 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dinuclear manganese complexes for water oxidation: evaluation of electronic effects and catalytic activity
Wael A. A. Arafa†a,
Markus D. Kärkäsa,
Bao-Lin Leea,
Torbjörn Åkermarka,
Rong-Zhen Liaoa,
Hans-Martin Berendsb,
Johannes Messingerb,
Per E. M. Siegbahna and
Björn Åkermark*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: bjorn.akermark@organ.su.se
bDepartment of Chemistry, Umeå University, Kemiskt Biologiskt Centrum (KBC), Linneaus Väg 6, SE-90187 Umeå, Sweden
First published on 27th January 2014
Abstract
During recent years significant progress has been made towards the realization of a sustainable and carbon-neutral energy economy. One promising approach is photochemical splitting of H2O into O2 and solar fuels, such as H2. However, the bottleneck in such artificial photosynthetic schemes is the H2O oxidation half reaction where more efficient catalysts are required that lower the kinetic barrier for this process. In particular catalysts based on earth-abundant metals are highly attractive compared to catalysts comprised of noble metals. We have now synthesized a library of dinuclear Mn2II,III catalysts for H2O oxidation and studied how the incorporation of different substituents affected the electronics and catalytic efficiency. It was found that the incorporation of a distal carboxyl group into the ligand scaffold resulted in a catalyst with increased catalytic activity, most likely because of the fact that the distal group is able to promote proton-coupled electron transfer (PCET) from the high-valent Mn species, thus facilitating O–O bond formation.
1. Introduction
The world's global energy consumption is primarily based on fossil fuels. However, the rapidly increasing global energy demand coupled to diminishing fossil fuel supplies call for the development of sustainable and carbon-neutral alternatives, with feedstock(s) that are environmentally friendly and abundant. There are already efficient solar cells for generation of electricity but in order for this to become truly useful, it is necessary that part of the energy can be stored. One attractive alternative would therefore be to store the energy in sunlight by the splitting of H2O into O2, protons and electrons (eqn (1)). The generated electrons can then be used to produce H2 or other solar fuels, thus allowing the energy to be stored in the form of chemical bonds. The production of solar fuels constitutes an attractive solution for a future sustainable energy economy.1–5Nature has since a long time figured out how the process of splitting of H2O can be realized and the natural photosynthetic machinery involves the synchronization of several complicated features, such as light-harvesting, charge separation and electron transfer. Both in natural and artificial photosynthesis, catalytic H2O oxidation is an essential part of the intricate photochemical process of solar to chemical energy conversion. Although deceptively simple, H2O oxidation (eqn (1)) is associated with a high thermodynamic potential (eqn (2)), a high kinetic barrier and requires a multitude of bond breaking and bond making events. This makes the H2O oxidation half reaction the pitfall for the construction of artificial devices for the production of solar fuels.6–8
| 2H2O → O2 + 4H+ + 4e− | (1) |
| E = 1.229 − (0.059·pH) V vs. NHE | (2) |
In the natural system, H2O oxidation is mediated by the oxygen-evolving complex (OEC) comprised of a Mn4Ca oxo-bridged cluster.9–12 This has been a source of inspiration for scientists and has resulted in a plethora of structural and functional model systems of the OEC.13 Although substantial progress has been made during the latest years in producing highly active and rugged water oxidation catalysts (WOCs), the essential question still remains: how to construct WOCs that are capable of operating under the harsh reaction conditions required to oxidize H2O and how to reproduce the high activity and stability displayed by the natural system. The majority of the developed artificial molecular WOCs are based on the scarce transition metals Ru14–27 and Ir.28–32 However, for large scale applications it is most likely necessary that the WOCs are based on earth-abundant elements. This prompts the examination and development of molecular WOCs based on low-cost first-row transition metals. Although there have been a couple of reports on WOCs comprised of Mn,33–42 Co,43–50 Fe51–54 and Cu,55–57 this research has not been as successful as with the rare metals Ru and Ir.
The incorporation of negatively charged groups into ligand frameworks has been shown to drastically reduce the redox potentials of the corresponding metal complexes.58,59 The electron-rich ligand environment helps to stabilize the metal center(s) in high oxidation states. We have previously reported the synthesis of Mn complexes housing ligands with benzylic amines.60,61 However, due to the labile benzylic positions, these complexes failed to mediate H2O oxidation.
Our group recently reported on the synthesis and development of the bio-inspired ligand 1 containing carboxylate, phenol and imidazole functionalities,33 which are all important elements in the natural system.62 Complexation of ligand 1 with Mn(OAc)2 yielded the dinuclear Mn2II,III complex 2, which in the solid state crystallized as the tetranuclear complex 2′ with loss of the bridging acetate ligand, thus resembling the Mn4Ca cluster in the OEC (Fig. 1).33 Maintaining a high redox flexibility of the active metal center(s) in the artificial WOCs constitutes a key parameter when designing synthetic WOCs, since the oxidation of H2O requires the collective transfer of four electrons. It was therefore vital that the negatively charged functional groups in the dinuclear Mn2II,III complex were shown to dramatically lower the redox potentials of the complex. By replacing the sensitive benzylic groups by imidazoles, along with the introduction of carboxylate moieties, it became possible for the dinuclear Mn2II,III complex 2 to promote catalytic H2O oxidation by use of the mild one-electron oxidant [Ru(bpy)3]3+ (bpy = 2,2′-bipyridine). In addition to being capable of mediating chemical H2O oxidation, photochemical H2O oxidation was also realized when using [Ru(bpy)3]2+-type complexes as photosensitizers. This has similarities to the natural photosynthetic system where light is employed to induce charge separation with subsequent H2O oxidation.
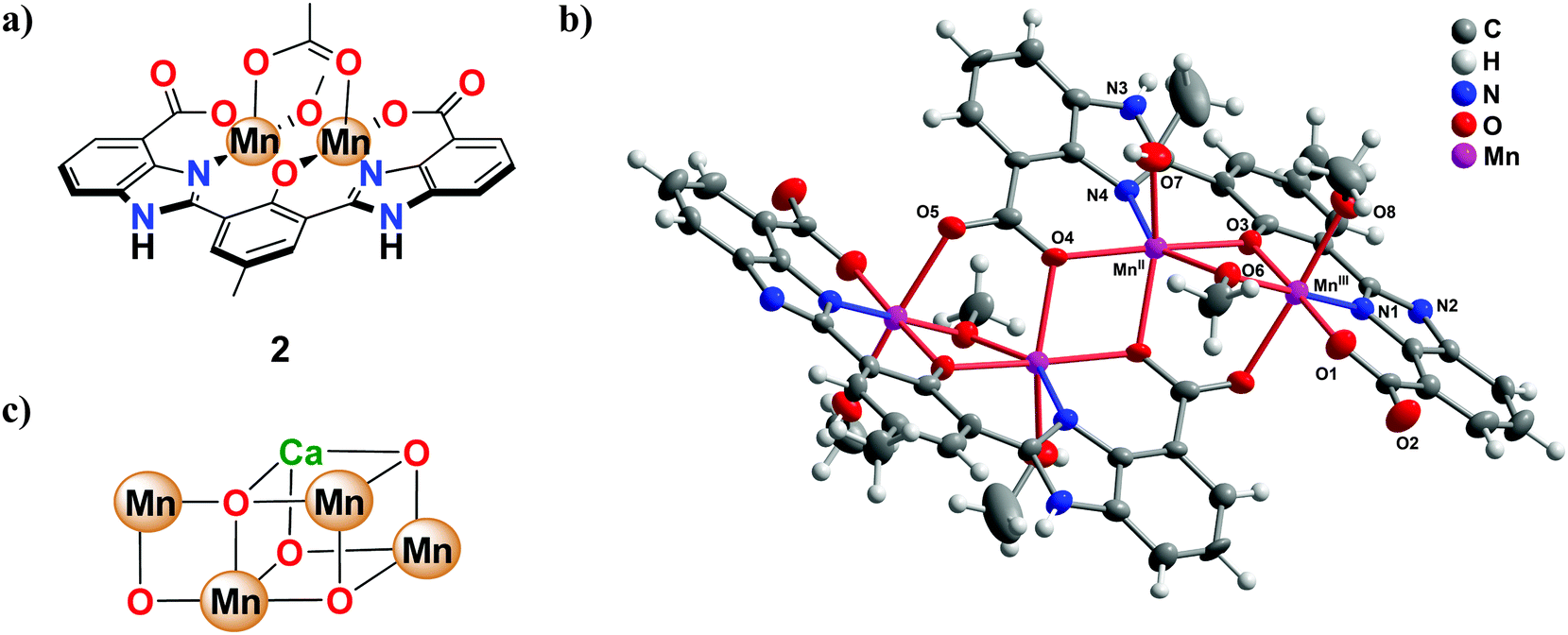 | ||
| Fig. 1 (a) Molecular structure of the dinuclear Mn2II,III complex 2, (b) crystal structure of the tetranuclear Mn complex 2′ generated from complex 2, and (c) representation of the CaMn4O5 active site in the OEC. | ||
Compared to heterogeneous WOCs, homogeneous molecular catalysts offer great advantages both in terms of kinetic studies and easy steric and electronic tuning of the catalysts. Thanks to the straightforward synthetic protocol associated with ligand 1 and complex 2, it was decided to study how the introduction of different functional groups into the ligand framework of 1 affected the catalytic activity and electronic properties of the corresponding dinuclear Mn complexes.
Here we report the synthesis and characterization of eight ligand scaffolds for dinuclear metal complexes, together with their corresponding dinuclear Mn2II,III counterparts. The developed Mn complexes contained a wide variety of different substituents and their electronic and catalytic features were systematically examined. Moreover, this study has established the impact of having a non-innocent distal group in these complexes to promote proton transfer processes during H2O oxidation catalysis. Within the library of the developed dinuclear Mn2II,III complexes, complex 6f possessing the distal carboxyl group was found to evolve O2 more efficiently than the other Mn complexes.
This effect could be due to both pre-orientation of the incoming H2O nucleophile and to hydrogen bonding to the aqua and hydroxide coordinated to the Mn center(s). Hydrogen bonding could facilitate proton-coupled electron transfer (PCET) and pre-orientation of H2O could facilitate nucleophilic attack on a high-valent Mn species, similar to the previously reported Co “Hangman” complexes.37,45 The incorporation of non-innocent distal groups into metal-based WOCs could thus be a general strategy for promoting catalytic H2O oxidation activity. In addition, the constructed dinuclear Mn2II,III WOCs have the advantage of being comprised of earth-abundant elements, which renders them especially attractive compared to noble metal-based WOCs.
2. Experimental section
2.1. Materials and methods
[Ru(bpy)3](PF6)3,63 5-((1,3-dioxoisoindolin-2-yl)methyl)-2-hydroxyisophthalaldehyde64 and 4′-methyl-2,2′-bipyridine-4-carboxylic acid65 were prepared according to a previously reported procedure. All other reagents including solvents were obtained from commercial suppliers and used directly without further purification. All solvents were dried by standard methods when needed. 1H and 13C NMR spectra were recorded at 400 MHz and at 100 MHz, respectively. Chemical shifts (δ) are reported in ppm, using the residual solvent peak ([D6]acetone δ(H) = 2.05 and δ(C) = 206.26; CDCl3δ(H) = 7.26 and δ(C) = 77.16; [D6]DMSO δ(H) = 2.50 and δ(C) = 39.52) as internal standard. Splitting patterns are denoted as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). High resolution mass spectra measurements were recorded on a Bruker Daltonics microTOF spectrometer with an electrospray ionizer. Elemental analyses were carried out at MEDAC Ltd, Chobham, Surrey, United Kingdom. IR spectra were recorded on a Perkin-Elmer Spectrum One spectrometer, using samples prepared as KBr discs.2.2. Electrochemistry
Electrochemical measurements were carried out using an Autolab potentiostat with a GPES electrochemical interface (Eco Chemie), using a glassy carbon disk (diameter 3 mm) as the working electrode, and a platinum spiral as a counter-electrode. The reference electrode was an Ag/AgCl electrode (3 M KCl aqueous solution) and the electrolyte used was a 0.1 M phosphate buffer (pH 7.2). All potentials are reported vs. NHE, using the [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple (E1/2 = 1.26 V vs. NHE66) as a reference.2.3. Oxygen evolution measurements
Oxygen evolution was measured by mass spectrometry (MS).67 Aqueous stock solutions were made of each catalyst (1 mM), containing K3PO4 (3 mM). The catalyst solutions used in the experiments were then made by diluting the stock solutions with phosphate buffer (0.1 M, pH 7.2) to the desired concentrations. The solutions were then deoxygenated by bubbling with N2 for at least 15 min before being used in the experiments.2.4. Chemical H2O oxidation with [Ru(bpy)3]3+
In a typical run [Ru(bpy)3](PF6)3 (3.4 mg, 3.4 μmol) was placed in the reaction chamber and the reaction chamber was evacuated using a rough pump. ∼35 mbar He was then introduced into the system. After a couple of minutes the catalyst solution (0.50 mL, 4.0 μM) was injected into the reaction chamber. The generated oxygen gas was then measured and recorded versus time by MS.2.5. Computational details
The geometry optimizations were performed at the B3LYP level68 of density functional theory as implemented in the Gaussian 09 package.69 The SDD70 pseudopotential was used to describe Mn, and the 6-31G(d,p) basis set was used for the C, N, O, H elements. Analytical frequency calculations were carried out at the same level of theory as the geometry optimization to obtain the Gibbs free energy corrections and to confirm the characteristics of the optimized structures. On the basis of these optimized geometries, single-point calculations were performed employing a larger basis set, where all elements, except Mn, were described by 6-311+G(2df,2p) at the B3LYP* (15% exact exchange) level.71 Solvation effects from the water solvent were calculated using the SMD72 continuum solvation model with the larger basis set at the B3LYP* level. The B3LYP*-D2 energies are reported, including dispersion corrections proposed by Grimme73 and Gibbs free energy corrections from B3LYP.2.6. EPR measurements
X-band (9 GHz) electron paramagnetic resonance (EPR) measurements were performed at temperatures between 5 and 30 K with a Bruker E500 ELEXYS spectrometer using a 4122SHQE or a 4116DM resonator. For cooling with liquid helium an Oxford Instruments ESR 900 cryostat was installed. Frozen solutions of the complexes were prepared in 0.2 M phosphate buffer (pH 7) and for solid state spectra the complex and potassium bromide were ground together. All samples were frozen in liquid nitrogen prior to EPR measurements. The modulation amplitude was 5 G and the microwave power was varied between 200 μW and 6 mW.2.7. Synthesis
4-Hydroxybenzoic acid (3a, 0.550 g, 4.00 mmol) and hexamethylenetetramine (4.50 g, 32.0 mmol) were dissolved in anhydrous CF3COOH (10 mL) and the reaction mixture was heated at 110 °C for 48 h. The reaction was cooled to room temperature before 4 M HCl (25 mL) was added to the reaction mixture. The precipitate was filtered, washed with H2O (3 × 10 mL) to afford the title compound (0.710 g, 86.2%). 1H NMR (400 MHz, [D6]acetone): δ = 12.21 ppm (br, 1H), 10.36 (s, 2H), 8.69 (s, 2H); 13C NMR (100 MHz, [D6]acetone): δ = 188.8, 160.0, 149.3, 122.0, 115.8, 113.1; HRMS (ESI) calcd for C9H5O5 [M–H+]−: 193.0142; found: 193.0145.
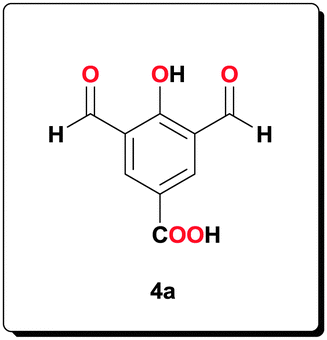
Compound 4a (1.10 g, 5.50 mmol) and 2-amino-3-nitrobenzoic acid (2.00 g, 11.0 mmol) were suspended in EtOH (50 mL). The suspension was heated until complete dissolution and then cooled to room temperature. A freshly prepared aqueous solution of Na2S2O4 (5.70 g, 32.8 mmol, 25 mL) was added and the reaction mixture was heated at 70 °C for 5 h. After the mixture had been allowed to cool to room temperature, a yellow precipitate was formed, which was filtered, washed with H2O (4 × 10 mL), acetone (4 × 10 mL) to afford the product (2.10 g, 83.9%). 1H NMR (400 MHz, [D6]DMSO): δ = 14.01 (br, 3H), 8.91 (s, 2H), 7.88 (d, J = 7.91 Hz, 2H), 7.76 (d, J = 7.47 Hz, 2H), 7.28 (t, J = 7.93 Hz, 2H); 13C NMR (100 MHz, [D6]DMSO): δ = 173.3, 167.7, 167.1, 154.7, 132.3, 123.0, 121.9, 121.0, 116.9, 114.3, 111.9; HRMS (ESI) calcd for C23H13N4O7 [M–H+]−: 457.0779; found: 457.0795.
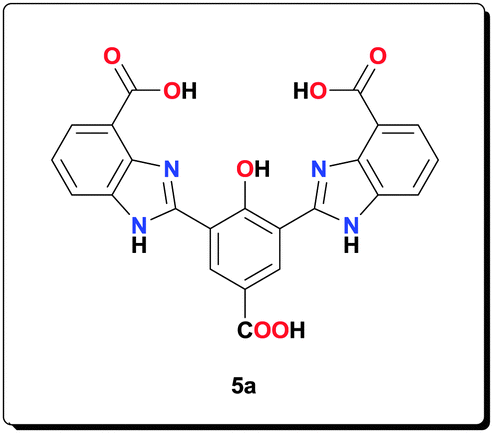
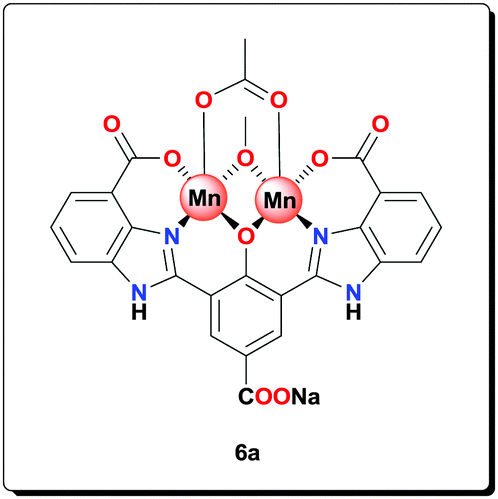
Mn(OAc)2·4H2O (0.153 g, 0.625 mmol) and NaOAc (0.209 g, 25.0 mmol) were added to a suspension of ligand 5a (0.114 g, 0.250 mmol) in MeOH (4 mL). The mixture was heated at 70 °C for 20 h and then centrifuged and washed with MeOH (3 × 5 mL), and dried under vacuum to afford the complex (0.140 g, 76.6%). HRMS (ESI) calcd for C26H17Mn2N4NaO10 [M+H+]+: 677.9598; found: 677.9593; anal. calcd for C26H22Mn2N4NaO13 [6a·3H2O]: C 42.70, H 3.03, N 7.66, Mn 15.02%; found: C 42.54, H 2.93, N 7.58, Mn 15.08%; IR (KBr): ν = 3428, 2926, 1563, 1497, 1474, 1403, 1385, 1320, 1260, 1020, 765, 676, 655, 619 cm−1.
4-Bromophenol (3b, 1.47 g, 8.49 mmol) and hexamethylenetetramine (9.52 g, 67.9 mmol) were dissolved in anhydrous CF3COOH (25 mL) and the yellow solution was heated at 110 °C for 48 h. After cooling to room temperature, the mixture was added to aqueous HCl (4 M, 50 mL) and stirred for 5 h. Filtration and washing with H2O (3 × 15 mL) afforded the product as a yellow solid (1.80 g, 92.3%). 1H NMR (400 MHz, [D6]acetone): δ = 11.71 (s, 1H), 10.28 (s, 2H), 8.19 (s, 2H); 13C NMR (100 MHz, [D6]acetone): δ = 192.5, 140.5, 126.0, 112.5; HRMS (ESI) calcd for C8H5BrNaO3 [M+Na+]+: 250.9314; found: 250.9310.

An aqueous solution of Na2S2O4 (6.09 g, 35.0 mmol, 6 mL) was added to an ethanolic solution containing 5-bromo-2-hydroxyisophthalaldehyde 4b (1.15 g, 5.00 mmol) and 2-amino-3-nitrobenzoic acid (1.82 g, 10.0 mmol). The resulting mixture was heated at 70 °C for 5 h and the resulting yellow precipitate was filtered and washed with H2O, acetone and finally dried under vacuum to give the product (2.00 g, 80.9%). 1H NMR (400 MHz, [D6]DMSO): δ = 13.64 (br, 2H), 8.58 (s, 2H), 7.97 (d, J = 7.56 Hz, 2H), 7.85 (d, J = 7.56 Hz, 2H), 7.33 (t, J = 7.56 Hz, 2H); 13C NMR (100 MHz, [D6]DMSO): δ = 166.8, 151.5, 141.1, 135.3, 133.9, 132.1, 124.6, 122.6, 122.1, 117.5, 115.4, 106.7; HRMS (ESI) calcd for C22H13BrN4NaO5 [M+Na+]+: 514.9962; found: 514.9967.
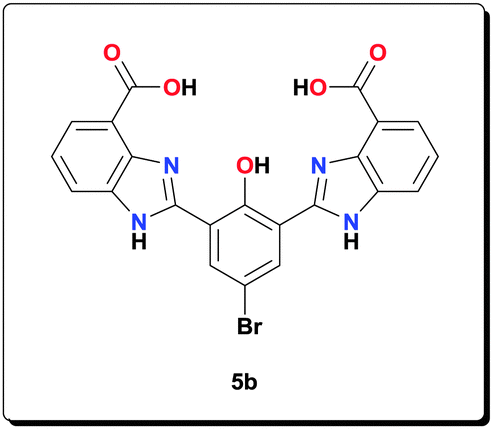
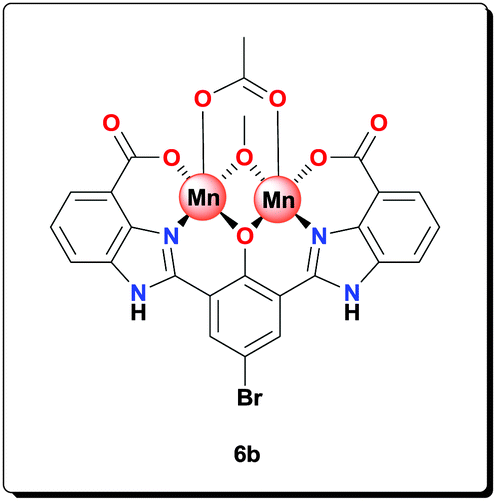
Ligand 5b (0.100 g, 0.203 mmol) was added to a solution of NaOAc (0.166 g, 2.03 mmol) and Mn(OAc)2·4H2O (0.124 g, 0.506 mmol) in MeOH (3 mL). The resulting mixture was then refluxed for 20 h. The precipitate formed was isolated by centrifugation, washed with MeOH (5 × 5 mL) and dried under vacuum to give the desired product (0.120 g, 79.4%). HRMS (ESI) calcd for C25H16BrMn2N4NaO8 [M+Na+]+: 711.8805; found: 711.8810; anal. calcd for C25H22Mn2N4O11 [6b·3H2O]: C 40.35, H 2.98, N 7.53, Mn 14.76%; found: C 40.43, H 3.05, N 7.49, Mn 14.71%; IR (KBr): ν = 3435, 2926, 1631, 1555, 1493, 1470, 1400, 1385, 1319, 1247, 1003, 765, 619 cm−1.
Biphenyl-4-ol (3c, 1.36 g, 8.00 mmol) and hexamethylenetetramine (22.4 g, 160 mmol) were placed in a round-bottomed flask which was purged with nitrogen. After addition of trifluoroacetic acid (48 mL), the mixture was refluxed at 110 °C for 72 h. The reaction mixture was combined with 4 M HCl (240 mL) in a 500 mL round-bottomed flask and stirred for 3 h. The yellow precipitate was collected by filtration, washed with H2O (35 mL) and dried under vacuum to yield the title compound as a yellow powder (1.67 g, 92.4%). 1H NMR (500 MHz, CDCl3): δ = 11.62 (s, 1H), 10.32 (s, 2H), 8.20 (s, 2H), 7.61–7.57 (m, 2H), 7.51–7.45 (m, 2H), 7.43–7.37 (m, 1H); 13C NMR (100 MHz, [D6]DMSO): δ = 192.2, 163.0, 138.2, 136.0, 133.7, 130.7, 129.3, 128.2, 126.8, 123.5; HRMS (ESI) calcd for C14H10NaO3 [M+Na+]+: 249.0522; found: 249.0532.
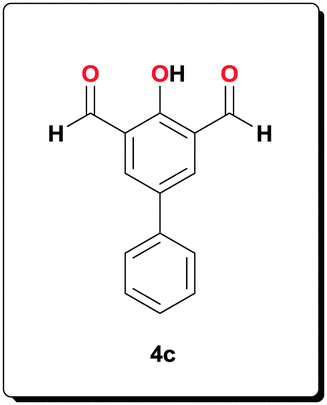
A solution of 4-hydroxybiphenyl-3,5-dicarbaldehyde 4c (1.50 g, 6.63 mmol) and 2-amino-3-nitrobenzoic acid (2.42 g, 13.3 mmol) in EtOH (53 mL) was prepared by heating the mixture to 70 °C. This orange solution was brought to r.t. before the addition of a freshly prepared solution of Na2S2O4 (85 wt%, 8.15 g, 39.8 mmol) in H2O (40 mL) after which the resulting red solution was heated for 5 h. Filtration and washing with EtOH (40 mL), H2O (75 mL) and Et2O, (100 mL) with subsequent drying in a vacuum yielded the desired ligand as a red powder (2.58 g, 79.1%). 1H NMR (500 MHz, [D6]DMSO): δ = 8.73 (s, 2H), 7.95 (d, J = 7.92 Hz, 2H), 7.81 (d, J = 7.58 Hz, 2H), 7.79 (d, J = 7.55 Hz, 2H), 7.51 (t, J = 7.55 Hz, 2H), 7.33 (t, J = 7.67 Hz, 3H); 13C NMR (100 MHz, [D6]DMSO): δ = 166.9, 157.1, 153.0, 141.5, 139.5, 134.3, 128.9, 128.7, 128.4, 126.6, 126.0, 124.2, 121.8, 116.1, 115.3; HRMS (ESI) calcd for C28H19N4O5 [M+H+]+: 491.1350; found: 491.1369.
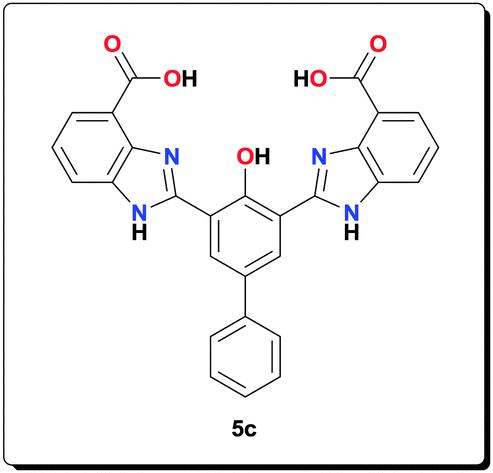
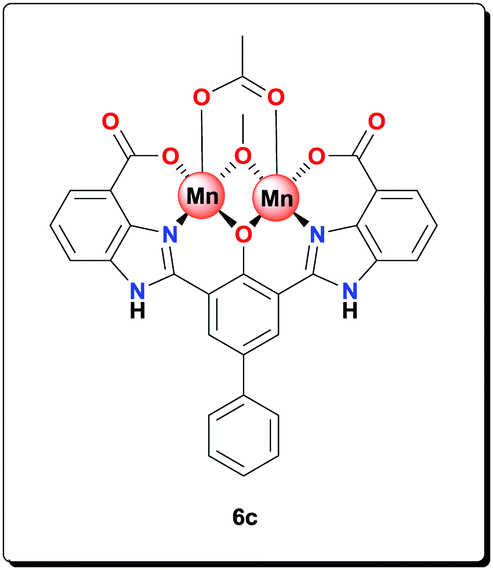
Ligand 5c (1.00 g, 2.04 mmol), NaOAc (1.67 g, 20.4 mmol) and Mn(OAc)2·4H2O (1.25 g, 5.10 mmol) were suspended in MeOH (40 mL) and refluxed for 24 h. The product was retrieved by centrifugation, washed with MeOH (4 × 45 mL) and dried under vacuum to give the complex as a yellow powder (1.20 g, 77.5%). HRMS (ESI) calcd for C30H18Mn2N4O8 [M–CH3OH+−OH]−: 671.9891; found: 671.9892; anal. calcd for C31H29Mn2N4O12 [6c·4H2O]: C 49.03, H 3.85, N 7.38, Mn 14.47%; found: C 48.51, H 3.42, N 7.22, Mn 14.63%; IR (KBr): ν = 3412, 3174, 2955, 2925, 2854, 1602, 1555, 1506, 1491, 1468, 1384, 1315, 1252, 1211, 1155, 1023, 1003, 892, 803, 763, 700, 662, 602, 496 cm−1.
4-Benzylphenol 3d (1.47 g, 8.00 mmol) and hexamethylenetetramine (22.4 g, 160 mmol) were placed in a round-bottomed flask which was purged with nitrogen. After the addition of trifluoroacetic acid (48 mL), the mixture was refluxed at 110 °C for 72 h. The reaction mixture was then combined with 4 M HCl (240 mL) in a 500 mL round-bottomed flask and stirred for 3 h. The yellow precipitate was collected by filtration, washed with H2O (500 mL) and dried under vacuum to yield the product as a yellow powder (1.49 g, 77.4%). 1H NMR (500 MHz, CDCl3): δ = 11.50 (s, 1H), 10.20 (s, 2H), 7.78 (s, 2H), 7.36–7.12 (m, 5H), 4.00 (s, 2H); 13C NMR (100 MHz, [D6]DMSO): δ = 192.2, 162.4, 139.7, 137.9, 133.4, 129.0, 128.9, 126.9, 123.3, 40.7; HRMS (ESI) calcd for C15H12NaO3 [M+Na+]+: 263.0679; found: 263.0681.
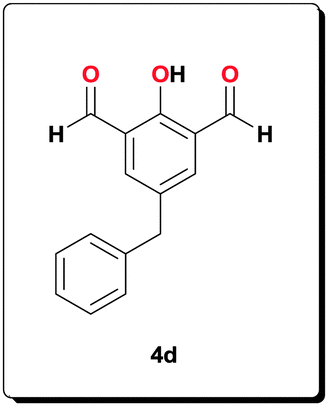
A solution of 5-benzyl-2-hydroxyisophthalaldehyde 4d (1.20 g, 5.00 mmol) and 2-amino-3-nitrobenzoic acid (1.82 g, 9.99 mmol) in EtOH (40 mL) was prepared by heating the mixture to 70 °C. This orange solution was brought to r.t. before addition of a freshly prepared solution of Na2S2O4 (85 wt%, 6.14 g, 30.0 mmol) in H2O (30 mL) and the resulting red solution was heated for 5 h. Filtration, and washing with EtOH (16 mL), H2O (25 mL) and Et2O (150 mL) with subsequent drying in a vacuum yielded the desired ligand as a red powder (2.05 g, 82.0%). 1H NMR (500 MHz, [D6]DMSO): δ = 8.38 (s, 2H), 7.95 (d, J = 7.89 Hz, 2H), 7.83 (d, J = 7.68 Hz, 2H), 7.43–7.29 (m, 6H), 7.25–7.18 (m, 1H), 4.05 (s, 2H); 13C NMR (100 MHz, [D6]DMSO): δ = 166.9, 152.5, 141.5, 134.5, 130.8, 128.7, 124.5, 122.0, 115.6, 115.2, 40.4; HRMS (ESI) calcd for C29H21N4O5 [M+H+]+: 505.1506; found: 505.1501.
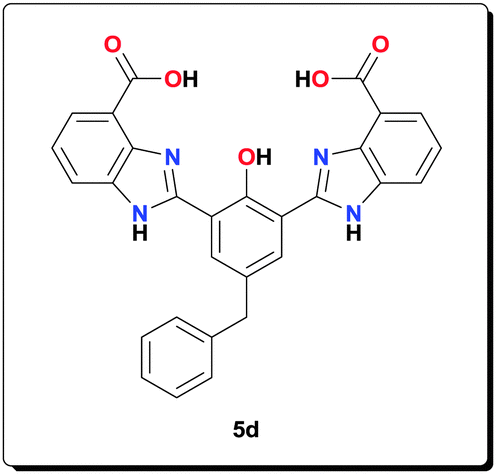
Ligand 5d (1.00 g, 1.98 mmol), NaOAc (1.62 g, 19.8 mmol) and Mn(OAc)2·4H2O (1.21 g, 4.94 mmol) were suspended in MeOH (40 mL) and refluxed for 24 h. The product was retrieved by centrifugation, washed with MeOH (4 × 45 mL) and dried under vacuum to yield a yellow powder (1.19 g, 77.7%). HRMS (ESI) calcd for C30H18Mn2N4O6 [M–CH3COOH–H+]−: 639.9993; found: 639.9999; anal. calcd for C32H31Mn2N4O12 [6d·4H2O]: C 49.69, H 4.04, N 7.24, Mn 14.21%; found: C 49.25, H 3.92, N 7.02, Mn 14.45%; IR (KBr): ν = 3426, 3190, 2924, 2854, 1603, 1556, 1495, 1473, 1385, 1319, 1251, 1211, 1155, 1052, 1015, 892, 765, 700, 662, 620, 536, 491 cm−1.

2-Amino-3-nitrobenzoic acid (0.360 g, 2.00 mmol) and 5-((1,3-dioxoisoindolin-2-yl)methyl)-2-hydroxyisophthalaldehyde64 (4e, 0.310 g, 1.00 mmol) were suspended in EtOH (30 mL). After addition of a freshly prepared aqueous solution of Na2S2O4 (1.23 g, 6.00 mmol, 6.0 mL), the reaction mixture was heated at 70 °C for 5 h. The resulting yellow precipitate was filtered and washed with H2O, acetone and finally dried under vacuum to afford the title compound (0.450 g, 78.5%). 1H NMR (400 MHz, [D6]DMSO): δ = 14.03 (bs, 2H), 8.37 (s, 2H), 7.95–7.84 (m, 6H), 7.80–7.75 (d, J = 7.47 Hz, 2H), 7.31 (t, J = 7.52 Hz, 2H), 4.82 (s, 2H); HRMS (ESI) calcd for C31H19N5NaO7 [M+Na+]+: 596.1177; found: 596.1170.
Hydrazine hydrate (98%, 1.00 g, 19.6 mmol) was added to a suspension of compound 5e′ (2.85 g, 4.98 mmol) in EtOH (20 mL). The mixture was gently heated at reflux for 24 h. The yellow precipitate was filtered, washed with EtOH (3 × 5 mL) and dried to afford the title compound (2.10 g, 95.3%). 1H NMR (400 MHz, [D6]DMSO): δ = 8.39 (s, 2H), 7.72–7.67 (m, 4H), 7.18 (t, J = 7.66 Hz, 2H), 4.01 (s, 2H); 13C NMR (100 MHz, [D6]DMSO): δ = 167.9, 154.9, 142.5, 141.0, 139.4, 131.2, 127.8, 125.1, 122.5, 120.4, 117.3, 112.9, 42.9; HRMS (ESI) calcd for C23H18N5O5 [M+H+]+: 444.1302; found: 444.1313.
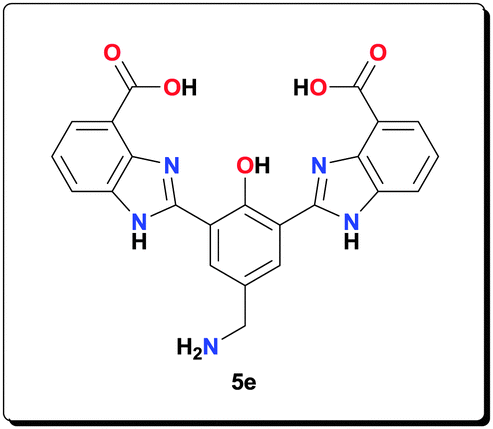
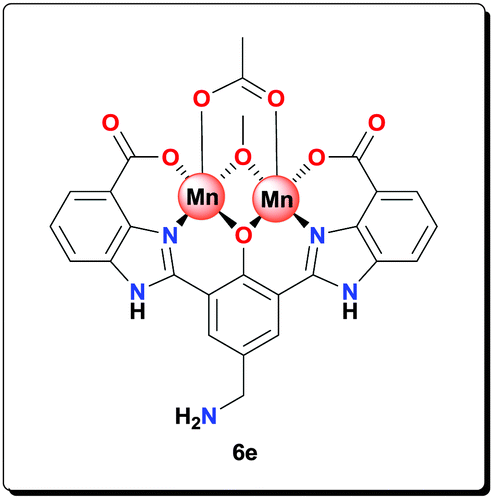
Ligand 5e (0.100 g, 0.226 mmol) was added to a solution of NaOAc (0.185 g, 2.26 mmol) in MeOH (3 mL). The resulting suspension was sonicated for 8 h to give a yellow gel. Mn(OAc)2·4H2O (0.138 g, 0.563 mmol) was added and the reaction mixture was refluxed for 20 h. The yellow precipitate formed was isolated by centrifugation, washed with MeOH (5 × 5 mL) and dried under vacuum to give the complex as a yellow solid (0.130 g, 83.3%). HRMS (ESI) calcd for C23H17N5NaO5 [M+Na+]+: 466.1122; found: 466.1125; anal. calcd for C27H26Mn2N5O10 [6e·CH3OH·H2O]: C 46.97, H 3.80, N 10.14, Mn 15.91%; found: C 46.89, H 3.85, N 10.02, Mn 15.83%; IR (KBr): ν = 3426, 3233, 1600, 1563, 1497, 1475, 1385, 1320, 1258, 1208, 1128, 1052, 1019, 893, 811, 764, 619 cm−1.
Compound 5e (0.770 g, 1.73 mmol) was dissolved in pyridine (20 mL) and the resulting solution was cooled to 0 °C. Adipoyl chloride (0.330 g, 1.81 mmol) was added dropwise to this cold solution and the reaction mixture was then stirred overnight at room temperature. Evaporation of the solvent under reduced pressure gave a crude product that was washed with aqueous 2 M HCl (2 × 10 mL) and Et2O (3 × 10 mL) to give the product (0.700 g, 85.9%). 1H NMR (400 MHz, [D6]DMSO): δ = 13.78 (br, 3H), 8.37 (s, 2H), 8.10–7.67 (m, 4H), 7.44 (s, 2H), 4.33 (s, 2H), 2.23 (s, 2H), 2.07 (s, 2H), 1.71–1.35 (m, 4H); 13C NMR (100 MHz, [D6]DMSO): δ = 174.4, 172.1, 166.3, 150.8, 150.3, 132.3, 131.6, 125.7, 123.7, 120.0, 116.2, 112.4, 41.7, 35.2, 33.5, 24.8, 24.3; HRMS (ESI) calcd for C29H25N5NaO8 [M+Na+]+: 594.1595; found: 594.1590.
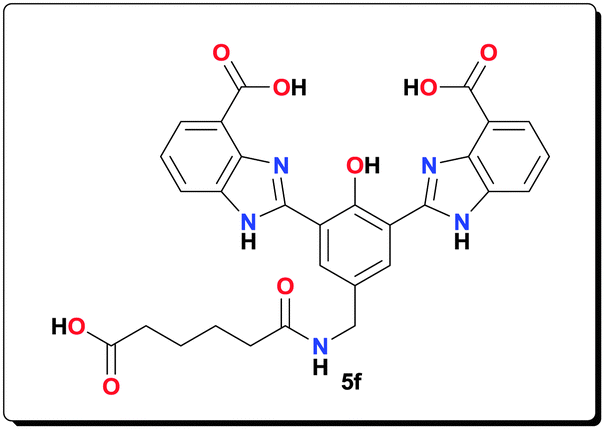
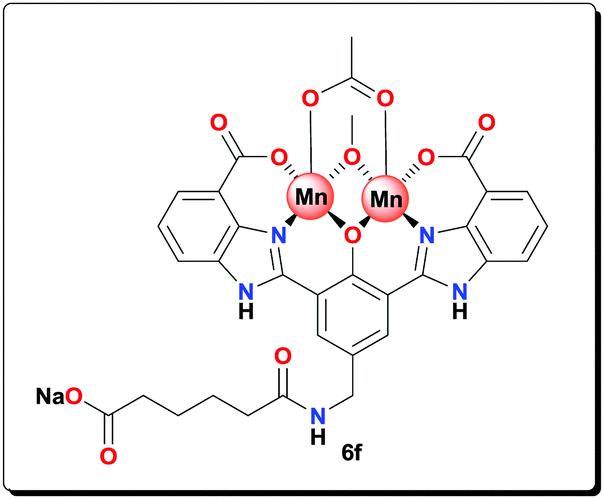
NaOAc (0.110 g, 1.32 mmol) and Mn(OAc)2·4H2O (0.080 g, 0.330 mmol) were added to a suspension of ligand 5f (0.080 g, 0.140 mmol) in MeOH (3 mL). The reaction mixture was then heated at 70 °C for 20 h. The yellow precipitate was filtered, washed with MeOH (3 × 10 mL), and dried under vacuum to afford the complex as a yellow powder (0.105 g, 90.7%). HRMS (ESI) calcd for C32H27Mn2N5O11 [M−Na+]−: 767.0474; found: 767.0470; anal. calcd for C32H31Mn2N5NaO13 [6f·2H2O]: C 46.50, H 3.78, N 8.47, Mn 13.29%; found: C 46.25, H 3.59, N 8.40, Mn 13.38%; IR (KBr): ν = 3426, 3191, 1603, 1564, 1502, 1400, 1383, 1263, 1003, 791, 751, 672, 658, 618, 495 cm−1.
Pivaloyl chloride (1.22 mL, 10.0 mmol) was carefully added to a nitrogen purged solution of compound 5e (0.443 g, 10.0 mmol) in pyridine (15 mL) at 0 °C. The resulting solution was stirred at room temperature overnight. Upon pouring the reaction mixture into crushed ice and acidification with conc. HCl, a yellow precipitate was formed, which was filtered, washed with H2O (2 × 10 mL), Et2O (3 × 10 mL), and dried under vacuum to yield the title compound (0.41 g, 77.8%). 1H NMR (400 MHz, [D6]DMSO): δ = 14.50 (br, 2H), 8.41 (s, 2H), 8.00 (d, J = 8.10 Hz, 2H), 7.93 (d, J = 7.62 Hz, 2H), 7.53 (t, J = 7.86 Hz, 2H), 4.34 (d, J = 5.29 Hz, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, [D6]DMSO): δ = 177.6, 166.0, 150.5, 150.2, 134.3, 132.7, 131.2, 126.3, 126.1, 124.5, 118.9, 116.5, 111.2, 42.1, 39.2, 27.5; HRMS (ESI) calcd for C28H25N5NaO6 [M+Na+]+: 550.1697; found: 550.1703.
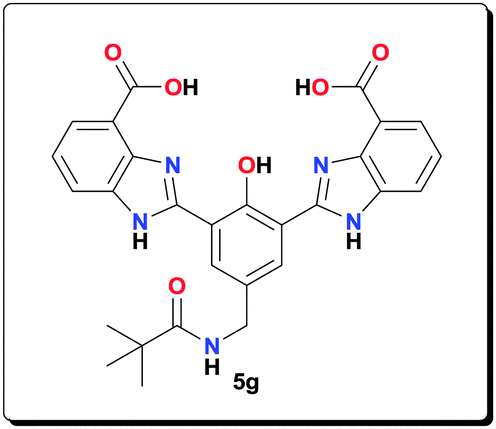
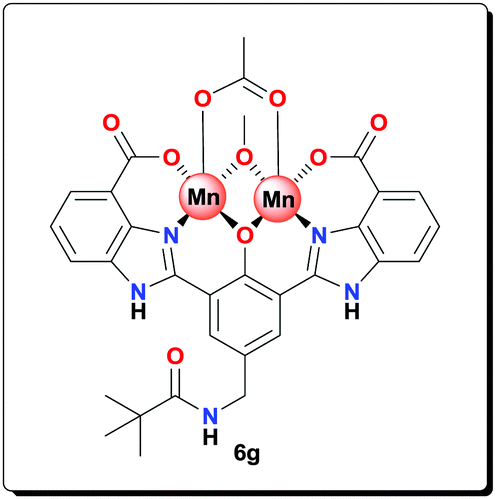
Ligand 5g (0.100 g, 0.190 mmol) was added to a solution containing Mn(OAc)2·4H2O (0.116 g, 0.474 mmol) and NaOAc (0.155 g, 1.90 mmol) in MeOH (4 mL). The reaction mixture was refluxed for 20 h and then centrifuged. The precipitate was washed with MeOH (5 × 5 mL) and dried under vacuum to afford complex 6g (0.123 g, 81.3%). HRMS (ESI) calcd for C31H28Mn2N5NaO9 [M+Na+]+: 747.0540; found: 747.0576; anal. calcd for C31H36Mn2N5O13 [6g·4H2O]: C 46.74, H 4.56, N 8.79, Mn 13.79%; found: C 46.77, H 4.50, N 8.74, Mn 13.83%; IR (KBr): ν = 3435, 1564, 1498, 1474, 1400, 1385, 1316, 1263, 1003, 765, 662 cm−1.
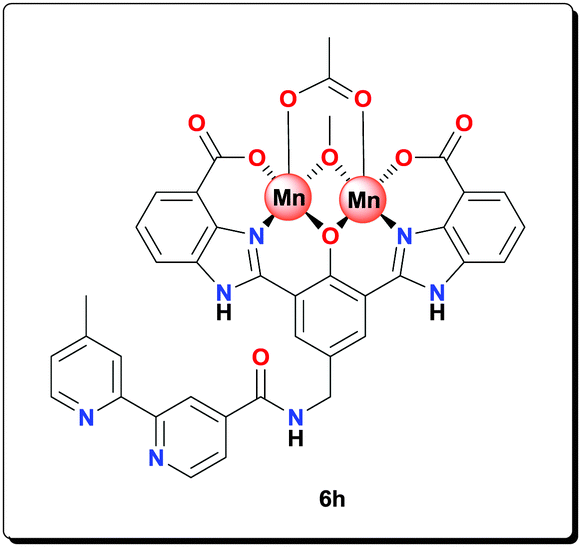
4′-Methyl-2,2′-bipyridine-4-carboxylic acid65 (0.150 g, 0.700 mmol) was refluxed in 10 mL of SOCl2 for 4 h. The excess of SOCl2 was removed under reduced pressure. The remaining residue (acid chloride) was dissolved in 2.0 mL of dry DMF. The solution was added dropwise to a solution of amine 5e (0.320 g, 0.720 mmol) and Et3N (0.40 mL) in dry DMF (10.0 mL). The resulting reaction mixture was heated at 100 °C for 24 h. After evaporation to dryness, the yellow precipitate was washed with H2O (3 × 10 mL), acetone (2 × 10 mL) and dried under vacuum to give the title compound (0.410 g, 89.1%). 1H NMR (400 MHz, [D6]DMSO): δ = 13.29 (br, 2H), 8.84 (s, 2H), 8.56 (s, 2H), 8.40 (s, 1H), 8.25 (s, 1H), 8.00–7.89 (m, 5H), 7.61–7.40 (m, 3H), 4.45 (s, 2H), 2.41 (s, 3H); 13C NMR (100 MHz, [D6]DMSO): δ = 166.8, 166.2, 156.4, 154.2, 151.6, 150.3, 150.0, 149.2, 148.3, 139.3, 131.1, 125.4, 125.0, 122.9, 122.4, 121.7, 121.3, 119.6, 118.2, 115.8, 42.5, 20.7; HRMS (ESI) calcd for C35H25N7NaO6 [M+Na+]+: 662.1759; found: 662.1760.
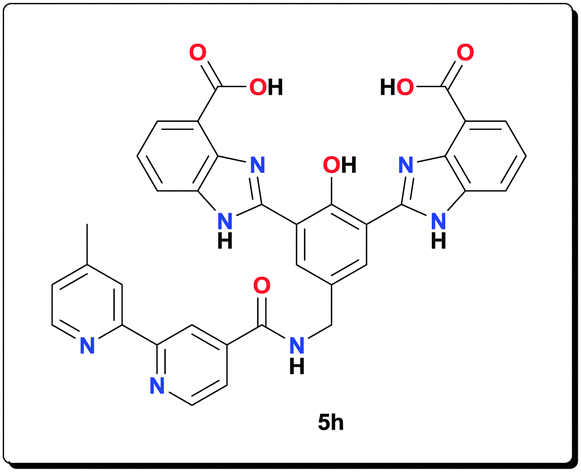
NaOAc (0.143 g, 1.75 mmol) and Mn(OAc)2·4H2O (0.107 g, 0.438 mmol) were added to a suspension of ligand 5h (0.112 g, 0.175 mmol) in MeOH (4 mL) and the reaction mixture was heated at 70 °C for 20 h. The resulting precipitate was centrifuged, washed with MeOH (3 × 5 mL) and dried under vacuum to afford the complex (0.125 g, 79.0%). HRMS (ESI) calcd for C38H28Mn2N7NaO9 [M+Na+]+: 859.0602; found: 859.0608; anal. calcd for C39H36Mn2N7O12 [6h·CH3OH·2H2O]: C 51.78, H 4.01, N 10.84, Mn 12.15%; found: C 51.69, H 4.05, N 10.80, Mn 12.11%; IR (KBr): ν = 3426, 1556, 1497, 1475, 1402, 1385, 1320, 1260, 1211, 1016, 765, 674, 660, 619, 562, 492 cm−1.
3. Results and discussion
3.1. Synthesis
To investigate how the electronics affected the Mn centers in WOC 2, eight different dinuclear Mn complexes 6a–h, depicted in Scheme 1, were synthesized, where a variety of substituents were introduced. These alterations would hence result in tuning of the corresponding Mn complexes and thereby affect the properties and reactivities. The synthesis of the functionalized ligand scaffolds involved the reductive cyclization reaction between the appropriate phenolic dialdehyde 4 and 2-amino-3-nitrobenzoic acid, with Na2S2O4 as the reducing agent.74 This allowed for easy access to a wide variety of ligands in good to excellent yields. The Mn complexes 6a–h were subsequently obtained by refluxing the ligands in the presence of Mn(OAc)2 and NaOAc. It is believed that all of the synthesized complexes were isolated in their Mn2II,III state, in conformity with complex 2. This is based on elemental analyses and high-resolution mass spectrometry (HRMS). However, extensive attempts to prove this by EPR have failed so far since we have been unable to observe the characteristic EPR signal for Mn2II,III complexes.75 Instead, a broad signal at around g = 2 was observed both in solution and in the solid state, which is typical for uncoupled MnII and reminiscent of the signal recently observed for an octahedral MnII complex.76 This signal decreased in intensity upon the addition of the one-electron oxidant [Ru(bpy)3]3+. It therefore seems possible that the complexes are in fact polymeric or at least tetranuclear, as indicated by the generated crystal structure of 2′ from complex 2. The formation of the tetranuclear complex 2′ indicates that the acetate ligand in complex 2, and probably also in complexes 6, is somewhat labile and we assume that on dissolution in phosphate buffer, the acetate ligand is rapidly replaced by phosphate to yield the active catalyst. On dissolution of the dinuclear Mn complexes in aqueous solutions containing PO43−, in the presence of air, new signals appeared, as detected by HRMS. These signals corresponded to the formation of Mn2III,III complexes, which are expected to be EPR silent. However, this will be studied in more detail in future work. | ||
| Scheme 1 Synthetic routes to ligands 5a–h and dinuclear Mn2II,III complexes 6a–h. | ||
3.2. Catalytic H2O oxidation by use of the mild one-electron oxidant [Ru(bpy)3]3+
A majority of the previously developed artificial WOCs require the use of the strong one-electron oxidant CeIV to drive H2O oxidation. However, this approach is not useful in solar fuel devices since the oxidant cannot be regenerated photochemically, using sunlight as the terminal energy source. For this to be realized, CeIV and related oxidants have to be substituted by photosensitizer-based oxidants, such as the well-studied [Ru(bpy)3]2+-type complexes (bpy = 2,2′-bipyridine).66,77 For this more preferable approach to be thermodynamically possible, the redox potential of the WOC has to be matched with that of the photosensitizer.Since the previously developed Mn WOC 2 could promote catalytic H2O oxidation with the mild one-electron oxidant [Ru(bpy)3]3+ (E (RuIII/RuII) = 1.26 vs. NHE66), either pre-generated or photochemically generated, the catalytic experiments for the newly developed complexes 6 were conducted as previously established; the [Ru(bpy)3]3+ oxidant was employed as the chemical oxidant in a buffered aqueous solution, under neutral conditions (0.1 M phosphate buffer, pH 7.2). The evolution of gaseous products was monitored and quantified by real-time mass spectrometry, a technique which we have previously used.67 In a typical reaction, [Ru(bpy)3]3+ was added to an aqueous solution containing the Mn complex, triggering immediate O2 evolution.
Several control experiments were conducted in order to verify that the observed O2 evolution was mediated by the investigated Mn complexes; (1) no O2 was generated when the Mn complex was omitted. The result was only degradation of the [Ru(bpy)3]3+ oxidant, without any evolution of O2. (2) To ensure that free Ru, possibly originating from the decomposition of the [Ru(bpy)3]3+ oxidant, did not react with the free ligand to generate an active catalyst, an aqueous solution containing the free ligand was added to [Ru(bpy)3]3+. However, this resulted in no observable O2 evolution. Collectively, this supports that the detected O2 formation was caused by the Mn complexes and not by any other unexpected catalytic process.
Fig. 2 and Table 1 show the O2 evolution activity of Mn complexes 6. It is evident that Mn complexes 6a–e all have similar O2 evolution activity, which highlights that the introduced substituents in 6a–e did not affect the catalytic activity to a large extent. The previously reported Mn complex 2 was also evaluated under these reaction conditions and was found to display comparable activity as catalysts 6a–e (not shown).
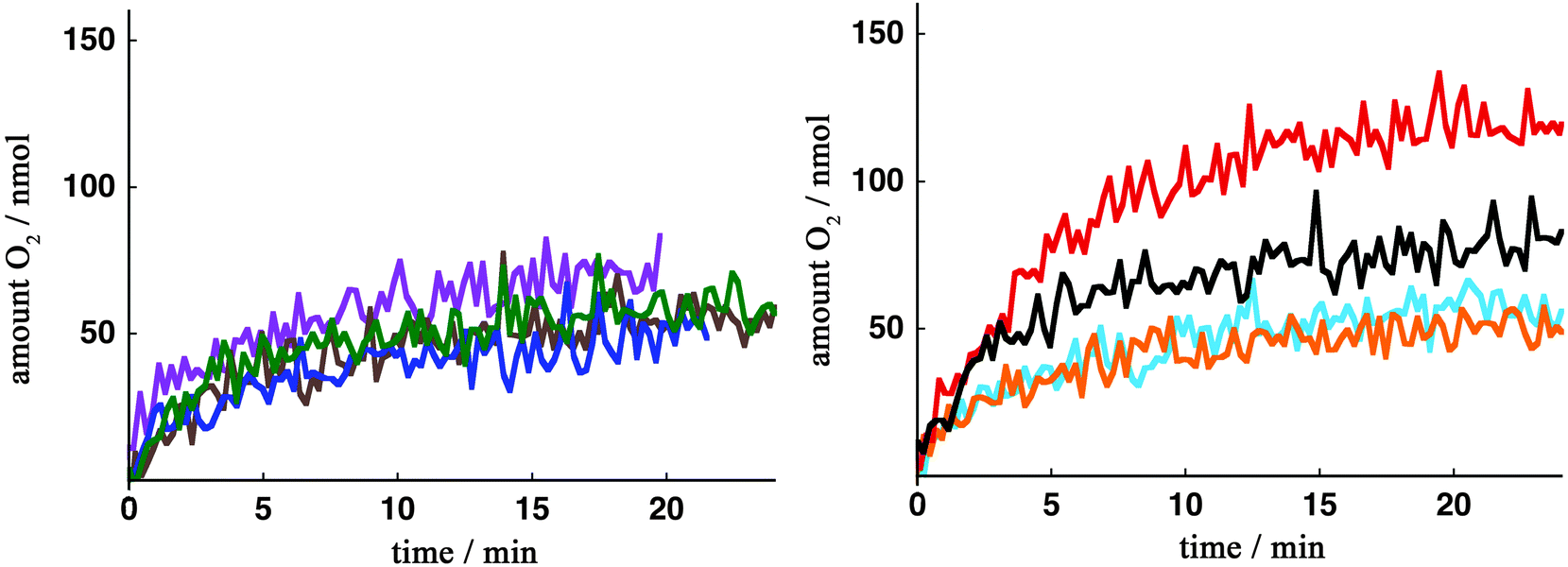 | ||
Fig. 2 O2 evolution kinetics by Mn complexes 6 as a function of time. Reaction conditions: an aqueous phosphate buffer solution (0.1 M, pH 7.2, 0.50 mL) containing the Mn complex (20 μM) was added to the oxidant [Ru(bpy)3](PF6)3 (3.4 mg, 3.4 μmol) and the evolved O2 was measured in real-time. 6a ( ), 6b ( ), 6b ( ), 6c ( ), 6c ( ), 6d ( ), 6d ( ), 6e ( ), 6e ( ), 6f ( ), 6f ( ), 6g ( ), 6g ( ), 6h (—). ), 6h (—). | ||
| Complex | Initial rate of O2 evolutionb (nmol O2 s−1) | Turnover number (mol O2 per mol catalyst) |
|---|---|---|
| a Reactions were carried out in aqueous buffered solutions (phosphate buffer; 0.1 M, pH 7.2, 0.50 mL) containing the catalyst (20 μM, 10 nmol) and the oxidant [Ru(bpy)3](PF6)3 (6.8 mM, 3.4 μmol). b Initial rates were obtained by linear fitting of the initial reaction (10–60 s). | ||
| 6a | 0.42 | 4.4 |
| 6b | 0.30 | 5 |
| 6c | 0.30 | 5 |
| 6d | 0.83 | 6.6 |
| 6e | 0.30 | 4.6 |
| 6f | 0.50 | 12 |
| 6g | 0.30 | 5 |
| 6h | 0.42 | 7 |
However, Mn complex 6f, housing the long aliphatic chain with a terminal carboxylate group, displayed higher catalytic activity than the other complexes. This effect could not be ascribed to significantly decreased redox potentials in complex 6f (vide infra), which would improve the O–O bond formation and subsequent O2 formation. To additionally probe how big influence the substituent in complex 6f had on the electronics, it was decided to synthesize the related Mn complex 6g. Although the two complexes 6f and 6g have electronic resemblance, they do not at all display the same catalytic activity towards oxidizing H2O. This highlights that there is a different reason as to why Mn complex 6f displays a higher catalytic activity and thus facilitates the oxidation of H2O. To further establish the catalytic importance of the substituent in complex 6f, we turned our attention to electrochemistry in the hope that this would reveal the intrinsic reason for the better efficiency of this complex.
It should also be noted that the bpy functionalized Mn complex 6h was found to be catalytically active. This complex is a precursor of fundamental importance and could potentially be used for the construction of coupled photosensitizer-WOC systems for incorporation into devices for solar to chemical energy conversion (Fig. 3).
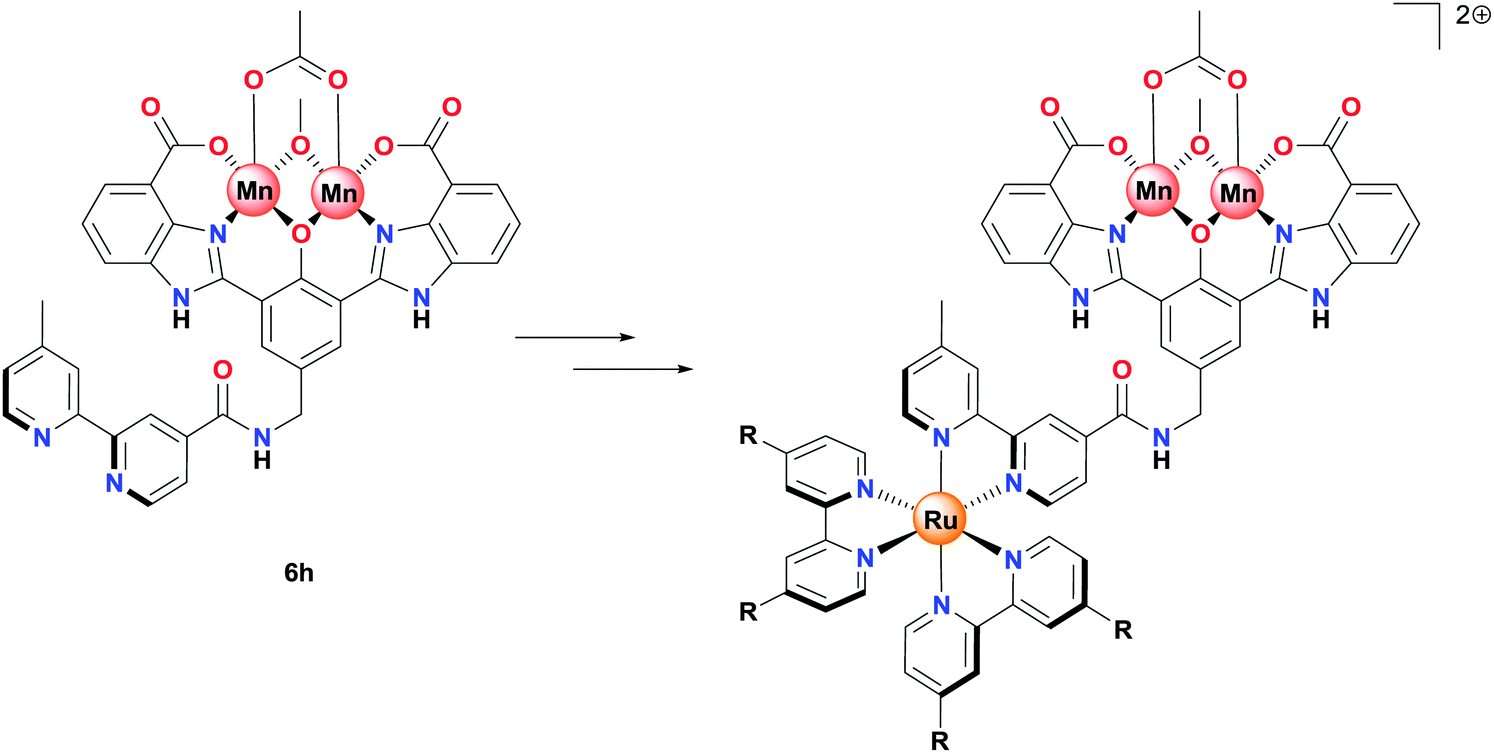 | ||
| Fig. 3 Schematic pathway from the dinuclear Mn2II,III complex 6h to a coupled Ru-Mn2 assembly for use in a solar fuel device. | ||
3.3. Electrochemical measurements
In order to assess the effect of the different substituents on the electronic properties of Mn complexes 6, it was decided to examine the electrochemical properties of the synthesized Mn complexes 6a,b and 6e–h. Electrochemical measurements were carried out at neutral conditions (pH 7.2), under similar conditions to the catalytic H2O oxidation experiments in order to obtain a deeper understanding of how the different substituents influenced the properties of the dinuclear Mn2II,III complexes. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were used to analyze the redox properties. Cyclic voltammograms revealed catalytic currents, assigned to electrochemical oxidation of H2O, for all of the studied Mn complexes 6 (Fig. 4). However, it was difficult to clearly assign the onset potentials due to overlapping redox processes in the onset potential region.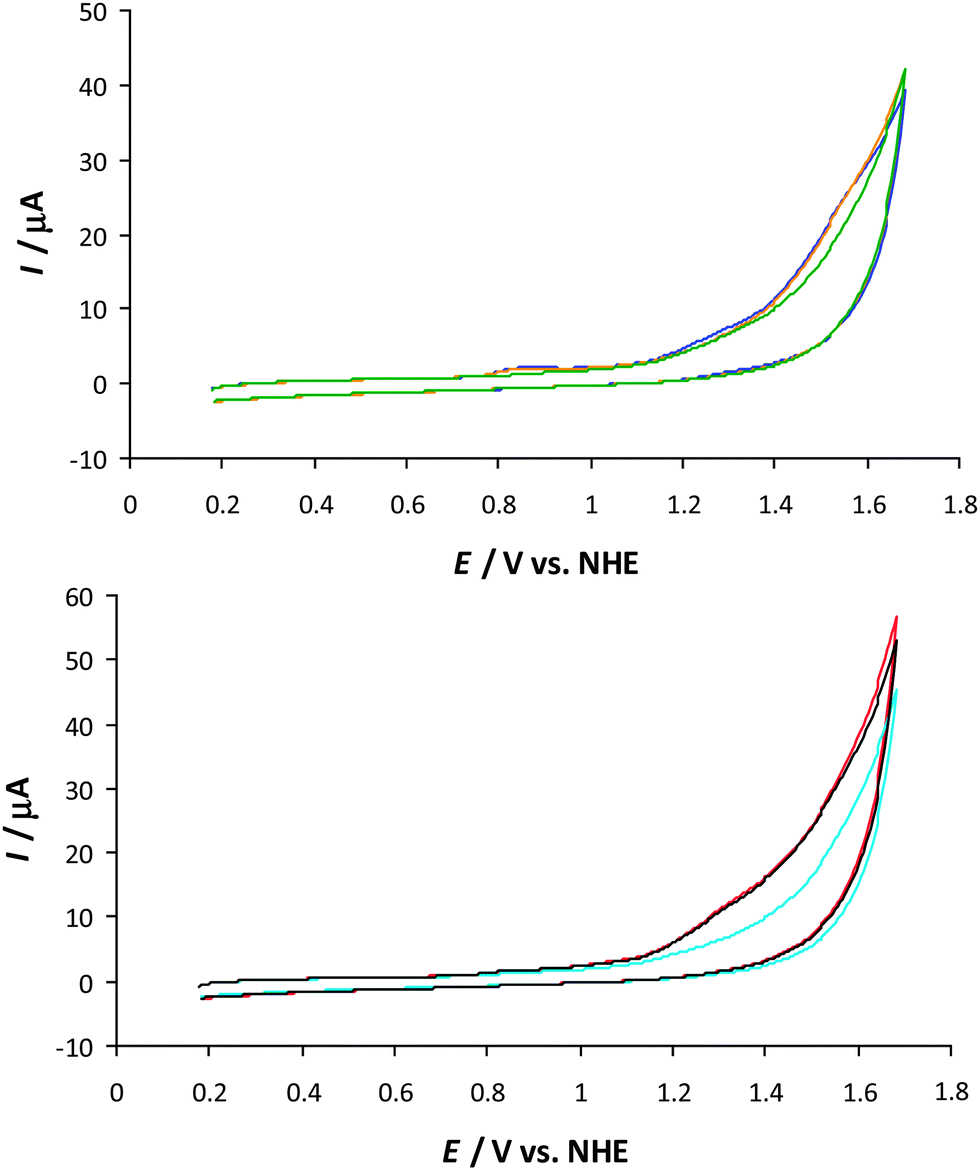 | ||
Fig. 4 Cyclic voltammograms of Mn complexes 6a–b,e and 6f–h in an aqueous phosphate buffer solution (pH 7.2). Conditions: voltammograms were recorded in an aqueous phosphate buffer solution (0.1 M, pH 7.2) containing the Mn complex (25 μM) with a scan rate of 0.1 V s−1. 6a ( ), 6b ( ), 6b ( ), 6e ( ), 6e ( ), 6f ( ), 6f ( ), 6g ( ), 6g ( ), 6h (—). ), 6h (—). | ||
Therefore, to gain further insight into the redox chemistry of the newly synthesized Mn complexes 6, DPV was carried out on the complexes. The differential pulse voltammograms of the dinuclear Mn complexes 6 are depicted in Fig. 5 and displayed several peaks. Within the developed library, the differential pulse voltammograms of complexes 6f and 6h displayed five discernible peaks in the region 0.50 < E < 1.30 V vs. NHE (Table 2). These peaks were subsequently assigned to the formal oxidations of Mn2II,III → Mn2III,III → Mn2III,IV → Mn2IV,IV → Mn2IV,V → Mn2V,V, in conformity with the previously reported result for a related dinuclear Mn complex.61 However, the other complexes displayed less than five redox peaks. This is probably related to the close potential gap, i.e. the narrow potential range for some of the redox couples, resulting in single observable two-electron redox peaks instead of two single-electron waves.
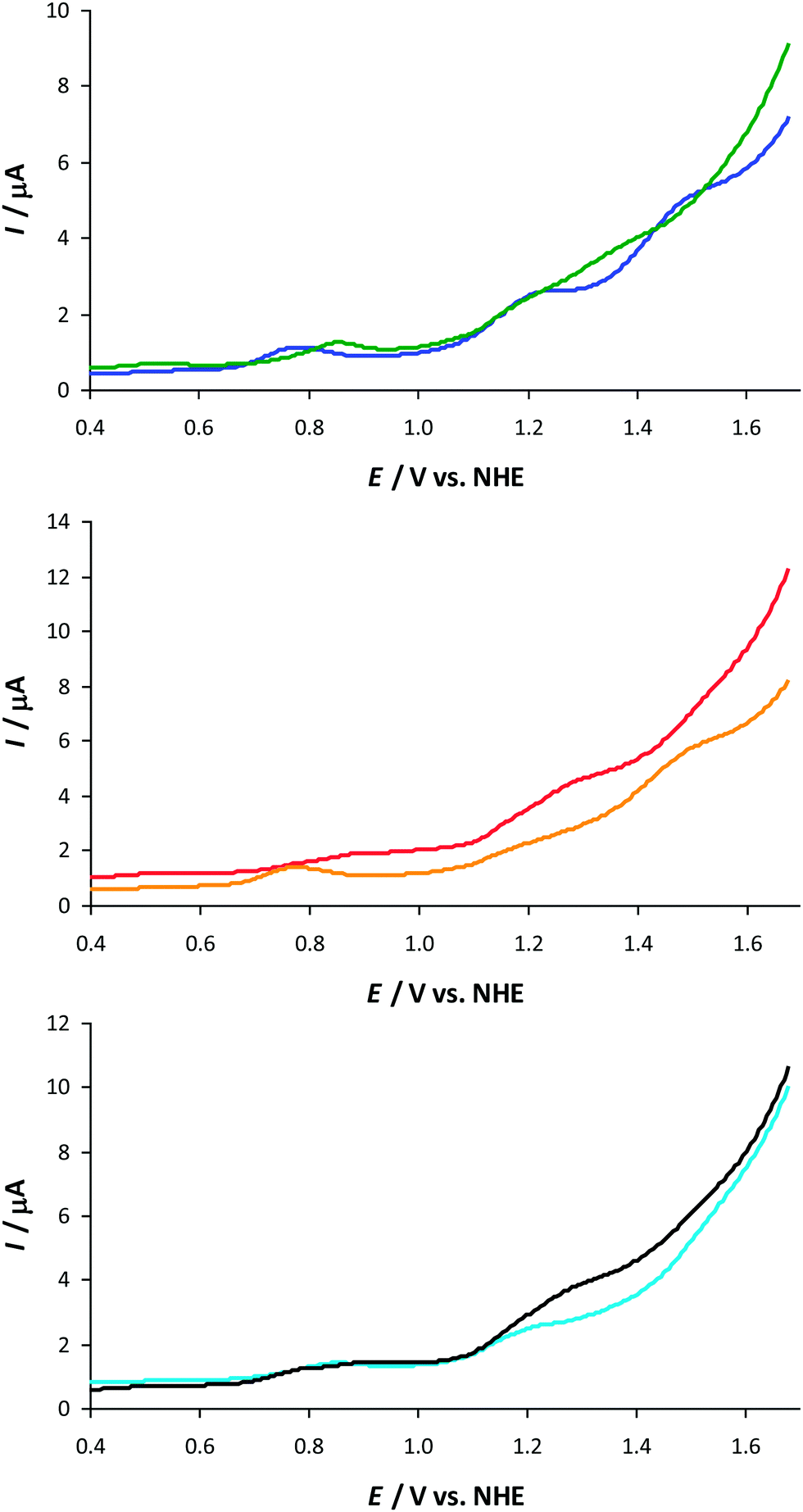 | ||
Fig. 5 Differential pulse voltammograms of Mn complexes 6a–b, 6e–f and 6g–h in an aqueous phosphate buffer solution (pH 7.2). Conditions: voltammograms were recorded in an aqueous phosphate buffer solution (0.1 M, pH 7.2) containing the Mn complex (25 μM) with a scan rate of 0.1 V s−1. 6a ( ), 6b ( ), 6b ( ), 6e ( ), 6e ( ), 6f ( ), 6f ( ), 6g ( ), 6g ( ), 6h (—). ), 6h (—). | ||
| Complex | Mn2III,III/Mn2II,III (V vs. NHE) | Mn2III,IV/Mn2III,III (V vs. NHE) | Mn2IV,IV/Mn2III,IV (V vs. NHE) | Mn2IV,V/Mn2IV,IV (V vs. NHE) | Mn2V,V/Mn2IV,V (V vs. NHE) |
|---|---|---|---|---|---|
| a Potentials were obtained from differential pulse voltammetry. Conditions: measured in an aqueous phosphate buffer solution (0.1 M, pH 7.2). Scan rate 0.1 V s−1, glassy carbon disk as a working electrode, a platinum spiral as a counter electrode and an Ag/AgCl electrode as a reference electrode. Potentials were converted to NHE by using the [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple as a standard (E1/2 = 1.26 V vs. NHE). b Possible two-electron redox process. | |||||
| 6a | 0.55 | 0.78b | — | 1.22b | — |
| 6b | 0.51 | 0.86b | — | 1.16 | 1.20 |
| 6e | — | 0.79b | — | 1.14 | 1.21 |
| 6f | 0.53 | 0.79 | 0.88 | 1.17 | 1.22 |
| 6g | 0.51 | 0.85b | — | 1.19b | — |
| 6h | 0.52 | 0.78 | 0.89 | 1.18 | 1.27 |
As is evident from these voltammograms and Table 2, the substituents in Mn complexes 6 did not cause any dramatic shift of the different redox processes in the complexes. This highlights that there must be another explanation for the increased efficiency of Mn complex 6f, containing the distal carboxyl group. Previous studies of so-called “Hangman” porphyrin and corrole complexes housing distal hydrogen bonding groups, such as carboxyl groups, have established the possibility of accessing metal complexes with increased affinity for promoting PCET reactions.37,45,78–81 There even exists an example of an Fe complex comprising a Hangman platform, which in the solid state exhibits a hydrogen bonded H2O molecule bound between a distal xanthene carboxylic acid and an Fe porphyrin hydroxide by two hydrogen bonds (Fig. 6).78 This suggested that the carboxyl group in 6f could perhaps also interact with the Mn centers. To confirm this hypothesis, quantum chemical calculations were performed on complex 6f.
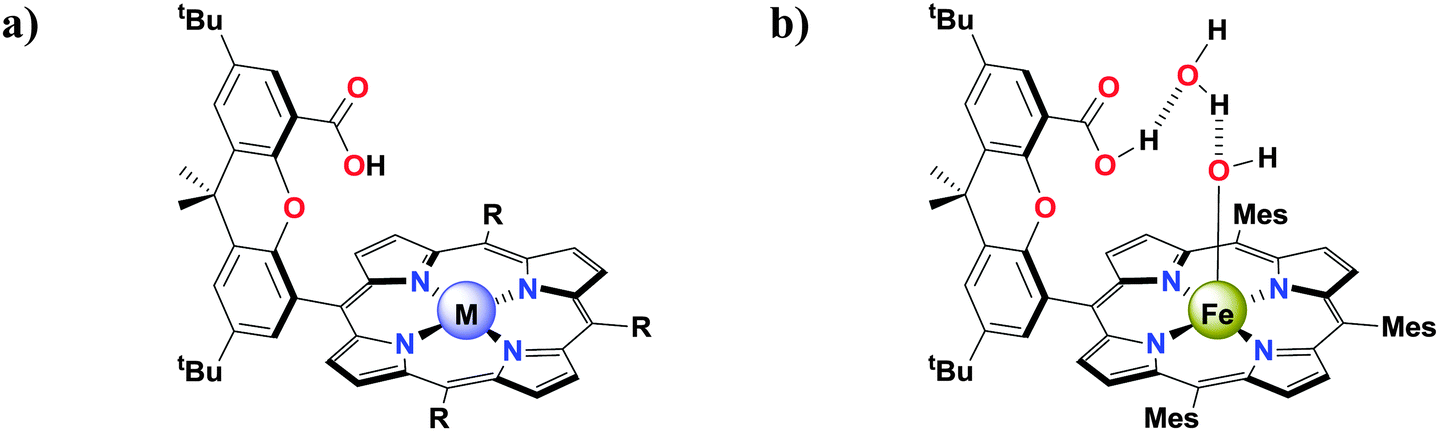 | ||
| Fig. 6 (a) Structure of a metal “Hangman” porphyrin housing a distal carboxyl group and (b) representation of the hydrogen bonded H2O molecule bound between a distal xanthene carboxylic acid and a Fe porphyrin hydroxide by two hydrogen bonds. | ||
3.4. Quantum chemical insight
DFT calculations were performed on the dinuclear complex 6f in the relevant formal Mn2IV,V state. The Mn2IV,V stage is believed to be capable of promoting O–O bond formation, as shown in the previous calculations on the dinuclear Mn complex [(H2O)(tpy)Mn(μ-O)2Mn(tpy)(OH2)]3+ (tpy = 2,2′;6′,2′′-terpyridine).82 The presence of one high-valent MnV (or MnIV–O˙) is also suggested in the OEC in the formation of O2.83–85 The optimized structures of the two low-energy isomers of complex 6f in its high-valent Mn2IV,V state (A and B) are depicted in Fig. 7.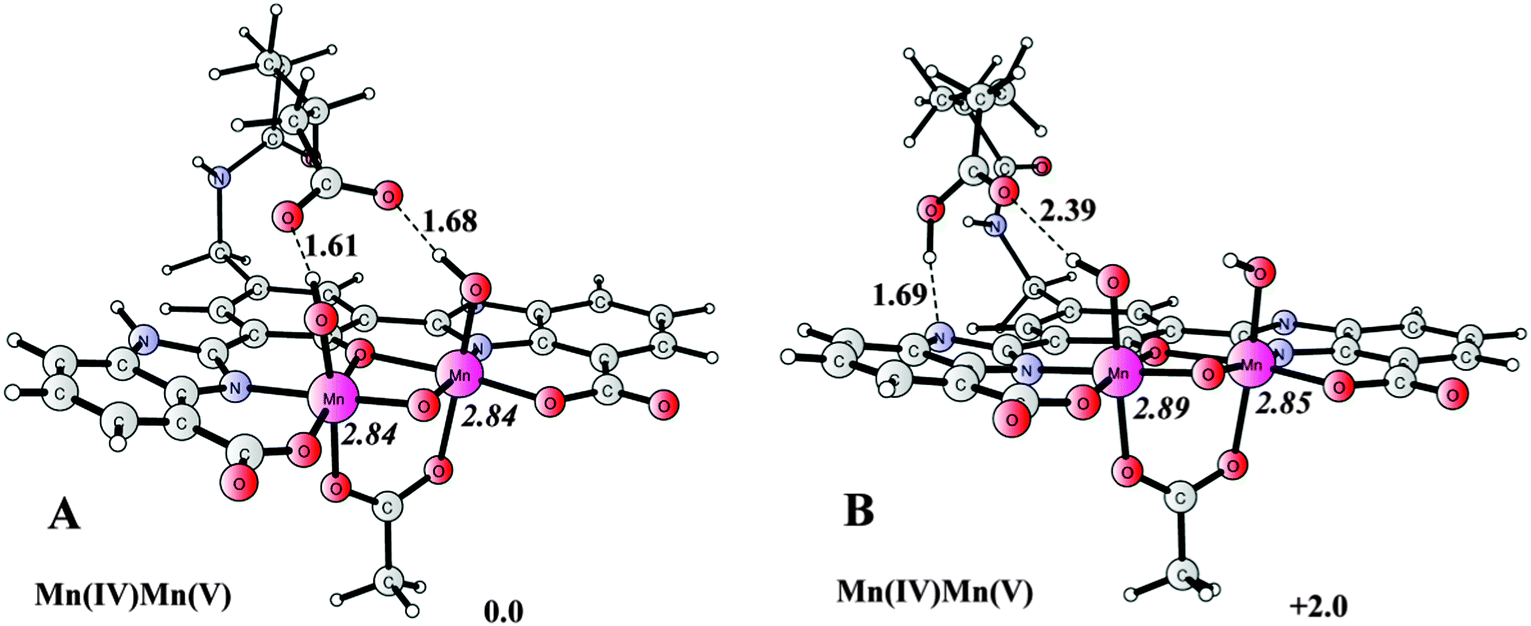 | ||
| Fig. 7 Optimized structures of the dinuclear Mn complex 6f in its formal Mn2IV,V state, showing the hydrogen bonding interaction between the distal carboxyl group and (a) the Mn bounded hydroxide(s), and (b) the imidazole moiety. Distances are given in Ångstrom, spin densities on Mn are given in italics, and relative energies are given in kcal mol−1. | ||
Both structures have a total charge of +0 and exist in the high spin octet state according to the calculations. The two Mn centers are bridged by an oxo group, an acetate, and the phenolate of the ligand. In addition, each Mn center also has a coordinated hydroxide ligand. Spin density analysis suggests that both metals are in fact in their MnIV state, with a coordinated ligand radical cation. This suggests that the ligand framework is redox-active, non-innocent, and highlights the intriguing properties of ligand scaffolds of this type.86 Redox-active ligands might thus be of importance in alleviating the metal centers from being heavily oxidized by storing redox equivalents at the ligand scaffolds during the oxidation of H2O.
In structure A of the oxidized complex 6f, the distal carboxyl group generates two hydrogen bonds to the two Mn coordinated hydroxide ligands. These two hydrogen bonds might thus influence the subsequent O–O bond forming step(s). This supports that the distal carboxylate group in the ligand framework of Mn complex 6f can accommodate a bridging hydroxide/aqua molecule to yield a unique hydrogen-bonded scaffold that might facilitate H2O oxidation and thus promote the crucial O–O bond formation through PCET. Multi-electron catalysis is at the heart of H2O oxidation and the ability of synchronizing proton and electron transfer events via PCET is fundamental in a plethora of biological and chemical reactions.87,88 PCET allows the charge of a system to remain unchanged, whereas non-proton coupled single electron transfer processes result in charge accumulation and high-energy intermediates. Detailed studies and a fundamental understanding of how to couple proton and electron transfer are at the frontier and might be the key to realizing more efficient WOCs.
An additional interesting feature of the ligand platforms in Mn complexes 6 is the presence of the imidazole motifs, which have the ability to function as proton transfer mediators. In structure B, the carboxyl group has abstracted a proton from the imidazole unit. Structure B was found to be 2.0 kcal mol−1 higher in energy than structure A. This hydrogen bonding capability of the distal carboxylate group renders it possible to also promote proton transfer reactions between the distal group and the protonated imidazole moiety. The alternative scenario for the enhanced activity of complex 6f is that the carboxyl group serves as a ligand to bridge the two metal centers. However, calculations revealed that the chain is not sufficient to accommodate this sort of coordination.
The possibility of including a non-innocent distal group within the ligand scaffolds in Mn complexes 6 is an interesting feature in these developed WOCs that has perhaps not received sufficient attention in H2O oxidation schemes. The key feature of incorporating a distal group that acts as a general base to stimulate a H2O molecule in undergoing nucleophilic attack on a high-valent metal-oxo species could be a general strategy for promoting catalytic H2O oxidation activity. This idea of hydrogen bonding as a means to increase the nucleophilicity of H2O is utilized in hydrolytic enzymes such as chymotrypsin. It has also been used by Nocera45,78,81 and ourselves37 to promote H2O oxidation and also recently in two theoretical papers.79,80 In those previous studies, rather elaborate rigid systems have been used. However, our present results suggest that even a carboxyl attached to a highly flexible chain will participate efficiently. Using this concept we will therefore try to prepare analogues to complex 6f, and also analogues with a different central ligand, using the synthetic versatility available for modifying the ligand frameworks of the complexes 6.
4. Conclusions
In the present work we have synthesized eight new dinuclear Mn2II,III complexes and examined the H2O oxidation process for these complexes. It was demonstrated that all of the developed Mn complexes were catalytically active in H2O oxidation. The complexes had sufficiently low redox potentials to allow for the use of the mild one-electron oxidant [Ru(bpy)3]3+ to drive H2O oxidation at neutral pH conditions.These studies have identified a potential general factor for obtaining WOCs that display increased catalytic efficiencies. Within the library of the synthesized Mn complexes, complex 6f containing the long aliphatic chain with a terminal carboxylate group as a distal group was found to exhibit a higher O2 evolution rate than the other catalysts. This observation did not originate from decreased redox potentials, which would result in more favourable thermodynamics for carrying out H2O oxidation.
Instead, the improved catalytic effect seems to result from pre-orientation of the incoming H2O nucleophile and hydrogen bonding between the distal carboxyl group and a high-valent Mn-hydroxy group. This hydrogen bonding interaction can subsequently promote PCET processes, which decrease the energy of the catalytic H2O oxidation process.
These findings highlight the importance of controlling both proton and electron transfer processes to promote O–O bond formation during H2O oxidation. In the natural photosynthetic system, PCET is prevalent and provides the key to efficient H2O oxidation catalysis. The present study provides a guide that might be helpful for designing more efficient WOCs in the future, which have the possibility of being incorporated into large scale systems for solar to chemical energy conversion.
Acknowledgements
Financial support from the Swedish Energy Agency (BÅ and JM), the Knut and Alice Wallenberg Foundation (Center of Molecular Catalysis at Stockholm University, Artificial Leaf Project Umeå), the Swedish Research Council (BÅ) and the Strong Research Environment Solar Fuels (Umeå University) is gratefully acknowledged. W. A. A. A would also like to thank the Egyptian Government for a PAROWN research fellowship.References
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 Search PubMed.
- R. F. Service, Science, 2011, 334, 925–927 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed.
- S. Dasgupta, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Soc. Rev., 2013, 42, 2213–2214 RSC.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821–825 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- C. S. Mullins and V. L. Pecoraro, Coord. Chem. Rev., 2008, 252, 416–443 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- F. Bozoglian, S. Romain, M. Z. Ertem, T. K. Todorova, C. Sens, J. Mola, M. Rodríguez, I. Romero, J. Benet-Buchholz, X. Fontrodona, C. J. Cramer, L. Gagliardi and A. Llobet, J. Am. Chem. Soc., 2009, 131, 15176–15187 CrossRef CAS PubMed.
- T. Wada, K. Tsuge and K. Tanaka, Angew. Chem., Int. Ed., 2000, 39, 1479–1482 CrossRef CAS.
- A. Lewandowska-Andralojc, D. E. Polyansky, R. Zong, R. P. Thummel and E. Fujita, Phys. Chem. Chem. Phys., 2013, 15, 14058–14068 RSC.
- Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- M. D. Kärkäs, E. V. Johnston, E. A. Karlsson, B.-L. Lee, T. Åkermark, M. Shariatgorji, L. Ilag, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Chem.–Eur. J., 2011, 17, 7953–7959 CrossRef PubMed.
- Z. Deng, H.-W. Tseng, R. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835–1848 CrossRef CAS PubMed.
- Y. Xu, L. Duan, L. Tong, B. Åkermark and L. Sun, Chem. Commun., 2010, 46, 6506–6508 RSC.
- Y. M. Badiei, D. E. Polyansky, J. T. Muckerman, D. J. Szalda, R. Haberdar, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2013, 52, 8845–8850 CrossRef CAS PubMed.
- M. Yagi, S. Tajima, M. Komi and H. Yamazaki, Dalton Trans., 2011, 40, 3802–3804 RSC.
- M. D. Kärkäs, T. Åkermark, E. V. Johnston, S. R. Karim, T. M. Laine, B.-L. Lee, T. Åkermark, T. Privalov and B. Åkermark, Angew. Chem., Int. Ed., 2012, 51, 11589–11593 CrossRef PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- M. D. Kärkäs, T. Åkermark, H. Chen, J. Sun and B. Åkermark, Angew. Chem., Int. Ed., 2013, 52, 4189–4193 CrossRef PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Chem. Commun., 2011, 47, 2712–2714 RSC.
- Z. Codolà, J. M. S. Cardoso, B. Royo, M. Costas and J. Lloret-Fillol, Chem.–Eur. J., 2013, 19, 7203–7213 CrossRef PubMed.
- A. Petronilho, M. Rahman, J. A. Woods, H. Al-Sayyed, H. Müller-Bunz, J. M. D. MacElroy, S. Bernhard and M. Albrecht, Dalton Trans., 2012, 41, 13074–13080 RSC.
- K. S. Joya, N. E. Subbaiyan, F. D'Souza and H. J. M. de Groot, Angew. Chem., Int. Ed., 2012, 51, 9601–9605 CrossRef CAS PubMed.
- U. Hintermair, S. A. Hashmi, M. Elimelech and R. H. Crabtree, J. Am. Chem. Soc., 2012, 134, 9785–9795 CrossRef CAS PubMed.
- E. A. Karlsson, B.-L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J.-E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS PubMed.
- Y. Gao, T. Åkermark, J. Liu, L. Sun and B. Åkermark, J. Am. Chem. Soc., 2009, 131, 8726–8727 CrossRef CAS PubMed.
- K. J. Young, M. K. Takase and G. W. Brudvig, Inorg. Chem., 2013, 52, 7615–7622 CrossRef CAS PubMed.
- Y. Gao, J. Liu, M. Wang, Y. Na, B. Åkermark and L. Sun, Tetrahedron, 2007, 63, 1987–1994 CrossRef CAS.
- H. Chen, R. Tagore, G. Olack, J. S. Vrettos, T.-C. Weng, J. Penner-Hahn, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 34–43 CrossRef CAS PubMed.
- A. K. Poulsen, A. Rompel and C. J. McKenzie, Angew. Chem., Int. Ed., 2005, 44, 6916–6920 CrossRef CAS PubMed.
- P. Kurz, Dalton Trans., 2009, 6103–6108 RSC.
- G. González-Riopedre, M. I. Fernández-García, A. M. González-Noya, M. Å. Vázquez-Fernández, M. R. Bermejo and M. Maneiro, Phys. Chem. Chem. Phys., 2011, 13, 18069–18077 RSC.
- E. A. Karlsson, B.-L. Lee, R.-Z. Liao, T. Åkermark, M. D. Kärkäs, V. Saavedra Becerril, P. E. M. Siegbahn, X. Zou, M. Abrahamsson and B. Åkermark, ChemPlusChem, 2014 DOI:10.1002/cplu.201402006.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC.
- D. K. Dogutan, R. McGuire Jr and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- M. Z. Ertem and C. J. Cramer, Dalton Trans., 2012, 41, 12213–12219 RSC.
- M. L. Rigsby, S. Mandal, W. Nam, L. C. Spencer, A. Llobet and S. S. Stahl, Chem. Sci., 2012, 3, 3058–3062 RSC.
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903–7907 CAS.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- Z. Codolà, I. Garcia-Bosch, F. Acuña-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem.–Eur. J., 2013, 19, 8042–8047 CrossRef PubMed.
- D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522–9531 CrossRef CAS PubMed.
- R.-Z. Liao, X.-C. Li and P. E. M. Siegbahn, Eur. J. Inorg. Chem., 2014, 728–741 CrossRef CAS.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- Z. Chen and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 700–703 CrossRef CAS PubMed.
- M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed.
- B.-L. Lee, M. D. Kärkäs, E. V. Johnston, A. K. Inge, L.-H. Tran, Y. Xu, Ö. Hansson, X. Zou and B. Åkermark, Eur. J. Inorg. Chem., 2010, 5462–5470 CrossRef CAS.
- T. Norrby, A. Börje, L. Hammarström, J. Alsins, K. Lashgari, R. Norrestam, J. Mårtensson, G. Stenhagen and B. Åkermark, Inorg. Chem., 1997, 36, 5850–5858 CrossRef CAS PubMed.
- M. F. Anderlund, J. Zheng, M. Ghiladi, M. Kritikos, E. Rivière, L. Sun, J.-J. Girerd and B. Åkermark, Inorg. Chem. Commun., 2006, 9, 1195–1198 CrossRef CAS.
- R. Lomoth, P. Huang, J. Zheng, L. Sun, L. Hammarström, B. Åkermark and S. Styring, Eur. J. Inorg. Chem., 2002, 2965–2974 CrossRef CAS.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- R. E. DeSimone and R. S. Drago, J. Am. Chem. Soc., 1970, 92, 2343–2352 CrossRef CAS.
- A. Johansson, M. Abrahamsson, A. Magnuson, P. Huang, J. Mårtensson, S. Styring, L. Hammarström, L. Sun and B. Åkermark, Inorg. Chem., 2003, 42, 7502–7511 CrossRef CAS PubMed.
- B. M. Peek, G. T. Ross, S. W. Edwards, G. J. Meyer, T. J. Meyer and B. W. Erickson, Int. J. Pept. Protein Res., 1991, 38, 114–123 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- Y. Xu, T. Åkermark, V. Gyollai, D. Zou, L. Eriksson, L. Duan, R. Zhang, B. Åkermark and L. Sun, Inorg. Chem., 2009, 48, 2717–2719 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al. Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- D. Yang, D. Fokas, J. Li, L. Yu and C. M. Baldino, Synthesis, 2005, 47–56 CrossRef.
- R. Lomoth, P. Huang, J. Zheng, L. Sun, L. Hammarström, B. Åkermark and S. Styring, Eur. J. Inorg. Chem., 2002, 2965–2974 CrossRef CAS.
- G. Berggren, A. Thapper, P. Huang, P. Kurz, L. Eriksson, S. Styring and M. F. Anderlund, Dalton Trans., 2009, 10044–10054 RSC.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- C.-Y. Yeh, C. J. Chang and D. G. Nocera, J. Am. Chem. Soc., 2001, 123, 1513–1514 CrossRef CAS PubMed.
- W. Lai, R. Cao, G. Dong, S. Shaik, J. Yao and H. Chen, J. Phys. Chem. Lett., 2012, 3, 2315–2319 CrossRef CAS PubMed.
- M. Z. Ertem and C. J. Cramer, Dalton Trans., 2012, 41, 12213–12219 RSC.
- R. McGuire Jr, D. K. Dogutan, T. S. Teets, J. Suntivich, Y. Shao-Horn and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC.
- M. Lundberg, M. R. A. Blomberg and P. E. M. Siegbahn, Inorg. Chem., 2004, 43, 264–274 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Chem.–Eur. J., 2006, 12, 9217–9227 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. Fecenko Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. Granville McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- O. S. Wenger, Chem.–Eur. J., 2011, 17, 11692–11702 CrossRef CAS PubMed.
Footnote |
| † Present address: Chemistry Department, Faculty of Science, Fayoum University, PO Box 63514, Fayoum, Egypt. |
| This journal is © the Owner Societies 2014 |