 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The reactions of Criegee intermediates with alkenes, ozone, and carbonyl oxides†
L.
Vereecken
*,
H.
Harder
and
A.
Novelli
Max Planck Institute for Chemistry, Hahn-Meitner-Weg 1, 55128 Mainz, Germany. E-mail: Luc.Vereecken@mpic.de
First published on 7th January 2014
Abstract
The reaction of Criegee intermediates with a number of coreactants is examined using theoretical methodologies, combining ROCCSD(T)//M06-2X quantum calculations with theoretical kinetic predictions of the rate coefficients. The reaction of CI with alkenes is found to depend strongly on the substitutions in the reactants, resulting in significant differences in the predicted rate coefficient as a function of the selected alkene and CI. Despite submerged barriers, these entropically disfavored reactions are not expected to affect CI chemistry. The reaction of H2COO + H2COO is found to be barrierless, with a rate coefficient nearing the collision limit, ≥4 × 10−11 cm3 molecule−1 s−1. The dominant reaction products are expected to be carbonyl compounds and an oxygen molecule, though chemically activated reactions may give rise to a plethora of different (per)acids and carbonyl compounds. CI + CI reactions are expected to be important only in laboratory environments with high CI concentrations. The reaction of H2COO with O3 was predicted to proceed through a pre-reactive complex and a submerged barrier, with a rate coefficient of 1 × 10−12 cm3 molecule−1 s−1. A study of the dominant CI reactions under experimental and atmospheric conditions shows that the latter reaction might affect CI chemistry.
Introduction
Carbonyl oxides, also known as Criegee intermediates (CI), are key intermediates in the ozonolysis of unsaturated compounds in the atmosphere. Despite being proposed as early as 1949,1,2 their characterisation has long been hampered by a lack of suitable detection methods, and until recently most experimental information was highly indirect. The ozonolysis reaction is complex, where the CI formed partially undergo chemically activated reactions, while the remainder is either formed with thermal energy content or is stabilized by collisional energy loss. This further complicates their study. The importance of CI in the atmosphere,3–7 not only as an intermediate in the ozonolysis but also as a source of OH, an oxidant for SO2 and NO2, and possibly other contributions to tropospheric chemistry, warrants extensive investigations of the chemistry of stabilized CI (SCI).Since the first detection of H2COO, the smallest CI, by Taatjes et al. in 2008,8 a large set of experimental data on CI and their chemistry has become available;9–18 we refer to the recent review paper by Taatjes et al.3 for a detailed discussion. Theoretical analysis has played a significant role in our understanding of CI, amplified further by the historical dearth of experimental data. The recent review by Vereecken and Francisco19 discusses the many available theory-based literature in detail. Since then, more relevant theoretical studies have been published, including work on the CI + O3 reaction20 and the CI + carbonyl reaction.21 Many reactions of CI remain poorly understood, but are essential for the interpretation of experimental data, both in field and laboratory. In this work, we study a number of reactions that are likely to affect laboratory work, and possibly contribute to CI chemistry in the atmosphere. The reactions studied were chosen based on the expected chemistry of the CI as determined by its complex 1,3-dipole character. The dominant contributing configurations in this wavefunction are shown below, in decreasing order of importance, and are critical in the understanding of CI chemistry:
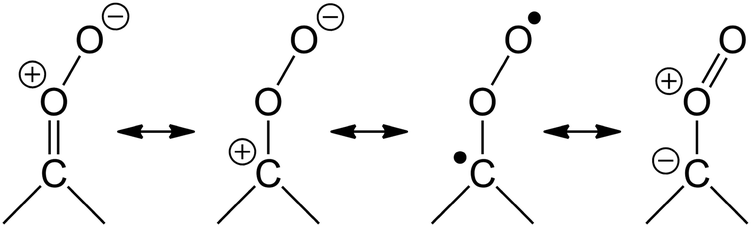
Contrary to older assumptions,22 CI are now known to be mostly zwitterionic14,18,19 with a relatively low contribution of biradical character. As a 1,3-bipole, it is expected to react similar to ozone, which has a similar zwitterionic structure with negatively charged outer oxygen atoms, and a positively charged central O-atom:

Ozonolysis of alkenes is a well-known process in the atmosphere, and CI are expected to display similar reactivity to olefines. Theoretical work by Crehuet et al.23 and Lan et al.24 showed these reactions to have lower barriers to reaction compared to the analogous ozonolysis; these reactions might therefore have higher rate coefficients and CI-initiated oxidation of alkenes could contribute even at lower concentrations of CI. This is particularly true in laboratory studies, where reactants are often used in higher concentrations than VOC and oxidants in the atmosphere;25 evidence for CI + olefines was already reported as early as 1967 by Story and Burgess.26
In their ground-breaking experiments on the infrared detection of CI, Su et al.14 observed fast H2COO loss on the order of microseconds; they proposed that this decay is governed by fast CI + CI reactions driven by their relatively high H2COO concentrations. We investigate the H2COO + H2COO reaction in this work. If this 1,3-dipole + 1,3-dipole reaction is fast, it is worthwhile to also examine the CI + O3 reaction. The analogous O3 + O3 reaction is known to be slow, but recent work by Kjaergaard et al.20 showed the CI + O3 reaction to be possibly barrierless. Computational difficulties prevented them from recommending a rate coefficient, and they propose a lower limit on the H2COO + O3 rate coefficient of 10−18 cm3 molecule−1 s−1.
The intent of this paper is to explore the listed set of SCI reactions and assess their possible relevance in the atmosphere and lab, rather than aiming at the most accurately possible characterization of a particular reaction. As such, the levels of theory chosen are not necessarily the highest levels of theory available, though they are expected to yield a good description of the reaction mechanism, and allow for a quantitative analysis that is sufficiently accurate for our current purposes. Improved calculations for the most critical of the reactions studied here are in progress, and experimental verification remains necessary. Extensive ESI† is available; tables and figures in the ESI† are numbered with an SI-prefix.
Methodology
a. Quantum chemical calculations
The geometries of all structures are optimized using the M06-2X density functional27 combined with the Dunning28 aug-cc-pVDZ or aug-cc-pVTZ basis set (denoted M06-2X/aVxZ, with x = D or T). Harmonic vibrational wavenumbers were obtained at the same level of theory, and scaled by a factor of 0.971.29,30 All M06-2X calculations used an ultrafine integration grid, and very tight SCF convergence criteria. For a subset of intermediate structures, the relative energies were improved by single point ROHF/UCCSD(T) calculations on the M06-2X geometries, employing aug-cc-pVDZ, aug-cc-pVTZ and aug-cc-pVQZ Dunning basis sets (denoted as CCSD(T)/aVxZ hereafter); extrapolation to an infinite basis set uses the Schwartz schemes proposed by Martin.31 The electronic wavefunctions of CI and O3 intrinsically have multi-reference character, where the dominant contributing Lewis structures are depicted above. Single-reference methodologies as employed here, however, are usually found to perform adequately.19 For CI, broken-symmetry32 unrestricted SCF wave functions converged to the dominant zwitterionic closed shell wavefunction, yielding the same results as for restricted spin singlet calculations. For ozone, open-shell singlet M06-2X calculations yield a spin density separation across the outer oxygen atoms, which implies a stronger contribution of the bi-radical component in the wavefunction; S2 remains <0.01 after annihilation of the first spin contaminant. For the reactions of CH2OO with CH2OO and O3, the subsequent chemistry involves singlet bi-radicals; these were likewise characterized using broken-symmetry SCF. As most of these bi-radicals have independent radical sites, it is expected that their singlet and triplet spin states have near-identical energies and properties; we therefore compared the optimized singlet and triplet bi-radical spin states, which serves as a further confirmation the singlet state was characterized correctly. For some of the biradical structures, we were unable to obtain open-shell ROHF wavefunctions for use in the CCSD(T) calculations, or were unable to get the CCSD calculations to converge owing to deficiencies in the ROHF wave function. For these structures, we don't report CCSD(T) results; note that singlet UHF are severely spin-contaminated, with S2 as high as 3 after annihilation of the first spin contaminant rather than the expected 0.0 value, and should not be used as a reference wavefunction. The T1 diagnostics, a measure of the multi-reference character of a compound, are listed in the ESI,† and differ significantly across the structures investigated, even for similar TS. This indicates that the multi-reference character of the wavefunction varies significantly and the use of single-reference methods might prove to be less accurate for some reactions. Generally, T1 values above 0.044 are considered to yield unreliable results.33All DFT calculations, and the ROCCSD(T) calculations for CI + CI and CI + O3 were done using Gaussian-09;34 ROHF/UCCSDT calculations for CI + alkene were performed using the Molpro-2010.1 program.35 The ball-and-stick models were generated using Jmol.36
b. Theoretical kinetic calculations
The rate coefficient for CI addition to alkenes, and for the addition of O3 to CH2OO was calculated using canonical transition state theory (TST), in a rigid rotor harmonic oscillator approximation. The reaction entropy change is driven predominantly by the loss in the TS of degrees of freedom for translation and rotation of the reactants, leading to a rigid TS; the TS remains sufficiently reactant-like to suggest that entropy changes related to skeleton vibrations and internal rotors of the reactant moieties remain comparatively small. The potential energy profile of a typical CI + alkene reaction is shown in Scheme 1, while that for CH2OO + O3 is shown in Scheme 4. In principle, such reaction schemes involving a pre-reactive complex ought to be treated using a two-transition state model, with an “outer” TS for the formation of the pre-reactive complex and an “inner” TS for the addition, preferably in a Master Equation paradigm describing the collisional energy exchange of the complexes competing against the energy-specific redissociation or addition. The outer TS is essentially the long-range attraction between the reactants, with a high rate coefficient near the collision limit for formation of the complexes. The inner TS, on the other hand, is very rigid, with energy-specific rate coefficients well below those for redissociation or collisional energy transfer, making it the rate limiting step. As tunneling is negligible, the pre-reactive complex thus has little impact on the overall reaction kinetics. For a protruding inner TS, the complex cancels out the rate equations, but for a submerged inner TS, the population at the inner TS may not have a Maxwell–Boltzmann shape below the high-pressure limit. However, as the TS considered in this work is not too deeply submerged, we opt to still use thermal partition functions to describe the TS populations as a first-order approximation. | ||
| Scheme 1 Stylized representation of the potential energy surface of the reaction of CI with alkenes, forming cyclic peroxides. The cyclo-addition TS can be above (dotted line) or below the energy level of the separated reactants. | ||
To improve the rate estimation for the CI + alkene reactions, we performed rate calculations for the reaction of ozone with alkenes (see Table SI-1, ESI†), for which extensive experimental rate data are available.37 As both the O3 and CI addition on alkenes share the 1,3-dipole cycloaddition mechanism, we can estimate the uncertainty on the rate predictions at the level of theory employed. The rate coefficients for CI + alkene rate coefficients are then scaled by the difference between the a priori predictions and the experimental observations for O3 + alkene; scaling factors are listed in Table SI-1 (ESI†).
The reaction of CH2OO + CH2OO was found to be barrierless. The reaction path was characterized by a large set of constrained geometry optimizations and frequency analyses along the minimum energy pathway at the M06-2X level of theory. The resulting energy profile and vibrational wavenumbers were used in micro-canonical variational transition state calculations to obtain the temperature-dependent rate coefficient, based on a rigid rotor harmonic oscillator approximation. Interpolation of the molecular characteristics between the characterized points along the reaction coordinate (RC) was done using cubic splines. The lowest predicted wavenumber, corresponding to a transitional degree of freedom, showed erratic behavior along the reaction coordinate due to numerical limitations in the optimization and frequency calculations; for this degree of freedom the wavenumber along the RC was smoothed following the profile of the other transitional degrees of freedom.
CI + alkenes
a. Reaction mechanism
The reaction mechanism of the reaction of carbonyl oxides with alkenes is similar to the ozonolysis of alkenes, i.e. they belong to the class of 1,3-dipolar cycloadditions, showing a concerted pericyclic cycloaddition, as shown in Scheme 1. Both reactions proceed through a pre-reactive complex, and pass over a cyclic transition state that shows 6-electron Huckel aromatic properties; this rigid TS structure is the cause of the low entropy of activation and concomitant low reaction rates found for this type of reactions.37 The cyclic peroxide formed in CI + alkene reactions is significantly more stable than the cyclotrioxalane (the “primary ozonide”, POZ) formed in the ozonolysis; the Evans–Polanyi principle suggests thus that a lower TS energy can be expected.b. Reaction barriers and rate coefficients
Table 1 summarizes the stability of the pre-reactive complex, and the energy of the lowest TS for a series of CI + alkene reactions, while Table 2 lists the predicted total rate coefficients at room temperature. The ESI† summarizes the reactivity trends (Fig. SI-1, ESI†), and lists more extensive tables for all TS and complex conformers (Table SI-2, ESI†). The CI and substrate compounds chosen play a smaller role in atmospheric chemistry, but serve as proxies for the larger biogenic terpenoids that constitute the bulk of the organic matter emitted into the atmosphere. The barrier heights for many of the CI + alkene reactions were found to be significantly lower than the corresponding ozonolysis reaction, in agreement with the work by Crehuet et al.23 and Lan et al.24 on H2COO. Many of the reaction barriers we obtained for larger VOC and CI are below the energy of the free reactants.| Reaction | Level of theory | Pre-reactive complex | Cycloaddition TS |
|---|---|---|---|
| Ethene + H2COO | M06-2X/aVDZ | −3.55 | −0.80 |
| CCSD(T)/aVTZ//M06-2X | −2.55 | 0.00 | |
| CCSD(T)/aVQZ//M06-2X | −2.18 | 0.63 | |
| Ethene + syn-CH3CHOO | M06-2X/aVDZ | −3.58 | 4.58 |
| CCSD(T)/aVTZ//M06-2X | 4.74 | ||
| Ethene + anti-CH3CHOO | M06-2X/aVDZ | −4.01 | 0.21 |
| CCSD(T)/aVTZ//M06-2X | 0.54 | ||
| Ethene + (CH3)2COO | M06-2X/aVDZ | −4.63 | 5.56 |
| CCSD(T)/aVTZ//M06-2X | 5.40 | ||
| Propene + H2COO | M06-2X/aVDZ | −5.33 | −2.85 |
| CCSD(T)/aVTZ//M06-2X | −1.55 | ||
| Propene + syn-CH3CHOO | M06-2X/aVDZ | −5.07 | 2.91 |
| CCSD(T)/aVTZ//M06-2X | 3.40 | ||
| Propene + anti-CH3CHOO | M06-2X/aVDZ | −6.39 | −1.79 |
| CCSD(T)/aVTZ//M06-2X | −1.25 | ||
| Propene + (CH3)2CHOO | M06-2X/aVDZ | −6.09 | 3.95 |
| trans-2-Butene + H2COO | M06-2X/aVDZ | −5.98 | −2.68 |
| CCSD(T)/aVTZ//M06-2X | −1.34 | ||
| trans-2-Butene + syn-CH3CHOO | M06-2X/aVDZ | −5.08 | 4.74 |
| CCSD(T)/aVTZ//M06-2X | 4.03 | ||
| trans-2-Butene + anti-CH3CHOO | M06-2X/aVDZ | −7.09 | −0.35 |
| CCSD(T)/aVTZ//M06-2X | −1.07 | ||
| trans-2-Butene + (CH3)2COO | M06-2X/aVDZ | −6.48 | 5.14 |
| cis-2-Butene + H2COO | M06-2X/aVDZ | −5.57 | −2.69 |
| CCSD(T)/aVTZ//M06-2X | −1.21 | ||
| cis-2-Butene + syn-CH3CHOO | M06-2X/aVDZ | −5.50 | 2.58 |
| CCSD(T)/aVTZ//M06-2X | 4.09 | ||
| cis-2-Butene + anti-CH3CHOO | M06-2X/aVDZ | −8.90 | −2.90 |
| CCSD(T)/aVTZ//M06-2X | −1.03 | ||
| cis-2-Butene + (CH3)2COO | M06-2X/aVDZ | −6.52 | 4.62 |
| 2,3-diMe-2-Butene + H2COO | M06-2X/aVDZ | −7.50 | −4.75 |
| 2,3-diMe-2-Butene + syn-CH3CHOO | M06-2X/aVDZ | −7.59 | 2.72 |
| 2,3-diMe-2-Butene + anti-CH3CHOO | M06-2X/aVDZ | −8.28 | −3.47 |
| 2,3-diMe-2-Butene + (CH3)2COO | M06-2X/aVDZ | −8.32 | 4.12 |
| Isoprene + H2COO | M06-2X/aVDZ | −6.25 | −1.71 |
CF3–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 + H2COO CH2 + H2COO |
M06-2X/aVDZ | −6.23 | −3.05 |
| CF3–CHCH2 + syn-CF3CHOO |
M06-2X/aVDZ | −5.90 | −3.31 |
| CF3–CHCH2 + anti-CF3CHOO |
M06-2X/aVDZ | −6.21 | −4.05 |
| Reaction | Level of theory | k(298 K) | k(298 K) scaled |
|---|---|---|---|
|
a Values for CF3CHCH2 considered unreliable (see text).
|
|||
| Ethene + H2COO | M06-2X/aVDZ | 6.24 × 10−14 | 2.13 × 10−14 |
| CCSD(T)/aVTZ//M06-2X | 1.62 × 10−14 | 8.75 × 10−15 | |
| CCSD(T)/aVQZ//M06-2X | 5.61 × 10−15 | 5.45 × 10−15 | |
| Ethene + syn-CH3CHOO | M06-2X/aVDZ | 1.52 × 10−18 | 5.20 × 10−19 |
| CCSD(T)/aVTZ//M06-2X | 1.17 × 10−18 | 6.32 × 10−19 | |
| Ethene + anti-CH3CHOO | M06-2X/aVDZ | 2.85 × 10−15 | 9.73 × 10−16 |
| CCSD(T)/aVTZ//M06-2X | 1.62 × 10−15 | 8.78 × 10−16 | |
| Ethene + (CH3)2COO | M06-2X/aVDZ | 3.28 × 10−19 | 1.12 × 10−19 |
| CCSD(T)/aVTZ//M06-2X | 4.35 × 10−19 | 2.35 × 10−19 | |
| Propene + H2COO | M06-2X/aVDZ | 2.37 × 10−14 | 1.54 × 10−14 |
| CCSD(T)/aVTZ//M06-2X | 2.63 × 10−15 | 1.93 × 10−15 | |
| Propene + syn-CH3CHOO | M06-2X/aVDZ | 6.56 × 10−18 | 4.26 × 10−18 |
| CCSD(T)/aVTZ//M06-2X | 2.83 × 10−18 | 2.08 × 10−18 | |
| Propene + anti-CH3CHOO | M06-2X/aVDZ | 3.04 × 10−14 | 1.98 × 10−14 |
| CCSD(T)/aVTZ//M06-2X | 1.21 × 10−14 | 8.93 × 10−15 | |
| Propene + (CH3)2COO | M06-2X/aVDZ | 6.22 × 10−19 | 4.04 × 10−19 |
| trans-2-Butene + H2COO | M06-2X/aVDZ | 1.74 × 10−13 | 1.87 × 10−14 |
| CCSD(T)/aVTZ//M06-2X | 1.34 × 10−14 | 2.00 × 10−15 | |
| trans-2-Butene + syn-CH3CHOO | M06-2X/aVDZ | 3.49 × 10−19 | 3.76 × 10−20 |
| CCSD(T)/aVTZ//M06-2X | 9.37 × 10−19 | 1.69 × 10−19 | |
| trans-2-Butene + anti-CH3CHOO | M06-2X/aVDZ | 2.93 × 10−15 | 3.16 × 10−16 |
| CCSD(T)/aVTZ//M06-2X | 9.41 × 10−15 | 1.40 × 10−15 | |
| trans-2-Butene + (CH3)2COO | M06-2X/aVDZ | 5.52 × 10−20 | 5.95 × 10−21 |
| cis-2-Butene + H2COO | M06-2X/aVDZ | 2.81 × 10−13 | 3.03 × 10−14 |
| CCSD(T)/aVTZ//M06-2X | 2.99 × 10−14 | 4.44 × 10−15 | |
| cis-2-Butene + syn-CH3CHOO | M06-2X/aVDZ | 4.68 × 10−18 | 5.03 × 10−19 |
| CCSD(T)/aVTZ//M06-2X | 4.82 × 10−19 | 7.15 × 10−20 | |
| cis-2-Butene + anti-CH3CHOO | M06-2X/aVDZ | 7.01 × 10−14 | 7.55 × 10−15 |
| CCSD(T)/aVTZ//M06-2X | 3.96 × 10−15 | 5.89 × 10−16 | |
| cis-2-Butene + (CH3)2COO | M06-2X/aVDZ | 9.67 × 10−20 | 1.04 × 10−20 |
| 2,3-diMe-2-Butene + H2COO | M06-2X/aVDZ | 1.59 × 10−12 | 7.96 × 10−14 |
| 2,3-diMe-2-Butene + syn-CH3CHOO | M06-2X/aVDZ | 1.29 × 10−18 | 6.47 × 10−20 |
| 2,3-diMe-2-Butene + anti-CH3CHOO | M06-2X/aVDZ | 7.49 × 10−14 | 3.75 × 10−15 |
| 2,3-diMe-2-Butene + (CH3)2COO | M06-2X/aVDZ | 3.90 × 10−20 | 1.95 × 10−21 |
| Isoprene + H2COO | M06-2X/aVDZ | 1.11 × 10−13 | 1.78 × 10−13 |
| CF3–CHCH2 + H2COO |
M06-2X/aVDZ | (1.76 × 10−13)a | |
| CF3–CHCH2 + syn-CF3CHOO |
M06-2X/aVDZ | (7.79 × 10−12)a | |
| CF3–CHCH2 + anti-CF3CHOO |
M06-2X/aVDZ | (2.85 × 10−11)a | |
c. Error analysis
The uncertainty on the predicted rate coefficients can be gauged by examining the rate coefficient of the ozonolysis reaction, a similar 1,3-dipole cycloaddition. Table SI-1 (ESI†) lists the predicted rate coefficients at the various levels of theory, compares them to the experimental rate coefficient,37 and presents the scaling factor needed to bring the predicted rate coefficient in agreement with experiment. In all cases, higher levels of theory show improved performance. For small alkenes, the agreement is generally good even at the M06-2X level of theory, with scaling factors of less than 2. For larger alkenes, the agreement deteriorates somewhat, with scaling factors between 1.6 and 20; still, this corresponds to an error on the barrier height of less than 2 kcal mol−1, and systematic improvement of the level of theory should improve these numbers further. For trifluoropropene, we find a large discrepancy of 2 orders of magnitude. We therefore consider the calculations using this compound unreliable, and the values are listed only for reference purposes.We should also consider that some of the reactions involve structures with a stronger multi-reference wavefunction than can be described by our chosen level of theory. The ESI† lists the T1 and/or D1 diagnostics for each of the structures, so that those most likely to be affected can be identified. Another source of error, which cannot be corrected for by comparison with ozonolysis, is the impact of the submerged barriers in many of these reactions. In all cases, we find rather low rate coefficients, well below the collision limit and the expected equilibration time between the pre-reactive complex and free reactants. As such, we feel that the reaction rate is likely not affected too much by our simplified kinetic treatment using only a single TS.
Overall, we estimate an uncertainty of an order of magnitude on the predicted rate coefficients, after scaling relative to the reference ozonolysis reaction. Improving the level of theory carries a significant computational cost; at this time, we prefer to wait for experimental evidence that the CI + alkene reaction can be relevant.
d. Discussion
We can distinguish three separate effects impacting the reaction rate of CI + alkene. First, for higher degrees of substitution on the alkenes, we find lower barriers to addition (e.g. CH2OO + ethene, propene, TME); this trend matches the well-known structure–activity relationship for ozonolysis. Second, and typically counteracting the first trend, we find that higher degrees of substitution on the alkene and/or CI result in strong steric hindrance between the substituents. This is illustrated clearly in the more detailed Table SI-2 (ESI†), where different orientations of the reactants can lead to strongly differing barrier heights by up to 4 kcal mol−1. This hindrance thus induces a pronounced regio- and stereo-selectivity in the reaction of CI + alkenes, and furthermore effectively inhibits reactions of highly substituted CI such as (CH3)2COO. Third, we see a clear effect of the relative stability of different alkenes and CI conformers, e.g. syn/anti-CH3CHOO or cis/trans-2-butene as listed in Table 3. The transition state already has some degree of peroxide character, where the different conformers are not separated by significant energy differences. The potential energy in the higher-energy conformer of the CI or alkene can then lead to lower relative energies of the TS, though the absolute energy remains comparable to the lower-energy reactant conformer transition state.| Compound | M06-2X/aVTZ | CCSD(T)/aVTZ//M06-2X |
|---|---|---|
| trans-2-Butene | 0.00 | 0.00 |
| cis-2-Butene | 1.23 | 0.99 |
| syn-CH3CHOO | 0.00 | 0.00 |
| anti-CH3CHOO | 3.49 | 3.48 |
The three interacting trends lead to widely differing rate coefficients. Generally, we conclude that towards alkenes, highly substituted CI will be non-reactive, CH2OO will be most reactive, and syn-CI are much less reactive than anti-CI.
CH2OO + O3
a. Reaction mechanism
Scheme 2 shows the PES of the reaction of CH2OO with O3 molecules, Table SI-3 (ESI†) lists the relative energies of the individual intermediate and TS conformers, and Fig. SI-2 (ESI†) shows the energy profile of the addition process. At large separation, the reaction of CH2OO with ozone molecules is governed by long-range electrostatic attraction between the partially charged atoms in the zwitterionic structures of both reactants. This results in the barrierless formation of a pre-reactive complex where the most stable complex conformer, with a stability of −3.2 kcal mol−1, has no less than three electrostatic interactions (see Fig. 1): the two negatively charged outer oxygen atoms of O3 interact with the positively charged CH2 moiety in CH2OO, while the positively charged central ozone oxygen interacts with the strongly negative outer oxygen of the CI. From this complex, a submerged TS for chain addition leads to the formation of a singlet ˙OOOCH2OO˙ bi-radical; two TS conformers were characterized. We have found no evidence for a direct cycloaddition channel forming cyc-O3CH2O2–, as proposed by Kjaergaard et al.20 Cycloaddition would involve the bonding of two negatively charged O-atoms from the reactants, which seems unlikely to proceed without a sizable barrier. Conversely, the chain addition pathway associating a negatively charged O-atom with the positively charged CH2 moiety seems to be the more plausible route mechanistically. The barrier for O3-addition from the complex is comparatively small, 0.35 kcal mol−1 at the CCSD(T)/aVTZ level of theory, but only 0.02 kcal mol−1 at the M06-2X level of theory and apparently non-existent at the B3LYP level of theory. We extrapolated the CCSD(T) energies from their aVDZ and aVTZ values to an infinite basis set (see Table SI-4, ESI†) using the aug-Schwartz4(DT) method described by Martin.31 The barrier height relative to the complex increased to 0.44 kcal mol−1, at an energy −2.5 kcal mol−1 below the free reactants. | ||
| Scheme 2 ZPE-corrected potential energy surface of the reaction of CI with ozone at the CCSD(T)/aVTZ//M06-2X level of theory (kcal mol−1). (a) At the CCSD(T)/aug-Schwartz(DT)//M06-2X level of theory; (b) estimated from the M06-2X level of theory; (c) estimated from CCSD(T)//CCSD(T) calculations by Kjaergaard et al.20 but not found at the M06-2X level of theory. | ||
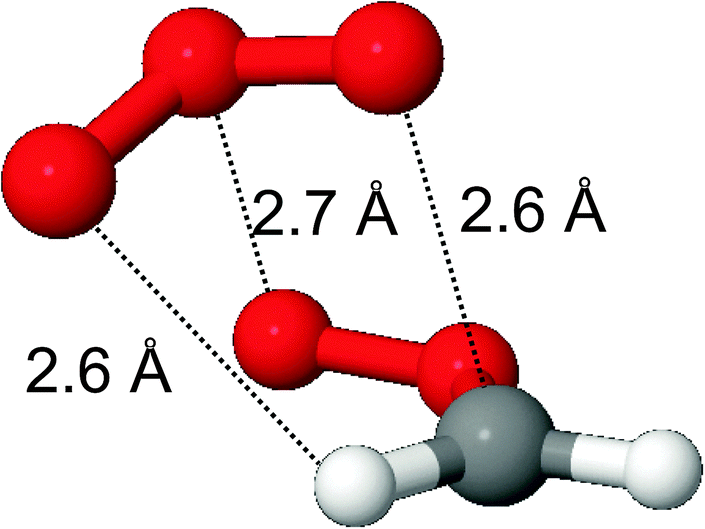 | ||
| Fig. 1 A pre-reactive complex formed in the reaction of H2COO with O3. | ||
The singlet ˙OOOCH2OO˙ bi-radical can cyclize to cyc-CH2O5– with a small energy barrier of 4 kcal mol−1. The entropically more favorable O2 elimination, however, forms triplet ˙OCH2OO˙ which in turn readily eliminates O2 to form formaldehyde. Kjaergaard et al.20 characterized a direct O2 elimination TS from cyc-CH2O5– forming triplet ˙OCH2OO˙; we were unable to reproduce this channel at the M06-2X/aVTZ level of theory, and all efforts led to formation of ˙OOOCH2OO˙, which in turn could not be found using B3LYP.
The quantum chemical calculations proved very challenging, and obtaining the lowest energy SCF wave function required explicit care. Several intermediates and TSs were characterized, only to find that these were artifacts of incorrect SCF convergence. Of particular note is closed-shell TS for chain-addition at 0.64 and 9.0 kcal mol−1 above the reactants. The main difference between the M06-2X results presented here and the PES by Kjaergaard et al.20 based on the older B3LYP functional is the stability of the singlet ˙OOOCH2OO˙ bi-radical. We prefer the ROCCSD(T)//M06-2X PES as it shows the expected features as already encountered for other CI reactions.4,19,21
b. Discussion
The internal energy afforded by the O3-addition on the CI carbon, combined with the low-lying TS for sequential O2 losses suggests that the product of the CI + O3 reaction will always be the carbonyl compound + 2O2, irrespective of the CI substituents. The key parameter is therefore the rate coefficient of this reaction. In the assumption that the pre-reactive complex quickly equilibrates with the free reactants, we predict an overall rate coefficient of 1.0 × 10−12 cm3 molecule−1 s−1 at room temperature. Especially for small CI, however, it seems likely that the reaction is in the low-pressure or fall-off regime, which would lower the overall rate coefficient somewhat due to a higher relative contribution of redissociation of the complex. This would also result in a weak pressure dependence. The rate coefficient shows a negative temperature dependence that can be described between 250–350 K by the Arrhenius expression k(T) = 3 × 10−14 exp(1068/T), again assuming that the inner TS for chain addition is the rate limiting step, i.e. where complex redissociation is significantly faster. The uncertainty on the rate coefficient is conservatively estimated at up to an order of magnitude, until higher-level calculations are available to confirm the PES found in this work, and until the addition/redissociation competition for the complex has been characterized.CH2OO + CH2OO
a. Reaction mechanism
Similar to the reaction of CI + O3, the CH2OO + CH2OO reaction is governed by strong electrostatic attraction at large reactant separation, driven by the zwitterionic character of the Criegee intermediates. The energy profile of cycloaddition, depicted in Fig. SI-3 (ESI†), is purely attractive, without evidence for a pre-reactive complex or TS. The approach initially occurs in-plane, with an elongated hexagonal shape. A CI + CI approach with the central O-atom pointing toward the coreactant CI leads to broad energy barriers, and is not competitive. At intermediate distances, the Criegee intermediates rotate to present the π-system to their co-reactant, such that at short distances a symmetric, simultaneous double head-to-tail addition occurs, forming two new C–O bonds. This highly exothermic reaction forms a six-membered cyclic biperoxide (see Scheme 3) at an M06-2X energy of ∼−100 kcal mol−1. We also characterized a cyclic –O2CH2CH2O2– intermediate, and examined part of its subsequent chemistry (see Scheme SI-1, ESI†). However, even after discounting the much lower exothermicity, such a head-to-head addition involves a high energy barrier of at least 10 kcal mol−1, owing to the need to overcome the electrostatic repulsion between similarly charged atoms. For similar reasons, we reject a carbon-to-carbon chain-addition mechanism. The cyc-O2CH2CH2O2– reaction branch is therefore likely superfluous.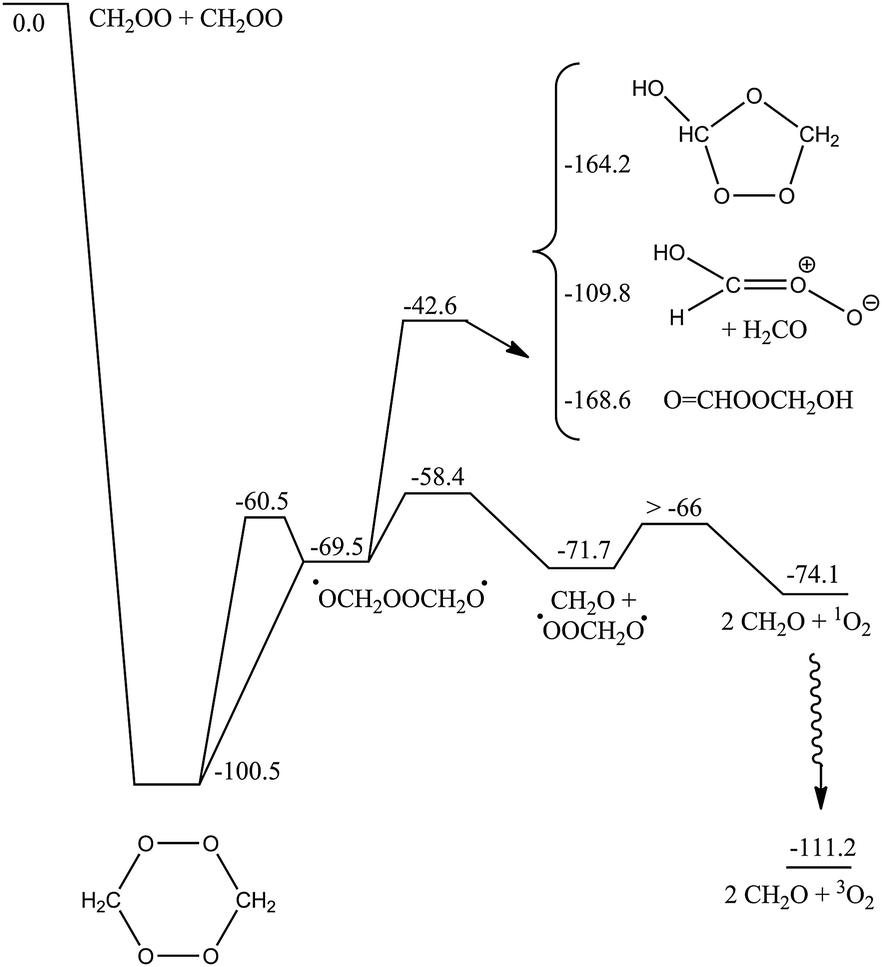 | ||
| Scheme 3 ZPE-corrected potential energy surface (kcal mol−1) of the CH2OO + CH2OO head-to-tail cycloaddition reaction at the M06-2X level of theory. All biradicals are in their singlet state, except 3O2. Partial CCSD(T)//M06-2X energies are available in the ESI,† as is a partial PES for head-to-head (cyclo)addition. | ||
The subsequent chemistry of the biperoxide, shown in Scheme 3, involves chemically activated breaking of the weaker O–O bonds, forming a singlet peroxide bisalkoxy radical ˙OCH2OOCH2O˙, which can eliminate CH2O, forming singlet ˙OOCH2O˙, which in turn can dissociate to CH2O + O2(1D) with an overall exothermicity of over 70 kcal mol−1. There are several higher-energy pathways accessible. Similar to the chemistry recently proposed21 for CI + carbonyl compounds, 1,5-H-migration reactions can form the OCHOOCH2OH intermediate. A 1,2-H-shift, followed by rapid cyclization or decomposition can form a hydroxy-substituted secondary ozonide, or an anti-hydroxy-CI + H2CO. The chemically activated intermediates, in turn, allow access to a rich chemistry forming acids, peracids and carbonyl compounds (see Scheme 4). In agreement with earlier work by Selçuki and Aviyente38 we find that syn-hydroxy-CI are unstable and isomerise to a peracid. CCSD(T)//M06-2X relative energies of a subset of structures are also available in Table SI-5 (ESI†).
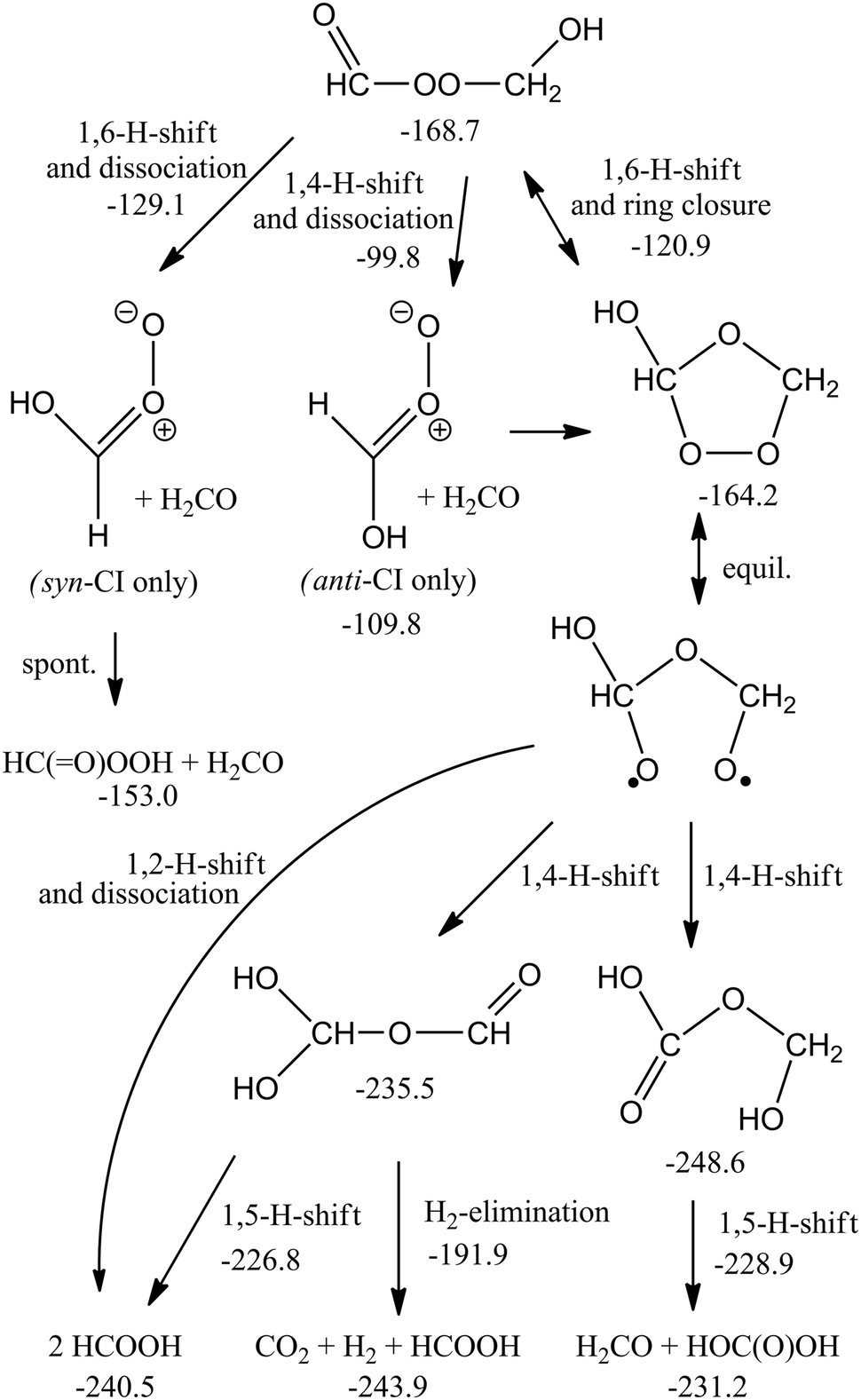 | ||
| Scheme 4 Reactions and products accessible through H-migration reactions in the ˙OCH2OOCH2O˙ biradical. Numbers indicate ZPE-corrected M06-2X/aug-cc-pVTZ relative energies (kcal mol−1) relative to the free reactants. | ||
b. Discussion
Variational microcanonical TST predictions of the rate coefficient yield k(300 K) = ∼3.8 × 10−11 cm3 molecule−1 s−1, constant within 20% over the 250 K to 350 K interval. The kinetic bottleneck is found at very large separations, ∼7–8 Å, where the electrostatic complex starts to form and becomes rigid. The predicted rate depends on the characteristics of the transitional degrees of freedom at these distances, as well as on the strength of the attractive potential at the M06-2X level of theory; we suspect that this rate coefficient is an underestimation. Overall, the uncertainty on the rate coefficient is estimated at a factor of 5. The recent direct IR-absorption measurements of Su et al.14 show a fast decay in the CH2OO absorption signal, which was also attributed to fast CI + CI reactions; our predictions for a fast reaction rate approaching the collision limit are thus supported by these observations.For smaller CI, the adduct will predominantly dissociate into carbonyl compounds + O2(1D). For larger CI, such as those formed from terpenoids, we should consider collisional stabilization of the cyclic biperoxide as well. The high nascent energy content could also enable other isomerisation or decomposition channels; examples include the formation of acids by internal H-migration as already proposed in the CI + SO2 and CI + carbonyl reactions.4,21Scheme 4 shows a set of reactions that are accessible through this H-migration; the intermediates formed have internal energies as high as 250 kcal mol−1, which ensure very fast isomerisation and/or dissociation even for very large CI. A Master Equation-based product analysis is deferred to a future publication, as is a study of the dependence of this chemistry on the CI substituents. Compared to the CI + carbonyl reactions recently studied by Jalan et al.,21 the isomerisation channels lie significantly lower compared to the free reactants, and contribution of these channels can be expected to be larger than for secondary ozonides, and remain relevant at higher pressures.
Box modeling of experimental reaction conditions
To illustrate the importance of the title reactions, we show the results of a modeling study of an ozonolysis experiment39 of trans-2-butene. 2-Butene (1.8 × 1015 molecules cm−3), ozone (1.0 × 1013 molecules cm−3), and propane (2.5 × 1016 molecules cm−3) were mixed in a flow tube with 1 atm of N2 carrier gas with 4% of O2, and allowed to react for 2.5 s prior to sampling through a steel nozzle into a low-pressure FAGE system (Fluorescence Assay by Gas Expansion) where the temporal formation of OH from syn-CH3CHOO decomposition was measured. A detailed description of this experimental study39 will be presented in separate publications; for our current purposes we are interested mainly in the prediction of the steady-state concentration of the CI, and specifically reactions that influence this concentration. The key result in this respect is that we are able to predict, quantitatively and without further fitting, the full time-dependent OH profile generated by CI decomposition as measured within the FAGE instrument, by applying the Master Chemical Mechanism (MCM 3.2)40,41 obtained from website http://mcm.leeds.ac.uk/MCM, supplemented with literature values and estimates for stabilized CI chemistry (Table 4). The quantitative agreement indicates that the CI concentration predicted in the flow tube at the sampling point is reproduced correctly by the model. The few additions to the MCM are the syn- and anti-specific reactions of CH3CHOO with carbonyl-,4,12 hydroxy-,4 and carboxylic compounds;4 their reaction with HO2 and RO2;4,19 the reaction of CI with O3, CI, and alkenes as characterized in this work; the IUPAC-recommended yield of 0.18 for stabilized CI;37 an estimated nascent 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 syn- to anti-ratio of stabilized CH3CHOO (in line with their relative energies); and a thermal decomposition rate of 3 s−1 for syn-CH3CHOO as obtained by Berndt et al.11 For the low-pressure section in the FAGE instruments, wall losses of OH were measured at 65 s−1, while wall loss of CI was estimated to be similar to that measured for HO2, 20 s−1. Fig. 2 shows the absolute contributions of CI sources and sinks in the 1 atm flow tube chemistry, while Fig. SI-4 (ESI†) shows the concentration–time profiles. We find that a multitude of reactions govern the predicted steady-state concentration of the CI, not only the unimolecular decomposition but also the reactions of CI with oxygenated intermediates, the reaction of CI + O3, and the CI + CI and CI + HO2/RO2 reactions. The relative contributions of these channels change over time, with e.g. O3 being important mostly at the reaction start when its concentration is still high, while the contributions of oxygenates increase as they are formed in the secondary chemistry. While the impact of each reaction on the CI concentration is limited, and subject to large uncertainties, it is also manifest that neglecting the summed contribution of these non-traditional CI chemical loss processes would lead to an incorrect prediction of the CI steady state concentration, and hence would induce a systematic disagreement between the kinetic model and experimental observations.
:1 syn- to anti-ratio of stabilized CH3CHOO (in line with their relative energies); and a thermal decomposition rate of 3 s−1 for syn-CH3CHOO as obtained by Berndt et al.11 For the low-pressure section in the FAGE instruments, wall losses of OH were measured at 65 s−1, while wall loss of CI was estimated to be similar to that measured for HO2, 20 s−1. Fig. 2 shows the absolute contributions of CI sources and sinks in the 1 atm flow tube chemistry, while Fig. SI-4 (ESI†) shows the concentration–time profiles. We find that a multitude of reactions govern the predicted steady-state concentration of the CI, not only the unimolecular decomposition but also the reactions of CI with oxygenated intermediates, the reaction of CI + O3, and the CI + CI and CI + HO2/RO2 reactions. The relative contributions of these channels change over time, with e.g. O3 being important mostly at the reaction start when its concentration is still high, while the contributions of oxygenates increase as they are formed in the secondary chemistry. While the impact of each reaction on the CI concentration is limited, and subject to large uncertainties, it is also manifest that neglecting the summed contribution of these non-traditional CI chemical loss processes would lead to an incorrect prediction of the CI steady state concentration, and hence would induce a systematic disagreement between the kinetic model and experimental observations.
| Coreactant | CI | k est(298 K) | Ref. |
|---|---|---|---|
| H2O | H2COO | 2 × 10−16 cm3 s−1 | 13, 42 |
| syn-CH3CHOO | 2 × 10−19 cm3 s−1 | 13, 42 | |
| anti-CH3CHOO | 1 × 10−14 cm3 s−1 | 13 | |
| (CH3)2COO | 2 × 10−18 cm3 s−1 | 13, 42 | |
| (H2O)2 | H2COO | 7 × 10−11 cm3 s−1 | 4, 43, 44 |
| syn-CH3CHOO | 3 × 10−14 cm3 s−1 | 4, 43, 44 | |
| anti-CH3CHOO | 5 × 10−11 cm3 s−1 | 4, 43, 44 | |
| (CH3)2COO | 8 × 10−16 cm3 s−1 | 4, 43, 44 | |
| Carbonyl compounds | All | 6 × 10−13 cm3 s−1 | 12 |
| Hydroxy compounds | All | 5 × 10−12 cm3 s−1 | 4 |
| Carboxylic acids | All | 5 × 10−12 cm3 s−1 | 4 |
| Olefins | see Table 2 | This work | |
| NO | All | 2 × 10−18 cm3 s−1 | 4 |
| NO2 | H2COO | 7 × 10−12 cm3 s−1 | 9 |
| syn-CH3CHOO | 2 × 10−12 cm3 s−1 | 13 | |
| anti-CH3CHOO | 2 × 10−12 cm3 s−1 | 13 | |
| (CH3)2COO | 4 × 10−12 cm3 s−1 | 4, 9, 13 | |
| SO2 | H2COO | 4 × 10−11 cm3 s−1 | 9 |
| syn-CH3CHOO | 2 × 10−11 cm3 s−1 | 13 | |
| anti-CH3CHOO | 7 × 10−11 cm3 s−1 | 13 | |
| (CH3)2COO | 4 × 10−11 cm3 s−1 | 4, 9, 13 | |
| O3 | All | 1 × 10−12 cm3 s−1 | This work |
| CO | All | 4 × 10−14 cm3 s−1 | 4 |
| OH | All | 5 × 10−12 cm3 s−1 | 4 |
| HO2 | All | 5 × 10−12 cm3 s−1 | 4 |
| RO2 | All | 5 × 10−12 cm3 s−1 | 4 |
| H2SO4 | All | 5 × 10−12 cm3 s−1 | 4 |
| Decomposition | All syn-CI | 3 s−1 | 11, 39 |
| CI + CI | All | 4 × 10−11 cm3 s−1 | This work |
 | ||
| Fig. 2 Absolute contribution of the reactions affecting the concentration of syn-CH3CHOO in the ozonolysis of trans-2-butene by at least 1%. Formation (positive contribution) via 2-butene + O3 is exclusive. Destruction reactions (negative contributions), shown cumulatively from zero downward in decreasing order of importance at the sample point at 2.5 s, are RO2 (red), carbonyl compounds (yellow), O3 (light blue), hydroxy compounds (dark blue), unimolecular decomposition to OH (cyan), HO2 (green), and CI + CI (black). | ||
Contributions under atmospheric conditions
In earlier work we analyzed the relative contribution of many coreactants in the atmosphere to the atmospheric fate of a set of CI intermediates, ranging from the simplest CH2OO to isoprene-derived SCI.4Table 4 shows an updated list of rate coefficients, where the main modifications compared to Vereecken et al.4 are (i) updated values of the CI-specific rate coefficients with H2O and (H2O)2, where we employ the relative rate predictions by Anglada et al.42 and Ryzhkov and Ariya43 scaled to match the absolute value for anti-CH3CHOO+H2O as measured by Taatjes et al.,13 (ii) the lumped rate coefficient of CI + carbonyl compounds was changed to 6 × 10−13 cm3 molecule−1 s−1 based on the measurements of Taatjes et al.,12 and (iii) inclusion of the CI + O3 reaction as studied in this work. Table 5 lists the predicted contributions of the various loss processes in different environments as defined earlier by Vereecken et al.4 Compared to these earlier estimates, the lower rate coefficient of reaction with water opens the possibility for higher contributions of other CI loss processes; we also find that our current prediction of the CI + O3 reaction rate makes this channel competitive.| Loss path | Boreal forest | Tropical forest | Mega city | Rural Europe |
|---|---|---|---|---|
| H2COO | ||||
| H2O | 0.01 | 0.01 | 0.01 | |
| (H2O)2 | 0.99 | 1.00 | 0.99 | 0.99 |
| anti-CH3CHOO | ||||
| H2O | 0.25 | 0.18 | 0.35 | 0.26 |
| (H2O)2 | 0.75 | 0.82 | 0.64 | 0.74 |
| Ester channel | 0.01 | |||
| syn-CH3CHOO | ||||
| H2O | 0.01 | 0.01 | 0.01 | |
| (H2O)2 | 0.54 | 0.81 | 0.15 | 0.58 |
| VHP channel | 0.22 | 0.14 | 0.16 | 0.26 |
| NO2 | 0.10 | 0.01 | ||
| SO2 | 0.05 | 0.20 | 0.02 | |
| O3 | 0.07 | 0.02 | 0.12 | 0.12 |
| Carbonyl compounds | 0.01 | 0.02 | ||
| Carboxylic acids | 0.04 | 0.01 | ||
| Hydroxyl-compounds | 0.06 | 0.01 | 0.25 | |
| (CH3)2COO | ||||
| H2O | 0.11 | 0.22 | 0.03 | 0.13 |
| (H2O)2 | 0.03 | 0.08 | 0.03 | |
| VHP channel | 0.43 | 0.54 | 0.19 | 0.52 |
| NO2 | 0.11 | 0.02 | ||
| SO2 | 0.10 | 0.22 | 0.05 | |
| O3 | 0.14 | 0.08 | 0.14 | 0.24 |
| Carbonyl compounds | 0.01 | 0.02 | 0.01 | |
| Carboxylic acids | 0.07 | 0.04 | ||
| Hydroxyl-compounds | 0.11 | 0.04 | 0.29 | |
Conclusions
In this work, we characterized the reactions of CI with alkenes, CH2OO, and O3. We find that the reaction of CI with olefines is highly dependent on substitution in the alkene and CI, with very large differences in the predicted rate coefficients. The predicted reaction rates indicate that the CI + alkene reaction will have a small to negligible influence on the CI chemistry, both in the atmosphere and in experimental work, except perhaps at the highest concentrations of alkenes. The reaction of CI + O3 was found to be relatively fast, proceeding through a pre-reactive complex and a submerged barrier, and leading to the formation of a carbonyl compound and oxygen molecules. The predicted rate coefficient carries a large uncertainty, but our current best estimate indicates that this reaction can influence both atmospheric CI chemistry and laboratory work. The CH2OO + CH2OO reaction is extremely fast, proceeding with a rate near the collision limit without evidence of an energy barrier. The low concentrations of CI in the atmosphere limit the impact of this reaction to experimental conditions where higher CI-concentrations can be generated.We recommend that both the CI + O3 reaction and the CI + CI reaction be further investigated by experimental means, and theoretically using even higher levels of methodologies. Particularly the CI loss by reaction with O3 in the atmosphere might affect the ambient concentration of CI, and hence reduce the impact of CI chemistry currently proposed for H2SO4 formation or the NOx cycle.
Acknowledgements
LV is supported by the Max Planck Graduate Center with the Johannes Gutenberg-Universität Mainz (MPGC).References
- R. Criegee and G. Wenner, Justus Liebigs Ann. Chem., 1949, 564, 9–15 CrossRef CAS.
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- C. A. Taatjes, D. E. Shallcross and C. Percival, Phys. Chem. Chem. Phys., 2013, 16, 1704–1718 RSC.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2012, 14, 14682–14695 RSC.
- G. Marston, Science, 2012, 335, 178–179 CrossRef CAS PubMed.
- L. Vereecken, Science, 2013, 340, 154–155 CrossRef CAS PubMed.
- C. Percival, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, D. O. Topping, D. Lowe, S. Utembe, A. Bacak, G. McFiggans, M. Cooke, A. T. Archibald, M. Jenkin, R. G. Derwent, I. Riipinen, D. Mok, E. P. F. Lee, J. Dyke, C. A. Taatjes and D. E. Shallcross, Faraday Discuss., 2013, 165, 45–73 RSC.
- C. A. Taatjes, G. Meloni, T. M. Selby, A. J. Trevitt, D. L. Osborn, C. J. Percival and D. E. Shallcross, J. Am. Chem. Soc., 2008, 130, 11883–11885 CrossRef CAS PubMed.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204–207 CrossRef CAS PubMed.
- R. L. Mauldin III, T. Berndt, M. Sipilä, P. Paasonen, T. Petäjä, S. Kim, T. Kurtén, F. Stratmann, V.-M. Kerminen and M. Kulmala, Nature, 2012, 488, 193–196 CrossRef PubMed.
- T. Berndt, T. Jokinen, R. L. Mauldin, T. Petäjä, H. Herrmann, H. Junninen, P. Paasonen, D. R. Worsnop and M. Sipilä, J. Phys. Chem. Lett., 2012, 3, 2892–2896 CrossRef CAS.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, E. P. F. Lee, J. M. Dyke, D. W. K. Mok, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2012, 14, 10391–10400 RSC.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177–180 CrossRef CAS PubMed.
- Y.-T. Su, Y.-H. Huang, H. A. Witek and Y.-P. Lee, Science, 2013, 340, 174–176 CrossRef CAS PubMed.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Am. Chem. Soc., 2012, 134, 20045–20048 CrossRef CAS PubMed.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Chem. Phys., 2013, 138, 244307 CrossRef PubMed.
- B. Ouyang, M. W. McLeod, R. L. Jones and W. J. Bloss, Phys. Chem. Chem. Phys., 2013, 15, 17070–17075 RSC.
- M. Nakajima and Y. Endo, J. Chem. Phys., 2013, 139, 101103 CrossRef PubMed.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- H. G. Kjaergaard, T. Kurtén, L. B. Nielsen, S. Jørgensen and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 2525–2529 CrossRef CAS.
- A. Jalan, J. W. Allen and W. H. Green, Phys. Chem. Chem. Phys., 2013, 15, 16841–16852 RSC.
- W. Sander, Angew. Chem., Int. Ed. Engl., 1990, 29, 344–354 CrossRef.
- R. Crehuet, J. M. Anglada, D. Cremer and J. M. Bofill, J. Phys. Chem. A, 2002, 106, 3917–3929 CrossRef CAS.
- Y. Lan, L. Zou, Y. Cao and K. N. Houk, J. Phys. Chem. A, 2011, 115, 13906–13920 CrossRef CAS PubMed.
- W. H. Bunnelle, Chem. Rev., 1991, 91, 335–362 CrossRef CAS.
- P. R. Story and J. R. Burgess, J. Am. Chem. Soc., 1967, 89, 5726–5727 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS PubMed.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS.
- J. Zheng, I. M. Alecu, B. J. Lynch, Y. Zhao and D. G. Truhlar, Database of Frequency Scale Factors for Electronic Model Chemistries, http://comp.chem.umn.edu/freqscale/index.html.
- J. M. L. Martin, Chem. Phys. Lett., 1996, 259, 669–678 CrossRef CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS PubMed.
- J. C. Rienstra-Kiracofe, W. D. Allen and H. F. Schaefer, J. Phys. Chem. A, 2000, 104, 9823–9840 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox and J. A. Pople, Gaussian 09, Revision B.01, Gaussian Inc., Wallington CT, 2010 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version 2010.1, a package of ab initio programs, molpro, 2010 Search PubMed.
- Jmol: an open-source Java viewer for chemical structures in 3D, http://www.jmol.org.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and IUPAC Subcommittee, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- C. Selçuki and V. Aviyente, THEOCHEM, 2000, 530, 97–107 CrossRef.
- A. Novelli, K. Hens, C. Tatum Ernest, K. Trawny, M. Rudolf, Z. Hosaynali-Beygi, H. Fischer, J. Williams, L. Vereecken, M. Martinez, J. Lelieveld, H. Harder, P. Paasonen, M. Sipilä, P. Keronen, T. Petäjä, T. Elste, C. Plass-Dülmer and D. Kubistin, Assessment of the role of stabilized Criegee intermediates in the OH radical measurements by LIF, Atmos. Chem. Mechanisms Conference, Davis, CA, USA, 2012.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- J. M. Anglada, J. González and M. Torrent-Sucarrat, Phys. Chem. Chem. Phys., 2011, 13, 13034–13045 RSC.
- A. B. Ryzhkov and P. A. Ariya, Phys. Chem. Chem. Phys., 2004, 6, 5042–5050 RSC.
- A. B. Ryzhkov and P. A. Ariya, Chem. Phys. Lett., 2003, 367, 423–429 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp54514h |
| This journal is © the Owner Societies 2014 |