 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insight into the potential energy landscape and relaxation pathways of photoexcited aniline from CASSCF and XMCQDPT2 electronic structure calculations†
Matthieu
Sala
a,
Oliver M.
Kirkby
b,
Stéphane
Guérin
a and
Helen H.
Fielding
*b
aLaboratoire Interdisciplinaire Carnot de Bourgogne UMR 5209 CNRS, Université de Bourgogne, BP 47870, F-21078 Dijon, France. E-mail: matthieu.sala@u-bourgogne.fr
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.h.fielding@ucl.ac.uk
First published on 2nd January 2014
Abstract
There have been a number of recent experimental investigations of the nonadiabatic relaxation dynamics of aniline following excitation to the first three singlet excited states, 11ππ*, 11π3s/πσ* and 21ππ*. Motivated by differences between the interpretations of experimental observations, we have employed CASSCF and XMCQDPT2 calculations to explore the potential energy landscape and relaxation pathways of photoexcited aniline. We find a new prefulvene-like MECI connecting the 11ππ* state with the GS in which the carbon-atom carrying the amino group is distorted out-of-plane. This suggests that excitation above the 11π3s/πσ* vertical excitation energy could be followed by electronic relaxation from the 11ππ* state to the ground-electronic state through this MECI. We find a MECI connecting the 11π3s/πσ* and 11ππ* states close to the local minimum on 11π3s/πσ* which suggests that photoexcitation to the 11π3s/πσ* state could be followed by relaxation to the 11ππ* state and to the dissociative component of the 11π3s/πσ* state. We also find evidence for a new pathway from the 21ππ* state to the ground electronic state that is likely to pass through a three-state conical intersection involving the 21ππ*, 11π3s/πσ* and 11ππ* states.
1 Introduction
Aniline is the simplest aromatic amine and, like all aromatic molecules containing OH and NH groups, it has a remarkably low fluorescence quantum yield attributed to highly efficient radiationless electronic relaxation pathways connecting the excited electronic states to the ground electronic state. Improving our understanding of the mechanism of electronic relaxation in small molecules like aniline is an important component of the toolkit required to design photostable materials from first principles.Aniline absorbs UV radiation around 282 nm (4.4 eV) and 230 nm (5.4 eV) nm.1–3 These bands arise from transitions from the ground electronic state to the two lowest lying 1ππ* states. There have been numerous experimental4–7 and theoretical8–22 studies of the ground electronic state, with particular focus on the pyramidal geometry of the amino group. There has been some controversy over the equilibrium geometry of the 11ππ* excited state. Configuration-interaction with single excitations (CIS) calculations predicted planar or pyramidal geometries with a smaller degree of pyramidalization than the ground state, depending on basis set.23–31 CASSCF calculations also predicted a pyramidal equilibrium geometry.32 Rotationally-resolved electronic spectra suggested a quasiplanar geometry for the 11ππ* excited state; however, this structure was a vibrationally averaged structure rather than the true minimum of the potential energy surface (PES).30 Early experimental investigations of the photochemistry and photophysics of aniline following excitation of the 11ππ* excited state focussed on fluorescence and intersystem crossing (ISC).33–36
Between the first two 1ππ* states, lies a 1π3s/πσ* state that has significant Rydberg character in the Franck–Condon (FC) region and evolves into a valence state with dissociative character along the N–H stretching coordinate.37–39 Worth et al. have predicted that two 3p Rydberg states also lie between the two 1ππ* states.40 There have been a number of experimental32,41–45 and theoretical32,40 investigations focussing on electronic relaxation pathways involving the 11π3s/πσ* state. Experiments observing the formation of H-atoms following photodissociation reported thresholds for fast H-atom production (a signature of NH bond fission on the 11πσ* surface) of 260 nm41 and 250 nm32 depending on whether they employed nanosecond or femtosecond lasers, respectively. Femtosecond pump–probe experiments have revealed four lifetimes: τ1 ≲ 100 fs, τ2 ∼ 100–400 fs, τ3 ∼ 0.4–3 ps, τ4 ≳ 80 ps.32,42–45 The fastest timescale, τ1, is only observed following excitation to the 21ππ* excited state and has been interpreted as relaxation back to the electronic ground-state43,44 or as decay through a series of conical intersections (CIs) to the 11πσ* PES.32,42τ2 has been interpreted as population transfer through a CI between the 11π3s/πσ* state and the 11ππ* state governing a competition between subsequent relaxation on the 11ππ* state and the dissociative 11πσ* component of the 11π3s/πσ* state,43,44 or the reverse process from 11ππ* to 11πσ*.32,42τ2 has also been proposed to arise from tunnelling through the barrier along the N–H stretching coordinate between the bound 11π3s and dissociative 11πσ* components of the 11π3s/πσ* state.45τ3 was observed only in time-resolved photoelectron spectroscopy experiments43–45 and was interpreted as motion on the 11π3s/πσ* PES43,44 or as intramolecular vibrational energy redistribution (IVR) in the 11ππ* state.45 The longest timescale, τ4, has been interpreted as decay from the 11ππ* state.
Motivated by the disagreements between the interpretations of these experiments, we have employed CASSCF and XMCQPDT2 calculations to explore the potential energy landscape and relaxation pathways of aniline following photoexcitation to the 11ππ*, 11π3s/πσ* and 21ππ* states. We find a new prefulvene-like minimum energy conical intersection (MECI) connecting the 11ππ* state with the GS which seems likely to be involved in non-radiative decay from 11ππ* to the ground-state. We find a MECI connecting the 11π3s/πσ* and 11ππ* states close to the local minimum on 11π3s/πσ* which suggests that excitation to 11π3s/πσ* is likely to be followed by relaxation to this MECI where population will subsequently be transferred both to 11ππ* (as observed in our earlier work43,44) and to the dissociative component of 11π3s/πσ* (as observed by Stavros et al.32). Finally, we find evidence for a new decay pathway connecting 21ππ* and the ground-state that passes through what seems likely to be a three-state CI involving 21ππ*, 11π3s/πσ* and 11ππ*. This supports our interpretation of our earlier experimental data43,44 and is consistent with the experimental observations of others.32,41,42
2 Computational details
Ground-state minima and excited state minima and TSs were optimized using the state specific complete active-space self-consistent field (SS-CASSCF) method. MECIs were optimized using state-averaged CASSCF (SA-CASSCF)46–48 with equal weighting given to the two states forming the CI.The different decay pathways relevant for the photochemistry of aniline were studied using linear interpolation in internal coordinates, relaxed potential scans and minimum energy path (MEP) calculations using the SA-CASSCF method. The MEP calculations were based on the intrinsic reaction coordinate (IRC) method.49,50 Although IRC calculations are often started at a TS, the IRC calculation performed in this work (Section 6) is started at the FC geometry using the gradient as the initial relaxation direction.
The CASSCF method is known to describe the multi-configurational nature of excited state electronic wavefunctions correctly, which is a crucial requirement for systems involving conical intersections, bond breaking or strong geometrical distortions. However, for the relatively small active spaces used in this work,51 it does not include dynamical correlation effects and therefore the energies may be inaccurate. To obtain more reliable energies, single-point extended multi-configuration quasi-degenerate second-order perturbation theory (XMCQDPT2) calculations52 were carried out at optimised stationary points and along decay pathways calculated using the CASSCF method. This protocol, termed MS-MR-PT2//CASSCF, is used because the XMCQDPT2 analytic gradient, required to optimize stationary points efficiently, is not available. This procedure53 has been used in previous investigations of the photochemistry of small organic molecule, including DNA bases.54–57
The XMCQDPT2 method is a new approach to second order multi-state multi-reference perturbation theory developed by Granovsky.52 It is an extension of the MCQDPT2 method58 designed to correct for some of the known deficiencies of the latter and other flavours of MS-MR-PT2, such as the problematic behaviour of the energies near conical intersections or avoided crossings. Since its implementation in the Firefly QC package,59 the XMCQDPT2 method has been applied to a number of problems of biological and photochemical interest.60–67 In addition, it was shown to compare favourably with respect to experimental observations and other high-level theoretical methods.68–70
In XMCQDPT2 calculations, it is important to include more CASSCF states in the model space spanned by the zero-order effective Hamiltonian than in the state averaged CASSCF stage.52,71,72 In all the XMCQDPT2 calculations reported here, the 30 lowest CASSCF states were included in the model space spanned by the XMCQDPT2 zero-order effective Hamiltonian. Test calculations at the ground state equilibrium and various distorted geometries were performed to check that this size of model space yielded converged energies. An Intruder State Avoidance (ISA) denominator shift of 0.02 was used in all the XMCQDPT2 calculations.
Two different active spaces were employed for the CASSCF calculations. CAS1 consists of 8 electrons in 7 orbitals: 3 occupied π orbitals and the corresponding 3 unoccupied π* orbitals (in the reference ground-state configuration) together with the occupied nitrogen lone-pair orbital. Since it does not include the 3s/σ* orbital, this active space cannot describe the 1π3s/πσ* state. CAS2 consists of 10 electrons in 9 orbitals: CAS1 augmented with a pair of σ and 3s/σ* orbitals centered on the amino group.† The 3s/σ* orbital has strong Rydberg character in the FC region and is needed to describe the low-lying 11π3s/πσ* state, which is known to play an important role in the photochemistry of aniline and many other substituted aromatic organic compounds.73,74 From now on, the 11π3s/πσ* state will be simply labelled as 1πσ*.
All the calculations performed with the CAS1 active space used the 6-311G** basis set whereas the calculations performed with the CAS2 active space use the 6-311++G** basis set augmented with two diffuse s functions and two sets of diffuse p functions on the nitrogen atom as well as a single diffuse s functions on each of the two amino hydrogen atoms. The exponents of the supplementary diffuse functions are determined in an even tempered manner by dividing the exponent of the most diffuse s and p functions already present in the 6-311++G** basis set by a factor of 3.0. Such an extension of the basis set was found to have a significant effect on the 1πσ* state vertical excitation energy and the height of the barrier to photodissociation, as has been found for pyrrole75,76 and phenol.77
For the MECI optimizations using the CAS1 active space, we found it necessary to use a quadratically convergent algorithm for the CASSCF wavefunction. The orbital rotation derivative contributions from the coupled perturbed multi-configurational self-consistent field (CP-MCSCF) equations were neglected in the MECI optimizations using the CAS2 active space.
Throughout this paper, the notation SAn-CASSCF is used to describe a state averaged CASSCF calculation using orbitals averaged over n electronic states.
The CASSCF optimizations were performed using the Gaussian 03 program package78 and the single point CASSCF and XMCQDPT2 calculations were performed using the Firefly QC package,59 which is based partially on the GAMESS (US) source code.79
3 Vertical and adiabatic excitation energies
The equilibrium geometries of the ground and 11ππ* states, optimized using the CAS1 active space, are shown in Fig. 2. Both have Cs symmetry and a pyramidal arrangement around the nitrogen atom.The focus of this paper is the four lowest lying singlet electronic states, which we label GS, 1ππ*, 1πσ* and 2ππ*, in order of increasing energy.37,39 The vertical excitation energies computed using the two active spaces described in Section 2 are reported in Table 1 and compared with previous calculations and experimental values.
| A′′1ππ* | A′1πσ* | A′2ππ* | A′′2πσ* | |
|---|---|---|---|---|
| a This work. b From ref. 38, assigned as the 0–0 transition. | ||||
| CAS1a | 4.81(4.59) | — | 7.38 | — |
| CAS1/XMCQDPT2a | 4.28(4.02) | — | 5.39 | — |
| CAS2a | 4.81(4.59) | 4.90 | 7.36 | 6.19 |
| CAS2/XMCQDPT2a | 4.22(3.96) | 4.74 | 5.25 | 6.25 |
| SAC-CI37 | 4.20 | 4.53 | 5.34 | 6.39 |
| MS-CASPT239 | 4.33 | 4.85 | 5.54 | 6.28 |
| CR-EOM-CCSD(T)40 | 4.21 | 4.69 | 5.42 | — |
| Exp. | 4.41(4.22)3 | 4.60b | 5.423 | — |
Generally, the calculation of excitation energies using multi-reference perturbation techniques require that all states of interest are described uniformly well at the CASSCF level. This requirement often implies the use of state averaged CASSCF orbitals since state specific CASSCF orbitals can be quite different for the different electronic states of interest. This is specially true when the states of interest have a different nature. For instance, Stavros et al.32 have shown that the 1ππ* state arises from a local excitation within the phenyl ring whereas the 2ππ* state has a significant charge transfer character. In addition the 1πσ* state has a significant 3s Rydberg character at the FC geometry. In this case, a state specific CASSCF calculation leads to a significant valence–Rydberg mixing whereas a state averaged CASSCF calculation yields orbitals with a well defined character. It was therefore necessary to use state averaged CASSCF orbitals for a well balanced description of the electronic states of interest at the XMCQDPT2 level of theory.
The CAS1 active space can not describe the 1πσ* state and is used to compute the two first ππ* state excitation energies at the SA3-CASSCF and XMCQDPT2 levels of theory. The CASSCF 1ππ* vertical excitation energy is in reasonable agreement with the experimental value. In contrast, the 2ππ* vertical excitation energy is grossly overestimated with an error of 1.96 eV. This is not unexpected as the CASSCF method has a well known propensity to overestimate excitation energies to excited states with significant charge transfer character. The XMCQDPT2 calculation using the same CASSCF wavefunction yields excitation energies in good agreement with previous calculations and experimental observations.
In addition to the 1πσ* state, the CAS2 active space generates a second low-lying π3s/πσ* state, hereafter denoted 2πσ*, which was calculated at 6.39 and 6.28 eV respectively in previous work.37,39 Because of the large overestimation of the 2ππ* state, the CASSCF method wrongly predicts the 2πσ* state below the 2ππ* state. The correct ordering is however recovered at the XMCQDPT2 level of theory. SA5-CASSCF and XMCQDPT2 vertical excitation energies using the CAS2 active space are presented in Table 1. These results show that the corrections due to the inclusion of the dynamical correlation energy are less important for the πσ* states than for the ππ* states. Overall, the XMCQDPT2 method provides accurate and well balanced results for the vertical excitation energies of the electronic states of interest.
Finally, we note that the CR-EOM-CCSD(T) calculations of Worth et al.40 predicted the 3pz and 3py states to lie at 5.31 and 5.42 eV respectively, between the 1πσ* and 2ππ* states which is in disagreement with previous high level calculations.37,39 We have performed a calculation at the XMCQDPT2 level of theory using the CAS2 active space augmented with the two corresponding 3p Rydberg orbitals. This calculation predicted both states to lie slightly above the 2ππ* state, at 5.58 and 5.62 eV respectively. These states are not considered further in this paper.
4 Photochemistry after excitation to the 1ππ* state
In this section, we consider the decay pathways of aniline after excitation to the 1ππ* state, at energies below the vertical excitation energy of the 1πσ* state.In ref. 32, the 1ππ* state was described as arising from a local excitation within the phenyl ring, and the analogy with the S1 state of benzene was pointed out. Indeed, the photochemistry of aniline after excitation to the 1ππ* state shares many similarities with that of benzene. Following excitation with wavelengths >245 nm, benzene fluoresces with a quantum yield of approximately 0.2 and the remaining population decays via ISC to a low lying triplet state. These two processes take place on the timescale of 10–100 ns.80,81 At wavelengths <245 nm (corresponding to >3000 cm−1 above the S1 ← S0 0–0 transition) the fluorescence is quenched and the molecule quickly decays to the ground state by internal conversion (IC) via the so-called prefulvene conical intersection.82–90 This conical intersection occurs at a geometry characterized by a strong out-of-plane distortion of one of the carbon atoms, accompanied by a strong reduction of the distance between carbon atoms either side of the one raised out of plane to generate the crossing. The activation energy needed to reach the prefulvene deactivation channel has been shown theoretically to be the consequence of the presence of a potential barrier on the pathway connecting the FC region and the MECI on the S1 PES. Conical intersections between the first excited ππ* state and the ground state with similar geometries have been described in many other aromatic molecules, for example, pyrazine91 and phenol.92
In aniline, following excitation to the 1ππ* state at wavelengths >270 nm, the fluorescence quantum yields are comparable to those of benzene and single vibronic level lifetimes ranging from 3 to 9 ns have been reported.33,34 Whereas in benzene the six carbon atoms are equivalent, the presence of the amino group in aniline breaks this symmetry and the C1, C2, C3 and C4 carbon atoms (see Fig. 1) are not equivalent. As we show below, this gives rise to the presence of four distinct S1(1ππ*)/GS prefulvene-like MECI points, labelled CIi1ππ*/GS (i = 1,2,3,4), where the superscript refers to the out-of-plane carbon atom (see Fig. 1). Although three of these (CI11ππ*/GS, CI21ππ*/GS and CI31ππ*/GS) have been reported before,32 to the best of our knowledge this is the first report of CI41ππ*/GS. It is worth noting that a similar prefulvene-like MECI has been found in phenol92 with an out-of-plane distortion of the carbon atom carrying the hydroxyl group. TSs have been found between the 1ππ* equilibrium geometry and CI11ππ*/GS and CI41ππ*/GS, indicating the presence of potential barriers along these pathways. Potential energy barriers along the pathway to the prefulvene MECI are known in benzene,82–90 pyrazine91 and phenol.92 The four MECI and two TS geometries, optimized using the CAS1 active space are presented in Fig. 3.
 | ||
| Fig. 1 The aniline molecule with the numbering scheme used throughout this paper. | ||
 | ||
| Fig. 2 Ground and 1ππ* state CASSCF optimized geometries. | ||
 | ||
| Fig. 3 Geometries of the four prefulvene-like MECIs and of the two TSs, TS11ππ* and TS41ππ*, optimized along the pathways leading to CI11ππ*/GS and CI41ππ*/GS, respectively. Optimizations were performed using the CAS1 active space. | ||
No TSs were found on the pathway connecting the 1ππ* equilibrium geometry and CI21ππ*/GS or CI31ππ*/GS. However, in both cases, the pathway crosses a very flat portion of the 1ππ* PES before reaching the MECI point. Both CASSCF and XMCQDPT2 energies of the four MECIs and two TSs are listed in Table 2. The XMCQDPT2 energies have been computed at the CASSCF optimized geometries. Because the degeneracy is lifted for XMCQDPT2 calculations at CASSCF optimized MECI points, the averaged energies of the two states and the magnitudes of the energy gaps between them are reported.
| CASSCF | XMCQDPT2 | |
|---|---|---|
| CI11ππ*/GS | 5.50 | 4.87(0.22) |
| CI21ππ*/GS | 5.38 | 4.76(0.07) |
| CI31ππ*/GS | 5.39 | 4.69(0.21) |
| CI41ππ*/GS | 5.26 | 4.55(0.46) |
| TS11ππ* | 5.57 | 5.10 |
| TS41ππ* | 5.35 | 4.96 |
These results show that, both at the CASSCF and XMCQDPT2 levels of theory, the CI41ππ*/GS MECI appears at slightly lower energy than the three others. Note that, the dynamical correlation energy correction is smaller at the TSs than at the MECIs. This indicates that the XMCQDPT2 barriers are larger than the CASSCF ones. Therefore, the lack of CASSCF TS on the pathways leading to CI21ππ*/GS and CI31ππ*/GS might be a consequence of the non uniform accuracy of the CASSCF method along the pathways.
Fig. 4 presents the pathway between the 1ππ* equilibrium geometry and the CI41ππ*/GS MECI geometries calculated at the SA2-CASSCF and XMCQDPT2 levels of theory. A single linearly interpolated scan between these two geometries would have overestimated the height of the potential energy barrier separating them so the scan was split into two parts: from the 1ππ* equilibrium geometry to TS41ππ* and from TS41ππ* to the CI41ππ*/GS MECI geometry.
 | ||
| Fig. 4 Ground (blue line with circles) and 1ππ* (red line with squares) state potential energy profiles along the linearly interpolated internal coordinate (LIIC) from the 1ππ* equilibrium geometry to the CI41ππ*/GS MECI computed at the (a) SA2-CASSCF and (b) XMCQDPT2 levels of theory. The vertical black dashed line marks the TS41ππ* position (see text for details). | ||
Our calculation shows that the energy required to open the prefulvene decay channel is higher than the vertical excitation energy of the 1πσ* state (Table 1) and therefore, processes taking place on the 1πσ* surface are likely to compete with the prefulvene decay channel. Indeed, Stavros et al.32 and Montero et al.42 reported evidence of decay through the prefulvene channel after excitation of the 2ππ* state. This decay pathway implies evolution on the 2ππ* PES towards a 2ππ*/1ππ* CI and subsequent relaxation on the 1ππ* PES towards the prefulvene CI. In this context, the new CI41ππ*/GS MECI reported here seems particularly relevant since its geometry is similar to the 2ππ*/1ππ* MECI. Both MECIs involve a strong out-of-plane distortion of the carbon atom carrying the amino group. Therefore, the direct pathway from the 2ππ*/1ππ* MECI to the CI41ππ*/GS MECI involves much less IVR than the corresponding pathways to the other prefulvene MECIs (see Section 6).
5 Photochemistry after excitation to the 1πσ* state
In this section, we study the decay pathways of aniline following excitation at wavelengths <269 nm, i.e. above the onset of the 1πσ* state.38In the FC region, the 1πσ* state PES has a quasi-bound minimum associated with strong Rydberg 3s character on the N atom. However, along the N–H stretching coordinate, the 1πσ* state acquires a valence σ* character and the PES becomes dissociative. The quasi-bound and dissociative components of the PES are separated by a significant barrier, the height of which has been estimated at 0.5 eV using the EOM-CCSD method.40 The 1πσ* state and its role in the electronic relaxation of photoexcited aniline has been the subject of a number of recent investigations.32,40–45
We have performed a relaxed potential energy scan along the N–H stretching coordinate on the 1πσ* PES, i.e. at each point, the N7–H14 bond length is kept fixed and the geometry of the rest of the molecule is optimized. The molecule is planar with C2v symmetry at the 1πσ* equilibrium geometry (see Fig. 6). Upon elongation of the N7–H14 bond, the symmetry is reduced to Cs′. Here, the Cs′ notation is used to distinguish the planar geometry (with non-equal N–H bond lengths), i.e. where the symmetry plane is the molecular plane, from the ground state pyramidal equilibrium geometry of Cs symmetry, where the symmetry plane is perpendicular to the phenyl ring and bisects the H–N–H angle of the amino group. In Cs′ symmetry, the 1πσ* state belongs to the A′ irreducible representation whereas the GS, 1ππ* and 2ππ* states belong to the A′ irreducible representation. In these calculations, the A′ and A′′ states have been computed separately.93 The geometries on the 1πσ* PES are optimized at the SS-CASSCF level of theory using the CAS2 active space. We note that in Cs′ symmetry, there is no valence–Rydberg mixing problem since the valence and Rydberg states have different symmetry. The A′ state energies at the optimized geometries have been computed at the SA3-CASSCF level of theory using the same active space. XMCQDPT2 calculations are also performed at the CASSCF optimized geometries and the results of both calculations are presented in Fig. 5.
 | ||
| Fig. 5 Relaxed potential energy scan along the N–H coordinate, computed at the (a) CASSCF and (b) XMCQDPT2 levels of theory using the CAS2 active space, showing the ground-state (blue circles), 1ππ* state (red squares), 1πσ* state (brown crosses), 2ππ* state (green diamonds). The geometries are optimized on the 1πσ* state at the SS-CASSCF level of theory using Cs′ symmetry. A zoom of the 1ππ* and 1πσ* states potential scans around the 1πσ* minimum is shown in panel (c). | ||
 | ||
| Fig. 6 Geometries of the 1πσ* state local minimum and TS as well as of the πσ*/ππ* and πσ*/GS MECIs. | ||
Interestingly, although the 1πσ* state is the second excited state at the FC geometry (see Table 1), from Fig. 5 it is clear that it lies below the 1ππ* state in the CASSCF relaxed scan. This is a strong indication that upon relaxation on the 1πσ* state from the FC geometry, the system must evolve toward a πσ*/1ππ* seam of conical intersections (see Fig. 7 below). At long N–H distance, the 1πσ* curve crosses the GS curve, indicating a competition between internal conversion to the ground state and N–H dissociation upon evolution on the dissociative part of the 1πσ* state. Both πσ*/1ππ* and πσ*/GS MECIs have been reported previously.32 Our CASSCF calculation yields a height of 0.58 eV for the barrier separating the quasi-bound well from the dissociative part on the 1πσ* curve, and an N–H bond strength of 3.37 eV, which is a significant underestimate compared to the experimental value of 3.92 eV reported by Ashfold and coworkers.41 The CASSCF optimized geometries of the 1πσ* state local minimum and TS, noted Minπσ* and TSπσ* respectively, as well as of the πσ*/ππ* and πσ*/GS MECIs, noted CIπσ*/1ππ* and CIπσ*/GS respectively, are presented in Fig. 6.
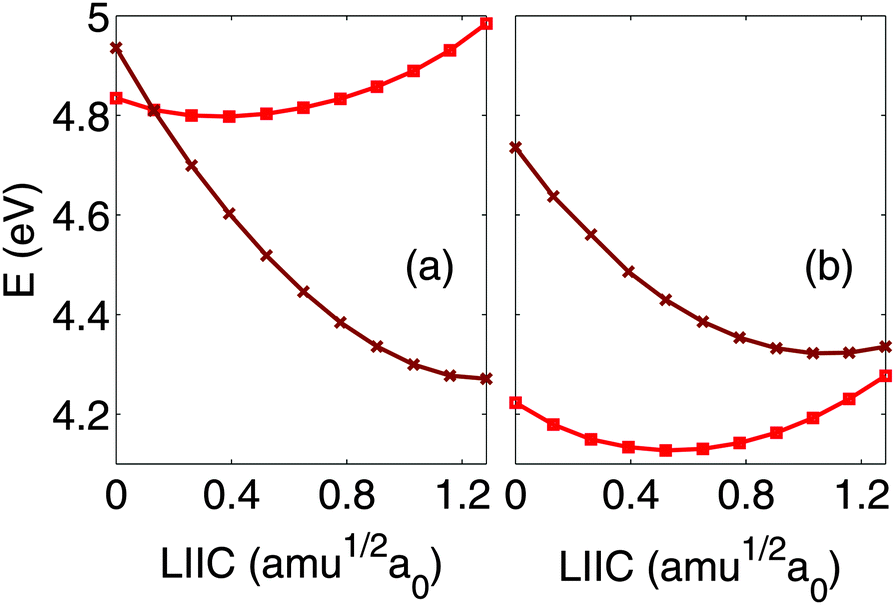 | ||
| Fig. 7 1ππ* (red squares) and 1πσ* (brown crosses) potential energy curves along the linearly interpolated internal coordinate between the FC and CASSCF optimized Minπσ* geometries, computed at the (a) SA5-CASSCF and (b) XMCQDPT2 levels of theory. | ||
At the Minπσ* geometry, the molecule is planar with a C2v symmetry point group and an N–H bond length of 1.01 Å. The TSπσ* and CIπσ*/GS geometries are also planar, with Cs′ symmetry and N–H bond lengths of 1.31 Å and 1.85 Å, respectively.
We now discuss the results obtained from the XMCQDPT2 calculation of the relaxed scan at the CASSCF optimized geometries, shown in Fig. 5(b). We obtain a barrier height of 0.41 eV, which is lower than the CASSCF value, and an N–H bond strength of 3.83 eV, in good agreement with the experimental value of 3.92 eV.41 As shown in Section 3, the inclusion of dynamical electron correlation has a greater effect on the 1ππ* state than on the 1πσ* state and for short N–H distances, the 1ππ* and 1πσ* states are almost degenerate with the 1ππ* state lying slightly below the 1πσ* state. As the N–H distance increases slightly, the 1πσ* crosses the 1ππ* curve close to its local minimum in the quasi-bound well. This supports the conclusion drawn from experimental observation of the relaxation dynamics following excitation close to or above the minimum of the 1πσ* state.43,44 This has interesting consequences regarding the vibronic structure of the molecule in this range of energies. In ref. 38, an experimental excitation energy of 4.60 eV was attributed to the 0–0 transition to the 1πσ* state. However, a conical intersection lying close to the 1πσ* local minimum points towards the existence of strongly vibronically coupled levels that cannot be attributed easily to vibrational levels of either 1ππ* or 1πσ* states, as discussed in ref. 94.
In order to gain further insight into the competition between relaxation to the 1ππ* state and relaxation to the dissociative part of the 1πσ* state, we have computed a linearly interpolated scan from the FC to the CASSCF optimized Minπσ* geometries (Fig. 7). State averaging over the five lowest states is used in this calculation. At the CASSCF level of theory, the πσ*/1ππ* CI appears very close to the FC geometry. However, the situation is quite different at the XMCQDPT2 level of theory and the CI appears at a geometry much closer to the Minπσ* geometry. This suggests that, after excitation to the 1πσ* state, the system will relax toward the CIπσ*/1ππ*, where it will either be transferred to the 1ππ* state and then decay on a very long (nanosecond) timescale, as discussed in the previous section, or stay on the 1πσ* state and evolve on the dissociative portion of the PES.
6 Photochemistry after excitation to the 2ππ* state
In this section, we study the decay pathway of aniline following excitation to the 2ππ* state. Experimentally, this corresponds to excitation with wavelengths <240 nm. A number of experimental studies have been directed towards unravelling 2ππ* electronic relaxation using a variety of techniques: time-resolved photoelectron spectroscopy,43,44 time-resolved photoionisation spectroscopy,42 time-resolved velocity-map imaging32 and H-atom (Rydberg) photofragment translational spectroscopy.41 In all these studies, ultrafast decays were observed, or inferred, and interpreted as either internal conversion to the ground state or H-atom loss on the 1πσ* surface. For example, Ashfold and coworkers recorded bi-modal total kinetic energy release (TKER) spectra at various excitation wavelengths in the range 240–220 nm.41 The fast H-atom component was attributed to dissociation on the 1πσ* surface after internal conversion via a 2ππ*/πσ* CI, or a 2ππ*/1ππ* CI with subsequent transfer to the 1πσ* surface via the πσ*/1ππ* CI. The slow H-atom component was attributed to dissociation from the ground-state following internal conversion from the 2ππ* state to the ground-state. Montero et al. recorded three different time constants in the same wavelength range, τ0 = 21 ± 5 fs, τ1 = 150 ± 30 fs and τ2 = 45 ± 2 ps.42 The τ0 constant was attributed to an internal conversion through a 2ππ*/1ππ* CI, with a subsequent competition between internal conversion to the 1πσ* state and dissociation, corresponding to the τ1 constant, or internal conversion to the ground state via the prefulvene CI discussed in Section 4, corresponding to the much larger τ2 constant. This large time constant would be the sign of a considerable intramolecular vibrational energy redistribution (IVR) in the 1ππ* state competing with internal conversion to the ground state.In terms of previous theoretical studies, a 2ππ*/1ππ* MECI was reported,32 at a geometry involving a strong out-of-plane deformation of the molecule.
Here, we report evidence for a new decay pathway leading to internal conversion to the ground state, mediated by three successive MECIs, two of which have not been reported before. The MEP connecting the 2ππ* state at the FC geometry to the ground state is presented in Fig. 8. The MEP calculation was performed in several steps and the geometry was constrained to Cs symmetry. We began by performing an IRC calculation, starting at the FC geometry on the 2ππ* state, using the CAS1 active space and a CASSCF wavefunction averaged over the 1ππ* and 2ππ* states. The initial relaxation direction was taken to be the gradient on the 2ππ* surface. The CAS1 active space is used to filter out the πσ* states, which greatly simplifies the IRC calculation. The IRC calculation was stopped at the 1ππ*/2ππ* crossing. Then, a single point was calculated at a geometry obtained by extrapolating the last IRC step. At this point, we checked that the 2ππ* state was lying below the 1ππ* state. Because the geometry was constrained to Cs symmetry, the symmetry of the 1ππ* and 2ππ* states (A′′ and A′ respectively) is conserved along the MEP. Therefore, the 1ππ* and 2ππ* states can be distinguished unambiguously by checking their respective wavefunctions. A second IRC calculation was then performed starting at this last geometry, using the CAS1 active space and a CASSCF wavefunction averaged over the 2ππ* and ground states.
 | ||
| Fig. 8 A′ Ground state (blue circles), A′′1ππ* (red squares), A′1πσ* (brown crosses), A′2ππ* (green diamonds) and A′′2πσ* (magenta triangles) potential energy profiles computed at the SA5-CASSCF (a) and XMCQDPT2 (b) levels of theory along the IRC scan on the A′2ππ* state. | ||
In the second stage, in order to include the πσ* states in our calculations, we performed single point SA-CASSCF and XMCQDPT2 calculations at the geometries obtained from the IRC calculations, using the CAS2 active space. The CASSCF wavefunction was averaged over the five lowest states. The results of these CASSCF and XMCQDPT2 calculations are displayed in Fig. 8(a) and (b), respectively. From now on, the intrinsic reaction coordinate will be noted qIRC.
First, we consider the CASSCF IRC scan shown in Fig. 8(a). As discussed in Section 3, the CASSCF method yields the wrong state ordering at the FC geometry, i.e. the 2πσ* state lies below the 2ππ* state. As relaxation on 2ππ* begins from the FC geometry, the 2ππ* state energy decreases and the 2πσ* state energy increases until the two curves cross around qIRC = 1.8. This 2ππ*/2πσ* CI is an artifact of the CASSCF method. Overall, the CASSCF MEP shows the existence of a barrierless relaxation pathway involving a strong out-of-plane deformation of the molecule, resulting in a boat conformation of the phenyl ring at the end of the IRC scan. Along this pathway, the 2ππ* state crosses, successively, the 1ππ*, 1πσ* and ground electronic states.
We now discuss the XMCQDPT2 MEP computed at the CASSCF optimized geometries shown in Fig. 8. As already discussed, the correction to the energies from the inclusion of dynamical electron correlation is quite large for the 2ππ* state. Upon relaxation, the energies of the 1ππ*, 1πσ* and 2ππ* states come close to one other around qIRC = 3. In this region, the three states lie within 0.25 eV of each other, which is a strong indication for the presence of a nearby three-state CI.95–97 In contrast to the CASSCF MEP, here the 1πσ* and 2ππ* states, both of A′ symmetry, form a narrowly avoided crossing rather than a true CI. This is, however, just an indication that the XMCQDPT2 pathway passes very close to the 1πσ*/2ππ* CI seam rather than crossing it as in the CASSCF calculation. The pseudo-diabatic assignment used to distinguish between the states is of little significance in the interaction region because the electronic characters of the 1πσ* and 2ππ* states are mixed; however, as we move away from the three-state CI region, the electronic character of both states become clear again. The same is true for the region where the 2ππ* state approaches the ground state at the end of the scan.
These calculations have unveiled three CIs that are relevant to the electronic relaxation of aniline following excitation to the 2ππ* state: 1πσ*/2ππ*, 1ππ*/2ππ* and GS/2ππ* CIs. We have optimized the three corresponding MECIs, labelled CI1πσ*/2ππ*, CI1ππ*/2ππ* and CIGS/2ππ*, at the CASSCF level of theory using the CAS2 active space for CI1πσ*/2ππ* and the CAS1 space for CI1ππ*/2ππ* and CIGS/2ππ*. The three resulting geometries are shown in Fig. 9. Although CI1ππ*/2ππ* has been reported previously,32 CI1πσ*/2ππ* and CI2ππ*/GS are reported here for the first time.
 | ||
| Fig. 9 Geometries of the three MECIs involving the 2ππ* state. | ||
In Fig. 10, we show the geometries of the molecule for four key points along the MEP: the FC geometry (qIRC = 0), the geometry of the approximate three-state CI (qIRC = 2.89) the geometry of the 1ππ*/2ππ* crossing (qIRC = 5.68) and the geometry corresponding to the GS/2ππ* crossing (qIRC = 12.55).
 | ||
| Fig. 10 Geometries at key points along the MEP (see text for details). | ||
Overall, the XMCQDPT2 decay pathway on the 2ππ* state surface connecting the equilibrium geometry to the CI2ππ*/GS is barrierless, although flatter than the CASSCF computed pathway. This suggests that the corrections due to the inclusion of dynamical electron correlation are greater in the FC region than in the region of the CI2ππ*/GS corresponding to a strongly distorted geometry. However, another possible explanation is that the geometries along the pathway are obtained via CASSCF partial optimizations, therefore they may be far from the true XMCQDPT2 MEP. Nonetheless, it is interesting to note that similar ring puckering decay pathways have been described in other simple aromatic organic molecules such as pyrrole98 or the purine DNA bases adenine55,99 and guanine.57 Moreover, the boat conformation of the phenyl ring, found at qIRC = 12.55 (see Fig. 10), is reminiscent of the Dewar form of benzene, which is know to be populated after excitation to the 2ππ* state of benzene.100–102 To investigate this further, we extrapolated from the CI at qIRC = 12.55, along the IRC until a geometry where the 2ππ* state is the ground state was found and then performed a geometry optimization. The resulting local minimum on the ground state PES (MinDew) is an aniline analogue of the Dewar benzene isomer (Fig. 11). We have also located a saddle point (TSDew) connecting this local minimum to the global equilibrium geometry (Fig. 11).
 | ||
| Fig. 11 Geometries of the Dewar minimum and TS on the ground state PES. | ||
The energies of the Dewar minimum and TS relative to the global minimum energy, computed at the SA2-CASSCF and XMCQDPT2 level of theory using the CAS1 active space, are presented in Table 3. The SA2-CASSCF energies of the CI2ππ*/GS points, are also reported. They show that the Dewar form of aniline is associated with a local minimum on the ground state PES, separated from the global minimum by a barrier with a height evaluated at 0.57 eV. Interestingly, the Dewar form of aniline might provide a possible experimental probe of the ring puckering decay pathway discussed above, i.e. an experiment aiming at its detection after excitation to the 2ππ* state would provide a test of the relevance of this decay path in the photochemistry of aniline.
| SA2-CASSCF | XMCQDPT2 | |
|---|---|---|
| MinDew | 3.97 | 3.54 |
| TSDew | 4.60 | 4.11 |
| CIMECI2ππ*/GS | 4.79 | — |
| CIIRC2ππ*/GS | 5.21 | — |
In addition to the ring-puckering decay pathway discussed above, the potential energy curves in Fig. 8 suggest two alternative decay pathways. At the three-state CI, the molecule can relax to the 1ππ* or 1πσ* states. In both cases, it is clear from Fig. 8 that the molecule can then relax directly to the FC region. From the FC region, the molecule can then decay to the ground state via the prefulvene CI1ππ*/GS shown in Fig. 3 or the CIπσ*/GS shown in Fig. 6, or dissociate on the 1πσ* PES. In order to find out if other direct decay pathways to the ground state exist, we performed linearly interpolated scans from the CI1ππ*/2ππ* to the prefulvene CI1ππ*/GS and from the CI1πσ*/2ππ* to the CI1πσ*/GS respectively, using the CAS2 active space. For the former, the orbitals were averaged over the five lowest states while for the latter, the inclusion of a sixth state was found necessary for a well balanced description of the states of interest along the pathway. Both calculations are presented in Fig. 12.
 | ||
| Fig. 12 Linearly interpolated scans (a) from the CI1ππ*/2ππ* to the prefulvene CI1ππ*/GS and (b) from the CI1πσ*/2ππ* to the CI1πσ*/GS computed at the SA5-CASSCF and SA6-CASSCF level of theory, respectively, using the CAS2 active space. | ||
In both cases, the initial geometries are not taken to be the MECI structures of CI1ππ*/2ππ* and CI1πσ*/2ππ*, but rather the CI points reached in the CASSCF IRC pathway of Fig. 8(a). The scan in Fig. 12(a) shows a rather flat but barrierless pathway connecting the CI1ππ*/2ππ* with the prefulvene CI1ππ*/GS. Interestingly, a similar barrierless pathway connecting the S2/S1 CI to the prefulvene S1/S0 CI in benzene has been calculated.103,104 However, Fig. 8(a) shows that the path connecting the CI1ππ*/2ππ* with the FC geometry is steeper. Therefore, it seems most likely that, if the molecule relaxes to the 1ππ* state, it will then relax to the FC region where it can relax further via the prefulvene CI1ππ*/GS or via the N–H dissociation path on the 1πσ* state after crossing the CIπσ*/1ππ*, as has been proposed previously.41,42
The scan of Fig. 12(b) shows a steep pathway connecting the CI1πσ*/2ππ* to the CI1πσ*/GS, suggesting the possibility of very efficient relaxation to the ground state or dissociation on the 1πσ* PES. A barrier is seen on the 1πσ* potential energy profile. However this barrier appears much smaller than the barrier on the relaxed N–H potential scan shown in Fig. 5, and should therefore be easily overcome by the wavepacket upon relaxation on the 1πσ* state after crossing the CI1πσ*/2ππ*.
It is not possible from this work to infer which decay pathway will be preferred by the molecule, after excitation to the 2ππ* state. It is known that nuclear dynamics around CIs do not only depend on the location of the MECI point but also on the topography of the extended CI seam. Specifically, different decay pathways can be preferred depending on which region of the CI seam is reached.105–107 Therefore, an extended mapping of the various CI seams connecting the electronic states of interest in this work would provide valuable information on the competition between the different accessible decay channels. Direct dynamics techniques108,109 can also be used to follow the relaxation pathway of the molecule directly. The conclusions drawn in this work indicate that both approaches should use post-CASSCF electronic structure calculations to reach the level of accuracy necessary for a reliable description of the relaxation of aniline after electronic excitation.
7 Summary
CASSCF and XMCQPDT2 calculations have been employed to determine key geometries and pathways on the potential energy landscape of the four lowest lying electronic singlet states of aniline, S0(GS), 1ππ*, 1πσ* and 2ππ*. We have located four prefulvene-like MECIs connecting the 1ππ* state with GS. The lowest energy MECI, in which the carbon-atom carrying the amino group is distorted out-of-plane, is reported here for the first time. It seems likely that this MECI is involved in non-radiative decay from 1ππ* to the ground-state, although the energy of this MECI lies above the vertical excitation energy to 1πσ* and will only be accessible at higher excitation energies. We have found a MECI connecting 1πσ* and 1ππ* states close to the local minimum on 1πσ*. We determine that excitation to 1πσ* is likely to be followed by relaxation to this MECI where population will subsequently be transferred both to 1ππ* (as observed in our earlier work43,44) and to the dissociative component of 1πσ* (as observed by Stavros et al.32). We also find evidence for a new decay pathway connecting 2ππ* and the ground-state that passes through a three-state CI involving 2ππ*, 1πσ* and 1ππ*. From this three-state CI, population may be transferred on the lower portion of the 2ππ* state with subsequent relaxation towards the CI2ππ*/GS, or on the 1πσ* or 1ππ* states, with subsequent relaxation towards the CI1πσ*/GS or prefulvene CI1ππ*/GS, respectively. This scheme supports our interpretation of our earlier data43,44 and is consistent with the observations of others.32,41,42 Overall, our calculations are in agreement with all experimental observations.Acknowledgements
Calculations were performed using HPC resources from DSI-CCUB (Université de Bourgogne). The authors acknowledge support from the European Marie Curie Initial Training Network Grant No. GA-ITN-214962-FASTQUAST for research funding. M. S. and S. G. acknowledge support from the Conseil Régional de Bourgogne. The authors are grateful to G. A. Worth and M. A. Robb for their comments on the manuscript.Notes and references
- K. Kimura, H. Tsubomura and S. Nagakura, Bull. Chem. Soc. Jpn., 1964, 37, 1336 CrossRef CAS.
- K. Kimura and S. Nagakura, Mol. Phys., 1965, 9, 117 CrossRef CAS.
- B. N. Rajasekhar, A. Veeraiah, K. Sunanda and B. N. Jagatap, J. Chem. Phys., 2013, 139, 064303 CrossRef CAS PubMed.
- D. G. Lister, J. K. Tyler, J. H. Hog and N. W. Larsen, J. Mol. Struct., 1974, 23, 253 CrossRef CAS.
- M. Fukuyo, K. Hirotsu and T. Higuchi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 640 CrossRef.
- G. Roussy and A. Nonat, J. Mol. Spectrosc., 1986, 118, 180 CrossRef.
- G. Schultz, G. Portalone, F. Ramondo, A. Domenicano and I. Hargittai, Struct. Chem., 1996, 7, 59 CrossRef CAS.
- A. Wolf, U. Voets and H.-H. Schmidtke, Theor. Chim. Acta, 1980, 54, 229 CrossRef CAS.
- Z. Niu, K. M. Dunn and J. E. Boggs, Mol. Phys., 1985, 55, 421 CrossRef CAS.
- J. E. Boggs and Z. Niu, J. Comput. Chem., 1985, 6, 46 CrossRef CAS.
- C. W. Bock, P. George and M. Trachtman, Theor. Chim. Acta, 1986, 69, 235 CrossRef CAS.
- A.-D. Gorse and M. Pesquer, THEOCHEM, 1993, 281, 21 CrossRef.
- Y. Wang, S. Saebo and C. U. Pittman Jr, THEOCHEM, 1993, 281, 91 CrossRef.
- M. Castellà-Ventura and E. Kassab, Spectrochim. Acta, Part A, 1994, 50, 69 CrossRef.
- J. Sponer and P. Hobza, Int. J. Quantum Chem., 1996, 57, 959 CrossRef CAS.
- O. Bludský, J. Sponer, J. Leszczynski, V. Spirko and P. Hobza, J. Chem. Phys., 1996, 105, 11042 CrossRef PubMed.
- I. López-Tocón, J. C. Otero, M. Becucci, G. Pietraperzia and E. Castellucci, Chem. Phys., 1999, 249, 113 CrossRef.
- M. A. Palafox, J. L. N. nez and M. Gil, THEOCHEM, 2002, 593, 101 CrossRef.
- P. M. Wojciechowski, W. Zierkiewicz, D. Michalska and P. Hobza, J. Chem. Phys., 2003, 118, 10900 CrossRef CAS PubMed.
- M. A. Palafox, M. Gill, N. J. Nunez, V. K. Rastogi, L. Mittal and R. Sharma, Int. J. Quantum Chem., 2005, 103, 394 CrossRef CAS.
- V. Hänninen and L. Halonen, J. Chem. Phys., 2007, 126, 064309 CrossRef PubMed.
- I. V. Alabugin, M. Manoharan, M. Buck and R. J. Clark, THEOCHEM, 2007, 813, 21 CrossRef CAS PubMed.
- J. C. Jiang and C. E. Lin, THEOCHEM, 1997, 392, 181 CAS.
- W. B. Tzeng, K. Narayanan, K. C. Shieh and C. C. Tung, THEOCHEM, 1998, 428, 231 CrossRef CAS.
- I. López-Tocón, R. G. della Valle, M. Becucci, E. Castellucci and J. C. Otero, Chem. Phys. Lett., 2000, 327, 45 CrossRef.
- D. M. Upadhyay, M. K. Shukla and P. C. Mishra, THEOCHEM, 2000, 531, 249 CrossRef CAS.
- K. Pirowska, P. Kolekand, J. Goclon and J. Najbar, Chem. Phys. Lett., 2004, 387, 165 CrossRef CAS PubMed.
- G. Piani, M. Pasquini, I. López-Tocón, G. Pietraperzia, M. Becucci and E. Castellucci, Chem. Phys., 2006, 330, 138 CrossRef CAS PubMed.
- E. Drougas, J. G. Philis and A. M. Kosmas, THEOCHEM, 2006, 758, 17 CrossRef CAS PubMed.
- W. E. Sainclair and D. W. Pratt, J. Chem. Phys., 1996, 105, 7942 CrossRef PubMed.
- L. Santos, E. Martinez, B. Ballesteros and J. Sanchez, Spectrochim. Acta, Part A, 2000, 56, 1905 CrossRef.
- G. M. Roberts, C. A. Williams, J. D. Young, S. Ullrich, M. J. Paterson and V. G. Stavros, J. Am. Chem. Soc., 2012, 134, 12578 CrossRef CAS PubMed.
- H. V. Weyssenhoff and F. Kraus, J. Chem. Phys., 1971, 54, 2387 CrossRef PubMed.
- R. Scheps, D. Florida and S. A. Rice, J. Chem. Phys., 1973, 61, 1730 CrossRef PubMed.
- J. L. Knee and P. M. Johnson, J. Chem. Phys., 1984, 80, 13 CrossRef CAS PubMed.
- B. Kim, C. P. Schick and P. M. Weber, J. Chem. Phys., 1995, 103, 6903 CrossRef CAS PubMed.
- Y. Honda, M. Hada, M. Ehara and H. Nakatsuji, J. Chem. Phys., 2002, 117, 2045 CrossRef CAS PubMed.
- T. Ebata, C. Minejima and N. Mikami, J. Phys. Chem. A, 2002, 106, 11068 CrossRef.
- X.-J. Hou, P. Quan, T. Höltzl, T. Veszprémi and M. T. Nguyen, J. Phys. Chem. A, 2005, 109, 10396 CrossRef CAS PubMed.
- F. Wang, S. P. Neville, R. Wang and G. A. Worth, J. Phys. Chem. A, 2013, 117, 7298 CrossRef CAS PubMed.
- G. A. King, T. A. A. Oliver and M. N. R. Ashfold, J. Chem. Phys., 2010, 132, 214307 CrossRef PubMed.
- R. Montero, A. P. Conde, V. Ovejas, R. Martínez, F. Castaño and A. Longarte, J. Chem. Phys., 2011, 135, 054308 CrossRef PubMed.
- R. Spesyvtsev, O. M. Kirkby, M. Vacher and H. H. Fielding, Phys. Chem. Chem. Phys., 2012, 14, 9942 RSC.
- R. Spesyvtsev, O. M. Kirkby and H. H. Fielding, Faraday Discuss., 2012, 157, 165 RSC.
- J. O. F. Thompson, R. A. Livingstone and D. Townsend, J. Chem. Phys., 2013, 139, 034316 CrossRef PubMed.
- I. N. Ragazos, M. A. Robb, F. Bernardi and M. Olivucci, Chem. Phys. Lett., 1992, 197, 217 CrossRef CAS.
- M. J. Bearpark, M. A. Robb and H. B. Schlegel, Chem. Phys. Lett., 1994, 223, 269 CrossRef CAS.
- F. Bernardi, M. A. Robb and M. Olivucci, Chem. Soc. Rev., 1996, 25, 321 RSC.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- In theory, increasing the size of the active space can allow us to recover a part of the dynamical electronic correlation energy. However, in most cases, this strategy requires an active space too large to be computationally practical.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- It is noteworthy that optimizations of excited state minima and conical intersections of similar compounds at the (MS-)MR-PT2 level of theory were performed in previous studies using numerical optimization procedures.110–113 The resulting structures and energetics were compared to CASSCF predictions. Overall, this studies suggested that, while it is known that the MS-MR-PT2//CASSCF standard procedure can yield dramatically different results than full CASSCF calculations, both in term of energetics and of mechanistic picture of the process under investigation, full MS-MR-PT2 calculations (both optimizations and potential energy scans) are not expected to significantly alter the conclusions obtained by MS-MR-PT2//CASSCF calculations. We mention, however, that in particularly tricky cases where the ordering of the states is very different from CASSCF to MS-MR-PT2, a full MS-MR-PT2 study can be the only solution to obtain reliable results.114 In addition, single reference methods such as CC2115 and ADC(2)116 can be used to map excited state potential energy surfaces in regions free of strong multi-reference character. These methods are known to provide excitation energies and excited state minima of better quality than CASSCF.117.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, Chem.–Eur. J., 2006, 12, 6559 CrossRef PubMed.
- L. Blancafort, J. Am. Chem. Soc., 2006, 128, 210 CrossRef CAS PubMed.
- M. Merchán and L. Serrano-Andrés, J. Am. Chem. Soc., 2003, 125, 8108 CrossRef PubMed.
- L. Serrano-Andrés, M. Merchán and A. C. Borin, J. Am. Chem. Soc., 2008, 130, 2473 CrossRef PubMed.
- H. Nakano, J. Chem. Phys., 1993, 99, 7983 CrossRef CAS PubMed.
- A. A. Granovsky, Firefly version 7.1.G, http://classic.chem.msu.su/gran/firefly/index.html Search PubMed.
- E. Epifanovsky, I. Polyakov, B. Grigorenko, A. Nemukhin and A. I. Krylov, J. Chem. Phys., 2010, 132, 115104 CrossRef CAS PubMed.
- I. V. Polyakov, B. L. Grigorenko, E. M. Epifanovsky, A. I. Krylov and A. V. Nemukhin, J. Chem. Theory Comput., 2010, 6, 2377 CrossRef CAS.
- B. L. Grigorenko, A. V. Nemukhin, D. I. Morozov, I. V. Polyakov, K. B. Bravaya and A. I. Krylov, J. Chem. Theory Comput., 2011, 8, 1912 CrossRef.
- B. L. Grigorenko, A. V. Nemukhin, I. V. Polyakov and A. I. Krylov, J. Phys. Chem. Lett., 2013, 4, 1743 CrossRef CAS.
- P. M. Kozlowski, T. Kamachi, M. Kumar, T. Nakayama and K. Yoshizawa, J. Phys. Chem. B, 2010, 114, 5928 CrossRef CAS PubMed.
- A. Udvarhelyi and T. Domratcheva, J. Photochem. Photobiol., A, 2011, 87, 554 CAS.
- I. Ioffe, A. L. Dobryakov, A. A. Granovsky, N. P. Ernsting and J. L. P. Lustres, ChemPhysChem, 2011, 12, 1860 CrossRef CAS PubMed.
- A. V. Bochenkova and L. H. Andersen, Faraday Discuss., 2013, 163, 297 RSC.
- K. Kornobis, N. Kumar, B. M. Wong, P. Lodowski, M. Jaworska, T. Andruni, K. Ruud and P. M. Kozlowski, J. Phys. Chem. A, 2011, 115, 1280 CrossRef CAS PubMed.
- S. Gozem, M. Huntress, I. Schapiro, R. Lindh, A. A. Granovsky, C. Angeli and M. Olivucci, J. Chem. Theory Comput., 2012, 8, 4069 CrossRef CAS.
- S. Gozem, F. Melaccio, R. Lindh, A. I. Krylov, A. A. Granovsky, C. Angeli and M. Olivucci, J. Chem. Theory Comput., 2013, 9, 4495 CrossRef CAS.
- F. Berndt, I. Ioffe, A. A. Granovsky, R. Mahrwald, S. Tannert, S. A. Kovalenko and N. P. Ernsting, J. Photochem. Photobiol., A, 2012, 234, 164 CrossRef CAS PubMed.
- A. L. Dobryakov, I. Ioffe, A. A. Granovsky, N. P. Ernsting and S. A. Kovalenko, J. Chem. Phys., 2012, 137, 244505 CrossRef CAS PubMed.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093 RSC.
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, Phys. Chem. Chem. Phys., 2010, 12, 1218 RSC.
- V. Vallet, Z. Lan, S. Mahapatra, A. L. Sobolewski and W. Domcke, Faraday Discuss., 2004, 127, 283 RSC.
- V. Vallet, Z. Lan, S. Mahapatra, A. L. Sobolewski and W. Domcke, J. Chem. Phys., 2005, 123, 144307 CrossRef PubMed.
- Z. Lan, W. Domcke, V. Vallet, A. L. Sobolewski and S. Mahapatra, J. Chem. Phys., 2005, 122, 224315 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- T. A. Stephenson, P. L. Radloff and S. A. Rice, J. Chem. Phys., 1984, 81, 1060 CrossRef CAS PubMed.
- T. A. Stephenson and S. A. Rice, J. Chem. Phys., 1984, 81, 1073 CrossRef CAS PubMed.
- I. J. Palmer, I. N. Ragazos, F. Bernardi, M. Olivucci and M. A. Robb, J. Am. Chem. Soc., 1993, 115, 673 CrossRef CAS.
- G. A. Worth, J. Photochem. Photobiol., A, 2007, 190, 190 CrossRef CAS PubMed.
- G. A. Worth, R. E. Carley and H. H. Fielding, Chem. Phys., 2007, 338, 220 CrossRef CAS PubMed.
- B. Lasorne, F. Silicia, M. J. Bearpark, M. A. Robb, G. A. Worth and L. Blancafort, J. Chem. Phys., 2008, 128, 124307 CrossRef PubMed.
- B. Lasorne, M. J. Bearpark, M. A. Robb and G. A. Worth, J. Phys. Chem. A, 2008, 112, 13017 CrossRef CAS PubMed.
- D. S. N. Parker, R. S. Minns, T. J. Penfold, G. A. Worth and H. H. Fielding, Chem. Phys. Lett., 2009, 469, 43 CrossRef CAS PubMed.
- T. J. Penfold and G. A. Worth, J. Chem. Phys., 2009, 131, 064303 CrossRef PubMed.
- R. S. Minns, D. S. N. Parker, T. J. Penfold, G. A. Worth and H. H. Fielding, Phys. Chem. Chem. Phys., 2010, 12, 15607 RSC.
- T. J. Penfold, R. Spesyvtsev, O. M. Kirkby, R. S. Minns, D. S. N. Parker, H. H. Fielding and G. A. Worth, J. Chem. Phys., 2012, 137, 204310 CrossRef CAS PubMed.
- A. L. Sobolewski, C. Woywod and W. Domcke, J. Chem. Phys., 1992, 98, 5627 CrossRef PubMed.
- O. P. J. Vieuxmaire, Z. Lan, A. L. Sobolewski and W. Domcke, J. Chem. Phys., 2008, 129, 224307 CrossRef PubMed.
- The 1ππ* and 2ππ* states are bound and their energies increase significantly with the N–H bond length. At large N–H bond length, 1ππ* and 2ππ* state potential energy curves cross that of high lying dissociative states, including a number of states dominated by doubly excited configurations. This situation leads to considerable difficulty in keeping track of the states of interest and to convergence problems. Computing separately the A′ and A′′ states was found to greatly simplify the calculation since only crossings implying states of the same symmetry remained to be dealt with.
- H. Köppel, W. Domcke and L. S. Cederbaum, Adv. Chem. Phys., 1984, 57, 59 Search PubMed.
- S. Matsika and D. R. Yarkony, J. Chem. Phys., 2002, 117, 6907 CrossRef CAS PubMed.
- S. Han and D. R. Yarkony, J. Chem. Phys., 2003, 119, 11561 CrossRef CAS PubMed.
- M. S. Schuurman and D. R. Yarkony, J. Phys. Chem. B, 2006, 110, 19031 CrossRef CAS PubMed.
- M. Barbatti, M. Vazdar, A. J. A. Aquino, M. Eckert-Maksić and H. Lischka, J. Chem. Phys., 2006, 125, 164323 CrossRef PubMed.
- S. Perun, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2005, 127, 6257 CrossRef CAS PubMed.
- H. Ward and J. Wishnok, J. Am. Chem. Soc., 1968, 90, 5353 CrossRef CAS.
- D. Bryce-Smith, A. Gilbert and D. A. Robinson, Angew. Chem., Int. Ed., 1971, 10, 745 CrossRef CAS.
- A. L. Sobolewski, J. Chem. Phys., 1990, 93, 6433 CrossRef CAS PubMed.
- A. Toniolo, A. L. Thompson and T. J. Martínez, Chem. Phys, 2004, 304, 133 CrossRef CAS PubMed.
- A. L. Thompson and T. J. Martínez, Faraday Discuss., 2011, 150, 293 RSC.
- B. Lasorne, M. J. Bearpark, M. A. Robb and G. A. Worth, J. Phys. Chem. A, 2008, 112, 13017 CrossRef CAS PubMed.
- D. Mendive-Tapia, B. Lasorne, G. A. Worth, M. J. Bearpark and M. A. Robb, Phys. Chem. Chem. Phys., 2010, 12, 15725 RSC.
- J. J. Serrano-Pérez, F. de Vleeschouwer, F. de Proft, D. Mendive-Tapia, M. J. Bearpark and M. A. Robb, J. Org. Chem., 2013, 78, 1874 CrossRef PubMed.
- G. A. Worth, M. A. Robb and B. Lasorne, Mol. Phys., 2008, 106, 2077 CrossRef CAS.
- M. Barbatti, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 620 CrossRef CAS.
- C. S. Page and M. Olivucci, J. Comput. Chem., 2003, 24, 298 CrossRef CAS PubMed.
- L. Serrano-Andrés, M. Merchán and R. Lindh, J. Chem. Phys., 2005, 122, 104107 CrossRef PubMed.
- B. G. Levine, J. D. Coe and T. J. Martínez, J. Phys. Chem. B, 2008, 112, 405 CrossRef CAS PubMed.
- M. Huix-Rotllant, D. Siri and N. Ferré, Phys. Chem. Chem. Phys., 2013, 15, 19293 RSC.
- I. N. Ioffe and A. A. Granovsky, J. Chem. Theory Comput., 2013, 9, 4973 CrossRef CAS.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409 CrossRef CAS.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395 CrossRef CAS.
- N. O. C. Winter, N. K. Graf, S. Leutwyler and C. Hättig, Phys. Chem. Chem. Phys., 2013, 15, 6623 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Containing figures showing the orbitals, the negative imaginary frequency normal modes for the transition states and the branching plane vectors for the minimum energy conical intersections. The Cartesian coordinates and selected geometrical parameters for all the stationary points optimized in this work are also reported. See DOI: 10.1039/c3cp54418d |
| This journal is © the Owner Societies 2014 |