 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Local interactions influence the fibrillation kinetics, structure and dynamics of Aβ(1–40) but leave the general fibril structure unchanged†
Juliane
Adler
a,
Holger A.
Scheidt
a,
Martin
Krüger
b,
Lars
Thomas
a and
Daniel
Huster
*ac
aInstitute of Medical Physics and Biophysics, University of Leipzig, D-04107 Leipzig, Germany. E-mail: daniel.huster@medizin.uni-leipzig.de
bInstitute of Anatomy, University of Leipzig, D-04103 Leipzig, Germany
cDepartment of Chemical Sciences, Tata Institute of Fundamental Research, Colaba, Mumbai 400 005, India
First published on 4th March 2014
Abstract
A series of peptide mutants was studied to understand the influence of local physical interactions on the fibril formation mechanism of amyloid β (Aβ)(1–40). In the peptide variants, the well-known hydrophobic contact between residues phenylalanine 19 and leucine 34 was rationally modified. In single site mutations, residue phenylalanine 19 was replaced by amino acids that introduce higher structural flexibility by a glycine mutation or restrict the backbone flexibility by introduction of proline. Next, the aromatic phenylalanine was replaced by tyrosine or tryptophan, respectively, to probe the influence of additional hydrogen bond forming capacity in the fibril interior. Furthermore, negatively charged glutamate or positively charged lysine was introduced to probe the influence of electrostatics. In double mutants, the hydrophobic contact was replaced by a putative salt bridge (glutamate and lysine) or two electrostatically repelling lysine residues. The influence of these mutations on the fibrillation kinetics and morphology, cross-β structure as well as the local structure and dynamics was probed using fluorescence, transmission electron microscopy, X-ray diffraction, and solid-state NMR spectroscopy. While the fibrillation kinetics and the local structure and dynamics of the peptide variants were influenced by the introduction of these local fields, the overall morphology and cross-β structure of the fibrils remained very robust against all the probed interactions. Overall, 7 out of the 8 mutated peptides formed fibrils of very similar morphology compared to the wildtype. However, characteristic local structural and dynamical changes indicate that amyloid fibrils show an astonishing ability to respond to local perturbations but overall show a very homogenous mesoscopic organization.
Introduction
Amyloid fibrils are insoluble protein aggregates found in various tissues, which are responsible for the development of numerous serious diseases including Alzheimer's, Huntington's, and Parkinson's diseases.1 Although very different proteins with a unique amino acid sequence cause all these diseases, there is a prevailing structural motif that unifies all generated protein fibrils, which is the cross-β structure.1,2 This structure shows an astonishing similarity between protein fibrils of very different sequence and origin. A characteristic fingerprint of the cross-β structure is the X-ray diffraction pattern of any protein fibrils, featuring a strong meridional reflex at about 4.8 Å, corresponding to the hydrogen bonding distance between β-strands and a more diffuse 10–11 Å reflection on the equator reflecting the intersheet distance in the fibril.3 The fact that many if not all proteins can form such structures suggests that amyloid fibril formation represents a rather universal process that follows uniform principles and the comparison to synthetic polymers suggests itself.4 Although these principles are not well understood at the moment, the fact that the large structural variety of soluble proteins is converted into a single prevailing structural motif in all amyloids suggests a uniformity of this process following relatively distinct physicochemical laws.One approach to address this problem is to study peptides with a slightly altered amino acid sequence to probe the influence of local interactions on the fibrillation pathway and the structure and dynamics of the resulting fibrils. By the introduction of mutations into a peptide structure known to form fibrils, the various electrostatic and hydrophobic forces that govern the fibrillation pathway and lead to the amyloid stability are altered and the impact of single mutations on the thermodynamics of the amyloid formation can be investigated.4 By far the best studied systems are the amyloid β (Aβ) fibrils.5 Mostly from NMR structures, there is a relatively clear model of the structure6–13 and dynamics14 of these fibrils as well as of the transient oligomeric15,16 and protofibrillar17,18 intermediates. Several point mutations on the Aβ sequence are known to occur in nature, which typically lead to an early onset of Alzheimer's disease. Very prominent mutations found in nature involve the Italian (E22K), the Dutch (E22Q), the Arctic (E22G), and the Iowa mutation (D23N), which all lead to fibrils of higher toxicity but also altered physicochemical properties.19 It has been demonstrated that certain point mutations show variable rates of aggregation but overall similar aggregate morphologies.20 For the Iowa mutation, recently a significant structural alteration was found: in contrast to the in-register parallel β-sheet conformation adopted by wildtype (WT) Aβ(1–40), fibrils grown from peptides carrying the Iowa mutation show an antiparallel β-sheet structure.21
Systematic alanine22 and proline23 scanning mutagenesis of Aβ(1–40) has been carried out by Wetzel et al. to study the thermodynamic stability of these mutants. The analysis of these artificial mutants revealed that the fibril elongation equilibrium of the Ala mutants was destabilized by up to ∼4 kJ mol−1. The Pro mutagenesis indicated three contiguous segments in the core of the sequence, which were highly sensitive to Pro replacement with a thermodynamic destabilization of up to ∼13 kJ mol−1. However, the surprising result of this study was that not a single mutation could completely destabilize the fibril architecture, indicating the robustness of the cross-β structure of Aβ(1–40).
The contribution of electrostatic interactions to the stability of insulin fibrils has also been studied in pH dependent measurements that vary the charge state of the histidine residues in the fibrils.24 It was shown that the stability of the insulin fibrils is sensitive to the net electrostatics of the polypeptide chain and the formation of ion-pairing.
From these examples, it is obvious that local interactions can influence the thermodynamic stability, fibrillation stability, and the structure and dynamics of the amyloid fibrils. R. Wetzel suggested that the packed cross-β structure of amyloid fibrils represents a robust 3D array, which is stabilized by covalent bonds, electrostatic bonds (i.e. the hydrogen bonds of the β-sheet), and packing interactions such as hydrophobic contacts of the side chains.4 In order to investigate the contribution of individual physical interactions to the fibrillation kinetics, the structure and the dynamics of Aβ(1–40), we studied a series of Aβ peptides carrying several rationally chosen mutations each introducing a specific local physical interaction. These mutations do not provide any biological relevance, but allow assessing physicochemical aspects of fibril formation because they should alter the regular interaction patterns of the molecule during fibrillation and allow us to investigate the robustness of the cross-β structure of Aβ. The mutations were introduced at amino acid position phenylalanine 19 (F19) and leucine 34 (L34), which have been confirmed as a hydrophobic contact in many studies.6,8,9,15,25 This well established hydrophobic contact is challenged in our mutants.
An overview of the mutations studied here is found in Scheme 1. The first six peptide variants show a single mutation at position 19. This phenylalanine residue was replaced by a glycine in MI or a proline in MII to observe the effect of maximal or minimal conformational flexibility, respectively. The next two mutations replaced the phenylalanine by a negatively charged glutamate (MIII) or a positively charged lysine (MIV) to study the effect of fixed charges inside the hydrophobic fibril interior. Mutations MV and MVI replaced the phenylalanine by aromatic tyrosine or tryptophan aromatic ring structures that in addition allow for altered hydrogen bonding. The last two alterations of the Aβ(1–40) structure involved double mutations of F19 and L34 to lysine and glutamate (MVII) and to lysine and lysine (MVIII), which replace the hydrophobic contact by a salt bridge or two equally charged residues repelling each other.
 | ||
| Scheme 1 Overview of the Aβ(1–40) mutation studies in this work. Schematic model of the mutations and the location of the reporting amino acids V18, F20, A21, and G33 colored in red. The hydrophobic contact that was mutated in this work is highlighted in green. | ||
We used fluorescence and solid-state NMR spectroscopy, X-ray diffraction, and transmission electron microscopy (TEM) to investigate the influence of these local physical forces on the fibrillation kinetics, structure, and dynamics of the Aβ peptides. While many of these parameters are quite significantly altered for the mutated Aβ peptides, the general structure and morphology of these aggregates remain largely unchanged underlining the robustness of the 3D arrangement of Aβ fibrils.
Results
Fibrillation kinetics and general fibril morphology
Fibrils of the Aβ mutants were grown under standard conditions6 (peptide concentration of 1 mg ml−1 in phosphate buffer (150 mM NaCl), pH 7.4, continuously shaken). The kinetics of the fibrillation was followed by recording ThT fluorescence spectra. A plot of the intensity of the fluorescence maximum as a function of time provides a characteristic time, at which half of the maximal ThT fluorescence intensity is reached. This characteristic time can be taken as a measure for the kinetics of the fibrillation process of the Aβ mutants. The characteristic times for the Aβ fibril formation (tchar) are shown in Table 1. Indeed, quite substantial differences in the fibrillation kinetics of the different mutants were observed. For the WT Aβ(1–40) peptides, at a concentration of 1 mg ml−1, a characteristic fibrillation time of 9.6 h was determined. The values measured for the F19G and the F19K mutants correspond approximately to this value. The fibrillation kinetics of the F19W mutant and also the F19K L34K double mutant are moderately increased. The F19Y and F19E mutants show much slower fibrillation kinetics with significantly increased fibrillation half life times and the slowest kinetics was observed for the F19P mutant. The only mutated Aβ(1–40) peptide that did not form any fibrils within two months of observation was the double mutant F19K L34E.| Aβ Sample | Cuvette exps. | Well plate experiments | |
|---|---|---|---|
| t char/h | t char/h | t lag/h | |
| a Not determined because of dehydration of the sample. | |||
| WT | 9.6 ± 1.0 | 5.5 ± 0.1 | 4.6 ± 0.1 |
| F19G | 11.3 ± 1.0 | 31.7 ± 0.1 | 27.9 ± 0.1 |
| F19P | 100.0 ± 10.0 | —a | —a |
| F19E | 46.2 ± 5.0 | 94.8 ± 1.1 | 74.7 ± 1.3 |
| F19K | 8.2 ± 1.0 | 9.9 ± 0.1 | 7.9 ± 0.2 |
| F19Y | 21.6 ± 2.0 | 17.6 ± 0.1 | 12.3 ± 0.2 |
| F19W | 16.7 ± 1.0 | 23.9 ± 0.1 | 18.5 ± 0.1 |
| F19K L34E | >1500 | —a | —a |
| F19K L34K | 14.6 ± 1.0 | 21.7 ± 0.1 | 16.9 ± 0.2 |
We also used a 96 well plate format and recorded ThT fluorescence using a microplate reader. The fluorescence intensity curves as a function of time are shown in the ESI† (Fig. S1). The determined lag times (tlag) and tchar values are also given in Table 1. Due to dehydration of the samples, the experiment needed to be terminated after 130 h, which is not sufficient for the F19P mutant to finish fibrillation. The tchar values determined in this experiment are comparable to the cuvette experiments for most mutants except for F19G and F19E.
The morphology of the fibrils grown from the different Aβ(1–40) peptides was studied using standard transmission electron microscopy (TEM). Electron micrographs of all peptide variants are shown in Fig. 1. This plot confirms that fibrils of comparable morphology were grown for all peptide variants except for the F19K L34E double mutant. Compared to the WT Aβ(1–40) peptides, the F19P and F19Y mutants showed the shortest fibrils, while the fibrils formed by the other mutated peptides are mainly long, straight and sometimes twisted around each other (Fig. 1D). The fibrils formed from the double mutant F19K L34K appear to be somewhat denser than the others (Fig. 1G). We analyzed the diameter of the fibrils and reported these diameters in Table S1 (ESI†). While the WT fibrils comprise a diameter of 10.0 ± 1.6 nm, the smallest fibrils were grown for the F19G mutant (diameter 8.2 ± 1.3 nm) and the F19K mutant formed the thickest fibrils with a diameter of 12.6 ± 2.0 nm. The double mutant F19K L34E did not form fibrils and a TEM image revealed small and amorphous aggregates (Fig. S2, ESI†).
 | ||
| Fig. 1 Transmission electron micrographs of the fibrils prepared from the different Aβ(1–40) peptides investigated in this study. (A) WT; (B) F19G mutant; (C) F19P mutant; (D) F19E mutant; (E) F19K mutant; (F) F19Y mutant; (G) F19W mutant (H) F19K L34K double mutant. The scale bar represents 250 nm. | ||
Influence of the perturbations on the local structure of the Aβ(1–40) fibrils
Local secondary structure in the Aβ(1–40) peptide backbone can be well studied by 13C MAS NMR spectroscopy.6–12 Due to the selective labeling scheme of our peptide variants, well resolved one dimensional 13C CP MAS NMR spectra could be measured for the samples of WT and mutated Aβ(1–40) peptides. One dimensional 13C CPMAS NMR spectra are displayed in Fig. S3 (ESI†). The NMR lines are relatively broad suggesting moderate polymorphism in the local Aβ structure.10,13 Some residues show rather drastic chemical shift alterations for the different Aβ(1–40) mutants. For unambiguous assignment, two dimensional 13C–13C correlation spectra were acquired. An example of such a correlation spectrum is shown in Fig. 2. This plot shows an overlay of the 2D spectra of the WT and the F19K Aβ(1–40) mutant, where the most drastic chemical shift change was observed for residue F20. The Cα signal of F20 shows a downfield shift of 5.5 ppm, while the Cβ signal shifts upfield by about 3.1 ppm. In addition, local structure changes occur such that a weak cross peak between the Cα atoms of V18 and A21 can be observed.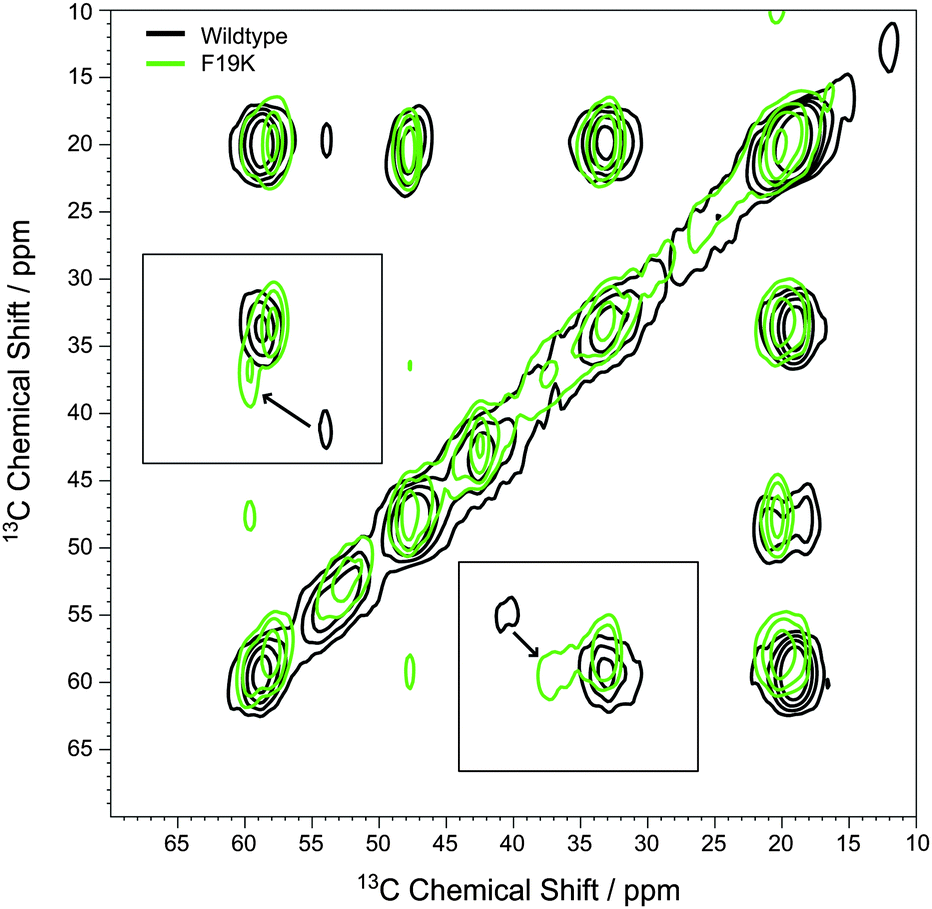 | ||
| Fig. 2 Exemplary 13C–13C PDSD spectra of the WT of Aβ(1–40) (black contours) and the Aβ(1–40) F19K mutant (green contours), measured at 30 °C using a mixing time of 100 ms and a MAS frequency of 7 kHz. The arrows indicate the very pronounced chemical shift change of the Cα/Cβ signals of F20. | ||
To summarize all chemical shift data, Fig. 3 shows the 13C NMR chemical shift differences of fibrils of a given Aβ mutant with respect to WT for all Aβ(1–40) variants studied except the F19K L34E mutation. The response of the four labeled amino acids to the local perturbations introduced by the respective mutation is quite different. For instance, V18, which is directly neighboring the mutation site F19, only shows very moderate chemical shift alterations irrespective of any mutation in this study. In contrast, F20, which is also directly neighboring F19, undergoes quite dramatic chemical shift changes. In particular in the F19K mutation the Cα signal of F20 is shifted more than 5 ppm downfield. The Cβ signal in turn shifts upfield by ≥2.5 ppm for all variants except F19Y. Clearly, significant local structure changes are induced by the mutated position 19. In contrast, residue A21 shows again a comparably moderate response to the introduced mutations, except in the F19P mutant, where the Cβ signal shifts upfield by 3.2 ppm. G33 shows a moderate response to most mutations, with the exception of F19E and F19Y, where downfield shifts of 2.3 and 5.2 ppm are observed, respectively.
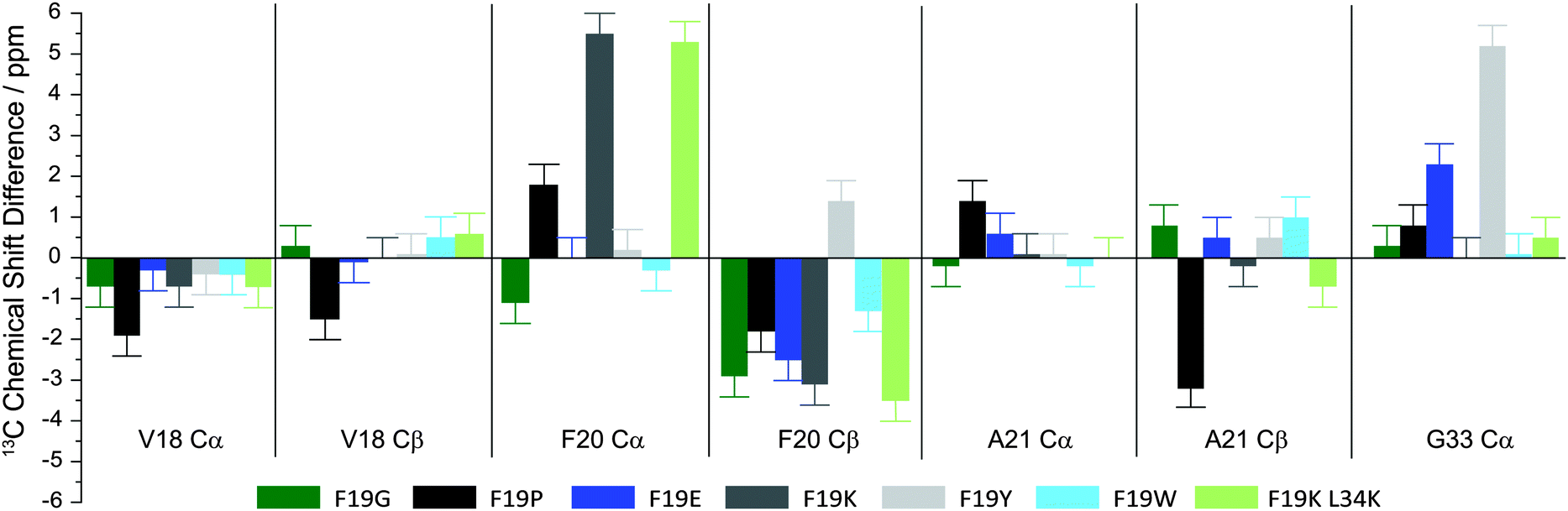 | ||
| Fig. 3 Overview of the observed 13C chemical shift differences of the labeled sites in the fibrils of various Aβ(1–40) mutants in comparison to the WT according to the color scheme given below. Chemical shifts were determined from 13C CPMAS and 13C–13C PDSP experiments. Bars represent the following peptide fibrils from left to right: F19G, F19P, F19E, F19K, F19Y, F19W, F19K L34K. | ||
We also acquired and detected 15N CPMAS NMR spectra of the various fibrils grown. The 15N CPMAS NMR spectra of all Aβ peptides are displayed in Fig. S4 (ESI†). Fig. 4 provides an overview of the 15N chemical shift perturbations of the respective variants in comparison to WT Aβ(1–40). These chemical shifts were measured in 13C–15N correlation spectra recorded using double CP experiments.26 As can be seen from the plot, the 15N chemical shift alterations of the mutated Aβ(1–40) peptides are also quite significant. In comparison to the 13C chemical shift results, all labeled amino acids show a strong response to the mutations, while the most moderate effects are observed for G33. However, the pattern of 15N chemical shift changes for the different Aβ(1–40) variants is relatively inconclusive with both upfield and downfield chemical shift changes for the same mutants. A summary of all isotropic 13C and 15N chemical shifts for all investigated Aβ(1–40) variants is given in Table S2 (ESI†).
 | ||
| Fig. 4 Overview of the observed 15N chemical shift differences of the labeled sites in the fibrils of various Aβ(1–40) mutants in comparison to the WT according to the color scheme given below. Chemical shifts were determined from 15N CPMAS and 13C–15N correlation experiments. Bars represent the following peptide fibrils from left to right: F19G, F19P, F19E, F19K, F19Y, F19W, F19K L34K. | ||
We compared the chemical shifts of our WT fibrils with literature data for agitated fibrils (Table S3, ESI†).7,9 The best agreement was found with the fibrils from the Tycko lab7 (within 0.5 ppm for the 13C chemical shifts). The fibrils reported by Bertini et al.9 represent a slightly different polymorph as larger 13C chemical shift differences of up to 1.7 ppm have been observed.
The isotopic chemical shifts of 13Cα and 13Cβ strongly depend on the secondary structure of a specific residue.27–29 As the external referencing could introduce small errors in the determination of absolute chemical shifts, we analyzed the Cα–Cβ chemical shift difference, which does not depend on external referencing.30Fig. 5 shows a plot of the chemical shift differences and the assignment of the chemical shift difference to specific local secondary structure. Note that for G33 only the absolute Cα chemical shift is plotted, as glycine lacks a side chain. For the WT Aβ(1–40) fibrils, chemical shifts are measured for the four labeled amino acids that are all in agreement with the β-sheet structure in accordance with all current models of Aβ(1–40) fibrils.6–12 However, for the mutations studied here, several local secondary structure alterations in the fibrils are detected. Most prominent are these alterations for residue F20. While the mutations F19Y and F19G display chemical shifts for F20 that are in agreement with the β-sheet structure, the F19E and F19P mutants show a tendency towards local random coil structure. Further, the F19K mutant and the F19K L34K double mutant show chemical shifts that indicate α-helical structure. But as V18 and A21 show β-sheet structure, it is unlikely that a hydrogen bonded helical loop exists for K19 and F20. Rather, the chemical shift data indicate local torsion angles that are different from local β-sheet structure. More moderate are the local structure changes of A21, where basically all mutants show β-sheet structure for this residue except F19P, which has a tendency for random coil structure. Similarly, the local structure of G33 is a β-sheet for all Aβ(1–40) mutants except F19Y, which shows α-helical structure. As already suggested from the very moderate chemical shift changes, residue V18 shows a local β-sheet structure for all Aβ(1–40) mutants investigated in this study.
 | ||
| Fig. 5 Overview of the local secondary structures of the labeled amino acids V18, F20, A21, and G33 in the WT and mutated Aβ(1–40) peptide fibrils as derived from the isotropic chemical shift measurement. Reference values for α-helix, random coil, and β-sheet structure are given as grey, blue, and green bars, respectively. Experimental data points are plotted as Cα–Cβ chemical shift differences, which is independent of external referencing.30 | ||
Influence of the perturbations on the local fluctuations of the Aβ(1–40) fibrils
Given the partially drastic local structure changes in the mutated Aβ(1–40) fibrils, one can assume that these alterations result in local packing differences of the fibrils and thus in modifications of the molecular dynamics of the residues involved in the structural changes. Therefore, we investigated the motional amplitude of the C–H bond vectors in the labeled amino acids of the mutated Aβ(1–40) fibrils. Using the DIPSHIFT separated local field experiment,31 the motionally averaged dipolar coupling of any given 1H–13C bond vector was determined.32 As the dipolar coupling decreases with increasing motional amplitude, an order parameter was defined as the ratio of the motionally averaged and the full dipolar coupling in the absence of any motion. A fully rigid C–H-bond would exhibit the maximal dipolar coupling strength of 22.8 kHz corresponding to an order parameter of one, while a value of zero for the order parameter corresponds to fully isotropic motion expressed by a vanishing dipolar coupling. Molecular motions with a given amplitude lead to partial averaging of the dipolar coupling and can be characterized by a specific order parameter. The 1H–13C order parameters sample all motions with correlation times shorter than ∼10 μs (1/δCH = 43.8 μs).33Values for the 1H–13C order parameters are presented in Table S2 (ESI†) and Fig. 6 displays the order parameters for the signals of the labeled amino acids in WT and the mutated Aβ(1–40) fibrils. As expected and in agreement with literature data,14 relatively high order parameters have been found throughout the fibrils. The backbone and most aliphatic side chains are quite rigid, while the F20 ring and the methyl group of V18 undergo large amplitude motions. Phenylalanine rings are known to undergo ring flips34 and methyl groups rotate fast.35 Methyl group rotation averages the order parameter to a value of 0.33. Experimental order parameters lower than 0.33 for the methyl groups indicate further averaging due to fluctuations of the C–C bond.
 | ||
| Fig. 6 1H–13C order parameters of the individual segments of the labeled amino acids V18, F20, A21, and G33 in amyloid fibrils of the WT and mutated Aβ(1–40) peptides. Note that the signals from the CH3 groups of V18 Cγ and A21 Cβ cannot be distinguished in the DIPSHIFT experiments due to resolution constraints. | ||
There are quite strong differences in the order parameters of the studied amino acids for the different mutations. Compared to the WT, the F19G, the F19K L34K double mutants show increased order parameters. This is also true for F19K with the exception of the Cα order parameter of F20. The variants F19Y, F19W, and F19P show rather similar order parameters to the WT, with the exception of V18 Cβ in the F19P mutant. F19E shows similar order parameters to the WT. Very interesting are the drastic order alterations of the phenylalanine ring, for which the order drastically increases for the F19G and the F19K variants.
Mesoscopic structure of mutated Aβ(1–40) fibrils
X-ray diffraction studies were carried out for all Aβ fibrils studied here and typical diffraction patterns are shown in Fig. S5 (ESI†). All Aβ peptides show two distinct reflexes at ∼9.2–11 and ∼4.7 Å characteristic of a cross-β structure. No significant differences were observed between the fibrils formed by the WT and most of the mutated Aβ peptides, indicating that the global fibrillar architecture was not influenced by the local perturbations introduced. However, the F19P variant showed more diffraction peaks in the region of 4.7 Å indicating less homogeneous fibrillar structures.Discussion
Proteins are known to adopt many different stable three dimensional structures to carry out their specific biological functions. However, conditions can be found under which most if not all proteins can convert into an even more stable fibrillar structure.1,2 Interestingly, these protein fibrils always show the characteristic fingerprint of a cross-β structure, which appears to be a very general structural motif in protein misfolding. Here, we have investigated how local physical forces can interfere with the fibrillation kinetics, the general morphology, and the local structure and dynamics of the fibrils formed from the perhaps most classical fibril forming peptide, the Aβ(1–40). To this end, the well described hydrophobic contact between F19 and L34 between opposing peptide segments6,8,9,15,25 was systematically mutated (see Scheme 1) and the response of the surrounding residues studied. Both mutated residues are localized in the stable β-strands of the Aβ(1–40) fibrils.Overall, our findings suggest that the cross-β structure of Aβ(1–40) is very robust against local fields and interactions. With the exception of MVII, all mutated peptides formed fibrils that all showed a similar fibril morphology in the electron micrographs and the characteristic signature of the cross-β structural motif, in which the β-strands are organized perpendicular to the fibril axis.36
In the glycine mutant (F19G) structural flexibility was probed. As this residue has the smallest side chain, structural flexibility is high and the amino acid permits for several backbone torsion angle pairs without steric hindrance.37 Although Gly is much less hydrophobic than Phe,38 it is frequently found in β-strands.39 The F19G mutant showed similar fibrillation kinetics to the WT and only minor local structure alterations. In the 13C NMR spectra, some moderate upfield shifts were observed for Phe but overall, the chemical shifts of the surrounding residues are in agreement with β-sheet structure. However, there are several significant dynamics changes. Compared to the WT, order parameters increase to unity for almost all Cα sites and also the side chains receive significant ordering. The most drastic order increase is observed for the aromatic ring of F20. These local order changes are in agreement with a denser packing of the fibril interior that causes motional amplitudes to decrease. Gly has a much smaller van der Waals volume (48 Å3) than Phe (135 Å3),40 but the space made available by this mutation is occupied by denser packing of the peptides in the fibril and not by an increase in the motional amplitudes of the surrounding residues. However, to accommodate this structural adjustment, only small structural alterations of the surrounding residues are necessary.
In the proline mutant (F19P), the influence of a very restricted amino acid side chain was probed. It is well known that proline is underrepresented in β-sheet structures and in particular in β-sheets that pack against each other.41 Consequently, proline is least compatible with β-sheet structure and rarely found in amyloid fibrils.42 The Ramachandran diagram of proline only allows for two backbone conformations.37 As the cross-β structure of the Aβ(1–40) fibrils is conserved for this mutation, it is likely that P19 is also in trans conformation. The fibrillation kinetics of the F19P mutant was severely decreased with a characteristic time, which exceeded that of the WT by a factor of >10. Nevertheless, fibrils of very comparable morphology were formed as revealed from the TEM images. However, the X-ray diffraction pattern showed more peaks in the 4.7 Å region, indicating less homogeneous structures. The covalently linked proline side chain strongly constrains the preceding residue.37 Indeed, both the Cα and Cβ NMR signals of V18 move upfield, which represents an inconclusive chemical shift change with regard to structural interpretation. However, both F20 and A21 show chemical shifts that better agree with a local random coil structure. The Pro variant also showed interesting dynamical variation compared to the WT. While the aliphatic signals of A21 and G33 Cα are relatively similar as in the WT, a very significant decrease in the order parameter is observed for F20 and V18. The Val Cα order parameter decreased from 0.94 in the WT to 0.81 in the mutant and for Cβ from 0.81 to 0.64. Similar for Phe Cα, which is 0.90 in the WT and only 0.83 in the mutant. Although Pro has a larger van der Waals volume (90 Å3) than Gly, it is still much smaller than the Phe residue in the WT. Nevertheless, due to the structural constraints of the Pro residue a denser packing of the fibril is prevented and the free volume has to be taken up by larger amplitude motions of the directly neighboring residues V18 and F20. The F19P mutation was also investigated in a Pro scan on Aβ(1–40).23 In agreement with our results, fibrillation kinetics was severely slowed down and the thermodynamic stability decreased by about 13 kJ mol−1. In spite of this destabilization, the amyloid structure that was formed remained relatively unperturbed.23
In the tyrosine mutant (F19Y), we probed if the capability of the residue to form hydrogen bonds has an influence on the fibrillation kinetics and the local structure and dynamics of Aβ(1–40) fibrils. Tyrosine has a relatively similar side chain to Phe with an additional hydroxyl group in the para position. Accordingly, its van der Waals volume (141 Å3) is only slightly larger than Phe and both Tyr and Phe have high probability to be found in β-sheets. In spite of these similarities, the characteristic time as well as the lag time for fibrillation of this mutant were about 2–3 times as long as for the WT. But the general morphology and cross-β structure of this mutant agreed with that of the WT. On an atomic level, some smaller structural alterations were found as indicated by the chemical shift changes. 13C chemical shifts remained relatively similar in this mutant except for G33 Cα, which shifted 5.2 ppm downfield. This can be taken as an indication that the presence of the hydroxyl group causes some structural alteration on the opposing L34, which is sensed by the neighboring G33. As Tyr has a significantly lower hydrophobicity than Phe,38 the hydrophobic contact between F19 and L34 is perturbed by this mutation. The local dynamics is identical to the WT with the notable exception of the F19 ring, which has an order parameter of 0.38 in the WT and 0.25 in the Tyr mutant. Although changes in the 15N NMR spectra were observed, there is not clear indication that the Tyr side chain is involved in hydrogen binding although the low dielectric constant of the fibril interior would make hydrogen bonds more favorable than in water.43
The tryptophan mutant (F19W) introduced the largest side chain into position 19 (van der Waals volume of 163 Å3) and also allowed for the formation of a hydrogen bond in the core of the fibril. Trp is the most hydrophobic residue among all natural amino acids.38 Nevertheless; this mutant showed the most negligible 13C chemical shift perturbations. Also, the order parameters were very similar to the WT suggesting only minor alterations in the local structure and dynamics. Nevertheless, this mutant showed a slower fibrillation kinetics as the WT. 15N chemical shift changes were more significant, which could be an indication for altered intermolecular hydrogen bonding within the fibrils. The Trp mutant also allowed estimating the local dielectric constant within the Aβ fibrils to a value of around 5 from the fluorescence maximum (Fig. S6, ESI†). Both the F19Y and F19W mutations replaced the Phe ring by other aromatic side chains, the indole ring of Trp mimics the Phe side chain best.
In the glutamate mutant (F19E), the influence of electrostatics on the fibril formation was investigated. The negatively charged Glu residue has the lowest probability to be located in a β-strand39 and exhibits a van der Waals volume of 109 Å3. As the interior of the Aβ(1–40) fibrils is not hydrated and harbors almost exclusively hydrophobic residues, one would expect an unfavorable solvation free energy of the charged residue to be located in the low ε environment of the fibril interior to compete with the tendency of the molecule to form fibrils. Indeed, the characteristic fibrillation time as well as the lag time are much longer than for the WT, but the fibrils formed by this mutant show again very similar morphology and the characteristic cross-β structure. Also in terms of local structure and dynamics, this variant showed the least striking character. Most chemical shifts remained close to the WT, only the Cβ signal of Phe showed an upfield shift of 2.5 ppm and the Cα signal of Gly a downfield shift of 2.3 ppm compared to the WT. Also the order parameters were relatively similar to the WT. Overall, it seems that the negatively charged side chain does not perturb the local structure and dynamics of the Aβ(1–40) fibrils much and only slows down the kinetics of fibril formation. To assess the energetic contribution from the solvation energy of the glutamate,44 we used the dielectric constant of 5 estimated for the interior of the fibril as determined from the fluorescence measurement of the F19W mutant (Fig. S6, ESI†). With this value, we can estimate an unfavorable solvation free energy of ∼75 kJ mol−1 for transferring the glutamate residue from aqueous buffer into the hydrophobic interior of the fibril.44 This rough estimate by far exceeds the thermodynamic destabilization that has been found for Ala and Pro scans of Aβ(1–40), for which values of no more than ∼13 kJ mol−1 were measured. This suggests that the side chain of the Glu residue may not be localized in the fibril core.
Similarly, the influence of electrostatics on Aβ fibril properties was probed in the lysine mutant (F19K). The positively charged Lys is about as likely to be found in a β-strand as Gly or Pro. The van der Waals volume of this amino acid is equal to that of Phe (135 Å3). Similar to the negatively charged Glu, an unfavorable solvation free energy of ∼60 kJ mol−1 would arise from burying the charged Lys side chain in the hydrophobic fibril interior. Regardless, fibrils of the same morphology and cross-β structure were formed with the same kinetics as observed for the WT Aβ(1–40). With regard to local structure changes, F20 undergoes a tremendous chemical shift change in this mutant, where the Cα signal shifts downfield by 5.5 ppm and the Cβ signal shifts upfield by 3.1 ppm. These chemical shift changes indicate strong alterations in the local geometry with torsion angles that are in agreement with an α-helix. Dynamically, the most drastic changes are observed also for F20. Further, G33 and V18 Cβ become more ordered and also the F20 ring increases its order parameter from 0.38 in the WT to 0.54 in this mutant. One could speculate that a cation–π interaction45 between K19 and F20 is responsible for these characteristic structural and dynamical changes in that mutant. For such a scenario, the K19 side chain would have to rotate out of the fibril interior or the Phe ring of F20 would have to move into the fibril interior. The observed increase in segmental order in particular for the F20 ring would be in agreement with such a scenario.
Next, in the double lysine mutant (F19K, L34K), we checked if the introduced electrostatic repulsion could prevent fibril formation of Aβ(1–40). Much to our surprise, fibrils of similar morphology and the typical cross-β structure formed with only somewhat slower kinetics. As discussed above, the Phe and Lys residues have a very similar van der Waals volume and also Leu is only slightly smaller (124 Å3) than Lys. However, quite significant repulsive forces should work against fibril formation. In addition to the solvation energy of two Lys residues, which amounts to ∼120 kJ mol−1 according to our estimate, a contribution from the electrostatic repulsion of the two positive charges has to be considered. To estimate this contribution, we consider a distance of 9 Å between the charges (as estimated from an NMR structural model of Aβ(1–40) fibrils9) and a dielectric constant of 5 in the fibril interior. With these numbers, an unfavorable Coulomb energy of ∼31 kJ mol−1 arises. So overall, from the double mutation an unfavorable free energy of ∼150 kJ mol−1 has to be counterbalanced by the fibrillation. As is unlikely that such large energies could be generated one would have to assume that the Lys side chains are not buried in the interior of the fibrils, which is suggested by the large chemical shift changes. With regard to local structure, the structural changes in the double mutant resemble those in the F19K single mutant with the same pronounced chemical shift changes of F20. Similarly, the order parameters are high for almost all signals, however, the Phe ring now showed a lower order parameter than in the WT. Although hydrophobic interactions play a very significant role in Aβ stability,22 the F19K, F19E, and the double F19K L34K mutants show that a partial alteration of the hydrophobic character of the fibril interior is still tolerated, however, the charged side chains are likely to move out of the fibril core.
Finally, the double Lys/Glu mutant (F19K, L34E) was the only Aβ(1–40) variant that did not form fibrils under standard buffer conditions within a timeframe exceeding 2 months. Originally, the mutant was designed to probe if the hydrophobic contact of F19 and L34 could be replaced by a salt bridge between K19 and E34. Although the unfavorable solvation free energy could partially be compensated by Coulomb attraction, the Lys/Glu double mutant did not form any fibrils. It is likely that the mutation that provides an additional salt bridge increased the stability of the monomer. Thus, the free energy of the monomer is decreased and the barrier between the monomeric and the fibrillar state is more difficult to overcome. We changed the fibrillation conditions to buffers with higher salt concentrations to destabilize the Aβ monomer but we did not find any conditions, where the Lys/Glu double mutant could be converted into fibrils. It remains to be seen if the monomers of the double F19K L34E mutant are more stabilized than the WT Aβ monomers to create an energy barrier that renders fibrillation unlikely.
In our NMR measurements, the most significant chemical shift alterations were found for the 15N. Although more difficult to interpret, these chemical shift changes suggest alterations in the hydrogen bond pattern of the fibrils. Empirical correlations suggest that hydrogen bond length increases can result in larger isotropic 15N chemical shifts for Gly46 and quantum chemical shift calculations seem to confirm this trend.47 It is likely that the 15N chemical shift alterations observed for the Aβ mutants investigated here indicate such bond length changes. It has been reported that hydrogen bonds in an apolar environment can be very strong because the solvent cannot compete for the hydrogen bond.43 Therefore, alterations in the hydrogen bond network as suggested by the 15N chemical shift changes may contribute significantly to the thermodynamic stability of the mutants. Nevertheless, also such hydrogen bond length changes are tolerated by the robust fibril structure of Aβ.
Experimental
Peptide synthesis
Wildtype (WT) (DAEFRHDSGY EVHHQKL![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) F
F![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) EDVGSNKGA II
EDVGSNKGA II![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) LMVGGVV) and mutated Aβ(1–40) peptides were synthesized using the standard F-moc solid phase synthesis strategy. Each Aβ(1–40) peptide variant was U-13C, 15N-labeled in position V18, F20, A21, and G33 (shown underlined in the sequence above). Isotopically labeled amino acids were purchased from Euriso-top, Saarbrücken, D. Six single mutants were prepared, where F19 (shown in bold in the above sequence) was replaced by glycine, proline, glutamate, lysine, tyrosine, or tryptophan, respectively. In addition, two double mutants were prepared, where F19 was replaced by lysine and Leu34 (shown in bold in the above sequence) by either glutamate or lysine. An overview of the isotopic labeling scheme and the mutation pattern is given in Scheme 1.
LMVGGVV) and mutated Aβ(1–40) peptides were synthesized using the standard F-moc solid phase synthesis strategy. Each Aβ(1–40) peptide variant was U-13C, 15N-labeled in position V18, F20, A21, and G33 (shown underlined in the sequence above). Isotopically labeled amino acids were purchased from Euriso-top, Saarbrücken, D. Six single mutants were prepared, where F19 (shown in bold in the above sequence) was replaced by glycine, proline, glutamate, lysine, tyrosine, or tryptophan, respectively. In addition, two double mutants were prepared, where F19 was replaced by lysine and Leu34 (shown in bold in the above sequence) by either glutamate or lysine. An overview of the isotopic labeling scheme and the mutation pattern is given in Scheme 1.
Sample preparation
The lyophilized peptide was solubilized in 25 mM phosphate buffer (pH 9) containing 150 mM NaCl and 0.01% NaN3 to avoid bacterial and fungal growth at a concentration of 1 mg ml−1. The sample was mixed around every minute for about 5 minutes and then dialyzed against the same buffer but at pH 7.4. Dialysis was performed for 4 h, the dialysis solution was changed after 2 h. The MWCO of the dialysis tube was 1000. The peptide solutions were then incubated at 37 °C and shaken at 450 rpm for at least 10 days for fibrillation. For NMR measurements, fibril solutions were ultracentrifuged at 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 1 h at 4 °C. The pellets were lyophilized, rehydrated to 50 wt% H2O, homogenized by freezing the sample in liquid nitrogen and thawing it at 37 °C and centrifuged into 4 mm MAS rotors.
000 × g for 1 h at 4 °C. The pellets were lyophilized, rehydrated to 50 wt% H2O, homogenized by freezing the sample in liquid nitrogen and thawing it at 37 °C and centrifuged into 4 mm MAS rotors.
Fluorescence measurements
The fibrillation kinetics of Aβ(1–40) peptides and the mutants was studied by fluorescence spectroscopy. Thioflavin T (ThT) was used as a fluorescence dye that shows increase in fluorescence intensity at 482 nm when bound to fibrils exhibiting the cross-β structure.48 A ThT stock-solution of 2.5 mM in the sample buffer was prepared and stored at 4 °C in the dark. For each measurement, an aliquot of 16 μl of the peptide solution and 15 μl of the ThT stock solution was dissolved in 2 ml buffer solution. Fluorescence spectra were measured on a FluoroMax-2 (Jobin Yvon, Edison NJ, USA). The data points were fitted with the Finke–Watzky-2-step model.49 A characteristic time for fibrillation was defined, where the intensity of the fluorescence reaches its half maximum.Fibrillation kinetics experiments were also carried out in a 96-well plate format.50 Sample concentration was 1 mg ml−1 in phosphate buffer pH 9.2 containing 20 μM ThT and 150 mM NaCl. A volume of 150 μl was pipetted into the wells. The plate was loaded into a Tecan infinite M200 microplate reader (Tecan Group AG, Männedorf, Switzerland), the temperature was set to 37 °C and a shaking cycle of 5 minute shaking (1 mm shaking amplitude and 173.9 rpm) followed by a 5 min waiting time was applied. The excitation was set to 450 nm and the emission was measured at 485 nm. The fluorescence intensity was measured every 30 min for at least 3 days for each sample. The F19P sample did not complete fibrillation within 5 days, longer experiments could not be performed because of sample dehydration. Data were analyzed using standard procedures from the literature.51
For the estimation of the dielectric constant in the Aβ(1–40) fibril interior, tryptophan fluorescence was used. Tryptophan shows a characteristic fluorescence emission wavelength depending on the dielectric constant of the environment. For a calibration curve, pure tryptophan (5 μM concentration) was dissolved in 700 μl of the following solvents: H2O (ε = 78), DMSO (ε = 46.5), acetonitrile (ε = 37.5), methanol (ε = 33.8), ethanol (ε = 25.8), ethylacetate (ε = 13.1), and cyclohexane (ε = 2.0). The excitation wavelength was set to 290 nm and the emission was measured between 300 and 470 nm at 25 °C. The calibration curve was repeated with the Aβ(1–40) mutant MVI (F19W) in the same solvents. The measured Trp emission wavelengths showed similar values (Fig. S6, ESI†). Finally, the measurement was carried out with Aβ(1–40) F19W fibrils, where 4 μl of the fibril solution was dissolved in 700 μl of the fibrillation buffer to yield a dielectric constant for the fibril interior of ε ≈ 5.
Transmission electron microscopy (TEM)
The general morphology of all fibrils obtained was determined by transmission electron microscopy (TEM). Fibril solutions were diluted 1:20 with pure water. 1 μl droplets of this solution were applied on formvar coated copper grids, allowed to dry for about 2 hours and negatively stained with 1% uranyl acetate in pure water. Transmission electron micrographs were recorded using a Zeiss EM 900 microscope (Zeiss NTS, Oberkochen, D) at 80 kV.
X-ray diffraction measurements
For the X-ray diffraction measurements, the samples were mounted onto the goniometer head of the X-ray source (Rigaku copper rotating anode MM007 with 0.8 kW, Tokyo, Japan) using a nylon loop (Hampton Research, Aliso Viejo, CA, USA) or a litho loop (MiTeGen, Ithaca, NY, USA). X-ray diffraction signals were recorded using an image plate detector (Rigaku, Tokyo, Japan) with an exposure time of 180 s at room temperature.Solid-state NMR-spectroscopy
MAS NMR spectra were acquired on a Bruker 600 Avance III NMR spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) at a resonance frequency of 600.1 MHz for 1H, 150.9 MHz for 13C and 60.8 MHz for 15N. A double channel 4 mm MAS probe was used. The temperature was set to 30 °C for all NMR experiments. The typical 1H 90° excitation pulse length for CP MAS experiments was 4 μs, the CP contact time was 700 μs at a spin lock field of ∼50 Hz, and the relaxation delay was 2.5 s. 1H dipolar decoupling during acquisition with an rf amplitude of 65 Hz was applied using Spinal64. For peak assignment, two dimensional 13C–13C proton driven spin diffusion52 spectra with a mixing time of 100 ms were acquired. The MAS frequency was either 7 or 10 kHz. On average, 100 data points were acquired in the indirect dimension providing a resolution of 333 Hz. The 1H–13C dipolar couplings were measured in constant time DIPSHIFT experiments.31 The amplitudes of the dipolar dephasing over one rotor period provide a measure for the strength of the dipolar coupling. Homonuclear decoupling using the frequency switched Lee–Goldberg (FSLG)53 with an effective rf field of 80 kHz was applied during dipolar evolution. The MAS frequencies were 5 kHz for 13C DIPSHIFT experiments and 4 kHz for 15N DIPSHIFT experiments. The strength of the dipolar coupling was determined from numerical simulations using an increment of 1° for powder averaging. CH or NH order parameters were calculated as the ratio of the motionally averaged dipolar coupling and the rigid limit value determined for crystalline amino acids at low temperature.32,5413C–15N correlation spectra for assignment of the 15N chemical shifts were measured using the standard double CP MAS experiment.26 For these experiments, a 3.2 mm triple resonance MAS probe was used. The 1H 90° excitation pulses had a length of 4 μs, the CP contact times had a duration of 2 ms for 1H–15N and 5 ms for 13C–15N. Up to 26 increments were acquired in the indirect dimension.
Conclusions
We investigated the fibrillation kinetics as well as the local structure and dynamics of several single and double mutants of Aβ(1–40) peptides that introduced local physical forces. None of these peptide variants represent commonly found Aβ mutations in Alzheimer's disease55 and were solely selected on the basis of physicochemical considerations. The analysis showed that the local Aβ structure and dynamics was somewhat altered by weakening of the crucial hydrophobic contact. Nevertheless, with one exception, all investigated peptide variants formed stable fibrils with the known morphology. This suggests that the overall β-sheet structure of Aβ fibrils is very robust and only smaller local alterations in the U-shaped structure of the amyloid fibrils occur.6–12 Also, the dynamics of the investigated residues mostly showed minor alterations, which could be explained by looser or denser packing near the respective mutation. We conclude that single mutations cannot completely destabilize the Aβ structure in contrast to globular proteins where a single proline mutation can completely prevent folding.4 This is in agreement with recent work on mutated tau repeat peptides.56 However, we found one double mutation, for which fibril formation was completely abolished most likely due to the stabilization of the monomeric state by the introduction of a new salt bridge.Acknowledgements
The study was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG TRR 102, A6). J.A. would like to acknowledge Dr Christoph Parthier for support with the X-ray diffraction measurements.Notes and references
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Nat. Chem. Biol., 2009, 5, 15 CrossRef CAS PubMed.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. Blake, J. Mol. Biol., 1997, 273, 729 CrossRef CAS PubMed.
- R. Wetzel, S. Shivaprasad and A. D. Williams, Biochemistry, 2007, 46, 1 CrossRef CAS PubMed.
- R. Tycko, Q. Rev. Biophys., 2006, 39, 1 CrossRef CAS PubMed.
- A. K. Paravastu, R. D. Leapman, W. M. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742 CrossRef CAS PubMed.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342 CrossRef CAS PubMed.
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, J. Am. Chem. Soc., 2011, 133, 16013 CrossRef CAS PubMed.
- A. T. Petkova, R. D. Leapman, Z. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262 CrossRef CAS PubMed.
- J. P. Colletier, A. Laganowsky, M. Landau, M. Zhao, A. B. Soriaga, L. Goldschmidt, D. Flot, D. Cascio, M. R. Sawaya and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16938 CrossRef CAS PubMed.
- J. M. Lopez del Amo, M. Schmidt, U. Fink, M. Dasari, M. Fandrich and B. Reif, Angew. Chem., Int. Ed., 2012, 51, 6136 CrossRef CAS PubMed.
- J. X. Lu, W. Qiang, W. M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Cell., 2013, 154, 1257 CrossRef CAS PubMed.
- H. A. Scheidt, I. Morgado, S. Rothemund and D. Huster, J. Biol. Chem., 2012, 287, 2017 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561 CAS.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157 CAS.
- H. A. Scheidt, I. Morgado and D. Huster, J. Biol. Chem., 2012, 287, 22822 CrossRef CAS PubMed.
- H. A. Scheidt, I. Morgado, S. Rothemund, D. Huster and M. Fandrich, Angew. Chem., Int. Ed., 2011, 50, 2837 CrossRef CAS PubMed.
- K. Murakami, K. Irie, A. Morimoto, H. Ohigashi, M. Shindo, M. Nagao, T. Shimizu and T. Shirasawa, J. Biol. Chem., 2003, 278, 46179 CrossRef CAS PubMed.
- E. A. Yates, E. M. Cucco and J. Legleiter, ACS Chem. Neurosci., 2011, 2, 294 CrossRef CAS PubMed.
- W. Qiang, W. M. Yau, Y. Luo, M. P. Mattson and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4443 CrossRef CAS PubMed.
- A. D. Williams, S. Shivaprasad and R. Wetzel, J. Mol. Biol., 2006, 357, 1283 CrossRef CAS PubMed.
- A. D. Williams, E. Portelius, I. Kheterpal, J. T. Guo, K. D. Cook, Y. Xu and R. Wetzel, J. Mol. Biol., 2004, 335, 833 CrossRef CAS PubMed.
- S. L. Shammas, T. P. J. Knowles, A. J. Baldwin, C. E. MacPhee, M. E. Welland, C. M. Dobson and G. L. Devlin, Biophys. J., 2011, 100, 2783 CrossRef CAS PubMed.
- M. Chandrakesan, B. Sarkar, V. S. Mithu, R. Abhyankar, D. Bhowmik, S. Nag, B. Sahoo, R. Shah, S. Gurav, R. Banerjee, S. Dandekar, J. J. Jose, N. Sengupta, P. K. Madhu and S. Maiti, Chem. Phys., 2013, 422, 80 CrossRef CAS PubMed.
- J. H. Yang, F. C. Chou and D. L. Tzou, J. Magn. Reson., 2008, 195, 116 CrossRef CAS PubMed.
- S. Spera and A. Bax, J. Am. Chem. Soc., 1991, 113, 5490 CrossRef CAS.
- D. S. Wishart and B. D. Sykes, Methods Enzymol., 1994, 239, 363 CAS.
- Y. Wang and O. Jardetzky, Protein Sci., 2002, 11, 852 CrossRef CAS PubMed.
- S. Luca, D. V. Filippov, J. H. van Boom, H. Oschkinat, H. J. de Groot and M. Baldus, J. Biomol. NMR, 2001, 20, 325 CrossRef CAS.
- M. G. Munowitz, R. G. Griffin, G. Bodenhausen and T. H. Huang, J. Am. Chem. Soc., 1981, 103, 2529 CrossRef CAS.
- D. Huster, L. Xiao and M. Hong, Biochemistry, 2001, 40, 7662 CrossRef CAS PubMed.
- A. G. Palmer III, J. Williams and A. McDermott, J. Phys. Chem., 1996, 100, 13293 CrossRef.
- L. Mueller, M. H. Frey, A. L. Rockwell, L. M. Gierasch and S. J. Opella, Biochemistry, 1986, 25, 557 CrossRef CAS.
- K. Beshah, E. T. Olejniczak and R. G. Griffin, J. Chem. Phys., 1987, 86, 4730 CrossRef CAS PubMed.
- E. D. Eanes and G. G. Glenner, J. Histochem. Cytochem., 1968, 16, 673 CrossRef CAS PubMed.
- D. Ting, G. Wang, M. Shapovalov, R. Mitra, M. I. Jordan and R. L. Dunbrack, Jr., PLoS Comput. Biol., 2010, 6, e1000763 Search PubMed.
- W. C. Wimley and S. H. White, Nat. Struct. Biol., 1996, 3, 842 CrossRef CAS PubMed.
- D. Nelson and M. Cox, Lehninger Biochemie, Springer-Verlag, Berlin, Heidelberg, New York, 2001 Search PubMed.
- T. E. Creighton, Proteins. Structures and molecular properties, W. H. Freeman and Company, New York, 2nd edn, 1993 Search PubMed.
- C. Chothia and J. Janin, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 4146 CrossRef CAS.
- A. D. Williams, E. Portelius, I. Kheterpal, J. T. Guo, K. D. Cook, Y. Xu and R. Wetzel, J. Mol. Biol., 2004, 335, 833 CrossRef CAS PubMed.
- J. U. Bowie, Curr. Opin. Struct. Biol., 2011, 21, 42 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Press, London, 1992 Search PubMed.
- D. A. Dougherty, Science, 1996, 271, 163 CAS.
- S. Kuroki, S. Ando, I. Ando, A. Shoji, T. Ozaki and G. A. Webb, J. Mol. Struct., 1990, 240, 19 CrossRef CAS.
- X. P. Xu and D. A. Case, Biopolymers, 2002, 65, 408 CrossRef CAS PubMed.
- P. S. Vassar and C. F. Culling, Arch. Pathol., 1959, 68, 487 CAS.
- A. M. Morris, M. A. Watzky, J. N. Agar and R. G. Finke, Biochemistry, 2008, 47, 2413 CrossRef CAS PubMed.
- J. Seeliger, K. Estel, N. Erwin and R. Winter, Phys. Chem. Chem. Phys., 2013, 15, 8902 RSC.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036 CrossRef CAS PubMed.
- N. M. Szeverenyi, M. J. Sullivan and G. E. Maciel, J. Magn. Reson., 1982, 47, 462 CAS.
- A. Bielecki, A. C. Kolbert and M. H. Levitt, Chem. Phys. Lett., 1989, 155, 341 CrossRef CAS.
- P. Barré, O. Zschörnig, K. Arnold and D. Huster, Biochemistry, 2003, 42, 8377 CrossRef PubMed.
- D. J. Selkoe and M. B. Podlisny, Annu. Rev. Genomics Hum. Genet., 2002, 3, 67 CrossRef CAS PubMed.
- Y. Raz, J. Adler, A. Vogel, H. A. Scheidt, T. Häupl, B. Abel, D. Huster and Y. Miller, Phys. Chem. Chem. Phys., 2014 10.1039/c3cp54890b.
Footnote |
| † Electronic supplementary information (ESI) available: ThT fluorescence kinetics, TEM images and analysis of fibril morphology, NMR spectra, X-ray diffraction patterns, Trp fluorescence data, and chemical shift tables. See DOI: 10.1039/c3cp54501f |
| This journal is © the Owner Societies 2014 |