 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceParticle size dependence on oxygen reduction reaction activity of electrodeposited TaOx catalysts in acidic media
Jeongsuk
Seo
a,
Dongkyu
Cha
b,
Kazuhiro
Takanabe
c,
Jun
Kubota
ad and
Kazunari
Domen
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp
bAdvanced Nanofabrication, Imaging and Characterization Laboratory, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
cDivision of Chemical and Life Sciences and Engineering, KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), 4700 KAUST, Thuwal 23955-6900, Saudi Arabia
dElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8520, Japan
First published on 13th November 2013
Abstract
The size dependence of the oxygen reduction reaction activity was studied for TaOx nanoparticles electrodeposited on carbon black for application to polymer electrolyte fuel cells (PEFCs). Compared with a commercial Ta2O5 material, the ultrafine oxide nanoparticles exhibited a distinctively high onset potential different from that of the bulky oxide particles.
In the past few decades, there have been many attempts to replace Pt or Pt-based electrocatalysts with new non-precious materials applicable in polymer electrolyte fuel cells (PEFCs). In particular, various promising non-platinum candidates have been reported as oxygen reduction reaction (ORR) electrocatalysts with a high activity comparable to those of commercial Pt/C catalysts, such as N-coordinated Fe- and Co-based catalysts, and N-doped metal-free carbon-based catalysts.1–5 However, their superior catalytic activities for the ORR are not applicable to PEFCs, because the transition metal-based and N-doped catalysts are chemically unstable in acidic media, which means that their high activity cannot be maintained in long-term PEFC operation. Hence, based on their high durability in acidic atmospheres, several Ta-, Nb-, and Zr-based catalysts of groups IV and V have been researched as ORR electrocatalysts.6–15 Despite their high stability, their extremely poor electroconductivity resulted in a low activity for the ORR. Many approaches to overcome this limitation were investigated, such as nitride or partially oxynitride compounds achieved by doping with N or by varied heat treatments.6–8,10,12,16 Unfortunately, the improved activity has repeatedly resulted in no long-term performance benefit because these materials tend to oxidize under acidic PEFC conditions. In fact, oxides are commonly known as the most stable substances in acidic media.17 Pure group IV or V metal oxides without C or N doping are generally insulators in bulk structures. As a result, most bulk oxide catalysts have shown a very low current density for the ORR, even with a high onset potential.3,15–17
It is very difficult to control group IV or V oxides on the nanoscale using conventional methods such as impregnation. Primary particles of oxide catalysts smaller than 10 nm can be prepared, but these fine particles easily aggregate to form larger secondary particles during high-temperature preparation. We recently reported group IV and V oxide nanoparticles of TaOx, NbOx, and ZrOx supported on carbon black (CB) prepared by potentiostatic electrodeposition as ORR electrocatalysts.11,13,18 Electrodeposition has been used to control the size and distribution of metal particles on conductive substrates in various fields.19–21 Group IV and V metal precursors are not soluble in aqueous solutions because of precipitation reactions with H2O to form oxides or hydroxides. Thus, the electrodeposition of metal species was first attempted in non-aqueous metal ethoxide solutions at room temperature, and oxide nanoparticles were successfully deposited uniformly on CB. The ultrafine oxide nanoparticles without any doping showed excellent ORR activity, with a high onset potential that should not have occurred in bulky particles. The high current retention of the TaOx catalysts in the ORR after a long-term stability test in an aqueous sulfuric solution further supported the high stability of these oxide materials in acidic media.13
Group IV and V oxide nanoparticles formed by electrodeposition exhibited very different electrocatalytic behavior from that of common bulky oxide particles. The relationship between oxide nanoparticle size and electrocatalytic performance is not yet fully understood. Also, the effect of particle size on ORR activity requires a new perspective on group IV and V oxide nanoparticles. For metallic catalysts such as Pt, the increase in electrochemical surface area (ECSA) resulting from size reduction has typically been cited as the reason for improved electrocatalytic activity. However, for group IV and V oxide nanoparticles, the enhanced ORR activity resulting from size reduction should be attributed to increased electroconductivity and an increased number of active sites on the catalyst surface. In this study, we successfully prepared TaOx nanoparticles of different sizes by electrodeposition. The particle size of the deposited species was determined by varying deposition parameters such as the applied potential or deposition time, and/or by altering the deposition environment by changing the concentration of the primary precursor or electrolyte or by adjusting the deposition temperature. The particle size dependence of the ORR electrocatalytic activity of electrodeposited TaOx nanoparticles will be discussed. Furthermore, a critical factor controlling the particle size of oxide catalysts will be introduced after comparing various deposition conditions.
For the preparation of TaOx electrocatalysts, bare CB electrodes as conductive substrates for electrodeposition were first prepared using a CB powder with an average size of 30–40 nm (Vulcan-XC72). The CB powder was mixed with a Nafion ionomer solution (Aldrich 1100EW, 5 wt% in water–aliphatic alcohols) and isopropyl alcohol (IPA, 99.9%, Kanto Chemical). The composition ratio of the CB powder to the ionomer solution was fixed at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 by weight. The CB slurry was stirred and sonicated repeatedly to achieve good dispersion. Subsequently, the highly dispersed CB slurry was sprayed onto carbon paper (EC-TP1-060T, Toyo Corporation, treated with polytetrafluoroethylene (PTFE)) at 343 K with a CB loading of 0.5 mg cm−2. The sprayed bare CB electrodes were completely dried at 343 K.
:3 by weight. The CB slurry was stirred and sonicated repeatedly to achieve good dispersion. Subsequently, the highly dispersed CB slurry was sprayed onto carbon paper (EC-TP1-060T, Toyo Corporation, treated with polytetrafluoroethylene (PTFE)) at 343 K with a CB loading of 0.5 mg cm−2. The sprayed bare CB electrodes were completely dried at 343 K.
Potentiostatic electrodeposition of Ta species onto the bare CB electrode was performed in a three-electrode system using a potentiostat (HZ5000, Hokuto Denko). The bare CB electrode with an area of 1 cm2 was mounted as a working electrode. A carbon rod and an Ag/AgCl electrode (HX-R4, Hokuto Denko) were used as counter and reference electrodes, respectively. The carbon rod was selected to avoid Pt contamination. The three electrodes were immersed in a TaCl5 (Wako Chemicals) or a Ta(OC2H5)5 (99.9%, Wako Chemicals) ethanol solution with a supporting electrolyte of NaClO4 (99%, Sigma-Aldrich) at 298 K. Constant potentials were applied to the bare CB electrode for 10 s. After the electrodeposition, the electrodes were sufficiently washed by ethanol to remove the metal precursor residues, and completely dried at 298 K. Finally, the as-electrodeposited electrodes were heated under pure H2 flow at 523 K for 30 min at a ramp rate of 5 K min−1 to modify their surface. The effects of the H2 treatment on the ORR activity were discussed in our previous papers.11,13 The detailed conditions of electrodeposition are summarized in Table 1, with the resulting loading amount of Ta estimated using inductively coupled plasma-atomic emission spectrometry (ICP-AES).
Fig. 1 shows transmission electron microscope (TEM) and scanning TEM (STEM) images of TaOx electrocatalysts. The dark and bright circles in the TEM and STEM images, respectively, are TaOx particles electrodeposited on CB particles 30–40 nm in diameter. The TaOx particles seem to have different sizes in different samples. The diameters of several tens of nanoparticles were obtained for each sample from several TEM and STEM images, with the results shown in the histograms of Fig. 1. The average particle diameters and the standard deviations are shown in Table 1. The particle size increased monotonically from (a) to (d). Briefly, the TaCl5 precursor resulted in the formation of smaller particles compared to Ta(OEt)5, and the higher concentration of the supporting electrolyte caused the formation of larger TaOx particles under the same deposition parameters, applied potential and deposition time. At a positive potential of −0.3 VAg/AgCl, larger particles were deposited on CB than at −0.5 VAg/AgCl. The electrodeposition of TaOx in a TaCl5 ethanol solution is accompanied by H2 evolution from the HCl formed from TaCl5 and ethanol.13 This side reaction probably affects the formation of smaller TaOx particles. In any case, TaOx nanoparticles of different sizes were successfully electrodeposited on CB electrodes under different applied potentials and Ta plating baths. Although the detailed mechanism of electrodeposition has not been clear yet, the Ta species were considered to be reductively deposited on the CB surfaces with chemical adsorption and they were re-oxidized right after the exposure to air. Because the electrodeposition of Ta took place with H2 evolution, the Faradaic efficiency for the electrodeposition was about 60% for sample (a).
 | ||
| Fig. 1 TEM (a, c, d) and STEM (b) images and the particle size distribution of differently sized TaOx catalysts. (a)–(d) correspond to samples (a)–(d) in Table 1. | ||
Cyclic voltammograms (CVs) of glassy carbon (GC) electrodes in 20 mM TaCl5 and 20 mM Ta(OEt)5 ethanol solutions are shown in Fig. 2. As reported in our previous paper,13 for the TaCl5 solution, the shoulder peaks around −0.5 VAg/AgCl represent the reduction of Ta5+ to Ta4+ and Ta3+, resulting in the deposition of Ta-containing species on the CB. The high cathodic current at approximately −1.0 VAg/AgCl was mainly due to H2 evolution from the HCl byproduct. In the Ta(OEt)5 solution, there was no significant cathodic current at around −1.0 VAg/AgCl, indicating that H2 evolution from the HCl byproduct did not occur in this solution. The shoulder peaks corresponding to the reduction of Ta5+ were broad and unclear for the Ta(OEt)5 solution. This difference in electrochemical properties between TaCl5 and Ta(OEt)5 ethanol solutions resulted in different TaOx particle sizes.
 | ||
| Fig. 2 Cyclic voltammograms of Ta species on glassy carbon (GC) in non-aqueous solutions of TaCl5 in ethanol (solid red line) and a dilution of liquid Ta(OEt)5 in ethanol (dashed blue line) under Ar purging at 298 K. 20 mM NaClO4 was dissolved in the solutions as a supporting electrolyte. The potential was cycled over the range from −2 or −1 to 1 VAg/AgCl at a scan rate of 5 mV s−1. | ||
The ORR properties of TaOx electrocatalysts with various sizes in a 0.1 M H2SO4 solution were investigated, as shown in Fig. 3A. The measurements were carried out in a conventional single-vessel electrochemical cell with Ag/AgCl reference and carbon counter electrodes. The potential of 1.23 to 0.20 VRHE was linearly scanned in the cathodic direction with a scan rate of 5 mV s−1 at 298 K, and the current difference between O2- and Ar-purged atmospheres is shown in the vertical axis. Because of the difficulty of fabrication of the rotating disk electrode (RDE) using our electrocatalysts, we cannot evaluate kinetic current for ORR and the ORR properties of the catalysts on a carbon sheet were compared. The ORR results for a Ta metal plate (99.95%, Nilaco), the Ta2O5 powder sprayed on a bare CB electrode with a loading of 0.25 mg cm−2 (99.9%, 500 nm particle size, High Purity Chemicals), and a bare CB electrode are also shown in Fig. 3A. The electrodeposited TaOx electrocatalysts with particle sizes below 4.6 nm showed significant ORR currents, with onset potentials of 0.8–0.92 VRHE. The TaOx electrocatalyst with a particle size of 13.5 nm exhibits similar ORR activity to those of CB and the Ta2O5 powder. The Ta metal plate was covered with a thick passivation layer, so a negligible ORR current was expected. Voltammograms corrected to mass-specific ORR currents are also shown in Fig. 3B. The ORR activities clearly decreased with increasing particle size, up to 13.5 nm.
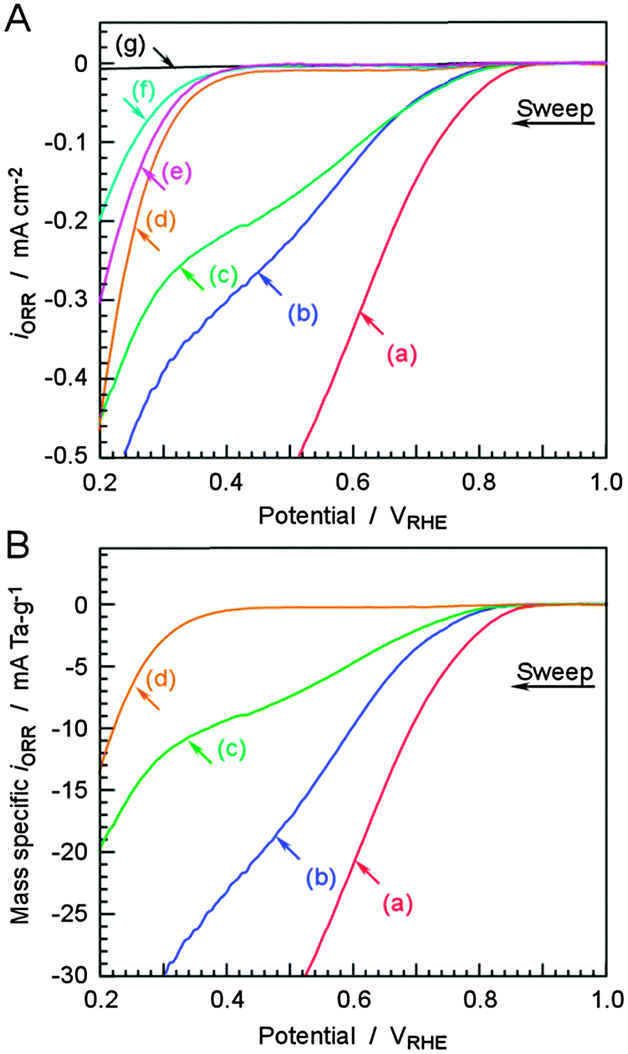 | ||
| Fig. 3 (A) Linear sweep voltammograms of differently sized TaOx nanoparticles deposited on CB electrodes in an average of 1.0 (a), 2.6 (b), 4.6 (c), and 13.5 (d) nm. The ORRs on the electrodeposited TaOx catalysts were compared with those on a bare CB electrode (as a blank, (e)), commercial Ta2O5 particles (500 nm, (f)), and a Ta metal plate (g). The potential was cathodically swept in the 0.1 M H2SO4 aqueous solution at a scan rate of 5 mV s−1 at 298 K. The current iORR indicates the difference in current density between Ar- and O2-saturated atmospheres. (B) Linear sweep voltammograms from (A) converted to a mass-specific current density. | ||
Mass-specific ORR currents at 0.6 VRHE and onset potentials at 2 μA cm−2 are plotted in Fig. 4. The negative shift in onset potential with increasing particle size was only 0.13 V, however, the current density decreased drastically. The current density fell to nearly zero for the largest TaOx nanoparticles, which were 13.5 nm in size, indicating that these particles are not useful electrocatalysts. The mass activities on the TaOx electrodes increased significantly as their size decreased. The electrochemical relationship between the particle size of metallic catalysts such as Pt or Pd and their activity has been generally discussed in an ECSA.22–24 In fact, to date, it has been difficult to determine the practical ECSA of non-platinum catalysts because of a lack of technical methods such as measuring the adsorption–desorption peak of H+ on the surface of a Pt catalyst. A more specific discussion of the ECSA was therefore unrealizable for the TaOx catalysts of different sizes. The density of molecularly adsorbed oxygen on TiN nanoparticles is known to depend on the particle size.12 This indicates that the number of active sites is not proportional to the geometric surface area of particles, and that we should consider the increased active site density per unit area with decreasing particle size. However, for TaOx electrocatalysts, the influence of the ultrafine particle size on the high activity is attributed to the increase of the electroconductivity of the particles different from the bulky oxides and the increase in the density of electrochemical active sites. Macagno et al. have reported that the donor density for a ferroelectric layer of the Ta2O5 film decreases with increasing thickness, and films thicker than 10 nm behave as pure insulators.25 In this work, the ORR current increased drastically for TaOx electrocatalysts smaller than 4.6 nm, indicating that the particle size is the most important factor determining the ORR activity of the oxide electrocatalysts.
 | ||
| Fig. 4 Dependence of the mass specific current at 0.6 VRHE and onset potential at 2 μA cm−2 on the average size of TaOx nanoparticles electrodeposited on CB. The error bars indicate the standard deviation of 1σ for each particle size. | ||
Conclusions
In summary, we introduced a new approach to determine the size dependence of the ORR activity of TaOx nanoparticles for use as electrocatalysts in PEFCs. TaOx nanoparticles of different sizes were successfully prepared by electrodeposition in non-aqueous TaCl5 or Ta(OEt)5 ethanol solutions at 298 K by varying deposition conditions such as the applied potential or the concentration of the plating bath. The electrodeposited TaOx nanoparticles exhibited size-dependent electrocatalytic performance, i.e. the smallest oxide particles have the highest ORR activity due to their improved electroconductivity and an increase of electrochemically active sites. Consequently, based on their high chemical stability under acidic conditions, these highly active oxide nanoparticles are proposed as potential non-platinum electrocatalysts for PEFC cathodes.This work was supported in part by the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST) of the Cabinet Office of Japan, the International Exchange Program of the A3 Foresight Program of the Japan Society for the Promotion of Science (JSPS), and the “Elements Strategy Initiative to Form Core Research Center” (since 2012) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Notes and references
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed.
- A. Ishihara, S. Doi, S. Mitsushima and K.-I. Ota, Electrochim. Acta, 2008, 53, 5442 CrossRef CAS PubMed.
- A. Ishihara, Y. Ohgi, K. Matsuzawa, S. Mitsushima and K.-I. Ota, Electrochim. Acta, 2010, 55, 8005 CrossRef CAS PubMed.
- Y. Ohgi, A. Ishihara, K. Matsuzawa, S. Mitsushima and K. Ota, J. Electrochem. Soc., 2010, 157, B885 CrossRef CAS PubMed.
- F. Yin, K. Takanabe, M. Katayama, J. Kubota and K. Domen, Electrochem. Commun., 2010, 12, 1177 CrossRef CAS PubMed.
- S. Isogai, R. Ohnishi, M. Katayama, J. Kubota, D. Y. Kim, S. Noda, D. Cha, K. Takanabe and K. Domen, Chem.–Asian J., 2012, 7, 286 CrossRef CAS PubMed.
- J. Seo, D. Cha, K. Takanabe, J. Kubota and K. Domen, Chem. Commun., 2012, 48, 9074 RSC.
- R. Ohnishi, K. Takanabe, M. Katayama, J. Kubota and K. Domen, J. Phys. Chem. C, 2013, 117, 496 CAS.
- J. Seo, L. Zhao, D. Cha, K. Takanabe, M. Katayama, J. Kubota and K. Domen, J. Phys. Chem. C, 2013, 117, 11635 CAS.
- T. Oh, J. Y. Kim, Y. Shin, M. Engelhard and K. S. Weil, J. Power Sources, 2011, 196, 6099 CrossRef CAS PubMed.
- J. Y. Kim, T.-K. Oh, Y. Shin, J. Bonnett and K. S. Weil, Int. J. Hydrogen Energy, 2011, 36, 4557 CrossRef CAS PubMed.
- Y. Liu, A. Ishihara, S. Mitsushima, N. Kamiya and K.-I. Ota, Electrochem. Solid-State Lett., 2005, 8, A400 CrossRef CAS PubMed.
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864 CrossRef CAS.
- J. Seo, D. Cha, K. Takanabe, J. Kubota and K. Domen, ACS Catal., 2013, 3, 2181 CrossRef CAS.
- S. Zein El Abedin, U. Welz-Biermann and F. Endres, Electrochem. Commun., 2005, 7, 941 CrossRef CAS PubMed.
- W. Simka, D. Puszczyk and G. Nawrat, Electrochim. Acta, 2009, 54, 5307 CrossRef CAS PubMed.
- J. Lee, J. Seo, K. Han and H. Kim, J. Power Sources, 2006, 163, 349 CrossRef CAS PubMed.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428 CrossRef CAS PubMed.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714 CrossRef CAS PubMed.
- W. Tang, H. Lin, A. Kleiman-Shwarsctein, G. D. Stucky and E. W. McFarland, J. Phys. Chem. C, 2008, 112, 10515 CAS.
- V. A. Macagno and J. W. Shchultze, J. Electroanal. Chem., 1984, 180, 157 CrossRef CAS.
| This journal is © the Owner Societies 2014 |