient than the one for the tin(IV) porphyrin (see below, especially Scheme 5).
Electrochemical reduction studies
As UV/Vis spectroscopy suggested structural changes at the porphyrin macrocycle during light-driven proton-reduction, we performed additional electrochemical and spectroelectro-chemical experiments to study the reduction processes of 1 in water in more detail.In the absence of water, the redox chemistry of tin(IV) porphyrins is known to be ligand-based with the metal retaining its SnIV oxidation state during both reduction and oxidation processes. In organic solvents, it has been shown that a maximum of five different oxidation states might be accessible (Scheme 4), for which reversible electrochemistry has been reported.10,11 In contrast to these reports for organic solvents, we already showed above that both the electrochemical oxidations and reductions of 1 in an aqueous electrolyte are irreversible (Fig. 3).
If the irreversible reduction process was related to the transformation of the tin(IV) porphyrin to the chlorin species, two electrons and two protons have to be transferred to the porphyrin macrocycle (see Scheme 5 and eqn (3)). Thus, the reduction potential for the reaction of the tin porphyrin (SnP) to the chlorin (SnC) is expected to be pH-dependent. An analysis of the pH-dependence of the reduction potentials of SnTPPC in aqueous buffer solutions should therefore provide information about the mechanism of the reduction process:
| SnP + 2H+ + 2e− → SnC | (3) |
At first, reductive CVs (and DPVs, see ESI,† Fig. S11) of porphyrin 1 were recorded in aqueous phosphate buffer at pH 5, 6, 7 and 8 using a hanging-mercury-drop working electrode. For all four pH-values, irreversible reduction waves could be detected, but these were found at different potentials. The events are complicated and their overall shapes and current magnitudes also change for different pH-values (Fig. 8). Nevertheless, it is clearly visible that the onset of the reduction event shifts to lower potentials for higher pH values of the electrolyte. This indicates that the reduction process is coupled to a proton-consuming reaction as it becomes more facile for higher [H+].
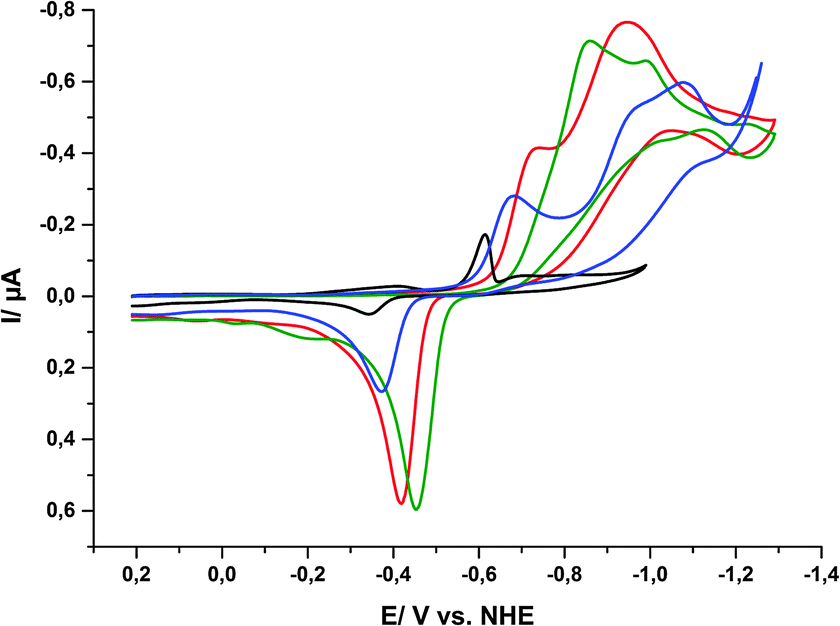 | ||
| Fig. 8 Cyclic voltammograms of 1 in aqueous phosphate buffers of different pH values ([1] = 0.5 mM, total phosphate concentration 0.5 M): pH 5 (black), pH 6 (blue), pH 7 (red) and pH 8 (green). | ||
For the first reduction wave between −600 and −900 mV, we could observe a strong pH-dependence with a slope of ΔE/ΔpH = −60 mV pH−1. For the second reduction wave the behaviour is not that straightforward as the shapes of the CVs vary significantly, thus Ered,2 decreases from pH 6 to pH 7, but increases again from pH 7 to pH 8. Finally, the re-oxidation peak most likely caused by tin amalgam formation between −300 and −500 mV is also pH-dependent as the re-oxidation potentials increase with increasing pH.
By formulating the Nernst equation for the reduction reaction (3), the pH-dependence of Ered for the conversion of 1 into the chlorin can be formulated as
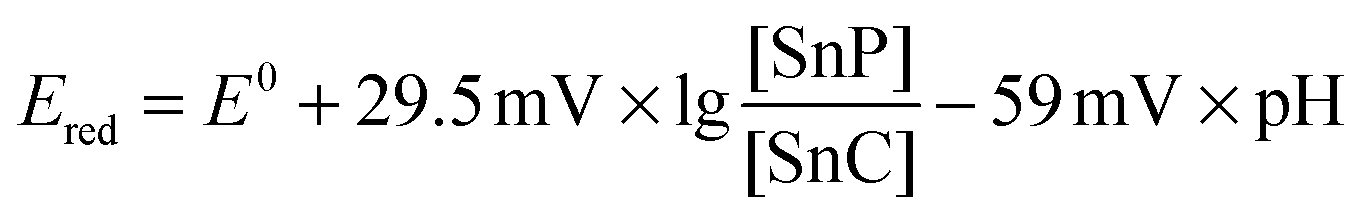 | (4) |
The expected slope of ΔE/ΔpH perfectly fits the −60 mV pH−1 observed for the first reduction wave. These observations give reason to assume that the first reduction process at −0.73 V (pH 7) is a proton-coupled reduction of 1 either of a +2e−/+2H+ or a +1e−/+1H+ type. For the +2e−/+2H+-case, the reduction would result in the formation of the chlorin, and thus the second process at −0.95 V (pH 7) would then correspond to the transfer of additional electrons to the chlorin ring system.
To assign the redox events, we carried out electrolysis of neutral solutions of 1 at −0.8 V or −1.05 V at pH 7 (using a mercury-pool working electrode) and followed the reactions by UV/Vis absorption spectroscopy.
The electrolysis of a neutral solution of 1 at the first reduction potential (−0.8 V vs. NHE) causes no changes in the absorption spectra. If the first reduction wave originates from the transfer of one electron to the porphyrin macrocycle resulting in an anionic π radical (Scheme 4), a decrease in Soret band intensity and the appearance of two new broad absorption bands centred at about 605 nm and 875 nm should be observed.40 The reason that we were not able to detect such changes could be due to the measurement conditions. To be able to measure samples taken from the electrolysed solution, small volumes of electrolytes were taken, diluted with the corresponding buffer and measured in a cuvette. This procedure takes some time for which the reactive π radicals might not be stable.
In contrast, the electrochemical reduction at the second reduction potential (−1.05 V) results in a colour change of the solution from purple to turquoise. In the corresponding UV/Vis spectra, the formation of the characteristic chlorin Q-band at 625 nm is very pronounced while the other Q-bands at 556 and 596 nm decrease (Fig. 9). The Soret band also decreases, but only in a very small amount.
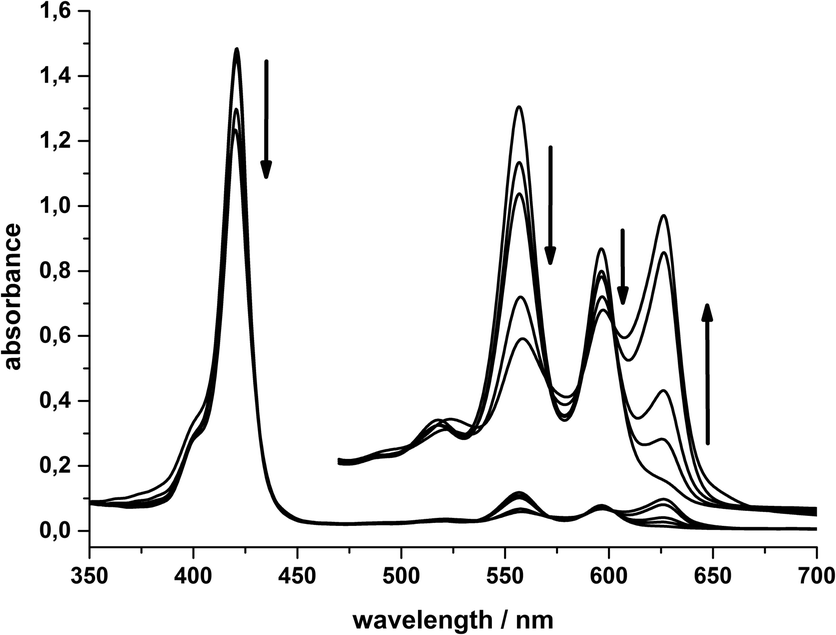 | ||
| Fig. 9 Changes in the absorption spectra of 10 μM SnTPPC 1 (Q-bands 100 μM) during the electrolysis of a 0.5 mM solution of SnTPPC in phosphate buffer (0.5 M, pH 7) at −1.05 V using a mercury-pool-electrode. Spectra after 0, 10, 20, 60 and 120 minutes of electrolysis time are shown. | ||
These spectral changes are virtually identical to those found during the light-driven hydrogen formation reactions and indicate that the formation of a significant amount of chlorin species is an important event both for the photochemical and electrochemical reduction of 1 in water.
Discussion of porphyrin reduction
In the previous sections we could show that the reduction of 1 in water is a complicated process, because porphyrins are non-innocent ligands in aqueous solution.We could observe chlorin formation during photochemical and also during electrochemical experiments. Both reductive quenchers we used (TEOA, EDTA) are able to transfer one electron to the excited state of porphyrin 1 and the single-electron-reduced species should be able to drive proton-reduction (Scheme 2). The reduction potentials we determined by cyclic voltammetry are both pH-dependent, and the electrochemical chlorin formation could be detected by electrolysis of a neutral solution of 1 at the second reduction potential (Ered,2 = −0.95 V vs. NHE).
Fuhrhop and Krüger suggested that the active species for H2-evolution are the phlorin and chlorinphlorin species of the tin(IV) porphyrins.9 As the chlorin macrocycle can also take part in such addition reactions, hydrogen evolution is not stopped by chlorin formation, as we also observed in our studies. As a speciality of this chemistry, the phlorin intermediates can thus not only act as electrons but also as hydride (H−) transfer reagents in the sequence (see Scheme 5, dashed reaction arrows).
Consequently, the reaction sequence for light-driven proton-reduction is much more complicated for SnTPPC than for the established photosensitiser Ru, as various reduced and protonated species relevant for the reaction can occur in parallel in solution. For [Ru(bpy)3]2+, such protonation steps do not take place and the photosensitiser acts as a pure electron agent with the central metal ion changing between the Ru2+ and Ru3+ oxidation states.
Photocatalytic water-oxidation
Wang et al.8 observed water-oxidation to O2 in a system containing SnTPPS as a photosensitiser, ruthenium dioxide RuO2 as a water-oxidation catalyst and [CoIII(NH3)5Cl]2+ as an irreversible electron acceptor. These results could not be reproduced by us. Using SnTPPC (or SnTPPS) as a photosensitiser in such a system gave no detectable amount of oxygen after irradiation.One reason for the missing oxygen evolution could be side reactions of the porphyrin π cationic radical species. After disproportionation from two of these species, substitution of hydroxide to the dicationic porphyrin could lead to the isoporphyrin species, a reaction which is known to occur for porphyrins in aqueous media.9,29,37 As the isoporphyrin complex should show different absorption bands than the tin(IV) porphyrin, we measured absorption spectra during irradiation of mixtures or SnTPPC, RuO2 and CoIII or peroxodisulfate S2O82− as an oxidative quencher8,41 in neutral phosphate buffer. No changes in the absorption bands could be detected, indicating that no electron transfer (and also no side reactions leading to the isoporphyrin species) occurred. At this time we can give no explanation for this as oxidative quenching reactions should be thermodynamically possible considering the redox potentials shown in Fig. 3. Further investigations are needed here, but so far we find that SnTPPC can and least not be used in systems for light-driven water oxidation where [Ru(bpy)3]2+ has been established as a suitable dye.
Implications for artificial and natural photosyntheses
In natural photosynthesis, the excited states of the pigments P680* and P700* both react with electron acceptor molecules, yielding the oxidized forms P680+ and P700+ as important intermediates. Many systems for artificial photocatalytic water-splitting are based on this kind of mechanism, which is for example imitated by the reactions of [Ru(bpy)3]2+ in System 2, where oxidative quenching leads to an oxidation of the central ruthenium atom forming Ru3+ which is a water-stable species. In the case of tin(IV) porphyrins, oxidative quenching would lead to π cationic radicals, which are very reactive in aqueous solution and might result in the decomposition of the porphyrin dye.In contrast, the presented proton-reduction systems are initiated by the reductive quenching of the excited state of the tin(IV) porphyrin, leading to π anionic radicals. Our results implicate that the π anionic ligands are non-innocent in water, but react further via proton-coupled disproportionation reactions resulting in chlorin formation (Scheme 5). While the reactions that follow reductive quenching do not inhibit hydrogen production (and might instead rather be needed for it, see Scheme 5), oxidative quenching and its consecutive reactions at the porphyrin macrocycle seem to be non-productive. Clues for this claim are the following observations: (a) for System 2, the addition of an oxidative quencher (MV2+) led to less hydrogen production using SnTPPC as a photosensitiser, (b) water-oxidation to O2 as reported by Wang et al.8 could not be confirmed despite multiple attempts. From the poor performance of 1 in System 2 and the lack of photocatalytic oxygen-formation in the Wang-system, we conclude that tin(IV) porphyrins might not be suitable for artificial photosynthetic systems involving oxidative quenching steps of the photosensitiser.
Overall, we showed that side reactions of the prophyrin ring system dominate the redox chemistry of 1 in aqueous solution both for photochemical and electrochemical processes. In nature, such side reactions do hardly occur for the chlorophyll molecules in the reaction centers of PS I and II. Instead, the pigments just cycle back and forth between their initial and their one-electron oxidized forms. Looking at the recent X-ray structure of PS II from Umena et al.,5 where it was possible to locate (among many other important details) thousands of water molecules for the first time, it is striking to find that the dye molecules of P680 and the electron transfer chain seem to be carefully isolated from water molecules within the protein matrix (see ESI,† Fig. S12). On the other hand, the site of water-oxidation, the oxygen evolving complex (OEC), is surrounded by water and substrate molecules (see ESI,† Fig. S12). In this way nature apparently prevents side reactions of the oxidised chlorophyll molecules with water, while substrate availability is ensured for the OEC.
Conclusions
We presented the syntheses and characterization of the bio-inspired water soluble tin(IV) porphyrin 1 as well as a working photocatalytic proton-reduction system using porphyrin 1 as a photosensitiser. We carried out a comprehensive characterisation of the tin(IV) porphyrin to show that it can be achieved in a defined and pure form via a simple two-step synthesis. Spectroscopic and electrochemical studies were performed in neutral aqueous solution – the same conditions where we screened activity in light-driven reactions.The tin(IV) porphyrin SnTPPC is able to act as a photosensitiser during photocatalytic proton-reduction in two different systems, one based on a reductive mechanism and the other one based on an oxidative quenching mechanism. The system based on the reductive quenching mechanism (System 1) generally resulted in much higher hydrogen evolution rates for the photosensitiser SnTPPC and the amount of hydrogen could be improved by an extensive optimization study of the system. The commonly used photosensitiser [Ru(bpy)3]2+ is not able to catalyse the generation of hydrogen in this system.
We could show that the formation of a tin(IV) chlorin species can be detected during photoinduced proton reduction as well as during electrochemical reduction of the tin(IV) porphyrin. This reaction at the macrocycle occurs after reductive quenching or electrochemical reduction of the porphyrin in water, followed by a disproportionation reaction and proton additions. The reduced porphyrin ligand is thus “non-innocent” in water.
Our results imply that aqueous artificial systems based on oxidative quenching mechanisms are not suitable for tin(IV) porphyrins as photosensitisers. This offers an interesting parallel to natural photosynthesis, where the chlorophyll molecules of the pigments P680 and P700 are placed inside the hydrophobic membrane to prevent them from unwanted side reactions.
In future studies we will focus on the understanding of the quenching process of the porphyrins` excited states. Until now, it is unclear whether the singlet or the triplet excited states are involved in the initial quenching processes. Additionally, the reactions at the porphyrin macrocycle following oxidative quenching need further investigation to possibly understand the absence of oxygen evolution with tin(IV) porphyrins as photosensitisers in photocatalytic water-oxidation systems.
A better understanding of this very interesting, promising and – in comparison to the often-used ruthenium complexes – very cheap class of dyes could then pave a way to their use in artificial photosynthesis.
Experimental
Materials
Chemicals were purchased in the highest available quality and used without further purification. All solvents for UV/Vis or fluorescence measurements were of spectroscopic grade. Deionised water (>10 MΩ cm) was used for all experiments.Instrumentation
NMR-spectroscopy was carried out using a Bruker AVANCE 400 Puls Fourier transform spectrometer with a 1H-resonance frequency of 400.13 MHz. A Genesis FT-IR-spectrometer (ATI Mattson) enabled us to determine vibrational spectra of samples compressed in discs of spectroscopy grade KBr. UV/Vis spectra were recorded on a Varian Cary 5000 spectrometer or an Agilent Specord S 300 VIS with solutions of the compounds in 10 mm quartz cuvets. Fluorescence spectra were measured using a Horiba Jobin Yvon Fluoromax-4 spectrofluorometer or a PerkinElmer LS 55 Luminescence Spectrometer with samples in 10 mm quartz cuvets. A Bruker Biflex III instrument was used to measure MALDI-TOF-MS of samples prepared in a 4-chloro-α-cyanocinnamic acid (Cl-CCA) matrix. Elemental analyses were obtained using a Euro Vector Euro EA 3000 elemental analyser. The chloride content of samples was determined by argentometric titration. Electrochemical analyses were performed using an ECO Chemie PGSTAT30 potentiostat and a Metrohm 663 VA stand. The three-electrode setup consisted of glassy carbon working/counter electrodes and a Ag/AgCl-reference (filled with 3 M KCl, +0.21 V vs. NHE). Other measurements were performed using a Metrohm 797 VA Computrace control. The three-electrode setup consisted of a mercury Multi Mode electrode (MME) from Metrohm (or a glassy carbon electrode) as a working electrode, a platinum electrode as a counter electrode and a Ag/AgCl-reference electrode (filled with 3 M KCl, +0.21 V vs. NHE). Electrolysis experiments were performed using an Ametek VersaSTAT4 Potentiostat and an EG&G electrolysis cell. A mercury pool was used as a working electrode, a platinum electrode as a counter electrode (separated over a salt bridge filled with 3 M KCl) and a Ag/AgCl-reference electrode (filled with 3 M KCl, +0.21 V vs. NHE).Hydrogen evolution studies were performed using a HP 6890 Series GC System equipped with an 8 ft × 1/8 in a 5 Å molecular sieve column (Supelco) using argon as a carrier gas. Photocatalytic experiments were carried out with reaction mixtures in 20 mL septum vials. The reactants were weighed in the vial as solids, before the vessel was capped with a gas-tight septum. The argon-flushed buffer solution (5 mL) was then added and the vial was irradiated using a slide projector equipped with a 250 W halogen bulb and a 85 mm f2.7 lens (no cut-off filter was used). The vial was always positioned at the same distance from the light-source (illuminance in front of the vial was always 7 × 104 l×) and the solution was stirred during irradiation. In a typical experiment, gas samples of the headspace were taken and injected to the GC using a HP 7694 headspace sampler equipped with a 1 mL sample-loop after one hour of irradiation. (During the time dependent measurements, 100 μL samples of the vial's headspace were taken with a syringe and injected directly.) Inside the headspace sampler, the vial is first pressurized with the carrier gas and the sample loop is flushed and filled until the pressure in the vial is again at atmospheric pressure. With pressurization of the vial, the headspace volume gets higher about an uncertain amount. For calculations of the total amount of evolved hydrogen, we assumed a headspace volume of the non-pressurized vial. That is why the calculated amounts of hydrogen may be too low. The gases, H2, O2 and N2, were monitored using a thermal conductivity detector. Calibrations for H2 were carried out by the injection of different volumina of a hydrogen gas mixture (7.49 v% H2 in argon). We estimate the error margin for the rate determination to be in the range of ± 25%.
Synthesis of 5,10,15,20-tetrakis(p-carboxyphenyl)porphyrine (TPPC) was carried out similar to literature procedures29,30 with slight modifications. Pyrrole (8.3 mL, 120 mmol) was added to a solution of 4-formylbenzoic acid (18.0 g, 120 mmol) in 500 mL of propionic acid and the solution was heated to reflux for 3 h. Cooling the resulting mixture to 4 °C for 18 h yielded a purple precipitate, which was filtered off, washed with methanol (5× 5 mL) and water (2 × 10 mL) and dried. The final product was obtained after recrystallisation from MeOH–CHCl3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and drying in vacuo. Yield: 6.6 g (27%). 1H-NMR (400 MHz, DMSO-d6): δ = 12.88 (s, 4 H, O-H), 8.79 (s, 8 H, pyrrole-H), 8.33 (d, 3J = 8.2 Hz, 8 H, ortho-H), 8.27 (d, 3J = 8.2 Hz, 8 H, meta-H), −2.98 (s, 2 H, N-H) ppm. 13C-NMR (100 MHz, DMSO-d6): δ = 167.6 (s, COOH), 145.9 (s, ipso-C), 134.9 (s, ortho-C), 131.0 (s, para-C), 128.4 (s, meta-C), 119.8 (s, meso-C), ppm. The signals for the quaternary C
:1) and drying in vacuo. Yield: 6.6 g (27%). 1H-NMR (400 MHz, DMSO-d6): δ = 12.88 (s, 4 H, O-H), 8.79 (s, 8 H, pyrrole-H), 8.33 (d, 3J = 8.2 Hz, 8 H, ortho-H), 8.27 (d, 3J = 8.2 Hz, 8 H, meta-H), −2.98 (s, 2 H, N-H) ppm. 13C-NMR (100 MHz, DMSO-d6): δ = 167.6 (s, COOH), 145.9 (s, ipso-C), 134.9 (s, ortho-C), 131.0 (s, para-C), 128.4 (s, meta-C), 119.8 (s, meso-C), ppm. The signals for the quaternary C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and pyrrole-C carbon atoms were not observed. IR (KBr): ν = 3021 (C–H-val. arom.), 1695 (CO-val.), 1605, 1565, 1507 (CC-val.), 1405 (CN-val.), 965 (C–H-def. pyrrole), 797 (C–H-def. arom.) cm−1. UV/Vis (phosphate puffer, pH = 7): λ (ε [L mol−1 cm−1]) = 414 (2.60 × 105), 526 (9.13 × 103), 566 (8.29 × 103), 595 (4.57 × 103), 652 (4.65 × 105) nm. Analysis calculated for C48H30N4O8·2H2O (%): C, 71.72; H, 3.47; N, 6.43. Found: C, 71.27; H, 4.72; N, 6.43.
N and pyrrole-C carbon atoms were not observed. IR (KBr): ν = 3021 (C–H-val. arom.), 1695 (CO-val.), 1605, 1565, 1507 (CC-val.), 1405 (CN-val.), 965 (C–H-def. pyrrole), 797 (C–H-def. arom.) cm−1. UV/Vis (phosphate puffer, pH = 7): λ (ε [L mol−1 cm−1]) = 414 (2.60 × 105), 526 (9.13 × 103), 566 (8.29 × 103), 595 (4.57 × 103), 652 (4.65 × 105) nm. Analysis calculated for C48H30N4O8·2H2O (%): C, 71.72; H, 3.47; N, 6.43. Found: C, 71.27; H, 4.72; N, 6.43.
For the synthesis of dichlorido-5,10,15,20-tetrakis(p-carboxyphenyl)-porphyrinato-tin(IV) (1), a solution of TPPC (1.29 g, 1.63 mmol) in pyridine (50 mL) was prepared and SnCl2·2H2O (730 mg, 3.25 mmol) was added as a solid. The mixture was heated to reflux for 2 h in the dark. Now diethylether (250 mL) was added to the cold solution. After 12 h at 4 °C in the dark, a purple precipitate was obtained, which was filtered off, washed with ether (4 × 20 mL), CH2Cl2 (4 × 20 mL) water (4 × 20 mL) and again ether (4 × 20 mL) and dried in vacuo. Yield: 1.2 g (75%). Further purification could be achieved by gently stirring a suspension of 80 mg of the crude product in 2 M hydrochloric acid (12 mL), followed by filtration and drying of the solid in vacuo. 1H-NMR (400 MHz, DMSO-d6): δ = 13.30 (s, 4 H, O-H), 9.27 (s, 8 H, pyrrole-H), 8.44 (d, 3J = 8 Hz, ortho-H), 8.39 (d, 3J = 8 Hz, meta-H) ppm. 13C-NMR (100 MHz, DMSO-d6): δ = 167.7 (s, COOH), 145.8 (s, ipso-C), 144.2 (s, CN), 135.3 (s, ortho-C), 133.8 (s, pyrrole-C), 131.7 (s, para-C) 128.8 (s, meta-C), 120.9 (meso-C) ppm. IR (KBr): ν = 3064 (C–H-val. arom.), 1716 (CO-val.), 1608, 1571, 1484 (CC-val.), 1450 (CN-val.), 1015 (C–H-def. pyrrole), 786 (C–H-def. arom.) cm−1. UV/Vis (phosphate puffer, pH = 7): λ (ε [L mol−1 cm−1]) = 421 (1.68 × 105), 517 (2.59 × 103), 566 (6.80 × 103), 596 (4.75 × 103) nm. MALDI-TOF-MS (Cl-CCA): m/z = 908 ([M-2Cl]+), 943 ([M-Cl]+), 1114 ([M-2Cl+(Cl-CCA)]+), 1150 ([M-Cl+(Cl-CCA)]+), 1320 ([M-2Cl+2(Cl-CCA)]+), see also ESI,† Fig. S13. Analysis calculated for C48H28N4O8SnCl2 (%): C, 58.92; H, 2.89; N, 5.73; Cl−, 7.47. Found: C, 58.76; H, 2.83; N, 5.82; Cl−, 6.30.
Acknowledgements
We would like to thank the research groups at the Institute for Inorganic Chemistry at the Christian-Albrechts-University Kiel, where a number of the presented experiments were carried out and many people supported our work. At ALU Freiburg, we would like to thank Dr Harald Scherer for measuring the NMR spectra and for his valuable advice concerning 119Sn-NMR. Financial support from the Deutsche Forschungsgemeinschaft (DFG, project KU 2885/1-1), the Fonds der Chemischen Industrie (Liebig Fellowship PhK) and the states of Schleswig-Holstein and Baden-Württemberg is gratefully acknowledged.References
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15 CAS.
- N. Armaroli and V. Balzani, Energy for a sustainable world. From the oil age to a sun-powered future, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- J. Barber, Chem. Soc. Rev., 2008, 38, 185 RSC.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- A. Ben-Shem, F. Frolow and N. Nelson, Nature, 2003, 426, 630–635 CrossRef CAS PubMed.
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074 Search PubMed.
- S. Wang, K. Enomoto, S. Tanizaki, I. Tabata, K. Hisada, T. Hori and S. Okubayashi, Sen'i Gakkaishi, 2003, 59, 48–52 CrossRef CAS.
- W. Krüger and J.-H. Fuhrhop, Angew. Chem., Int. Ed. Engl., 1982, 21, 131 CrossRef.
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Amsterdam, 2003 Search PubMed.
- J. H. Furhop, K. M. Kadish and D. G. Davis, J. Am. Chem. Soc., 1973, 95, 5140–5147 CrossRef CAS.
- D. P. Arnold and J. Blok, Coord. Chem. Rev., 2004, 248, 299–319 CrossRef CAS PubMed.
- A. Harriman, M. C. Richoux and P. Neta, J. Phys. Chem., 1983, 87, 4957–4965 CrossRef CAS.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- D. Fermín and N. Eugster, in N4-Macrocyclic Metal Complexes, ed. J. H. Zagal, F. Bedioui and J.-P. Dodelet, Springer, New York, 2006, pp. 517–574 Search PubMed.
- A. Harada, K. Shiotsuki, H. Fukushima, H. Yamaguchi and M. Kamachi, Inorg. Chem., 1995, 34, 1070–1076 CrossRef CAS.
- E. B. Fleischer, J. M. Palmer, T. S. Srivastava and A. Chatterjee, J. Am. Chem. Soc., 1971, 93, 3162–3167 CrossRef CAS.
- (a) J. C. Pommier, M. Dabadie and A. Rzama, C. R. Acad. Sci. Paris C, 1980, 291, 65–67 CAS; (b) O. Herrmann, S. H. Mehdi and A. Corsini, Can. J. Chem., 1978, 56, 1084–1087 CrossRef CAS.
- (a) A. Harada, H. Yamaguchi, K. Okamoto, H. Fukushima, K. Shiotsuki and M. Kamachi, Photochem. Photobiol., 1999, 70, 298–302 CrossRef CAS; (b) F. R. Longo, M. G. Finarelli and J. B. Kim, J. Heterocycl. Chem., 1969, 6, 927–931 CrossRef CAS.
- P. Rothemund and A. R. Menotti, J. Am. Chem. Soc., 1948, 70, 1808–1812 CrossRef CAS.
- M. Maiti, B. R. Danger and R. P. Steer, J. Phys. Chem. A, 2009, 113, 11318–11326 CrossRef CAS PubMed.
- D. P. Arnold and J. P. Bartley, Inorg. Chem., 1994, 33, 1486–1490 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- K. Kalyanasundaram and M. Neumann-Spallart, J. Phys. Chem., 1982, 86, 5163–5169 CrossRef CAS.
- A. W. Adamson, J. Chem. Educ., 1983, 60, 797 CrossRef CAS.
- H. Hennig and D. Rehorek, Photochemische und photokatalytische Reaktionen von Koordinationsverbindungen, Teubner, Stuttgart, 1988 Search PubMed.
- D. Rehm and A. Weller, Ber. Bunsen-Ges., 1969, 73, 834–839 CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 2006 Search PubMed.
- A. Harriman, G. Porter and P. Walters, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 1335 RSC.
- J. M. Ribo, J. Crusats, J.-A. Farrera and M. L. Valero, J. Chem. Soc., Chem. Commun., 1994, 681 RSC.
- H.-L. Ma and W.-J. Jin, Spectrochim. Acta, Part A, 2008, 71, 153–160 CrossRef PubMed.
- G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer, Wiley-VCH, New York, Weinheim, Germany, Cambridge, 1993 Search PubMed.
- F. Rappaport and B. A. Diner, Coord. Chem. Rev., 2008, 252, 259–272 CrossRef CAS PubMed.
- L. Taiz and E. Zeiger, Plant physiology, Sinauer, Sunderland, Massachusetts, 5th edn, 2010 Search PubMed.
- K. Kalyanasundaram, J. Kiwi and M. Grätzel, Helv. Chim. Acta, 1978, 61, 2720–2730 CrossRef CAS.
- H. A. Pray, C. E. Schweickert and B. H. Minnich, Ind. Eng. Chem., 1952, 44, 1146–1151 CrossRef CAS.
- A. Harriman and P. Walters, Inorg. Chim. Acta, 1984, 83, 151–154 CrossRef CAS.
- C. Debaig-Valade, O. Bagno, J. C. Pommier and J. Joussot-Dubien, Photochem. Photobiol., 1981, 33, 899–902 CrossRef CAS.
- U. Eisner, J. Chem. Soc., 1957, 3461–3469 RSC.
- K. M. Kadish, P. Tagliatesta, Y. Hu, Y. J. Deng, X. H. Mu and L. Y. Bao, Inorg. Chem., 1991, 30, 3737–3743 CrossRef CAS.
- N. D. Morris and T. Mallouk, J. Am. Chem. Soc., 2002, 124, 11114–11121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectroscopic and electrochemical data. See DOI: 10.1039/c3cp55023k |
| This journal is © the Owner Societies 2014 |