 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural factors influencing the intramolecular charge transfer and photoinduced electron transfer in tetrapyrazinoporphyrazines†
Veronika
Novakova
*a,
Petr
Hladík
b,
Tereza
Filandrová
b,
Ivana
Zajícová
b,
Veronika
Krepsová
b,
Miroslav
Miletin
b,
Juraj
Lenčo
cd and
Petr
Zimcik
*b
aDepartment of Biophysics and Physical Chemistry, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, 50005, Hradec Kralove, Czech Republic. E-mail: veronika.novakova@faf.cuni.cz; Tel: +420 495067380
bDepartment of Pharmaceutical Chemistry and Drug Control, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, 50005, Hradec Kralove, Czech Republic. E-mail: petr.zimcik@faf.cuni.cz; Web: http://portal.faf.cuni.cz/Groups/Azaphthalocyanine-group/ Tel: +420 495067257
cDepartment of Biochemical Sciences, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, 50005, Hradec Kralove, Czech Republic
dInstitute of Molecular Pathology, Faculty of Military Health Sciences, University of Defence, Trebesska 1575, 500 01 Hradec Králové, Czech Republic
First published on 28th January 2014
Abstract
A series of unsymmetrical tetrapyrazinoporphyrazines (TPyzPzs) from the group of azaphthalocyanines with one peripherally attached amino substituent (donor) were synthesized, and their photophysical properties (fluorescence quantum yield and singlet oxygen quantum yield) were determined. The synthesized TPyzPzs were expected to undergo intramolecular charge transfer (ICT) as the main pathway for deactivating their excited states. Several structural factors were found to play a critical role in ICT efficiency. The substituent in the ortho position to the donor center significantly influences the ICT, with tert-butylsulfanyl and butoxy substituents inducing the strongest ICTs, whereas chloro, methyl, phenyl, and hydrogen substituents in this position reduce the efficiency. The strength of the donor positively influences the ICT efficiency and correlates well with the oxidation potential of the amines used as the substituents on the TPyzPz as follows: n-butylamine < N,N-diethylamine < aniline < phenothiazine. The ICT (with conjugated donors and acceptors) in the TPyzPz also proved to be much stronger than a photoinduced electron transfer in which the donor and the acceptor are connected through an aliphatic linker.
Introduction
Photoinduced electron transfer (PET) is one of the numerous pathways for deactivating the excited states of molecules and has been widely used to develop diverse sensors based on the ON–OFF switching of this highly efficient quenching mechanism. A PET requires the presence of donor and acceptor molecules/moieties that can be fully separated as individual species in solution1,2 or connected through an aliphatic (non-conjugated) linker.3,4 A radical cation on the donor and a radical anion on the acceptor are formed after excitation. Another deactivation process that is used for a design of the sensors is intramolecular (or internal) charge transfer (ICT)5 where charge transfer states between fully conjugated donors and acceptors are formed after excitation.6 In this article, we use the acronym PET to indicate non-conjugated linkers and the acronym ICT to refer to the fully conjugated connection.Tetrapyrazinoporphyrazines (TPyzPzs) are the aza-analogs of phthalocyanines (Pcs) in which the benzene rings are replaced with pyrazines. Interest in their synthesis and photophysical properties has increased during the last decade because of their strong absorption and emission in the red region of the visible spectrum, which is desirable for biological applications.7–9 Moreover, the presence of additional nitrogen atoms in the macrocyclic core of TPyzPzs imparts it with remarkable electron-deficient properties,10,11 which cause the macrocycle to behave as an electron acceptor. In addition, PET12 or ICT13 may occur after combination with a donor. The ICT process, in particular, is efficient in TPyzPzs and has recently been used to develop red-emitting pH or metal–cation sensors.14,15 However, since the first observation of this excited-state quenching in TPyzPzs, no systematic study has focused on determining the structural factors that influence the efficiency of these two deactivation processes.12 The only study of the structural factors was recently published and suggested that the ICT is influenced by the electron-deficient properties of the macrocycle core.16 Therefore, in this study, we aim to scrutinize the impact of structural modifications in close proximity to the donor on the efficiency of an ICT and to compare ICT and PET in TPyzPzs to allow rational design of sensors based on the TPyzPz macrocycle.
Experimental
Synthesis
Details regarding the synthesis of both precursors (substituted pyrazine-2,3-dicarbonitriles) and TPyzPzs and their full characterization are described in the ESI† and are discussed in the Results section. All samples were re-purified using preparative TLC for the photophysical measurements (both ΦF and ΦΔ) to ensure that they were highly pure.Fluorescence
The fluorescence spectra were obtained using an AMINCO Bowman Series 2 luminescence spectrometer. Emission spectra were corrected for instrument response. The fluorescence quantum yields (ΦF) were determined in THF via the comparative method using unsubstituted zinc phthalocyanine (ZnPc, Sigma-Aldrich) as a reference (ΦF = 0.32 in THF17). Both the reference and sample were excited at 595 nm. The absorbance at the excitation wavelength was held below 0.015, and the absorbance at the Q-band maximum was held below 0.05 to limit the inner filter effect. The value of ΦF was calculated using eqn (1):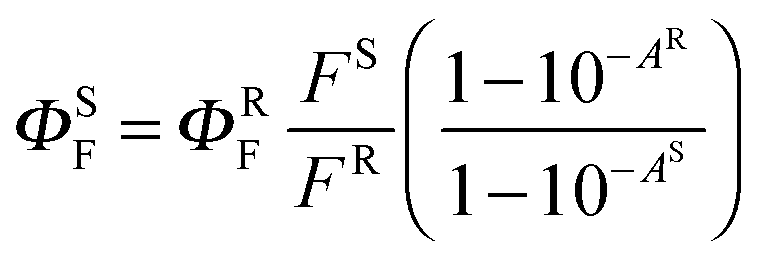 | (1) |
Determination of the singlet oxygen production
The quantum yields of the singlet oxygen (ΦΔ) were determined in THF according to a previously published procedure18 using the decomposition of a chemical trap 1,3-diphenylisobenzofuran (DPBF) with ZnPc as a reference (ΦΔ = 0.53 in THF19). The detailed procedure was as follows: 2.5 mL of a DPBF stock solution in THF (5 × 10−5 M) was transferred into a 10 × 10 mm quartz optical cell and was saturated with oxygen for 1 min. A stock solution of the tested compound in THF (typically 30 μL) was then added to achieve an absorbance of the final solution in the Q-band maximum of approximately 0.1. The solution was stirred and irradiated using a xenon lamp (100 W, ozone-free XE DC short-arc lamp, Newport). The incident light was filtered through a water filter (6 cm) and an OG530 cut-off filter (Newport) to remove the heat and the light below 523 nm, respectively. Decrease of DPBF in solution with irradiation time was monitored at 414 nm. All experiments were performed in triplicate, and the data presented herein represent the mean of the three experiments (estimated error: ±10%).Electrochemical measurements
The electrochemical measurements were performed at room temperature using an Autolab PGSTAT101 potentiostat (Metrohm, Czech Republic). The three-electrode experimental setup consisted of a Pt working electrode, a Pt counter electrode, and an Ag/AgCl reference electrode separated from the bulk solution by an integrated salt bridge. Samples (typically 1 × 10−3 M) were dissolved in 0.1 M tetrabutylammonium hexafluorophosphate in anhydrous acetonitrile (Sigma-Aldrich) as a supporting electrolyte. The dissolved oxygen was removed via vigorous purging with N2 gas for 5 min. All values in Table 2 are referenced to an SCE with ferrocene as the internal standard (half-wave potential E1/2(Fc/Fc+) = +0.38 V vs. SCE20).Results
Synthesis
The studied TPyzPzs were synthesized from corresponding precursors, i.e., substituted pyrazine-2,3-dicarbonitriles (1–8). The precursors 1–8 were prepared via the nucleophilic substitution of chlorine atoms in 5-chloro-6-phenylpyrazine-2,3-dicarbonitrile, 5-chloro-6-methylpyrazine-2,3-dicarbonitrile, or 5,6-dichloropyrazine-2,3-dicarbonitrile. With two exceptions, the reactions proceeded with a high yield (Scheme 1) after the starting material was mixed with an excess of the nucleophile (amine or thiolate). First, the extremely low nucleophilicity of nitrogen in phenothiazine prohibited the direct substitution and synthesis of 4, but the reaction required the addition of CsF to form the highly reactive phenothiazin-10-ide. Similar attempts to synthesize a precursor substituted with diphenylamine were unsuccessful. The reaction indicated no trace of a product (5-diphenylamino-6-phenylpyrazine-2,3-dicarbonitrile) despite a number of bases and conditions being attempted (e.g., NaOH, NaH, K2CO3, CsF, DBU in DMF or THF). Second, the isolation of compound 6, which is an intermediate in the synthesis of 7, proved to be difficult due to the similar Rf values of 6 in all mobile phases tested with both the starting material and the side product 5,6-dibutoxypyrazine-2,3-dicarbonitrile. Consequently, only a small quantity was isolated for analytical purposes. The precursor 7 (with different retention properties on silica) was synthesized in one pot with a satisfactory yield of 30% without isolation of the intermediate 6.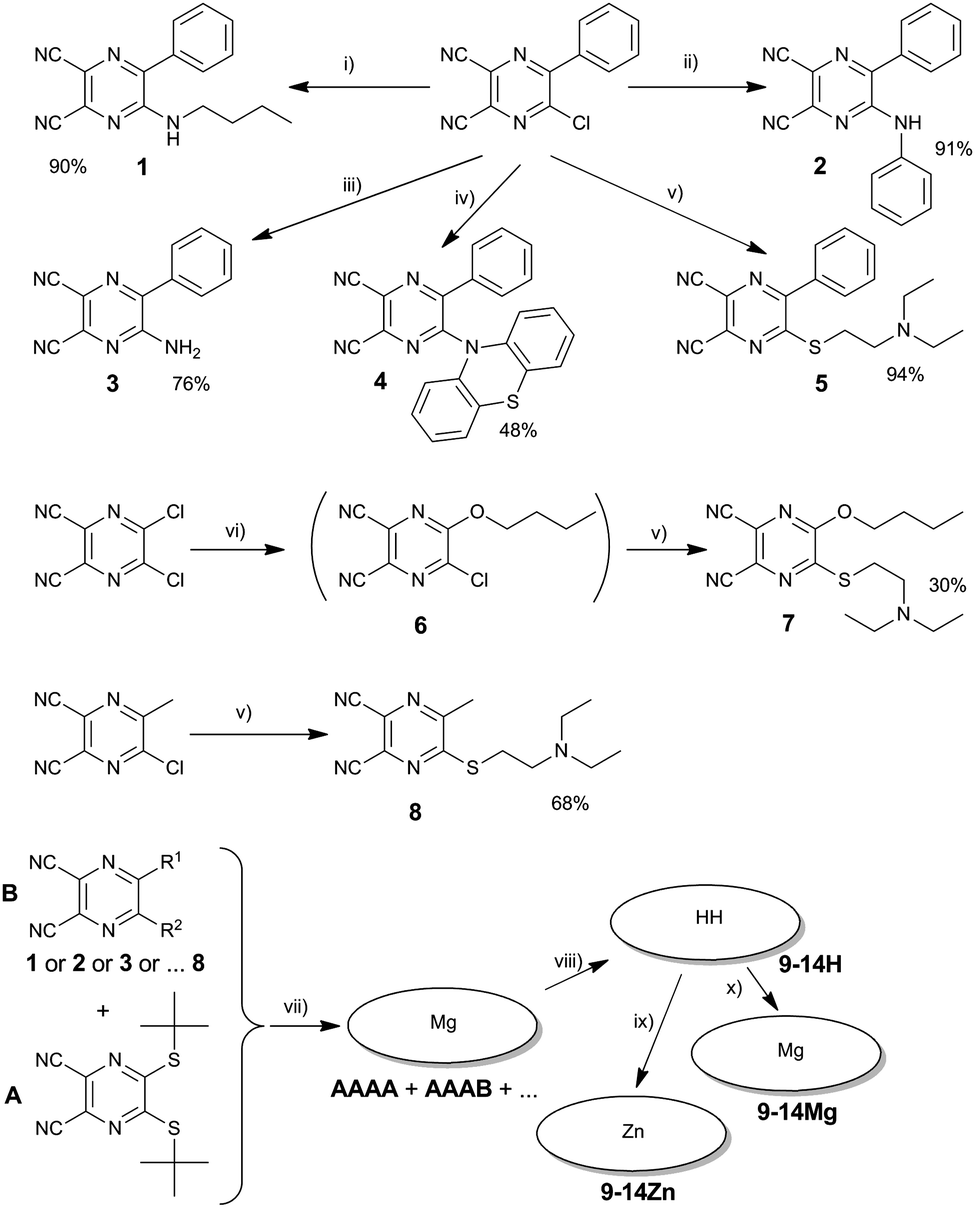 | ||
| Scheme 1 Synthetic procedures: (i) n-butylamine, THF, rt, 2 h. (ii) Aniline, THF, reflux, 8 h. (iii) NH4OH, THF, rt, 4 h. (iv) Phenothiazine, CsF, anhydr. DMF, rt, 5 h. (v) 2-Diethylaminoethanethiol hydrochloride, aq. NaOH, THF, rt, 15 min. (vi) n-Butanol aq. NaOH, −10 °C, 5 min. (vii) Anhydr. n-butanol, Mg, reflux, ∼6 h. (vii) p-Toluenesulfonic acid, THF, rt, 2 h, isolation of congeners. (ix) Anhydr. Zn(CH3COO)2, pyridine, reflux, 2 h. (x) Anhydr. Mg(CH3COO)2, pyridine, reflux, 2 h. | ||
Some of the studied TPyzPzs were prepared via published procedures (see ESI†). The rest were synthesized (Fig. 1 and Scheme 1) via a mixed cyclotetramerization of 3 eq. of 5,6-bis(tert-butylsulfanyl)pyrazine-2,3-dicarbonitrile (precursor A) with 1 eq. of one of the aforementioned precursors (1–8, precursor B). The reaction initiated by magnesium butoxide yielded a statistical mixture of magnesium complexes with six congeners (AAAA, AAAB, ABAB, AABB, ABBB, and BBBB). Treatment of the mixture with p-toluenesulfonic acid converted the magnesium complexes to metal-free derivatives that typically allow an easier isolation of the required AAAB-type congener.13–15 Zinc and magnesium were complexed to the center of the metal-free TPyzPz after the successful isolation of the AAAB-type congener using either zinc acetate or magnesium acetate, respectively. A cyclotetramerization reaction with compound 3 proceeded as well, but the resulting mixture of congeners was inseparable due to strong tailing on the TLC and overlapping of the fractions. For that reason, the corresponding TPyzPz could not be isolated.
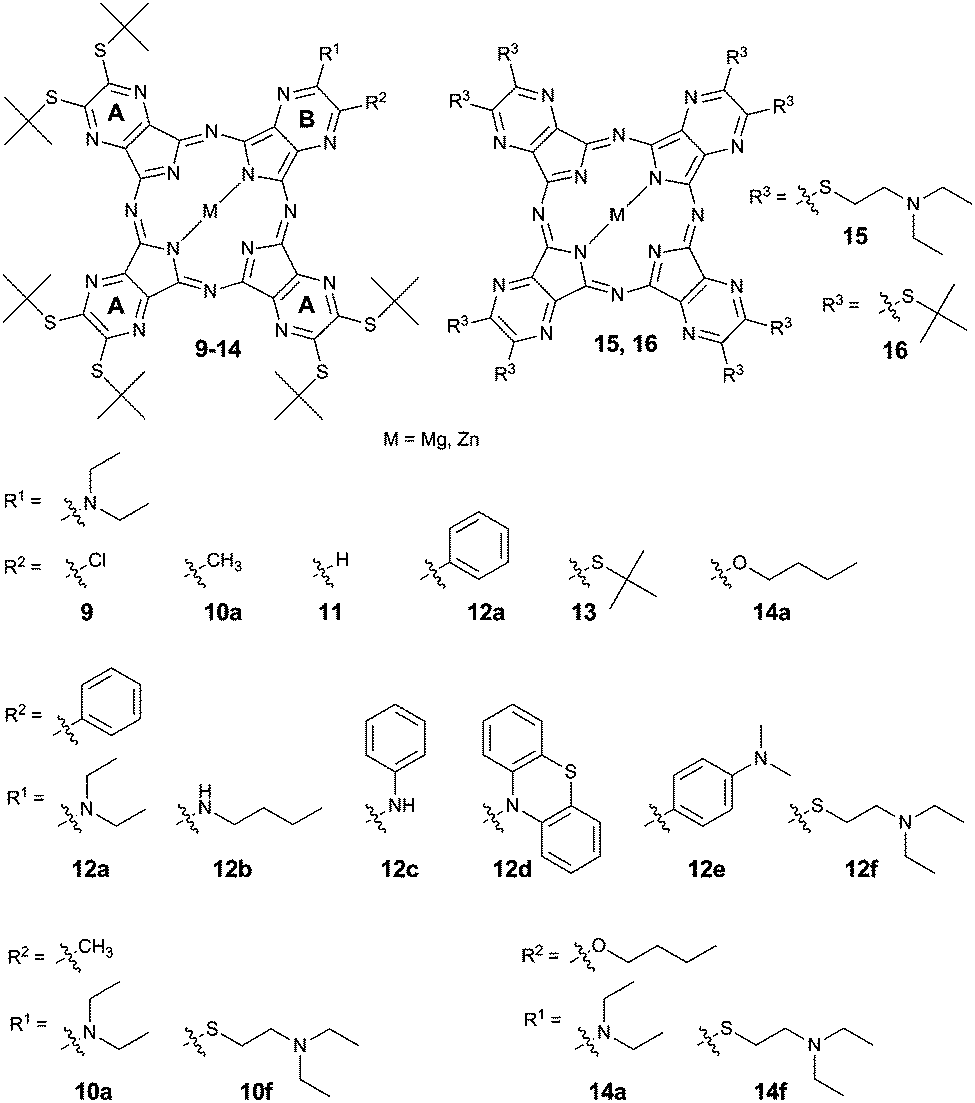 | ||
| Fig. 1 Structures of studied TPyzPzs. Substituent R1 contains always a donor group. | ||
Absorption spectra
Absorption spectra of 9–16 (both Mg and Zn complexes) in THF exhibited a shape typical of TPyzPzs with a high-energy B-band (∼360 nm) and a low energy Q-band (∼660 nm) (see ESI,† Fig. S2). The position of the Q-band varied slightly with the substitution pattern (Table 1); however, the bands of the magnesium complexes were systematically red-shifted by approximately 1–2 nm when compared with the bands of the zinc derivatives. The sharp Q-band with well-resolved vibrational bands indicated that no aggregation occurred at the tested concentrations (0.05–5 μM). Aggregation of the planar TPyzPz macrocycles can result in a misinterpretation of the photophysical results because it typically decreases both ΦF and ΦΔ independently of the presence or the absence of ICT or PET.| M | λ max | F max | Φ F | Φ Δ | Φ F + ΦΔ | |
|---|---|---|---|---|---|---|
| a Fluorescence data were corrected for instrument response. In the case of published data, the values were recalculated using original spectra. b Data from ref. 14. c Data from ref. 17. | ||||||
| 9 | Mg | 654 | 664 | 0.508 | 0.303 | 0.81 |
| Zn | 653 | 663 | 0.245 | 0.551 | 0.80 | |
| 10a | Mg | 652 | 667 | 0.428 | 0.356 | 0.78 |
| Zn | 651 | 667 | 0.175 | 0.647 | 0.82 | |
| 10f | Mg | 648 | 655 | 0.308 | 0.138 | 0.45 |
| Zn | 646 | 662 | 0.174 | 0.306 | 0.48 | |
| 11 | Mg | 662 | 674 | 0.447 | 0.316 | 0.76 |
| Zn | 660 | 673 | 0.202 | 0.623 | 0.83 | |
| 12a | Mg | 655 | 669 | 0.326 | 0.430 | 0.76 |
| Zn | 653 | 668 | 0.123 | 0.622 | 0.75 | |
| 12b | Mg | 658 | 668 | 0.497 | 0.348 | 0.85 |
| Zn | 656 | 668 | 0.210 | 0.582 | 0.79 | |
| 12c | Mg | 657 | 664 | 0.268 | 0.215 | 0.48 |
| Zn | 655 | 664 | 0.080 | 0.255 | 0.34 | |
| 12d | Mg | 654 | 661 | 0.038 | 0.036 | 0.07 |
| Zn | 652 | 660 | 0.014 | 0.043 | 0.06 | |
| 12e | Zn | 655 | 665 | 0.005 | 0.021 | 0.03 |
| 12f | Mg | 652 | 659 | 0.245 | 0.126 | 0.37 |
| Zn | 650 | 666 | 0.165 | 0.286 | 0.45 | |
| 13 | Mg | 653 | 672 | 0.092 | 0.128 | 0.22 |
| Zn | 651 | 666 | 0.018 | 0.190 | 0.21 | |
| 14a | Mg | 655 | 669 | 0.007 | 0.043 | 0.05 |
| Zn | 654 | 666 | 0.002 | 0.107 | 0.11 | |
| 14f | Mg | 648 | 653 | 0.178 | 0.143 | 0.32 |
| Zn | 646 | 652 | 0.130 | 0.344 | 0.47 | |
| 15 | Mg | 650 | 655 | 0.011 | 0.056 | 0.07 |
| Zn | 649 | 654 | 0.002 | 0.094 | 0.10 | |
| 16 | Mg | 651 | 659 | 0.468 | 0.30 | 0.77 |
| Zn | 649 | 657 | 0.299 | 0.55 | 0.85 | |
| Amine | 1st | 2nd | Pyrazine-2,3-dicarbonitrile | 1st | 2nd | 3rd | 4th |
|---|---|---|---|---|---|---|---|
| n-Butylamine | 1.70 | — | Compound 1 | 1.75 | 1.98 | — | — |
| N,N-Diethylamine | 1.14 | — | 5-Diethylamino-6-phenylpyrazine-2,3-dicarbonitrile | 1.63 | 1.80 | 1.96 | — |
| Aniline | 0.98 | — | Compound 2 | 1.50 | — | — | — |
| Phenothiazine | 0.60 | 0.91 | Compound 4 | 1.05 | 1.51 | 1.68 | 1.90 |
Fluorescence and singlet oxygen
The studied TPyzPzs all emitted red fluorescence after excitation. The signal intensity, however, varied strongly depending on the peripheral substitution (see below). The shapes of the emission spectra were typical for this type of compounds (Fig. 2), with only a small Stokes shift observed in the range of ∼6–13 nm. The excitation spectra were also recorded and matched the absorption spectra perfectly (Fig. 2), thereby demonstrating that only monomers were present. This result confirmed that bulky tert-butylsulfanyl substituents on 9–14 and 16 (or N,N-diethylaminoethylsulfanyl substituents in 15) fully inhibited the aggregation. The subsequently discussed changes in the quantum yields can therefore not be attributed to an aggregation phenomenon. The fluorescence quantum yields (ΦF) were determined in THF using a comparative method with ZnPc as the reference compound. The ΦF values (Table 1) of the zinc complexes varied from 0.30 (for compounds without a donor or those in which the ICT or PET was inefficient) to 0.002 (in compounds with highly efficient ICT or PET). In magnesium complexes, ΦF varied from 0.508 to 0.007. The magnesium complexes were always characterized by ΦF values that were greater than those of the corresponding zinc complexes, which is consistent with a number of previous observations of TPyzPzs and is explained by the heavy-atom effect.17,21 The heavy-atom effect supposes that substitution with heavier atoms increases the probability of intersystem crossing and thus decreases the photon emission from the first excited state.22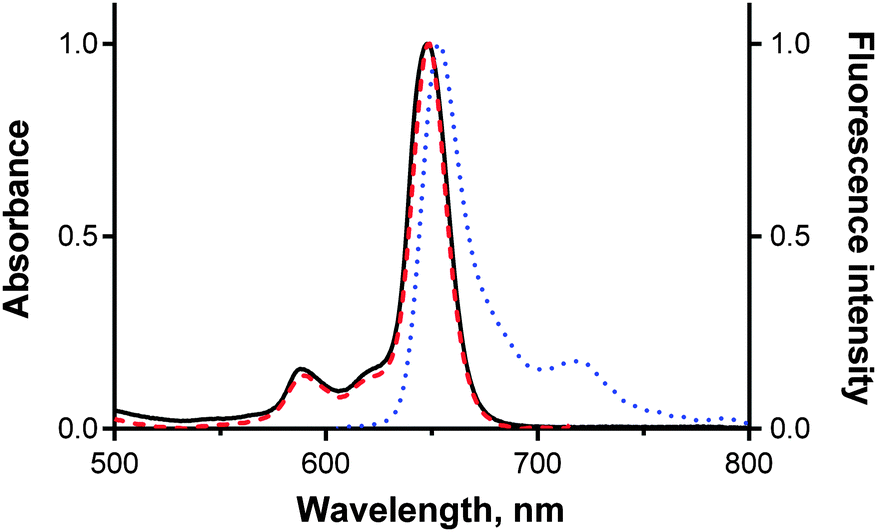 | ||
| Fig. 2 Normalized absorption (black, full line), fluorescence emission (blue, dotted line) and excitation (red, dashed line) spectra of compound 14fMg in THF. The spectra are typical for all studied TPyzPzs. Emission spectra were corrected for instrument response. | ||
Singlet oxygen production was determined via the chemical trapping of singlet oxygen by DPBF with ZnPc as the reference compound. Consistent with the heavy-atom effect, the ΦΔ values were greater for zinc complexes (0.647–0.021) than for the corresponding magnesium derivatives (0.356–0.036). The sum of ΦΔ + ΦF for both the zinc and magnesium complexes of one ligand yielded comparable values, indicating that similar competitive processes (PET or ICT) are responsible for the deactivation of the first excited states in both complexes. The only difference between the zinc and magnesium derivatives of the ligand was in the ratio between the intersystem crossing and photon emission, which is driven by the heavy-atom effect. Differences in the sum of ΦΔ + ΦF caused by the peripheral substituents are examined in detail in the Discussion section.
Electrochemical behavior
The electrochemistry of the studied amines and the corresponding pyrazine-2,3-dicarbonitriles was assessed via cyclic voltammetry in acetonitrile at room temperature with ferrocene as an internal standard. The experiments focused on the oxidation process to evaluate the electron-donating properties of the engaged nitrogen atoms. n-Butylamine, N,N-diethylamine, aniline, and phenothiazine underwent the first oxidation best expressed by the half-wave potentials E1/2 = 1.70, 1.14, 0.98, and 0.60 vs. SCE, respectively. A similar trend was observed in the corresponding series of pyrazine-2,3-dicarbonitriles (see Table 2). The process can be attributed solely to the oxidation of the attached nitrogens because 5-chloro-6-phenylpyrazine-2,3-dicarbonitrile—a similar compound without any donor amines—underwent no oxidation up to 2.8 V vs. SCE (see Fig. S6 in the ESI†). Chemically irreversible oxidations were observed in all cases except for phenothiazine and its corresponding pyrazine-2,3-dicarbonitrile 4, in which reversible oxidations occurred (Fig. 3 and Fig. S4 and S5 in the ESI†). Notably, the final TPyzPzs showed evidence of corresponding oxidation processes, although the appropriate half-wave potentials could not be properly determined. Nevertheless, the relationship observed in the amine series and the pyrazine-2,3-dicarbonitriles suggests that the oxidation feasibility of the peripheral donor nitrogens in TPyzPzs increases in the order 12b < 12a < 12c < 12d.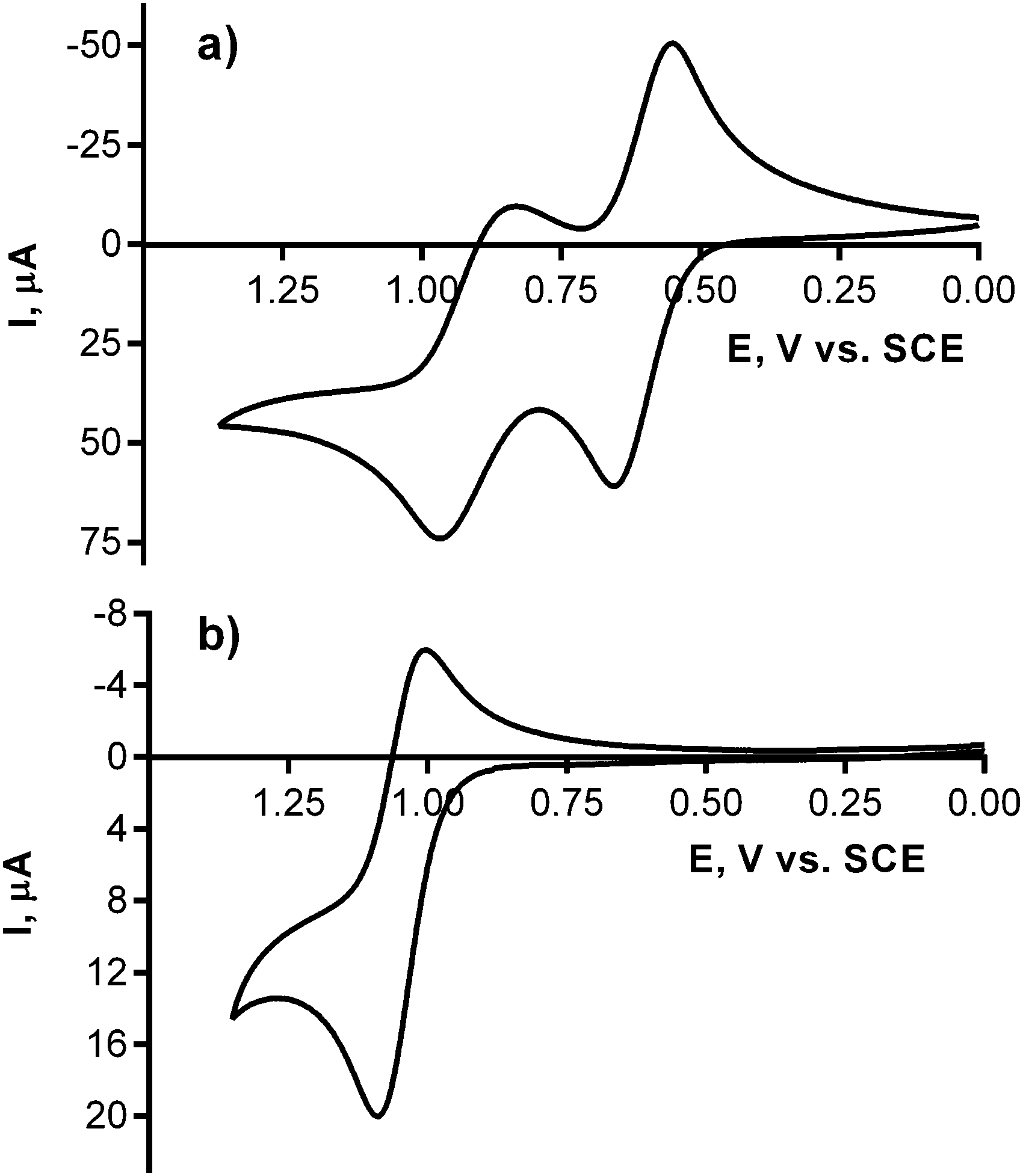 | ||
| Fig. 3 Cyclic voltammogram (acetonitrile, 100 mV s−1, 0.1 M tetrabutylammonium hexafluorophosphate, 25 °C) of phenothiazine (a) and corresponding pyrazin-2,3-dicarbonitrile 4 (b, only first oxidation presented). | ||
Discussion
A systematic observation of the low ΦΔ and ΦF values in TPyzPzs with alkylamino peripheral substituents led to a thorough investigation of this problem. Using a combination of experiments, including changes in the solvent polarity and acidity, cyclic voltammetry measurements, steady-state and time-resolved emission spectra, and transient absorption studies, we subsequently confirmed that ICT is the major deactivating pathway for the excited states in such derivatives (e.g. in compound 13 that was previously studied in detail).13 ICT was found to be competitive to both fluorescence and intersystem crossing. Quenching of the TPyzPz (or Pc) triplet states by oxygen in organic solvents (and in saturated oxygen) typically approaches 100%; therefore, the ΦΔ values can serve as indicators of intersystem crossing.23 The sum of ΦΔ and ΦF can thus quickly determine the ICT efficiency and can be used to compare a series of similar compounds.Little attention has been previously paid to the structural factors that can influence ICT. Substituents on or nearby the donor center were considered to have only a marginal effect, and research efforts were primarily aimed at determining the recognition properties of the donor moiety in sensing applications.15 If differences were observed, limited details were provided, with no further explanation.14 Hence, this study is focused on deeper investigation and discussion on some structural modifications in close proximity to the donor center. The study can be divided into three particular goals: (a) substituents in a position ortho to the donor center, (b) substituents on the donor center, (c) the influence of a conjugated × non-conjugated linker between the donor and the acceptor (i.e., a comparison between ICT and PET). TPyzPzs 9–15 were synthesized, and their photophysical properties were determined and compared in a selected series with compound 16, which does not contain any donor center. Both the magnesium and zinc complexes with differing ΦΔ-to-ΦF ratios were investigated. As documented in Table 1 and Fig. 4, the trends in ICT efficiency (expressed as the sum of ΦΔ and ΦF) were similar for both complexes.
 | ||
| Fig. 4 Sum of singlet oxygen quantum yield (ΦΔ, striped parts of the columns) and fluorescence quantum yield (ΦF, full parts of the columns) of studied TPyzPzs grouped according to the studied phenomenon: (a) effect of the ortho substituent, (b) effect of donor strength, (c) comparison of PET and ICT. Horizontal dotted line represents a mean (from Mg and Zn complex) value of the sum ΦΔ + ΦF for control complex 16 that has no donor center. | ||
The effect of a substituent in the ortho position has been recently described and used to develop new cation sensors without any serious discussion.14 As indicated in Fig. 4a, the presence of Cl (9Mg, 9Zn), CH3 (10aMg, 10aZn) or hydrogen (11Mg, 11Zn) ortho to the donor (the N,N-diethylamino substituent) eliminates any ICT, and the sum of ΦΔ and ΦF is fully comparable with compounds without any donor center (16Mg, 16Zn). The ICT is slightly less deactivated with phenyl (12aMg, 12aZn) as the ortho substituent. The strongest ICT is observed with alkylsulfanyl (13Mg, 13Zn) or alkoxy (14aMg, 14aZn) substituents in the ortho position. These effects were correlated with the steric or electronic effects (Hammett substituent constants σp and σm, see ESI†); however, these correlations were consistently tenuous and provided insufficient answers. The rationale behind this strong effect of the ortho substituent remains unexplained and, for future use, must only be taken empirically. However, the effect is clearly connected with the close proximity of the substituent to the donor. A strong ICT efficiency was achieved when a linker (benzene ring, 12eZn) was inserted between the donor and the acceptor (compare ΦΔ + ΦF (12eZn) = 0.03 with ΦΔ + ΦF (12aZn) = 0.75).
For further study, a substituent was fixed in the ortho position, and the substituents on the donor were altered by increasing their electron-donating properties (12a-dMg, 12a-dZn). Phenyl, as an ortho substituent, was selected due to its moderate deactivating effect on the ICT (see previous discussion), which allows an evaluation of the potentially positive effects of the electron-donating substituents on the donor. The electron-donating properties were estimated based on the electrochemical measurements of the ease of oxidation. The electron-donating properties of the amines alone or when attached as substituents on the pyrazine-2,3-dicarbonitrile increase in the order n-butylamine < N,N-diethylamine < aniline < phenothiazine, as deduced from the half-wave oxidation potentials in Table 2. This series correlates well with the ICT efficiency for compounds 12a-dMg and 12a-dZn (Fig. 4b). Therefore, the substituents on the donor clearly play an important role, and sufficiently strong donors (e.g., phenothiazine in 12dZn and 12dMg) can suppress the effect of a substituent in the ortho position (see previous discussion).
After the excitation, the charge transfer state is formed in ICT when the donor and the acceptor are in conjugation; however, the radical anion and the radical cation are formed in PET (no conjugation between the donor and the acceptor). A comparison of the efficiencies of these two processes in TPyzPzs is interesting because both principles have been recognized in other classes of molecules for the development of a number of sensors or activable compounds.5,24,25 In this study, PET was demonstrated in compounds with one N,N-diethylaminoethylsulfanyl substituent (10fMg, 10fZn, 12fMg, 12fZn, 14fMg, or 14fZn) or with eight such substituents (15Mg, 15Zn). The first important observation is that a substituent in the ortho position has no effect on the PET efficiency. All compounds from the 10f, 12f, 14f series with one donor center were characterized by a similar sum of ΦΔ and ΦF (approximately half the value of a compound without any donor—16Mg, 16Zn) irrespective of the adjacent substituent (Fig. 4c). Apparently, the longer distance eliminates the “ortho” effect. However, the overall PET efficiency is significantly below that of ICT, as demonstrated by a comparison with compound 12eZn, in which the donor (in conjugation) is approximately at the same distance from the acceptor and the “ortho effect” is limited (Fig. 4c). The sum of ΦΔ and ΦF for the compound 12eZn is approximately one order of magnitude less than those for compounds 10fMg, 10fZn, 12fMg, 12fZn, 14fMg, and 14fZn with PET. An intensification of the PET12 or ICT13 efficiency with an increasing number of donor centers has been demonstrated in the literature. Thus, the decrease in the ΦΔ and ΦF values in compounds 15Mg and 15Zn substituted with eight donor centers for PET was unsurprising. However, the interesting fact is that only the presence of eight donor centers in these compounds brought the PET efficiency closer to that of the ICT in 12eZn with one donor center.
Another experiment was performed to demonstrate the key role played by the lone pair on the donor nitrogen in the deactivation process of the PET in TPyzPzs. The fluorescence quantum yield of compound 14fZn in THF is 0.13. The addition of a small quantity of sulfuric acid (0.1% v/v) into this solution increased ΦF to 0.23, a value comparable to that of compound 16Zn with no donor center. An increase of the sulfuric acid concentration to 1% (v/v) or 2% (v/v) did not change the fluorescence intensity further, which indicates that the donor center was fully protonated after the first addition. A similar recovery of photophysical properties was noted for ICT in TPyzPzs15,26 or for PET in Pc after protonation of the donor.27–29
Conclusions
This work focused on the structural factors that influence the ICT or PET in TPyzPzs. A logically designed series of TPyzPzs containing primarily one donor center was synthesized to assess the influence of substitution on the efficiency of the deactivation processes. Several factors were recognized. A substituent in the position ortho to the donor center plays an important role in ICT for reasons that have heretofore not been fully revealed. Chloro, methyl, hydrogen, or phenyl substituents were found to inhibit efficient ICT processes, whereas alkylsulfanyl or alkoxy substituents supported the ICT. The effect of an ortho substituent can be eliminated through the introduction of a linker between the TPyzPz macrocycle and the donor. Strong electron donors can also suppress the “ortho effect.” The ICT efficiency depends significantly on the electron-donating properties of the donor, which may easily be characterized using its oxidation potential. Phenothiazine-10-yl was the strongest donor in the studied series. Conjugation of the donor with an acceptor (i.e., ICT) leads to a more efficient deactivation of the excited states in TPyzPzs than in the case of an aliphatic linker between the donor and the acceptor (i.e., PET). These factors should be further considered for the design of new fluorescence sensors based on the ON–OFF switching in TPyzPzs.Acknowledgements
The work was supported by the Czech Science Foundation (P207-11-1200). The authors would like to thank Jiří Kuneš and Zdeněk Kučera for NMR measurements.Notes and references
- M. E. Daraio, P. F. Aramendía and E. San Román, Chem. Phys. Lett., 1996, 250, 203–208 CrossRef CAS.
- M. E. Daraio, A. Völker, P. F. Aramendía and E. San Román, Langmuir, 1996, 12, 2932–2938 CrossRef CAS.
- I. Bruseghini, L. Fabbrizzi, M. Licchelli and A. Taglietti, Chem. Commun., 2002, 1348–1349 RSC.
- X. F. Zhang, J. Fluoresc., 2011, 21, 1559–1564 CrossRef CAS PubMed.
- Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104–109 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- P. Zimcik, M. Miletin, H. Radilova, V. Novakova, K. Kopecky, J. Svec and E. Rudolf, Photochem. Photobiol., 2010, 86, 168–175 CrossRef CAS PubMed.
- I. Manet, F. Manoli, M. P. Donzello, E. Viola, A. Masi, G. Andreano, G. Ricciardi, A. Rosa, L. Cellai, C. Ercolani and S. Monti, Inorg. Chem., 2013, 52, 321–328 CrossRef CAS PubMed.
- M. P. Donzello, D. Vittori, E. Viola, I. Manet, L. Mannina, L. Cellai, S. Monti and C. Ercolani, Inorg. Chem., 2011, 50, 7391–7402 CrossRef CAS PubMed.
- M. P. Donzello, Z. Ou, F. Monacelli, G. Ricciardi, C. Rizzoli, C. Ercolani and K. M. Kadish, Inorg. Chem., 2004, 43, 8626–8636 CrossRef CAS PubMed.
- M. P. Donzello, Z. Ou, D. Dini, M. Meneghetti, C. Ercolani and K. M. Kadish, Inorg. Chem., 2004, 43, 8637–8648 CrossRef CAS PubMed.
- P. Zimcik, M. Miletin, Z. Musil, K. Kopecky, L. Kubza and D. Brault, J. Photochem. Photobiol., A, 2006, 183, 59–69 CrossRef CAS PubMed.
- V. Novakova, P. Zimcik, M. Miletin, L. Vachova, K. Kopecky, K. Lang, P. Chábera and T. Polívka, Phys. Chem. Chem. Phys., 2010, 12, 2555–2563 RSC.
- V. Novakova, L. Lochman, I. Zajícová, K. Kopecky, M. Miletin, K. Lang, K. Kirakci and P. Zimcik, Chem.–Eur. J., 2013, 19, 5025–5028 CrossRef CAS PubMed.
- V. Novakova, M. Miletin, K. Kopecky and P. Zimcik, Chem.–Eur. J., 2011, 17, 14273–14282 CrossRef CAS PubMed.
- L. Vachova, V. Novakova, K. Kopecky, M. Miletina and P. Zimcik, Dalton Trans., 2012, 41, 11651–11656 RSC.
- P. Zimcik, V. Novakova, K. Kopecky, M. Miletin, R. Z. Uslu Kobak, E. Svandrlikova, L. Váchová and K. Lang, Inorg. Chem., 2012, 51, 4215–4223 CrossRef CAS PubMed.
- Z. Musil, P. Zimcik, M. Miletin, K. Kopecky, M. Link, P. Petrik and J. Schwarz, J. Porphyrins Phthalocyanines, 2006, 10, 122–131 CrossRef CAS.
- L. Kaestner, M. Cesson, K. Kassab, T. Christensen, P. D. Edminson, M. J. Cook, I. Chambrier and G. Jori, Photochem. Photobiol. Sci., 2003, 2, 660–667 CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- A. Tuhl, S. Makhseed, P. Zimcik, N. Al-Awadi, V. Novakova and J. Samuel, J. Porphyrins Phthalocyanines, 2012, 16, 817–825 CrossRef CAS.
- K. N. Solovyov and E. A. Borisevich, Phys.-Usp., 2005, 48, 231–253 CrossRef CAS PubMed.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS.
- H. He, J. Y. Liu and D. K. P. Ng, J. Porphyrins Phthalocyanines, 2013, 17, 99–103 CrossRef CAS.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- V. Novakova, E. H. Mørkved, M. Miletin and P. Zimcik, J. Porphyrins Phthalocyanines, 2010, 14, 582–591 CrossRef CAS.
- X. J. Jiang, P. C. Lo, S. L. Yeung, W. P. Fong and D. K. P. Ng, Chem. Commun., 2010, 46, 3188–3190 RSC.
- X. J. Jiang, P. C. Lo, Y. M. Tsang, S. L. Yeung, W. P. Fong and D. K. P. Ng, Chem.–Eur. J., 2010, 16, 4777–4783 CrossRef CAS PubMed.
- X. J. Jiang, S. L. Yeung, P. C. Lo, W. P. Fong and D. K. P. Ng, J. Med. Chem., 2011, 54, 320–330 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of studied compounds, correlations between Hammett substituent constants and photophysical parameters, cyclic voltammograms. See DOI: 10.1039/c3cp54731k |
| This journal is © the Owner Societies 2014 |