 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A combined experimental and theoretical study of reactions between the hydroxyl radical and oxygenated hydrocarbons relevant to astrochemical environments
R. J.
Shannon
a,
R. L.
Caravan
a,
M. A.
Blitz
ab and
D. E.
Heard
*ab
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: d.e.Heard@leeds.ac.uk
bNational Centre for Atmospheric Science, University of Leeds, Leeds, LS2 9JT, UK
First published on 2nd January 2014
Abstract
The kinetics of the reactions of the hydroxyl radical (OH) with acetone and dimethyl ether (DME) have been studied between 63–148 K and at a range of pressures using laser-flash photolysis coupled with laser induced fluorescence detection of OH in a pulsed Laval nozzle apparatus. For acetone, a large negative temperature dependence was observed, with the rate coefficient increasing from k1 = (1.6 ± 0.8) × 10−12 cm3 molecule−1 s−1 at 148 K to (1.0 ± 0.1) × 10−10 cm3 molecule−1 s−1 at 79 K, and also increasing with pressure. For DME, a similar behaviour was found, with the rate coefficient increasing from k2 = (3.1 ± 0.5) × 10−12 cm3 molecule−1 s−1 at 138 K to (1.7 ± 0.1) × 10−11 cm3 molecule−1 s−1 at 63 K, and also increasing with pressure. The temperature and pressure dependence of the experimental rate coefficients are rationalised for both reactions by the formation and subsequent stabilisation of a hydrogen bonded complex, with a non-zero rate coefficient extrapolated to zero pressure supportive of quantum mechanical tunnelling on the timescale of the experiments leading to products. In the case of DME, experiments performed in the presence of O2 provide additional evidence that the yield of the CH3OCH2 abstraction product, which can recycle OH in the presence of O2, is ≥50%. The experimental data are modelled using the MESMER (Master Equation Solver for Multi Energy Well Reactions) code which includes a treatment of quantum mechanical tunnelling, and uses energies and structures of transition states and complexes calculated by ab initio methods. Good agreement is seen between experiment and theory, with MESMER being able to reproduce for both reactions the temperature behaviour between ∼70–800 K and the pressure dependence observed at ∼80 K. At the limit of zero pressure, the model predicts a rate coefficient of ∼10−11 cm3 molecule−1 s−1 for the reaction of OH with acetone at 20 K, providing evidence that the reaction can proceed quickly in those regions of space where both species have been observed. The results and modelling build considerably on our previous experimental study performed under a much more limited range of conditions (Shannon et al., Phys. Chem. Chem. Phys., 2010, 12, 13511–13514).
1. Introduction
Rate coefficients are experimentally challenging to measure below ∼200 K owing to the condensation of at least one of the reagents at the walls of the reaction chamber, and Laval nozzle techniques have enjoyed considerable success for kinetic studies in this temperature regime.2–9 It is firmly established that many neutral–neutral reactions are fast at very low temperatures with some exhibiting rate coefficients close to the gas kinetic limit. The majority of experimental studies have involved barrierless reactions which possess an attractive potential energy surface for the approach of the reagents.10 The rate coefficients for such reactions are also normally significant at room temperature11 and typically display a weak increase in the rate coefficient as the temperature is lowered. However, recent work by Shannon et al. have shown that another class of reaction with an overall barrier to reaction can also be fast at low temperatures.1,5 This class of reaction involves hydrogen atom abstraction from small oxygenated volatile organic molecules (OVOCs) by the OH radical, with the formation of a hydrogen-bonded complex prior to abstraction facilitating a large increase in the reaction rate coefficients at low temperatures. In a previous publication,1 we reported the low temperature kinetic behaviour of two such systems, namely the reactions of OH with acetone and dimethyl ether (DME), but under a very limited range of low temperatures, and at a single pressure.Both of these reactions are important in combustion systems and in the Earth's atmosphere, with DME having been proposed as a potential diesel substitute.12 In addition, as shown in our earlier work for these reactions and for methyl ethyl ketone1 and also for methanol,5 reactions between the OH radical and OVOCs are potentially important in very low temperature extra-terrestrial environments such as star-forming regions and the interstellar medium. OH and several OVOCs have been observed by astronomers in cold, dense molecular clouds13 In particular OH, acetone and DME have all been observed in the Sagittarius B2 molecular cloud.14–16 Unfortunately, very few rate coefficients for reactions of OH with OVOCs have been measured below 200 K, and consequently the kinetic databases used for astrochemical modelling contain very little information about this type of reaction.17
Owing to their importance in both atmospheric and combustions systems, rate coefficients for the hydrogen-atom abstraction reactions of OH with both acetone (R1) and DME (R2) have been measured by a large number of groups at temperatures above 200 K,
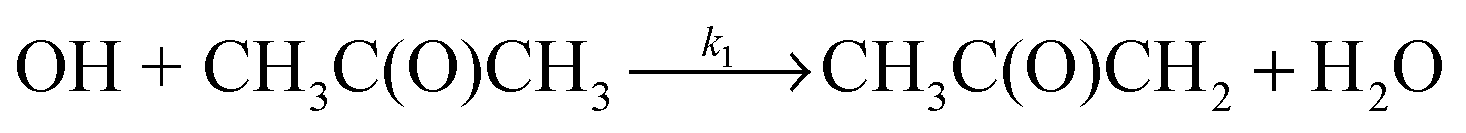 | (R1) |
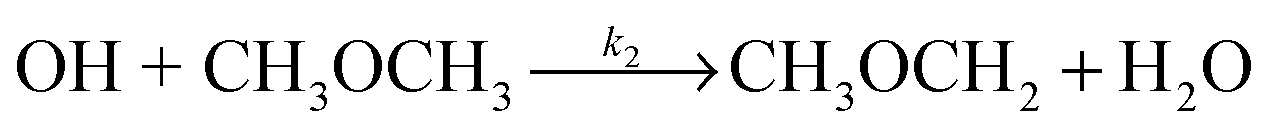 | (R2) |
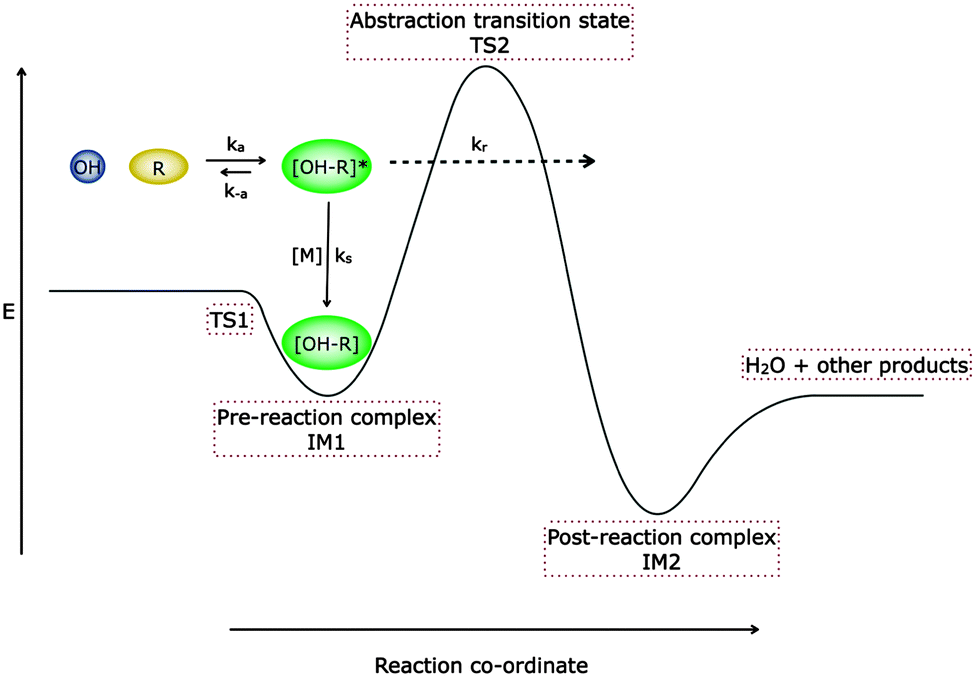 | ||
| Fig. 1 Schematic potential energy surface demonstrating the two transition state model of Greenwald et al.18,19 The potential energy surface represents the formation of a hydrogen bonded complex (IM1) with an associated microscopic rate coefficient, ka. The complex can either be stabilised (with a corresponding microscopic rate coefficient of ks[M] where [M] is the total gas density), re-dissociate back to reactants (with microscopic rate coefficient k−a), or form abstraction products via a post-reaction complex IM2 (with microscopic rate coefficient kr), either by surmounting or tunnelling through the barrier to reaction (TS2). | ||
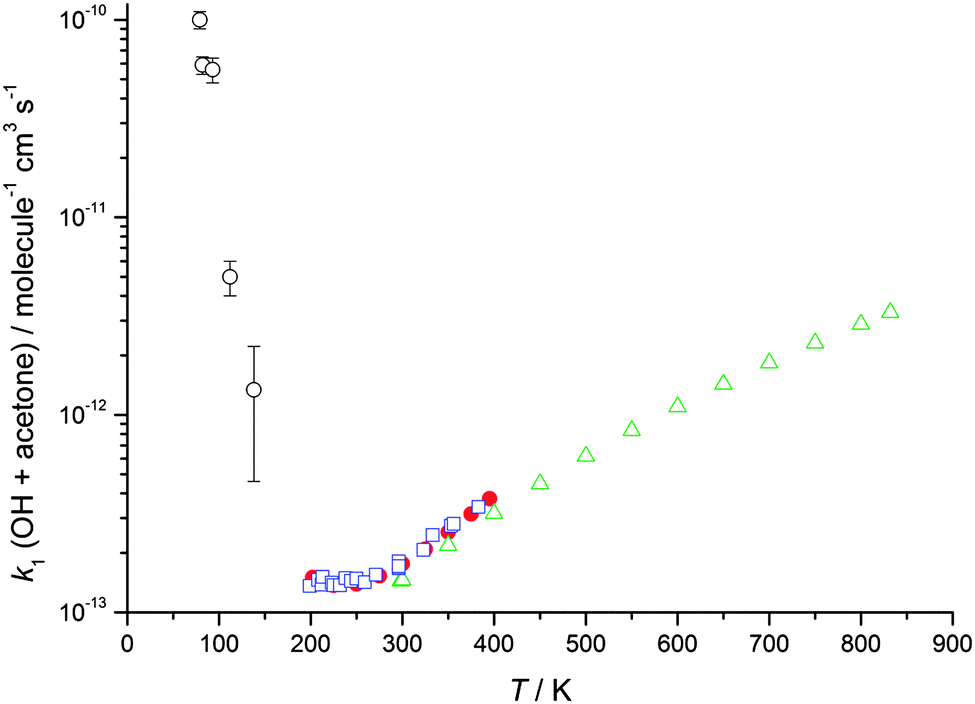 | ||
| Fig. 2 The temperature dependence of the experimentally determined rate coefficients for the reaction of OH with acetone. Open green triangles: Yamada et al.39 Filled red circles: Wollenhaupt et al.52 Open blue squares: Gierczak et al.53 Open black circles: this work (total density in N2 ranging from (5.1–17) × 1016 molecule cm−3). Error bars as for Table 1. | ||
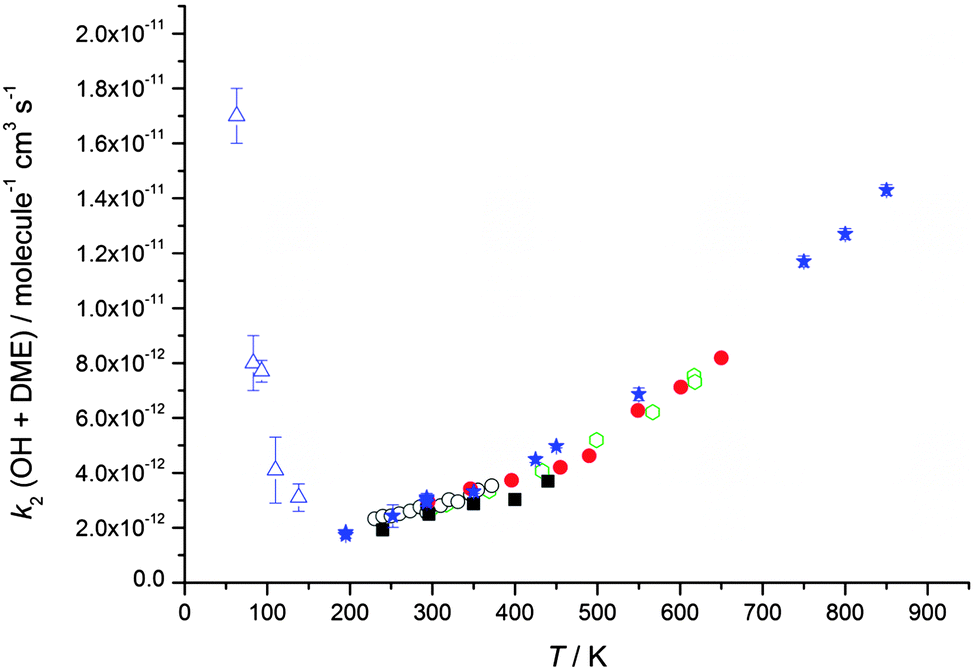 | ||
| Fig. 3 Temperature dependence of the experimentally determined rate coefficients for the reaction of OH with dimethyl ether (DME). Green open hexagons: Bonard et al.49 Red filled circles: Arif et al.48 Black open circles: Mellouki et al.47 Black filled squares: Wallington et al.54 Blue filled stars: Carr et al.32 Blue open triangles: this work (total density in N2 ranging from (3.4–9.1) × 1016 molecule cm−3). Error bars as for Table 2. | ||
In this paper, we report rate coefficients for the reactions of OH with acetone and DME determined over a much wider range of conditions compared with our previous study1 in order to further probe the role of the pre-reaction complex as well as quantum mechanical tunnelling.23 The extent of deviation from Arrhenius behaviour as well as the contribution of the two mechanisms suggested by Caralp et al.20 and Wallington and Kurylo21 to the overall rate coefficient are investigated through a series of kinetic measurements as a function of temperature and pressure using a pulsed Laval nozzle apparatus, and modelling using the MESMER (Master Equation Solver for Multi Energy Well Reactions) package.24
2. Experimental procedure
The Leeds pulsed Laval nozzle apparatus has been described in detail previously,5,25 and only a brief description will be given here. The reaction mixture consisting of the radical precursor, the reagent and the bath gas (N2) is admitted into a 1 cm3 stainless steel reservoir via two pulsed valves (Parker series 9) fired at 5 Hz repetition frequency with a pulse duration of ∼10 ms. This gas mixture is then expanded through a convergent–divergent Laval nozzle into a stainless steel reaction chamber held at low pressure. The reaction chamber is pumped by a Leybold RUVAC 251 roots pump backed by a Leybold D65B rotary pump and the pressure is recorded by two capacitance manometers (Leybold CTR 90 0–1000 Torr and 0–10 Torr). The pumping system is throttled with a specially adapted gate valve to allow control of the chamber pressure to ±0.01 Torr.Various mixtures of nitrogen (BOC, OFN, >99.998%), acetone (VWR, >99%), DME (Aldrich >99.9%), acetone-d6 (Eur-isotop, >99.8%), t-butyl-hydroperoxide ((CH3)3OOH = t-BuOOH, Aldrich, 70% wt aqueous solution) or oxygen (BOC, analytical grade) were flowed in to a 1 L stainless steel ballast tank via a set of calibrated mass flow controllers (MKS instruments) to enable thorough mixing. For experiments where t-BuOOH was used as the OH precursor, nitrogen was passed through a bubbler containing liquid t-BuOOH which was then entrained in the gas flow. Acetone, DME or acetone-d6 were purified using freeze–pump–thaw cycles on a vacuum line. The vapour pressures of acetone, DME or acetone-d6 were then admitted to an empty cylinder and were diluted with nitrogen and left overnight prior to use to ensure full mixing.
The temperature, density and pressure within the Laval nozzle expansion are all related to their stagnation, pre-expansion values by the Mach number, M. The stagnation pressure Po in the pre-expansion region of the reservoir and the impact pressure Pi were both measured, and are related to M by:25
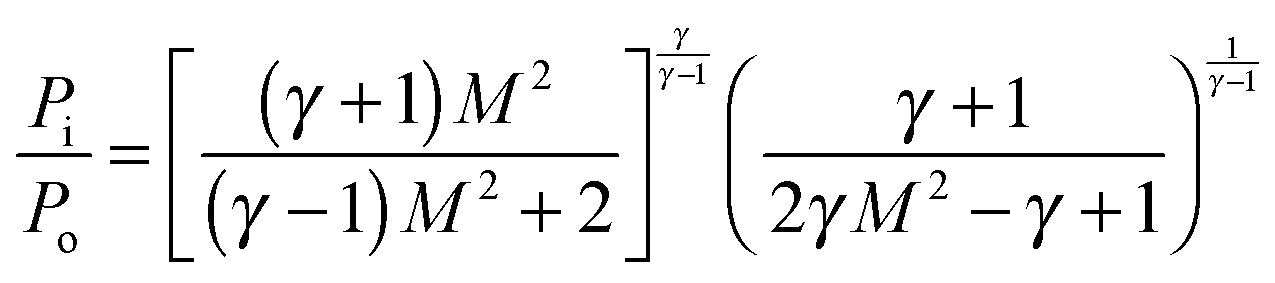 | (E1) |
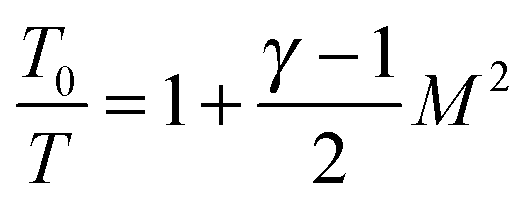 | (E2) |
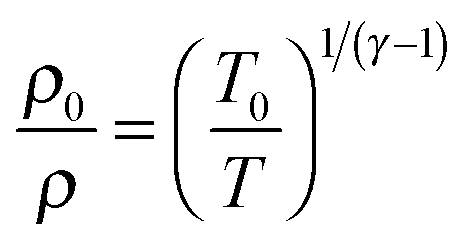 | (E3) |
OH radicals were generated using two methods. For the reaction of OH with acetone, the OH radicals were usually produced via 248 nm photolysis of acetone itself using a KrF excimer laser (Lambda Physik LPX200, 5 Hz) in the presence of oxygen:
| CH3C(O)CH3 + hν → CH3CO + CH3 | (R3) |
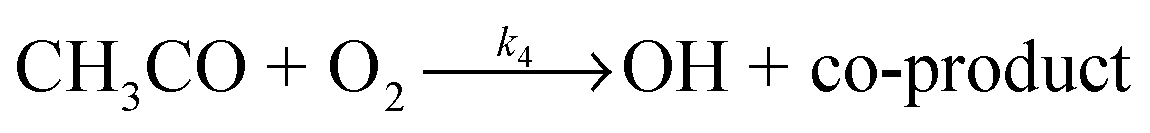 | (R4) |
| (CH3)3COOH + hν → (CH3)3CO + OH | (R5) |
When the reaction of the acetyl radical with O2(R4) was used as the source of OH, the temporal profile of the OH concentration, which is proportional to the fluorescence intensity, is given by:
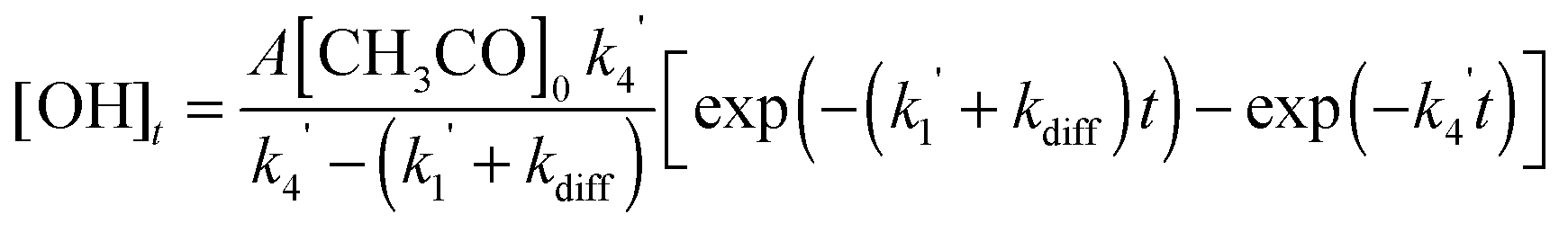 | (E4) |
The OH temporal profiles using t-BuOOH as the OH precursor were described well by a single exponential decay, and the following function was fitted to the data:
| [OH] = [OH]10 exp(−kX′·t) | (E5) |
3. Theoretical calculations
Rate coefficients were calculated using the energy grained master equation solver MESMER (Master Equation Solver for Multi Energy well Reactions).24 This package is an open source program which solves the chemical master equation for a reaction system in order to obtain phenomenological rate coefficients using an eigenvector/eigenvalue method. Given a potential energy surface with an arbitrary number of intermediates, the coupled differential equations describing both reactive and energy transfer processes are solved allowing the role of the pre-reaction complex to be considered explicitly. In this approach, the phase space of each isomer is lumped into energy grains of a set size and the time evolution of the grain populations are then calculated individually in order to obtain the time evolution for the concentration of each species. A grain size of 50 cm−1 was used, which is sufficiently small for convergence of the calculated rate coefficients above 60 K, although a smaller grain size of 10 cm−1 was used to ensure convergence below 60 K.Isoenergetic reaction rates between grains of different isomers are calculated using RRKM theory, whilst energy transfer between grains of the same species is modelled using an exponential down model parameterised by an energy independent 〈ΔEdown〉, the average energy transferred in a downward direction upon collision with a bath gas molecule. Once both reactive and collisional transition probabilities have been obtained from the RRKM and exponential down model expressions, the evolution of the grain populations is given by the matrix equation:24
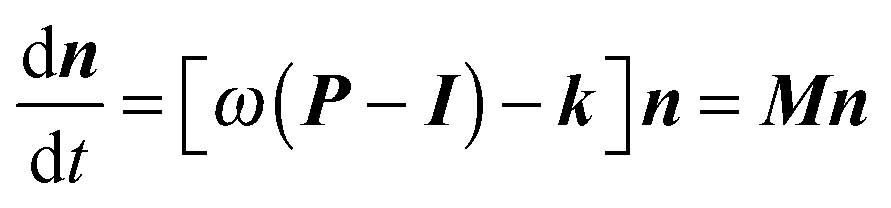 | (E6) |
 | (E7) |
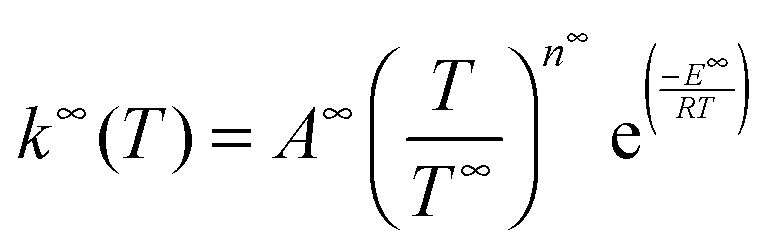 | (E8) |
For both the reactions of OH with acetone and DME there are already a number of potential energy surfaces in the literature and new surfaces have therefore not been calculated in the current work. For the reaction of OH with acetone, the surface used by Caralp et al.20 was employed, based on calculations by Henon et al.31 at the MP2/6-31G**//CCSD/6-311G** level of theory. For the reaction of OH with DME, the recent surface calculated by Carr et al.32 was employed, which used a full coupled cluster with complete basis set extrapolation FCC/CBS//CCSD/AVDZ level of theory. Further details regarding the optimisations for the geometric parameters and energies of the reactants, complexes, transition states and products are given in Carr et al.32
The calculated surfaces for both of these reactions identify more than one saddle point corresponding to transition states (TS2) for hydrogen abstraction. For the reaction of OH with DME,32 it was identified that these three separate saddle points were in fact different conformers of one distinct transition state, related by hindered internal rotations. As the emphasis of this work is upon the low temperature behaviour of these reactions, a potential energy surface consisting of a pre-reaction well with one transition state was used for both reactions, with the TS corresponding to the lowest energy ab initio calculated saddle point. In these calculations the final reaction step from IM2, see Fig. 1, to bimolecular products was not considered, with the reaction between IM1 and IM2 being treated as irreversible. It was found that this approximation introduced an error of less than 1% over the temperature range of our simulations.32
In order to calculate the molecular sums and densities of states, rigid rotor and harmonic oscillator approximations were used for all rotations and most vibrational frequencies, respectively. However, there are a number of low frequency vibrational normal modes present in many of the species of both PESs which are better described as hindered or free internal rotations. Hindrance potentials for these modes were calculated using the Gaussian 03 suite of ab initio software33 at the MPW1K/6-31+G(2d,2p) level of theory. The rotational potential was then calculated by incrementing the dihedral angle corresponding to rotation about 360° and at each increment a constrained geometry optimisation was performed. Some of the relaxed scans which incorporated the long-range hydrogen bonded interaction between the OH and the acetone failed to calculate potentials about a full 360°. Relaxed potentials were instead approximated for these modes through calculating a rigid potential, and then performing a constrained geometry optimisation at the value of the dihedral angle corresponding to the barrier to rotation. The rest of the potential was then scaled by the difference between rigid and relaxed barriers. For each hindered rotational mode a vector corresponding to the torsional coordinates was obtained and such vectors were projected from the Hessian for each species according to the methods described by Sharma et al.34 The resulting frequencies, obtained upon diagonalisation of the projected Hessian are then orthogonal to the torsional motions.
As these reactions involve a hydrogen transfer, tunnelling effects are expected to be significant, particularly at low temperatures. Tunnelling transmission coefficients were calculated and the RRKM rate coefficients correspondingly corrected. Tunnelling was treated via two methods, using the imaginary frequency of the transition state calculated from an Eckart potential according to the method of Miller,35 and using the semi classical Wentzel–Kramers–Brillouin (WKB) method.36 For the latter method, the change in potential energy along the entire reaction path was required and this was obtained by performing an intrinsic reaction co-ordinate (IRC) calculation at the MPW1K/6-31+G(2d,2p) level of theory.33 The IRC was subsequently refined through performing projected frequency calculations to obtain the zero point energy at a number of points along the potential and CCSD(T)/aug-cc-pVDZ calculations were also performed to obtain higher level electronic energies at these points. This approach together with a WKB tunnelling method gave good agreement with experiment in a previous study of the reaction of OH with acetone.1
The purpose of the modelling is to complement and fit the experimental results, rather than to attempt to calculate the rate coefficients a priori. Given the large amount of experimental data available both from the current work and in previous studies, a number of parameters were varied in order to fit the experimental data through minimising the goodness of fit parameter χ2:
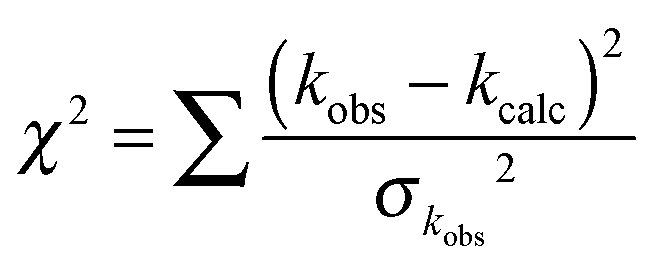 | (E9) |
4. Results
4.1. Measurement of rate coefficients
Rate coefficients for the reactions of OH with acetone and DME were experimentally measured for temperatures between 63 and 148 K and for total gas densities ranging from 3.2 × 1016 to 2.6 × 1017 molecule cm−3, and are listed in Tables 1 and 2 for acetone and DME, respectively. For acetone, the rate coefficients are plotted as a function of temperature and total density (for 80 and 146 K) in Fig. 2 and 4, respectively, with the corresponding data for DME plotted in Fig. 3 and 5 respectively.| Temperature/K | OH/OD precursor | Flow density/1016 molecule cm−3 | Rate coefficient, k1/molecule−1 cm3 s−1 |
|---|---|---|---|
| a Data presented in Fig. 2. b Data previously published in Shannon et al.1 c Reaction under study is OD + CD3COCD3 → CD3COCD2 + D2O. | |||
| 79 ± 3 | Acetyl/O2 | 17 ± 2 | (1.0 ± 0.1) × 10−10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
| 82 ± 4 | Acetyl/O2 | 3.2 ± 0.6 | (3.7 ± 0.4) × 10−11 |
| 82 ± 4 | Acetyl/O2 | 5.1 ± 0.5 | (5.9 ± 0.6) × 10−11a |
| 86 ± 3 | Acetyl/O2 | 6.5 ± 0.6 | (5.6 ± 0.6) × 10−11b |
| 83 ± 3 | Acetyl/O2 | 8.9 ± 0.6 | (7.6 ± 0.8) × 10−11 |
| 81 ± 4 | Acetyl/O2 | 13 ± 2 | (8.9 ± 0.9) × 10−11 |
| 93 ± 4 | Acetyl/O2 | 6.5 ± 0.5 | (5.6 ± 0.8) × 10−11a |
| 112 ± 9 | Acetyl/O2 | 6 ± 1 | (5 ± 1) × 10−12a,b |
| 138 ± 9 | t-BuOOH | 8 ± 1 | (1.3 ± 0.9) × 10−12a |
| 133 ± 10 | t-BuOOH | 13 ± 3 | (2.8 ± 0.6) × 10−12 |
| 148 ± 15 | t-BuOOH | 10 ± 1 | (1.6 ± 0.8) × 10−12 |
| 143 ± 15 | t-BuOOH | 18 ± 2 | (2.4 ± 1.1) × 10−12 |
| 146 ± 15 | t-BuOOH | 26 ± 4 | (3.5 ± 0.7) × 10−12 |
| 146 ± 15 | Acetyl/O2 | 26 ± 4 | (3 ± 1) × 10−12 |
| 93 ± 4 | d-Acetyl/O2 | 6.5 ± 0.5 | (6.8 ± 0.7) × 10−11c |
| 146 ± 15 | d-Acetyl/O2 | 26 ± 4 | (1.4 ± 1.3) × 10−12c |
| Temperature/K | Flow density/1016 molecule cm−3 | Rate coefficient, k2/molecule−1 cm3 s−1 |
|---|---|---|
| a Data presented in Fig. 3. b Data previously published in Shannon et al.1 | ||
| 63 ± 2 | 3.4 ± 0.3 | (1.7 ± 0.1) × 10−11a |
| 83 ± 3 | 6.1 ± 0.5 | (8 ± 1) × 10−12a,b |
| 81 ± 4 | 13 ± 2 | (1.6 ± 0.3) × 10−11 |
| 83 ± 3 | 8.9 ± 0.6 | (9 ± 1) × 10−12 |
| 79 ± 3 | 17 ± 2 | (1.9 ± 0.2) × 10−11 |
| 93 ± 4 | 6.5 ± 0.5 | (7.7 ± 0.4) × 10−12a,b |
| 110 ± 8 | 9.1 ± 1.4 | (4.1 ± 1.2) × 10−12a |
| 138 ± 9 | 8 ± 1 | (3.1 ± 0.5) × 10−12a |
| 133 ± 10 | 13 ± 3 | (3.0 ± 0.4) × 10−12 |
The results clearly demonstrate a dramatic, inverse temperature dependence of the rate coefficients of both reactions for temperatures below 200 K, consistent with our previous, initial study over a smaller range of temperature (86–112 K for acetone, 93–112 K for DME, both at a single density).23 We attribute the enhancement in the rate coefficients for OH loss to the presence of the pre-reaction complex (IM1) which is calculated to have a significant binding energy for both systems.31,32,39,40 We postulate that this complex enhances the rate coefficient either through providing a termolecular stabilisation channel or through enhancing the bimolecular abstraction channel by quantum mechanical tunnelling at low temperatures.23 The data recorded in this study over a wider range of temperature and pressure will enable the contribution from each mechanism to be investigated, which we did not report in our previous publication.
To explore the contribution from tunnelling, rate coefficients were measured for the reaction of OD with acetone-d6 at 93 and 146 K:
| OD + CD3C(O)CD3 → CD3C(O)CD2 + D2O | (R6) |
This system was chosen in preference to the reaction of OH with acetone-d6 or OD with undeuterated acetone since the acetone-d6 could be used both as an OD precursor in the presence of O2, via the reaction of CD3CO + O2, and the reactant. If tunnelling were the dominant mechanism, one would expect its rate to be significantly slower for a D-atom transfer compared to a H-atom. However, as can be seen in Table 1, at 93 K, and for very similar overall densities, the rate coefficients for (R1) and (R6) are similar (within error of each other), providing evidence that tunnelling through TS2 is not the dominant channel. At 146 K, the rate coefficient for (R6) is about a factor of 2 lower than for (R1), again for a very similar density, but the rate coefficients are subject to a considerably larger error (almost 100% for the case of the deuterated reaction (R6)), because the magnitude of the rate coefficient is close to the limit of what can be measured in the limited time-window of the pulsed Laval method. It is hence difficult to be certain of any change with deuteration at this temperature. No deuteration experiments were attempted for the reaction of OH with DME.
These results suggest that for the reaction of OH with acetone, the dominant influence for the large enhancement in the rate coefficient observed below 200 K is a Lindemann–Hinshelwood type mechanism in which the OH–acetone association complex (IM1) is being formed as a stabilised product. If such a mechanism were operating, it would be expected that the overall rate coefficient for OH loss (k1) would be proportional to the number of deactivating collisions between the N2 bath gas molecules and energised IM1 species, and hence the total N2 number density. Using two Laval nozzles, k1 was measured at a variety of [N2] for temperatures between 79–86 K and also 133–146 K, as shown in Fig. 4. There is a small variation in temperature as the total gas density changes for a given nozzle. At ∼80 K, there is an increase in the rate coefficient with [N2], consistent with the Lindemann–Hinshelwood mechanism for stabilisation of the OH–acetone complex. However, at ∼140 K the rate coefficient is less dependent upon pressure, although it must be noted that the uncertainties are considerably larger owing to the smaller rate coefficients. For the temperatures around 140 K, it was not possible to measure k1 at the lower densities used at ∼80 K, as the magnitude of k1 was already approaching ∼1 × 10−12 cm3 molecule−1 s−1, which is the lower limit for this technique, owing to the more limited (∼100 μs compared to ∼250 μs at 80 K) reaction time and amount of acetone which can be added and still maintain a stable flow. However, at ∼140 K, the data suggest that collisional stabilisation of the complex is unable to compete with its re-dissociation to reactants, and the measured rate coefficients (which are considerably smaller) are dominated by a pressure independent mechanism, such as tunnelling.
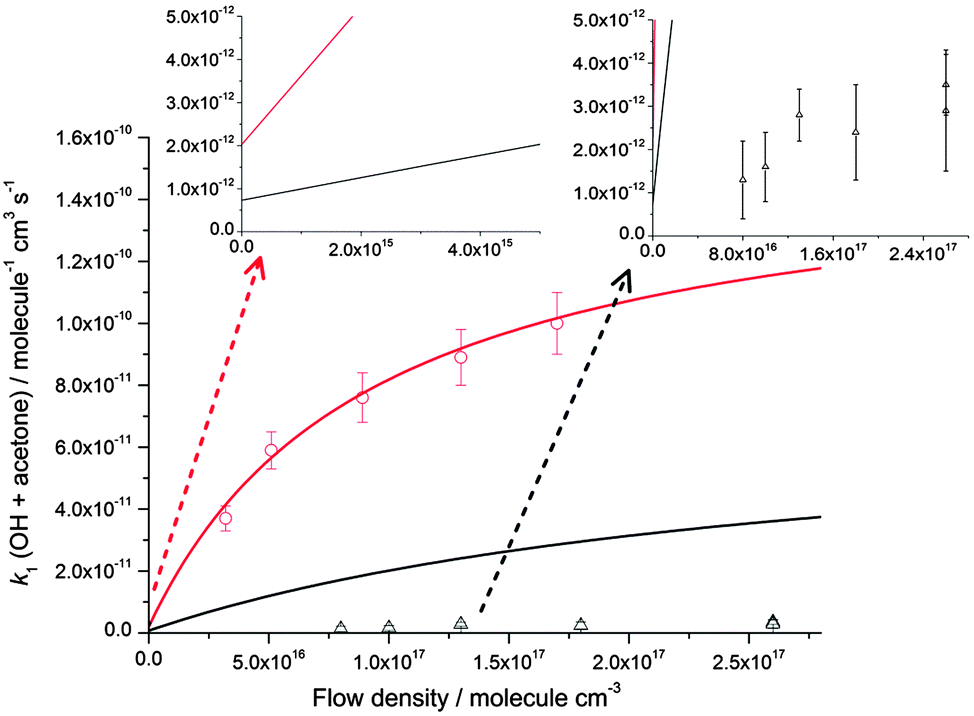 | ||
| Fig. 4 Pressure dependence of the rate coefficients for the reaction of OH with acetone at ∼80 K (red circles, the temperature varies between 79 ± 3 K and 83 ± 3 K for these points) and ∼140 K (black triangles, the temperature varies between 133 ± 10 K and 148 ± 15 K for these points) plotted alongside the MESMER master equation calculations at 80 K (red line) and 140 K (black line). The insets show non-zero intercepts calculated using MESMER at both 80 and 140 K and a vertically expanded version of the 140 K experimental data. The experimental error bars are as shown in Table 1. | ||
Rate coefficients for the reaction of OH with DME, k2, were also measured at four different total [N2] densities for the same nozzle for temperatures between 79–83 K, and are displayed in Fig. 5. k2 is seen to increase significantly with [N2], with the reaction appearing to be closer to the linear, low density limiting regime, compared to the reaction of OH with acetone (Fig. 4) at the same temperature.
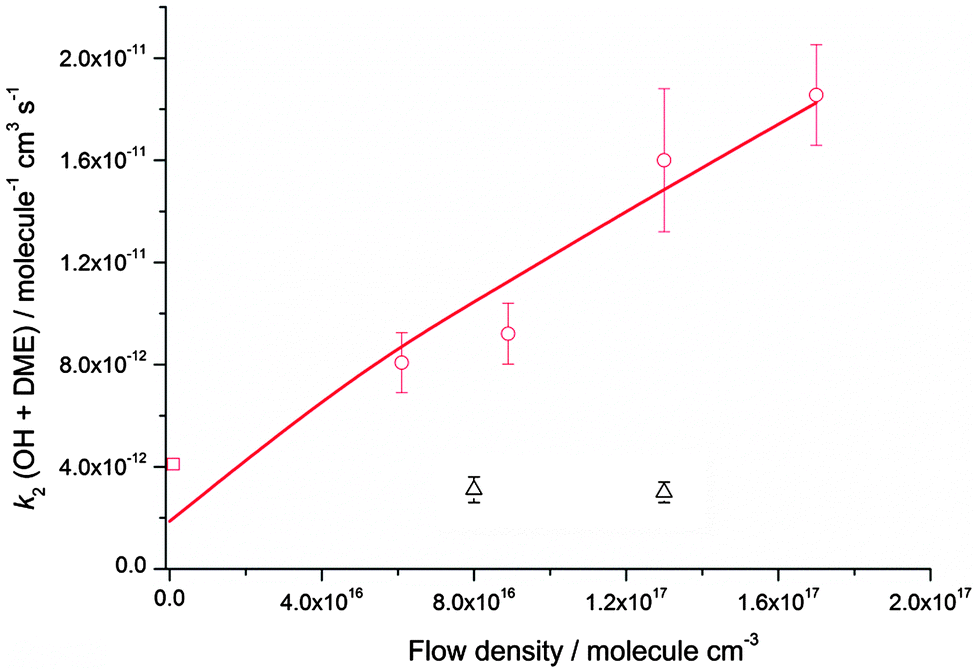 | ||
| Fig. 5 MESMER master equation calculations (red line, 80 K) together with experimentally determined values of k2(OH + DME) for a range of different total gas densities at a temperature of ∼80 K (red circles, the temperature varies between 79 ± 3 K and 83 ± 3 K) and 140 K (black triangles, the temperature varies between 133 ± 10 K and 138 ± 9 K). The rate coefficient shown at zero pressure (red square) is estimated using the OH recycling experiments in the presence of O2 performed at 93 ± 4 K, see text for details. The experimental errors bars are as given in Table 2. | ||
The dependence of both k1 and k2 upon [N2] at ∼80 K is evidence that in both cases, a stabilisation mechanism is dominating at this temperature and for the density range of the experiments performed. However, this does not exclude the possibility of a tunnelling mechanism dominating at even lower temperatures and/or densities. Indeed, inspection of Fig. 4 and 5 suggest that extrapolation to zero pressure may yield non-zero rate coefficients supportive of the presence of a bimolecular channel such as tunnelling. In a recent study of the reaction of OH with methanol at ∼80 K,5 the overall rate coefficient was observed to be independent of pressure, consistent with a tunnelling mechanism and supported by theoretical calculations. The tunnelling mechanism was further supported by observation of the CH3O radical product using LIF, consistent with a high calculated yield for this product under these conditions.
For the reaction of OH with DME:
 | (R2) |
| CH3OCH2 + O2 → CH3OCH2O2* | (R7) |
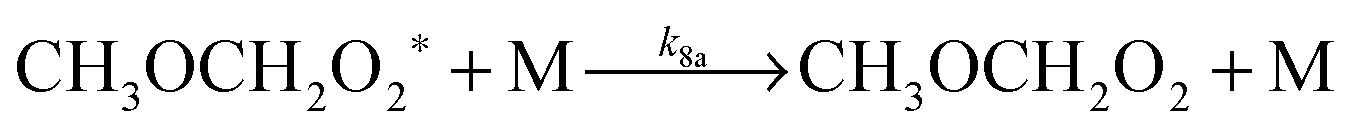 | (R8a) |
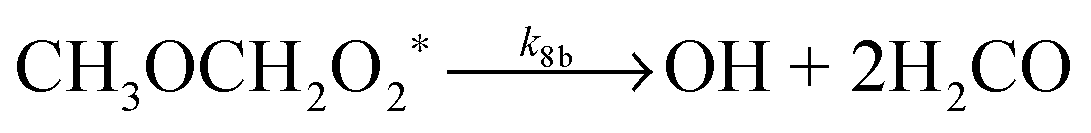 | (R8b) |
| k2′(no O2 present) = k2[CH3OCH3] | (E10) |
| k2′(O2 present) = k2(1 − α)[CH3OCH3] | (E11) |
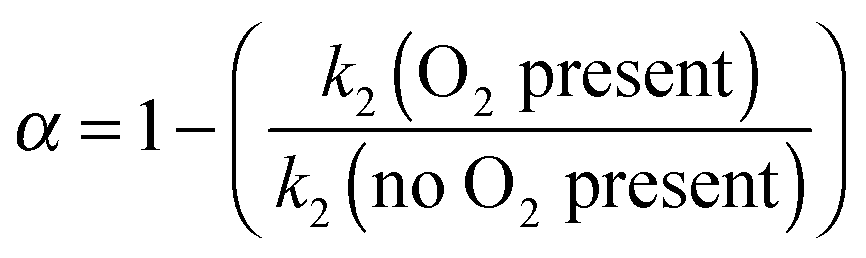 | (E12) |
At 93 K experiments were performed with 20% of the bath gas comprising O2, and the variation of k2′ with [CH3OCH3] both with and without the addition of O2 are shown in Fig. 6, for a total density in N2 of 6.5 × 1016 molecule cm−3. A clear reduction in the gradient is seen in the presence of O2, with k2(1 − α) (O2 present) = (8.0 ± 0.8) × 10−12 molecule−1 cm3 s−1 and k2 = (1.7 ± 0.1) × 10−11 molecule−1 cm3 s−1 (no O2 present). These results provide evidence that the CH3OCH2 radical is indeed formed as a direct product from the reaction of OH with DME at 93 K, for example via quantum mechanical tunnelling. Using the rate coefficients obtained in the presence and absence of O2, a yield of OH of α = 0.53+0.07−0.08 is obtained. If all the CH3OCH2 radicals were being recycled to form OH via reaction (R8b), then the yield of CH3OCH2 product from the abstraction reaction of OH with DME is about 50%. However, this is only a lower limit to the yield of CH3OCH2, as some of the initially formed CH3OCH2O2* will be collisionally stabilised to the peroxy radical CH3OCH2O2via reaction (R8a), and OH is therefore not recycled. If we assume this lower limit, namely that CH3OCH2O2* is not stabilized by collisions (the association reaction of CH3OCH2 with O2 is in the low-pressure regime), then multiplying the overall rate coefficient by the yield allows a lower limit for the rate coefficient in the limit of zero pressure to be determined as 0.53 × 7.7 × 10−12 = 4.1 × 10−12 cm3 molecule−1 s−1.44 This value has been added to Fig. 5 at zero pressure. At 93 K we did not perform the addition of O2 experiments as a function of total density of N2, and the branching ratio 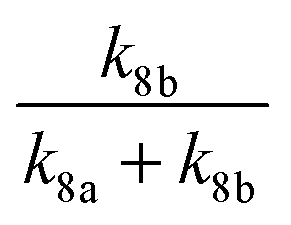 to form OH from CH3OCH2O2* is unknown experimentally under our conditions. Experiments performed at 80 K using the same methodology gave OH yields of 0.14 ± 0.08, 0.13 ± 0.12 and 0.10 ± 0.09 for total densities of 5.2, 8.7 and 16.4 × 1016 molecule cm−3 (N2), respectively, but the large uncertainties preclude any quantitative discussion of these yields. The significant yield of CH3OCH2via a tunnelling mechanism is consistent with the intercept of Fig. 5 from data measured at higher pressures which shows the pressure dependent contribution to the overall rate coefficient is significant.
to form OH from CH3OCH2O2* is unknown experimentally under our conditions. Experiments performed at 80 K using the same methodology gave OH yields of 0.14 ± 0.08, 0.13 ± 0.12 and 0.10 ± 0.09 for total densities of 5.2, 8.7 and 16.4 × 1016 molecule cm−3 (N2), respectively, but the large uncertainties preclude any quantitative discussion of these yields. The significant yield of CH3OCH2via a tunnelling mechanism is consistent with the intercept of Fig. 5 from data measured at higher pressures which shows the pressure dependent contribution to the overall rate coefficient is significant.
 | ||
| Fig. 6 Variation of the pseudo-first-order rate coefficient for the loss of OH radicals as a function of the density of DME in the presence of [O2] = 3 × 1015 molecule cm−3 (black open circles) and without O2 (open red triangles). The blue full lines are the weighted linear fits to the data, and the dashed green lines represent upper and lower 95% confidence limits. The gradients of the blue lines yield bimolecular rate coefficients k2 = (8.0 ± 0.8) × 10−12 molecule cm3 s−1 (in the presence of O2) and (1.7 ± 0.1) × 10−11 molecule cm3 s−1 (without O2). The total density of N2 = (6.5 ± 0.5) × 1016 molecule cm−3. | ||
4.2. MESMER calculation of rate coefficients
In order to complement the experimental results, rate coefficients were calculated for both reactions of OH with acetone and DME using the master equation software package MESMER.24 As described in Section 3, given the large amount of experimental data available in this study, a fitting methodology was employed in order to yield information about the kinetic parameters and mechanisms of the reactions. The effect of treating torsional modes explicitly was considered, with fits to experimental data carried out considering the vibrations as harmonic oscillators, and as hindered rotors, and the results compared.Due to the large number of experimental studies which have measured the rate coefficient k1 for the reaction of OH with acetone at temperatures above ∼200 K, only a subset of the available data set was used for the fitting process, as the focus of this current work is the lower temperature regime. The studies by both Gierczak et al.45 and Yamada et al.39 contained sufficient data points to span the 200–800 K temperature range, with no more than 100 K between each point. A total of 13 experimental rate coefficients from previous studies and the values from this work, were used to obtain the best-fit values of the energies of TS2, IM1 (see Fig. 1) as well as vibrational frequencies (including imaginary values corresponding to the tunnelling coordinate), and the A factor used in the ILT procedure. The optimised parameters from fitting to the experimental kinetic data are given in Table 3, together with ab initio calculations from previous studies.31,39,40 It is clear from inspection of Table 3 that the harmonic oscillator and hindered rotor models give significantly different results. Considering the energy of TS2 (OH/acetone reaction), the harmonic oscillator only model yields a value of 3.6 kJ mol−1, which is in relatively good agreement with the ab initio energy of 6.1 kJ mol−1 calculated by Yamada et al.39 at the CBS-QB3 level of theory. This finding is in agreement with a previous master equation study by Caralp et al.20 in which it was found necessary to use the barrier height obtained from the fits to experimental data rather than the larger barrier heights of 14.6 and 16.7 kJ mol−1 at the MP2/6-31G**//CCSD(T)/6-311G and B3LYP/6-311++G(d,p)//CCSD(T)/6-311++G(d,p) levels of theory calculated by Henon et al.31 and Vandenberk et al.,40 respectively. However, by treating some of the lower frequency vibrations as hindered rotations, the fitted barrier height is 13.0 kJ mol−1, in better agreement with the CCSD(T) calculations.
| Parameter | Harmonic oscillator | Hindered rotor | a | b | c |
|---|---|---|---|---|---|
| Energy of TS2/kJ mol−1 | 3.6 | 13.0 | 16.7 | 14.6 | 6.1 |
| Well depth energy of IM1/kJ mol−1 | −38.1 | −29.4 | −18.4 | −22.2 | −20.1 |
| Imaginary frequency of tunnelling mode/cm−1 | −541 | −1116 | −1250 | −1107 | −1293 |
| A factor for association reaction to form complex and used for ILT analysis/molecule−1 cm3 s−1 | 2.1 × 10−10 | 1.9 × 10−10 | — | — | — |
As seen from Table 3, again for the reaction of OH with acetone, both the harmonic oscillator and the hindered rotor models require the well depth for IM1 to be larger relative to the ab initio results in order to achieve the best fit with the experimental data. The well depth returned from the fits using the more rigorous hindered rotor model is closer to the ab initio well depths than its harmonic oscillator model counterpart. For both treatments of the vibrational modes, the amount by which the well depth of IM1 has to be increased to give the best fit with the experimental data is likely to be outside the error of the ab initio calculations. A more rigorous treatment of the anharmonic low frequency bending modes of IM1 would likely increase further the density of states of IM1 relative to the separated OH + acetone reactants, which would in turn reduce the rate coefficient for the re-dissociation of IM1 back to reactants. The well depth of IM1 that is required to give the best fit with the experimental data would then be closer to the ab initio values.
The rate coefficients calculated at zero pressure using MESMER with the hindered rotor model fits for the reaction of OH with acetone are shown as a function of temperature in Fig. 7, using both the Eckart and WKB methods of tunnelling, and a calculation without a tunnelling correction. As the experimental data above 200 K are in the low pressure limit, and the data obtained in this study have been shown to be in the fall-off regime, the experimental data obtained in this work are not plotted in Fig. 7. It can be seen that all treatments calculate the rate coefficients as increasing rapidly at very low temperatures, owing to the dominant role played by collisional stabilisation of the complex under those conditions. Indeed at the temperatures for the Laval experiments, the calculated rate coefficients for the different approaches are similar. However, for the experimental data at 200–400 K, the predicted behaviour is quite different. Using the Eckart model with a fitted imaginary frequency, good agreement can be achieved between experiment and theory over a wide range of temperatures despite the larger abstraction barrier height (13.0 kJ mol−1). However, from Fig. 7 it can be observed that the WKB tunnelling approach, which is similar in methodology to that used by Caralp et al.,20 for this reaction, significantly underestimates the experimental rate coefficients in the 200–400 K region. If no tunnelling correction is included, the experimental rate coefficients are under-estimated by an even greater amount in this region. The reasons for the poor agreement between experiment and theory using the WKB tunnelling method are not clear. Whilst the Eckart approach is more rudimentary, varying the imaginary frequency gives this method a certain degree of flexibility which could be accommodating any limitations in either the potential used for the WKB calculations or the lack of consideration of tunnelling paths orthogonal to the minimum energy path.
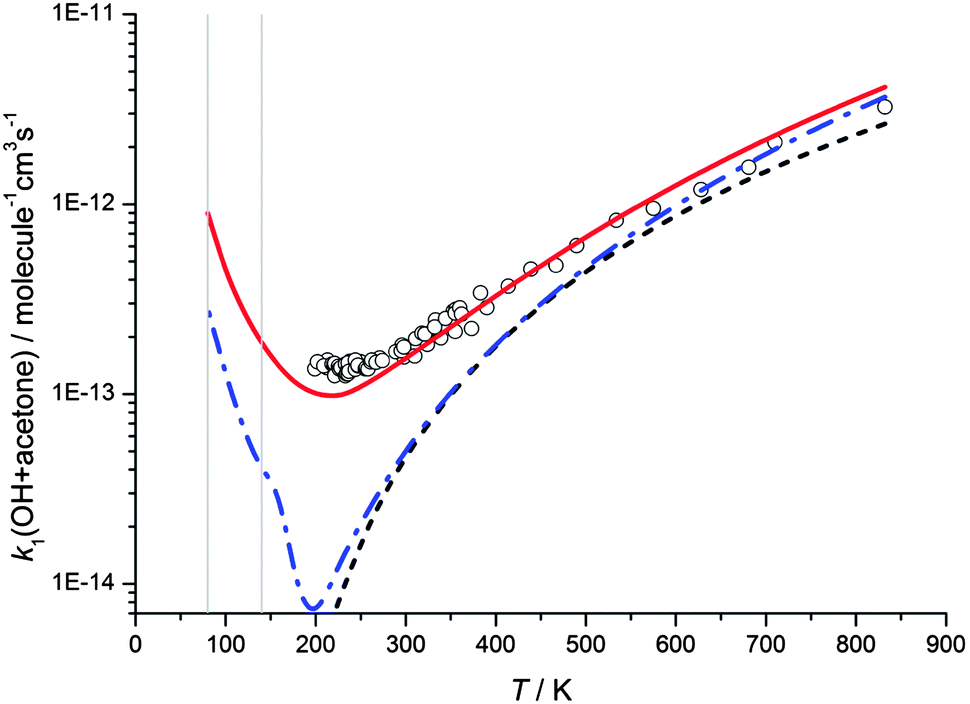 | ||
| Fig. 7 MESMER master equation calculated rate coefficients for k1(OH + acetone) versus temperature plotted alongside experimental data currently in the literature (references as for Fig. 2). The red full and blue dot/dash lines show master equation calculations from the hindered rotor model with transmission coefficients calculated using the Eckart and the WKB methods, respectively, whilst the black dashed line shows calculated rate coefficients without including a tunnelling correction. All MESMER master equation calculations are for the limit of zero pressure. The grey vertical lines are positioned at 80 and 140 K to aid comparison of the MESMER predicted rate coefficients with the experimentally obtained values listed in Table 1, which were measured at a range of pressures. | ||
The pressure dependence of the rate coefficients for the reaction of OH with acetone is well described by the master equation calculations at ∼80 K, as shown in Fig. 4, and although predicting a pressure dependence at 140 K, overestimates the magnitude of the rate coefficient significantly. The lack of agreement at 140 K is unclear, but may be due to an incorrect energy dependence of the density of states, which will have more impact in the calculations at higher energies. Another, more speculative reason may be that at 140 K the system is not as well described using statistical rate theory. The lifetime of the collision complex becomes shorter at higher temperatures, and consequently there might be insufficient time for complete intramolecular vibrational relaxation within the complex which is a central tenet of statistical rate theory, leading to a deviation between experiment and theory.
The master equation calculations show a non-zero y-axis intercept in the rate coefficient at zero pressure, corresponding to a pressure independent reaction channel to form products due to tunnelling. The gas densities experienced in molecular clouds are extremely low (typically 102–104 molecule cm−3),46 and hence the pressure independent channels are the relevant ones to explore the chemistry of these regions. In order to explore the temperature dependence of the pressure independent channel, master equation calculations were performed at a total density of 1 × 106 molecule cm−3 and down to 10 K, as shown in Fig. 8. Calculations were performed using both the Eckhart and WKB treatments of tunnelling, and at 10 K the predicted rate coefficients are both more than 100 times greater than the room temperature rate coefficient.
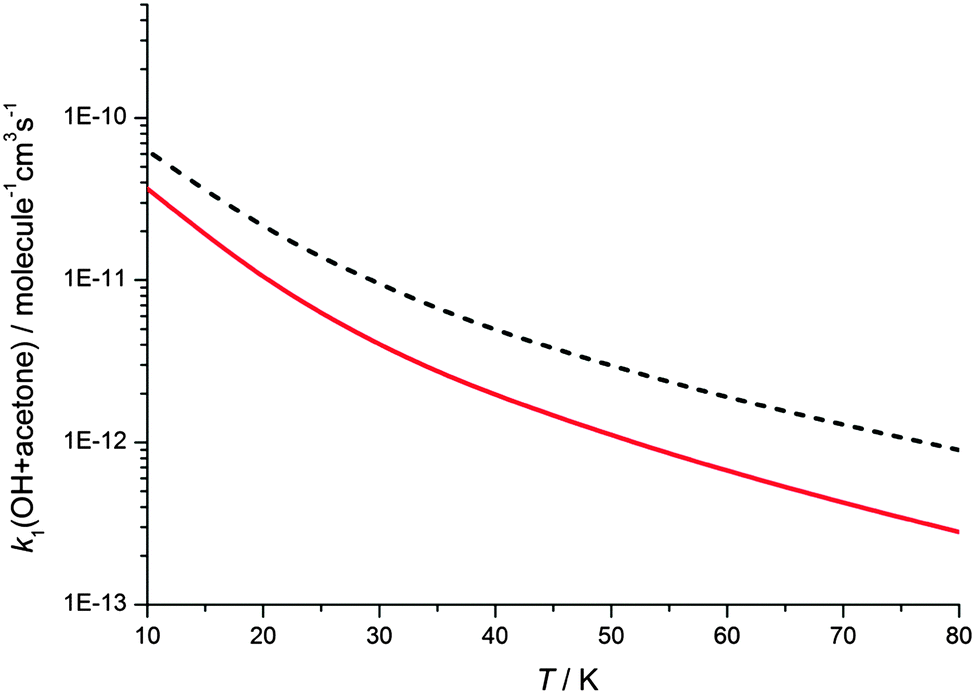 | ||
| Fig. 8 Rate coefficients k1(OH + acetone) calculated at a total density of 1 × 106 molecule cm−3 using MESMER. The black dashed line shows calculated rate coefficients using transmission coefficients from the Eckart treatment of tunnelling, whilst the solid red line uses transmission coefficients from the WKB method. See text for details. | ||
The calculated rate coefficients for the reaction of OH with DME as a function of temperature and pressure are shown in Fig. 5 and 9, respectively. Extensive master equation calculations using MESMER have recently been performed32 for this reaction in the range T = 200–1400 K in order to obtain an optimised set of fitted geometric and energetic parameters that best describe the kinetic data obtained in that study and also extensively reported in the literature at higher temperatures.47–51 The calculations showed the presence of a pre-reaction complex with a binding energy of ∼22 kJ mol−1, but no pressure dependence was experimentally observed, hence providing no direct evidence for stabilisation into the complex influencing the kinetics of this reaction under the conditions of that study. The MESMER calculations were able to model the experimental rate coefficients within 16% between 200–1400 K with a model using a single transition state.32 The optimised geometric and energetic parameters have been used in this study to extend the MESMER calculations to the lower temperature experimental conditions of the current work, with the exception of the well depth for IM1 (OH–DME), which was not constrained by the higher temperature fits. In order to best fit the low temperature data it was found that the binding energy needed to be increased to 29.8 kJ mol−1. From Fig. 5 and 9 it can be seen that MESMER is able to reproduce the temperature behaviour over a wide range of temperatures, and also predicts the pressure dependence well for T ∼ 80 K. Once again, the MESMER calculations predict that at zero-pressure there is a significant intercept corresponding to a pressure independent channel for the reaction of OH with DME, consistent with the experimental data demonstrating a yield of ≥50% for the CH3COCH2 abstraction product at this temperature.
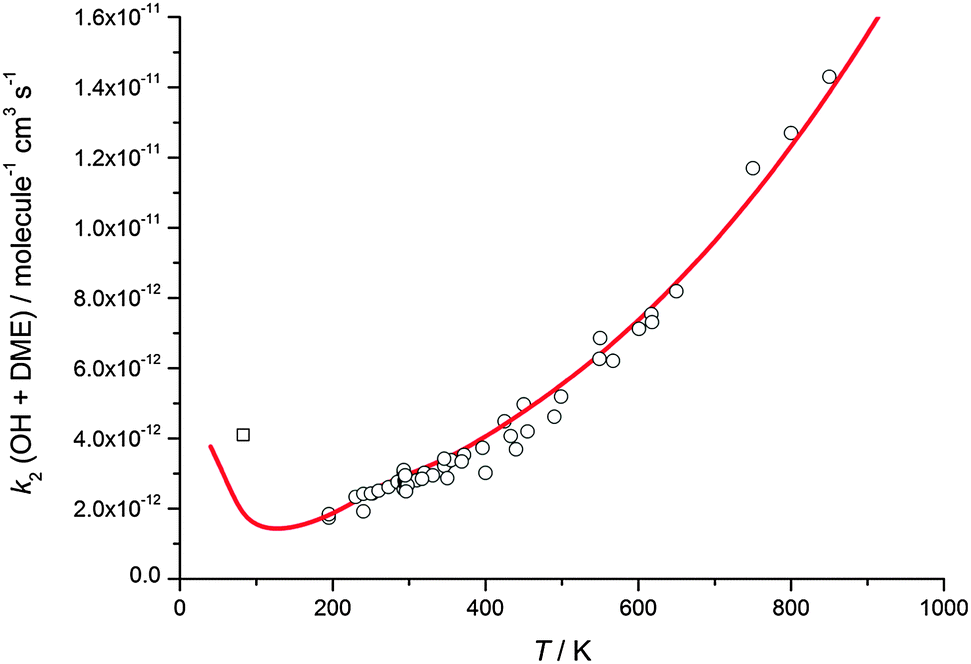 | ||
| Fig. 9 MESMER master equation rate coefficients for k2(OH + DME) versus temperature plotted alongside experimental data currently in the literature (references as for Fig. 3). The experimental data point (open square) is estimated from the OH recycling yield experiments in the presence of O2 at 93 K, as described in the text. | ||
5. Discussion
The rate coefficients below 150 K for both the reactions of OH with acetone and DME are significantly larger than observed previously at higher temperatures, and a pressure dependence provides evidence for an association channel with stabilisation into a weakly bound complex. At temperatures above 200 K the rate coefficients for both reactions increase with temperature and are pressure independent, consistent with reaction proceeding over an energy barrier. For the reaction of OH with DME, the rate coefficient shows significant curvature when plotted in Arrhenius form, and for the reaction of OH with acetone between 300 and 200 K the rate coefficients begin to become independent of temperature. In a previous study of the reaction of OH with methanol,5 a significant enhancement in the rate coefficient was also observed at very low temperatures. However, the reaction was found to be pressure independent, and observation of the CH3O radical reaction product demonstrated the role of tunnelling from the hydrogen-bonded complex through the activation barrier, supported by theoretical calculations.The two transition state model developed by Greenwald et al.,18,19 illustrated by the potential energy surface shown in Fig. 1, is useful to explore the role of the hydrogen-bonded pre-reactive complex at low temperatures and rationalise the enhancement in the rate constants at low temperatures. Using this two transition state model, simple expressions describing the high and low temperature limiting behaviour of system can be obtained. In the limit of low temperature, there is insufficient collision energy to access TS2 and so the transition state is located at TS1. The rate of dissociation of the complex becomes negligible compared to its rate of formation and the complex exhibits a significant lifetime and its concentration may become significant. Under these conditions the dominant bottleneck to reaction is TS1, and the observed rate coefficient is associated with the rate of formation of the complex IM1, which is limited by the capture rate coefficient for OH with the oxygenated VOC. The increased lifetime of IM1 will also enable stabilisation by collisions with the bath gas and facilitates product formation via tunnelling for these systems involving hydrogen atoms. Conversely in the limit of high temperature the complex rapidly dissociates back to reactants, and there is rapid equilibration between the reactants and the complex, and the rate determining step is crossing the barrier at TS2. The observed rate coefficient then reduces to the equilibrium constant for the formation of the complex multiplied by the rate coefficient for the complex crossing the barrier to reaction. The large increase in the observed rate constant below 200 K can be understood in terms of the controlling transition-state moving to from TS2 to TS1.1
Unlike the OH + methanol reaction for which no pressure dependence was observed at very low temperatures,5 the rate determining step in the reaction of OH with acetone and DME reactions under similar conditions is the formation of the stabilized pre-reactive complex, IM1, evidenced by observation of a pressure dependence. The MESMER modelling shows that despite the weak binding energies of these complexes (∼20 kJ mol−1), and the relatively low total densities of the N2 bath gas used (5–20 × 1016 molecule cm−3), collisional stabilization of the energized complex occurs readily on the timescale on the experimental timescale (up to 300 μs reaction time in the Laval expansion). Inspection of Fig. 4 and 5 shows that for a given pressure and temperature, the rate coefficients for the reaction of OH with acetone are closer to the high pressure limit than for DME. This behaviour can be readily understood as acetone is a larger molecule and hence the number of vibrational modes, and hence the density of states of the complex will be larger relative to that of the separated reagents, and so the lifetime of the complex will be longer. As was shown in Section 4.2 above, the best-fit parameters from comparison of the MESMER model with the experimental data included a well-depth for OH–acetone which was considerably deeper than calculated from ab initio methods, as a result of the methodology used in calculating a density of states which was too low. With increasingly sophisticated treatment of hindered rotational modes and vibrational anharmonicity,5 the calculated density of states of the complex would be expected to increase, resulting in an increase in the predicted rate coefficients using MESMER. Hence the well depth obtained from the best fit of the MESMER model to the experimental data would become closer to those values calculated with ab initio methods.
Although it is established that tunnelling is not the rate determining step in the reactions of OH with either acetone or DME, there is evidence from the non-zero intercepts of Fig. 4 and 5 at zero pressure of a bimolecular channel for both reactions that is pressure independent and due to tunnelling. As shown in Fig. 7, the MESMER calculations show that tunnelling is required in order to model the system successfully over the wide range of temperatures for which kinetic data are available. For the reaction of OH with DME there is additional evidence of the formation of the CH3OCH2 product, and hence for efficient quantum mechanical tunnelling.
In addition, the master equation can calculate the time dependence of any of the species, and in Fig. 10 these are shown for the OH reactant, the pre-reaction complex, IM1, and the post-reaction complex IM2 (the step from IM2 to bimolecular products was not considered in the simulations). The lifetime of the pre-reactive complex is considerable, peaking at around 100 μs. Once the stabilised complex is formed, on longer timescales it will eventually all be converted to products via quantum mechanical tunnelling, but does not have sufficient energy to convert back to the reagents. There is always competition at low temperatures between stabilization of IM1 and tunnelling through the barrier, and depending upon the density of states in IM1, the kinetic behaviour for reactions of OH with OVOCs will be at different points on the fall-off curve for a given combination of pressure and temperature. For the smaller methanol molecule studied previously the master equation calculated a rate coefficient for the loss of OH due to stabilisation of the complex to be ∼1 × 10−13 molecule−1 cm3 s−1 at 80 K and a similar total density, and the rate coefficient was dominated by tunnelling.5 However, as seen in Fig. 4 and 5 for the OH reactions with acetone and DME under similar conditions, the reaction rate coefficients are in the fall-off region with a dominant contribution to the rate coefficient from collisional stabilisation.
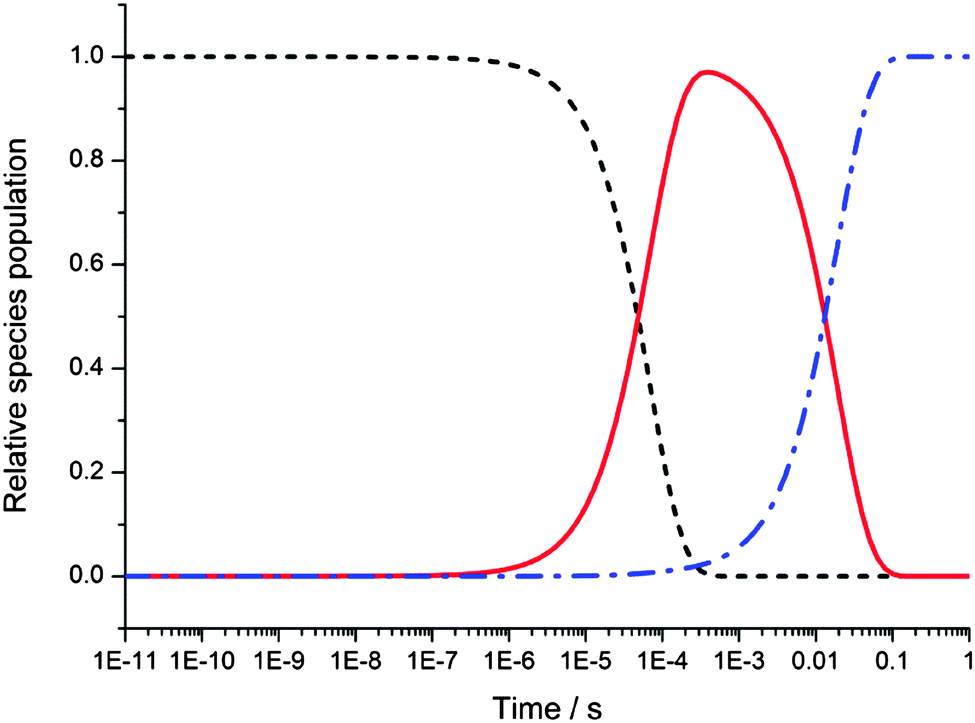 | ||
| Fig. 10 Time evolution of the concentrations of OH (black dashed line), IM1 (red full line) and IM2 (blue dot/dash line) for the reaction of OH with acetone calculated by MESMER at 80 K and a total gas density of [N2] = 6 × 1016 molecule cm−3. See text for details. | ||
6. Conclusions
The rate coefficients for the reactions of OH with acetone and dimethyl ether (DME) were studied between 63–148 K using a pulsed Laval nozzle apparatus. For both reactions, a marked inverse temperature dependence was observed, with rate coefficients considerably higher than those obtained previously at ∼200 K. For both reactions the rate coefficient was found to be pressure dependent at the very low temperatures used, in contrast to our previous study of the reaction of OH with methanol for which the rate coefficient was independent of pressure, and for which a tunnelling mechanism was shown to dominate. The pressure dependence suggests a Lindemann–Hinshelwood type mechanism operates for these reactions, with collisional stabilisation of the OH–OVOC pre-reaction complex resulting in the observed loss of OH radicals. For acetone, the rate coefficient at 93 K did not show a kinetic isotope effect, showing that tunnelling is not dominating the mechanism. However, for both reactions extrapolation of the pressure dependent rate coefficients to zero pressure shows an intercept which provides evidence for a contribution from a pressure independent bimolecular channel leading to abstraction products via tunnelling. In the case of the reaction of OH with DME, further evidence for tunnelling to form the abstraction product CH3OCH2 was obtained via measurement of a smaller rate coefficient in the presence of O2 due to recycling of OH from the reaction of CH3OCH2 with O2, and a lower limit for the yield of CH3OCH2 of ∼0.5 was obtained. The experimental measurements were supported by master equation calculations using the MESMER code which included a treatment of quantum mechanical tunnelling, and using optimised energies for the stationary points on the minimum energy path for reaction obtained by fitting to the available data over a wide range of temperatures. MESMER was able to reproduce the large increase in the rate coefficients at very low temperatures for both reactions as well as fitting the data at higher temperatures, and was able to model the pressure dependence well at very low temperatures. The calculations support the mechanism for these reactions of the formation of a pre-reactive complex (IM1) for which the dominant fate is collisional stabilisation, but with a contribution from tunnelling to form abstraction products. MESMER was also used to calculate the rate coefficients at 1 × 106 molecule−1 and at temperatures down to 10 K, typical of the molecular clouds or the interstellar medium, and showed that the rate coefficient for tunnelling was ∼10% of the collision rate. Therefore, similar to the reaction of OH with methanol for which tunnelling was dominant under the conditions in space, the reactions of OH with acetone and DME are also likely to be fast under these conditions, and may be important in such an environment. The reactions of OH with methanol, acetone, DME and also methyl ethyl ketone (MEK) all show similar behaviour, and we hypothesise that reactions of OH with oxygenated VOCs, for which a hydrogen-bonded pre-reactive complex is formed, will behave similarly at very low temperatures, and demonstrate a contribution from tunnelling.Acknowledgements
R.J.S. and R.L.C. acknowledge the Natural Environment Research Council for funding of PhD studentships. We would also like to thank Dr A. Goddard for technical assistance.References
- R. J. Shannon, S. Taylor, A. Goddard, M. A. Blitz and D. E. Heard, Phys. Chem. Chem. Phys., 2010, 12, 13511–13514 RSC.
- J. Bouwman, M. Fournier, I. R. Sims, S. R. Leone and K. R. Wilson, J. Phys. Chem. A, 2013, 117, 5093–5105 CrossRef CAS PubMed.
- H. Sabbah, L. Biennier, I. R. Sims, Y. Georgievskii, S. J. Klippenstein and I. W. M. Smith, Science, 2007, 317, 102–105 CrossRef CAS PubMed.
- E. Vöhringer-Martinez, B. Hansmann, H. Hernandez, J. S. Francisco, J. Troe and B. Abel, Science, 2007, 315, 497–501 CrossRef PubMed.
- R. J. Shannon, M. A. Blitz, A. Goddard and D. E. Heard, Nat. Chem., 2013, 5, 745–749 CrossRef CAS PubMed.
- J. Daranlot, M. Jorfi, C. Xie, A. Bergeat, M. Costes, P. Caubet, D. Xie, H. Guo, P. Honvault and K. M. Hickson, Science, 2011, 334, 1538–1541 CrossRef CAS PubMed.
- B. R. Rowe, G. Dupeyrat, J. B. Marquette and P. Gaucherel, J. Chem. Phys., 1984, 80, 4915–4921 CrossRef CAS PubMed.
- I. W. M. Smith and B. R. Rowe, Acc. Chem. Res., 2000, 33, 261–268 CrossRef CAS PubMed.
- I. W. M. Smith, Angew. Chem., Int. Ed., 2006, 45, 2842–2861 CrossRef CAS PubMed.
- I. W. M. Smith, E. Herbst and Q. Chang, Mon. Not. R. Astron. Soc., 2004, 350, 323–330 CrossRef CAS.
- I. W. Smith, A. M. Sage, N. M. Donahue, E. Herbst and D. Quan, Faraday Discuss., 2006, 133, 137–156 RSC.
- A. M. Rouhi, Amoco, Haldor Topsoe Develop Dimethyl Ether As Alternative Diesel Fuel, Chem. Eng. News, 1995, 73, 37–39 Search PubMed.
- I. W. M. Smith, A. M. Sage, N. M. Donahue, E. Herbst and D. Quan, Faraday Discuss., 2006, 133, 137–156 RSC.
- F. Combes, M. Gerin, A. Wootten, G. Wlodarczak, F. Clausset and P. J. Encrenaz, Astron. Astrophys., 1987, 180, L13–L16 CAS.
- D. L. Thacker, W. J. Wilson and A. H. Barrett, Astrophys. J., 1970, 161, L191–L197 Search PubMed.
- G. Winnewisser and F. F. Gardner, Astron. Astrophys., 1976, 48, 159–161 CAS.
- V. Wakelam, E. Herbst, J.-C. Loison, I. Smith, V. Chandrasekaran, B. Pavone, N. Adams, M.-C. Bacchus-Montabonel, A. Bergeat and K. Béroff, Astrophys. J., Suppl. Ser., 2012, 199, 21 CrossRef.
- E. E. Greenwald, S. W. North, Y. Georgievskii and S. J. Klippenstein, J. Phys. Chem. A, 2007, 111, 5582–5592 CrossRef CAS PubMed.
- E. E. Greenwald, S. W. North, Y. Georgievskii and S. J. Klippenstein, J. Phys. Chem. A, 2005, 109, 6031–6044 CrossRef CAS PubMed.
- F. Caralp, W. Forst, E. Henon, A. Bergeat and F. Bohr, Phys. Chem. Chem. Phys., 2006, 8, 1072–1078 RSC.
- T. J. Wallington and M. J. Kurylo, J. Phys. Chem., 1987, 91, 5050–5054 CrossRef CAS.
- I. W. M. Smith and A. R. Ravishankara, J. Phys. Chem. A, 2002, 106, 4798–4807 CrossRef CAS.
- R. J. Shannon, S. Taylor, A. Goddard, M. A. Blitz and D. E. Heard, Phys. Chem. Chem. Phys., 2010, 12, 13511–13514 RSC.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- S. E. Taylor, A. Goddard, M. A. Blitz, P. A. Cleary and D. E. Heard, Phys. Chem. Chem. Phys., 2008, 10, 422–437 RSC.
- S. A. Carr, M. T. Baeza-Romero, M. A. Blitz, B. J. S. Price and P. W. Seakins, Int. J. Chem. Kinet., 2008, 40, 504–514 CrossRef CAS.
- J. T. Bartis and B. Widom, J. Chem. Phys., 1974, 60, 3474–3482 CrossRef CAS PubMed.
- M. A. Blitz, K. J. Hughes, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2006, 110, 2996–3009 CrossRef CAS PubMed.
- J. A. Miller, S. J. Klippenstein, S. H. Robertson, M. J. Pilling and N. J. B. Green, Phys. Chem. Chem. Phys., 2009, 11, 1128–1137 RSC.
- S. Robertson, M. Pilling, D. Baulch and N. Green, J. Phys. Chem., 1995, 99, 13452–13460 CrossRef CAS.
- E. Henon, S. Canneaux, F. Bohr and S. Dobe, Phys. Chem. Chem. Phys., 2003, 5, 333–341 RSC.
- S. A. Carr, T. J. Still, M. Blitz, A. J. Eskola, M. J. Pilling, P. Seakins, R. Shannon, B. Wang and S. H. Robertson, J. Phys. Chem. A, 2013, 117, 11142–11154 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision D.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- S. Sharma, S. Raman and W. H. Green, J. Phys. Chem. A, 2010, 114, 5689–5701 CrossRef CAS PubMed.
- W. H. Miller, J. Am. Chem. Soc., 1979, 101, 6810–6814 CrossRef CAS.
- B. C. Garrett and D. G. Truhlar, J. Phys. Chem., 1979, 83, 2921–2926 CrossRef CAS.
- Y. Georgievskii and S. J. Klippenstein, J. Chem. Phys., 2005, 122, 194103 CrossRef PubMed.
- M. J. Pilling and S. H. Robertson, Annu. Rev. Phys. Chem., 2003, 54, 245–275 CrossRef CAS PubMed.
- T. Yamada, P. H. Taylor, A. Goumri and P. Marshall, J. Chem. Phys., 2003, 119, 10600–10606 CrossRef CAS PubMed.
- S. Vandenberk, L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2002, 4, 461–466 RSC.
- A. J. Eskola, S. A. Carr, M. A. Blitz, M. J. Pilling and P. W. Seakins, Chem. Phys. Lett., 2010, 487, 45–50 CrossRef CAS PubMed.
- J. Lockhart, M. A. Blitz, D. E. Heard, P. W. Seakins and R. J. Shannon, J. Phys. Chem. A, 2013, 117, 5407–5418 CrossRef CAS PubMed.
- D. R. Glowacki, J. Lockhart, M. A. Blitz, S. J. Klippenstein, M. J. Pilling, S. H. Robertson and P. W. Seakins, Science, 2012, 337, 1066–1069 CrossRef CAS PubMed.
- M. T. B. Romero, M. A. Blitz, D. E. Heard, M. J. Pilling, B. Price and P. W. Seakins, Chem. Phys. Lett., 2005, 408, 232–236 CrossRef CAS PubMed.
- T. Gierczak, M. K. Gilles, S. Bauerle and A. R. Ravishankara, J. Phys. Chem. A, 2003, 107, 5014–5020 CrossRef CAS.
- E. F. van Dishoeck, G. A. Blake, B. T. Draine and J. I. Lunine, The chemical evolution of protostellar and protoplanetary matter, in Protostars and Planets III, ed. E. H. Levy and J. I. Lunine, University of Arizona Press, 1993, pp. 163–241 Search PubMed.
- A. Mellouki, S. Teton and G. Lebras, Int. J. Chem. Kinet., 1995, 27, 791–805 CrossRef CAS.
- M. Arif, B. Dellinger and P. H. Taylor, J. Phys. Chem. A, 1997, 101, 2436–2441 CrossRef CAS.
- A. Bonard, V. Daele, J. L. Delfau and C. Vovelle, J. Phys. Chem. A, 2002, 106, 4384–4389 CrossRef CAS.
- R. D. Cook, D. F. Davidson and R. K. Hanson, J. Phys. Chem. A, 2009, 113, 9974–9980 CrossRef CAS PubMed.
- T. J. Wallington, P. Dagaut, R. Z. Liu and M. J. Kurylo, Int. J. Chem. Kinet., 1988, 20, 541–547 CrossRef CAS.
- M. Wollenhaupt, S. A. Carl, A. Horowitz and J. N. Crowley, J. Phys. Chem. A, 2000, 104, 2695–2705 CrossRef CAS.
- T. Gierczak, M. K. Gilles, S. Bauerle and A. R. Ravishankara, J. Phys. Chem. A, 2003, 107, 5014–5020 CrossRef CAS.
- T. J. Wallington, R. Z. Liu, P. Dagaut and M. J. Kurylo, Int. J. Chem. Kinet., 1988, 20, 41–49 CrossRef CAS.
- J. Sehested, T. E. Møgelberg, T. J. Wallington, E. W. Kaiser and O. J. Nielsen, J. Phys. Chem., 1996, 100, 17218–17225 CrossRef CAS.
| This journal is © the Owner Societies 2014 |