 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorescence polarization assay for the experimental validation of an in silico model of the chemokine CXCL8 binding to receptor-derived peptides†
Maria
Girrbach
ab,
Irene
Meliciani
c,
Björn
Waterkotte
bd,
Susann
Berthold
e,
Alisha
Oster
bd,
Fiona
Brurein
bd,
Timo
Strunk
c,
Parvesh
Wadhwani
f,
Sonja
Berensmeier
e,
Wolfgang
Wenzel
c and
Katja
Schmitz
*bdg
aKarlsruhe Institute of Technology, Centre for Functional Nanostructures, Haid-und-Neu-Straße 6, 76131 Karlsruhe, Germany
bKarlsruhe Institute of Technology, Institute of Functional Interfaces, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cKarlsruhe Institute of Technology, Institute of Nanotechnology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dKarlsruhe Institute of Technology, Institute of Organic Chemistry, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
eTechnische Universität München, Bioseparation Engineering Group, Boltzmannstr. 15, 85748 Garching, Germany
fKarlsruhe Institute of Technology, Institute of Biological Interfaces (IBG-2), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
gTechnical University of Darmstadt, Clemens-Schöpf-Institut für Organische Chemie und Biochemie, Alarich-Weiss-Str. 4, 64287 Darmstadt, Germany. E-mail: schmitz@biochemie.tu-darmstadt.de; Fax: +49-6151-16-75058; Tel: +49-6151-16-6964
First published on 5th March 2014
Abstract
Peptide based inhibitors of protein–protein interactions are of great interest in proteomics, structural biology and medicinal chemistry. Optimized inhibitors can be designed by experimental approaches or by computational prediction. Ideally, computational models are adjusted to the peptide–protein complex of interest according to experimental data obtained in specific binding experiments. The chemokine CXCL8 (interleukin-8) is an interesting target for drug discovery due to its role in inflammatory diseases. Given the available structural data and information on its receptor interactions it constitutes a basis for the rational design of inhibitor peptides. Starting from the reported structure of CXCL8 in complex with a peptide derived from its receptor CXCR1 we developed a computational docking procedure to estimate the changes in binding energy as a function of individual amino acid exchanges. This indicates whether the respective amino acid residue must be preserved or can be substituted to maintain or improve affinity, respectively. To validate and improve the assumptions made in this docking simulation we established a fluorescence polarization assay for receptor-derived peptides binding to CXCL8. A peptide library was tested comprising selected mutants characterized by docking simulations. A number of predictions regarding electrostatic interactions were confirmed by these experiments and it was revealed that the model needed to be corrected for backbone flexibility. Therefore, the assay presented here is a promising tool to systematically improve the computational model by iterative cycles of modeling, experimental validation and refinement of the algorithm, leading to a more reliable model and peptides with improved affinity.
Introduction
Peptides for inhibiting protein–protein interactions can be derived from the amino acid sequence of either of the interaction partners by systematic binding experiments performed by using fragments of the respective other protein. Thus, a minimal binding motif is defined which can be further optimized by identifying conserved residues and by performing other side chain modifications. Optimization can proceed either by experiments or by rational design and, ideally, by a combination of both to narrow the choice of molecules to be tested.Precise computational models with highly accurate predictions would help reduce the time and effort required to develop peptides targeting protein–protein interactions. However, several computational bottlenecks need to be overcome to arrive at reliable predictions. Estimation of the absolute binding energy requires large computational efforts even for small molecule ligands binding to structurally well characterized receptors. Even methods aiming at predicting relative binding energy changes have difficulties in balancing treatments of strong interactions (such as electrostatics, salt-bridges or hydrogen bonding) with solvation and entropic effects. The force fields used in docking algorithms are based on a number of assumptions to reduce the computational effort to predict the behavior of the binding partners. They also comprise empirical parameters that may have to be adjusted to the particular setting of individual receptor–ligand systems. Based on collections of experimental data from several databases, computational methods have been developed to predict the so-called hot spots, i.e. amino acid residues with a crucial contribution to the overall binding energy.1
In our work, we investigate a rather simple computational model based on the all-atoms free energy force field PFF02 that was initially developed for protein-structure prediction.2,3 In previous reports we found that the results obtained with this model compared favourably with the results from other methods publicly available on web servers such as FoldX,4 ROBETTA5 and KFC6 and to experimental data reported in the literature.7
However, for most protein–peptide-complexes experimental data are not readily available, and even the existing datasets may have been performed under different conditions making them difficult to compare. Therefore the development of computational models needs to be accompanied by actual binding experiments on the specific set of ligands and the respective protein target to test if the simplifying assumptions made in the model yield meaningful predictions.
In this study we investigated receptor derived peptides as ligands and prospective inhibitors of the inflammatory chemokine CXCL8 as a model system. CXCL8 (CXC-class chemokine 8; interleukin-8) triggers the chemotaxis of leukocytes to damaged or infected tissue.8,9 Its cognate receptors, CXCR1 and CXCR2, belonging to the family of G protein-coupled receptors (GPCRs) are expressed in different types of leukocytes.10 As a pro-inflammatory cytokine, CXCL8 plays an important role in autoimmune diseases and chronic inflammation. Like other CXC-chemokines with an ELR-motif at their N-termini, it induces angiogenesis and promotes the development of tumors and metastases.11 Therefore the binding of CXCL8 to its receptors is an important target in drug development and numerous approaches have been made to inhibit the CXCL8–receptor interaction.12–15 The structure and function of CXCL8 and its interactions with CXCL8 receptors have been thoroughly studied.16–25,31 As previously demonstrated, peptides derived from the sequence of the extracellular part of the human CXCL8 receptor26–28 or its rabbit homolog29 bind to CXCL8 and inhibit receptor recognition and the resulting cellular responses such as calcium influx or leukocyte migration. Peptides designed from segments of the receptors12,19,29 were subsequently shortened and modified according to experimental findings. Other inhibitor peptides were discovered by phage display to map epitopes of chemokine receptors30 or for chemokine binding domains.27 Due to the wealth of available data we chose the CXCL8/CXCR1–peptide system as a model to computationally predict peptides with improved affinity to CXCL8.
This model system bears the additional advantages that CXCL8 can be heterologously expressed in E. coli and purified in high yields,31–34 and structural data of CXCL8 with a 17 amino acid peptide derived from the N-terminal sequence of chemokine receptor CXCR1 are available in the literature.22
In this study we used the existing structural data to calculate the binding energies of a receptor-derived peptide to CXCL8.22 Changes in protein–peptide binding energies upon exchange of individual amino acids with alanine revealed hot-spots with large contributions to this interaction7 and identified amino acid exchanges that would increase the peptide's affinity to CXCL8. To validate and improve the model we further developed a fluorescence polarization assay to experimentally measure the affinity of receptor-derived peptides to the chemokine CXCL8.
The changes in binding energy in response to the mutation of selected residues in the receptor-derived peptides were quantified. Based on the comparison of computational and experimental data the computer model can be improved. Furthermore the assay can be employed to experimentally identify or test future inhibitory sequences, especially for the pharmaceutically relevant CXCL8/CXCR1–peptide system in an efficient manner.
Results and discussion
In silico full peptide exchange of an CXCL8–receptor–peptide complex
To identify peptides with an increased affinity to CXCL8, a full peptide scan was conducted based on the structural model of CXCL8 bound to the receptor-derived peptide H2N-MWDFDD-Ahx-MPPADEDYSP-COOH7 in which Ahx was replaced by glycine (see Fig. 1). In the course of the peptide scan, each amino acid residue of this peptide was successively replaced by each of the 20 standard proteinogenic amino acids. After docking the peptide mutant to the CXCL8 structure, the binding energy was estimated and the difference in the binding energy of the wildtype peptide–CXCL8 complex was calculated. The results are shown in a heatmap representation (Fig. 2). Most amino acid replacements led to an increase in binding energy and thereby a loss of affinity as indicated by positive ΔΔG values (shown in red in Fig. 2). In particular, incorporation of the basic amino acids histidine, lysine and arginine into the mainly negatively charged sequence led to a dramatic loss of affinity, indicating that salt bridges have a significant contribution to the overall binding energy. Correspondingly, an increase in affinity was predicted for the incorporation of additional residues of glutamic and aspartic acid (green squares in Fig. 2). Likewise, replacing the existing negatively charged residues in positions 3, 5, 6, 12, 13 and 14 with any but another negatively charged residue led to an increase in binding energy. Interestingly, a gain in affinity was predicted for most substitutions of the tryptophan residue in position 2 and the N-terminal methionine. There were several exchanges by individual non-polar residues with a predicted gain in affinity such as Gly7Ile or Pro10Phe (see Fig. 2). These exchanges are of particular interest for peptide improvement since they do not necessitate the introduction of further charges into the highly negatively charged peptide.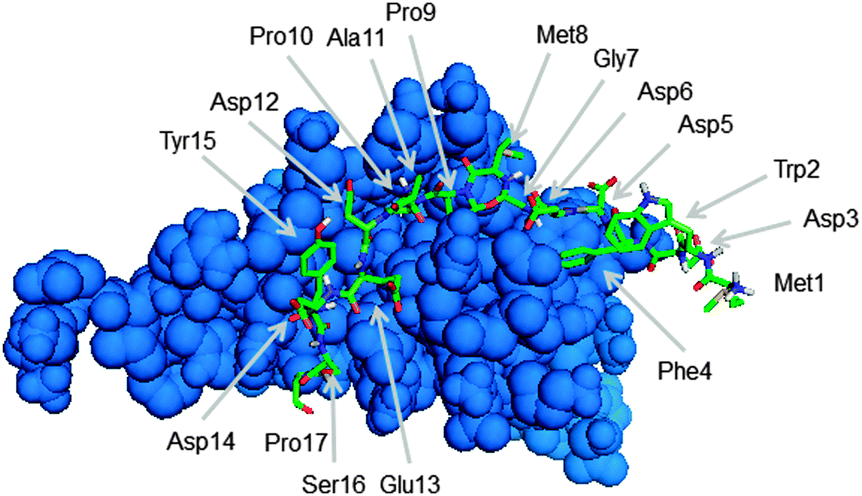 | ||
| Fig. 1 Structure of CXCL8 (blue) in complex with the receptor derived peptide H2N-MWDFDDGMPPADEDYSP-COOH (green stick model) used in this work. | ||
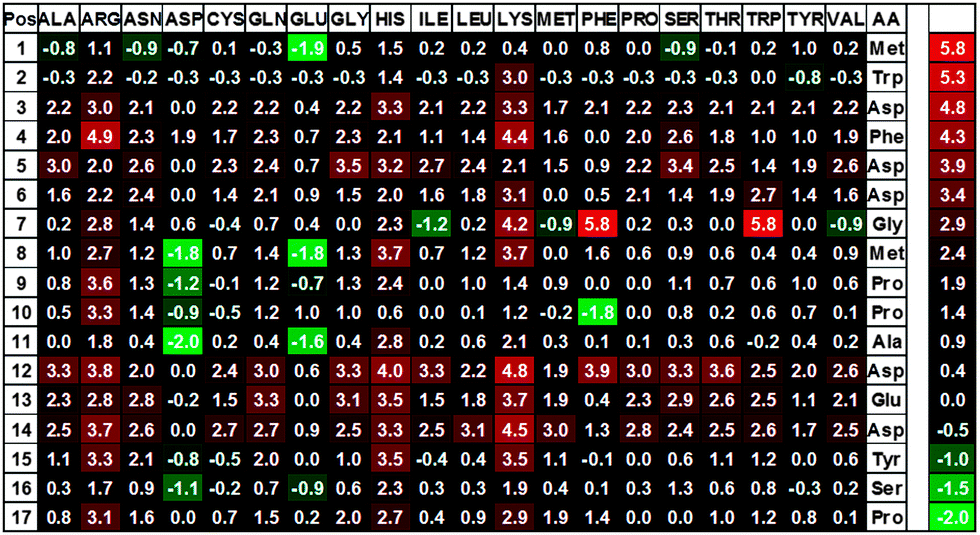 | ||
| Fig. 2 Results of docking receptor peptide mutants from the full peptide scan to CXCL8. Free binding enthalpy differences, ΔΔG, in kJ mol−1 are presented as a heat map with the positive values (ΔΔG > 0) corresponding to a loss in affinity represented in red, neutral mutations (ΔΔG ≈ 0) leading to no change in affinity shown in black and the negative values (ΔΔG < 0) indicating a gain of affinity colored in green. Positions in the receptor peptide are indicated as position number on the left column and amino acids in the top row. | ||
Development of an affinity assay for peptides binding to CXCL8
To validate these modeling results we measured the affinity of CXCL8 to selected peptides in a fluorescence polarization (FP) assay. In this type of assay, the smaller ligand molecules (here: the peptides) were fluorescently labeled and were incubated with varying concentrations of the larger protein to measure binding via the loss of peptide flexibility leading to an increase in the observed fluorescence polarization. This type of measurement has the advantage that it can be performed in equilibrium without the need to separate bound from free peptide or to immobilize the protein to a surface, which might lead to structural changes in the 72 aa small protein.To obtain large amounts of protein, CXCL8 was recombinantly expressed from bacteria following an established protocol35 with some modifications. Heating the bacterial lysate at 70 °C for 10–15 min as reported by Cheng et al.34 and optimizing the cation exchange chromatography step removed unwanted proteins from the lysate and obliterated the need for an additional affinity chromatography step. Preliminary fluorescence polarization experiments with carboxyfluorescein and a fluorescein-labeled unrelated peptide (Fluo-AVLPALP-COOH) demonstrated that there was no non-specific interaction of fluorescein or fluorescein-labeled peptides per se that would confer affinity on the fluorescein-labeled receptor derived peptides (see Fig. S2, ESI†).
To test whether the molecular weight difference between CXCL8 (72 aa) and the fluorescently labeled receptor peptide (17 aa) was sufficient to detect the difference between bound and free peptide in fluorescence polarization, we synthesized the peptide reported by Attwood and Skelton (H2N-MWDFDD-Ahx-MPPADEDYSP-COOH)22,26 on solid phase and attached a carboxyfluorescein as a fluorescent label to the N-terminus.
The first fluorescence polarization experiments in phosphate buffer at physiological pH yielded 67 (millipolarization units) mPU for the free peptide and 164 mPU for the receptor ligand complex at 94 μM CXCL8 (see Fig. S1, ESI†). The binding isotherm indicated that saturation was not yet reached at this concentration.
In preliminary experiments to optimize peptide concentration, the free peptide showed increased polarization values of over 180 mPU at low concentrations (10–20 nM) while at higher concentrations (100 nM) polarization values of around 80 mPU were recorded. This effect was attributed to peptide adsorption to the walls of the microtiter plate. Different buffer supplements were tested as recommended by Moerke36 and the most effective suppression of non-specific binding was observed upon the addition of 0.1% of Triton X-100 (see Fig. S3, ESI†).
We also found that polarization values depended on the salt concentration of the buffer (see Fig. S4, ESI†). Therefore we decided to use a dilute PBS-buffer system with 25 mM phosphate and 35 mM NaCl. The average polarization measured for the free wildtype peptide was 88 mPU and the polarization for the peptide–protein-complex reached a maximum of 227 mPU so that an appropriate dynamic range was available.
Several experiments have been conducted with CXCL8 and receptor peptides demonstrating that information regarding the influence of individual amino acids of the chemokine or the receptor on affinity can be obtained from this system. For this purpose, competition assays with radioligands,12,26 NMR-measurements19,22,23 and fluorescence quenching experiments29 have been performed. To elucidate the mechanism of CXCL8 binding to its receptor NMR-experiments are well suited since the contribution of each individual amino acid can be estimated from the shift of NMR peaks upon ligand addition.
To test the suitability of peptide mutants as protein ligands, each of these mutants needs to be tested individually. This type of systematic screening was performed by Attwood et al. to develop an optimized peptide with a Ki of 7 μM based on the CXCR1 N-terminal sequence,12,26 which was later used by Skelton for their NMR studies22 and in the study presented here. Atwood et al. used a radioligand assay in which the unlabeled peptides competed with radioactively labeled CXCL8 for CXCR1 receptors in membrane preparations from leukocytes.12 In more recent work, the Harris group introduced receptor peptides derived from consensus sequences in the extracellular domains of different chemokine receptors that were identified by alignment analysis and that exhibited weak binding affinities (10 μM to 1 mM) to a range of different chemokines.27 Affinities were also estimated by surface plasmon resonance (SPR) spectroscopy on immobilized chemokines.27 Houimel and Mazzucchelli derived the epitopes of known antibodies against CXCL8 receptors from phage-display libraries and demonstrated that these hexapeptides could inhibit the binding of CXCL8 to its receptor in a competitive radioligand assay.30 However, no reports have described the binding of inhibitor peptides to chemokines directly by fluorescence polarization, although the binding of fluorescently labeled chemokines to their cognate receptors has been studied using this technique.37,38
The advantage of fluorescence polarization is that the amount of the protein–peptide complex can be estimated in equilibrium without the need for separation of the unbound protein, which might disturb the equilibrium. Furthermore, there is no requirement to immobilize the protein as in SPR-approaches. Immobilization can lead to a decrease in affinity if the binding site is blocked or occluded in the immobilization process. The only requirements to allow the use of low concentrations of peptide in fluorescence polarization are (1) the need to fluorescently label the peptide of interest to obtain a stronger signal than that of intrinsic aromatic residues and (2) that the fluorophore itself does not show any binding preferences. An additional advantage over radioligand binding is that no safety and waste disposal issues occur when fluorescently labeled peptides are employed.
Fluorescence polarization to validate computational predictions
To test the predictions made by the in silico model we used 30 custom-made fluorescein-labeled peptides representing all alanine exchange mutations of the CXCL8 receptor peptide. We selected positions of the full peptide scan representing mutations that led to a predicted increase, a decrease or no significant change in binding energy, respectively. The wildtype peptide was also included to calculate differences in Kd or binding free energy. Fig. 3 shows the binding isotherms of the receptor peptide mutants with the highest (D12K) and lowest (G7L) measured Kd values. Free binding enthalpies were calculated based on these values using the relationshipΔG = RT ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kd Kd | (1) |
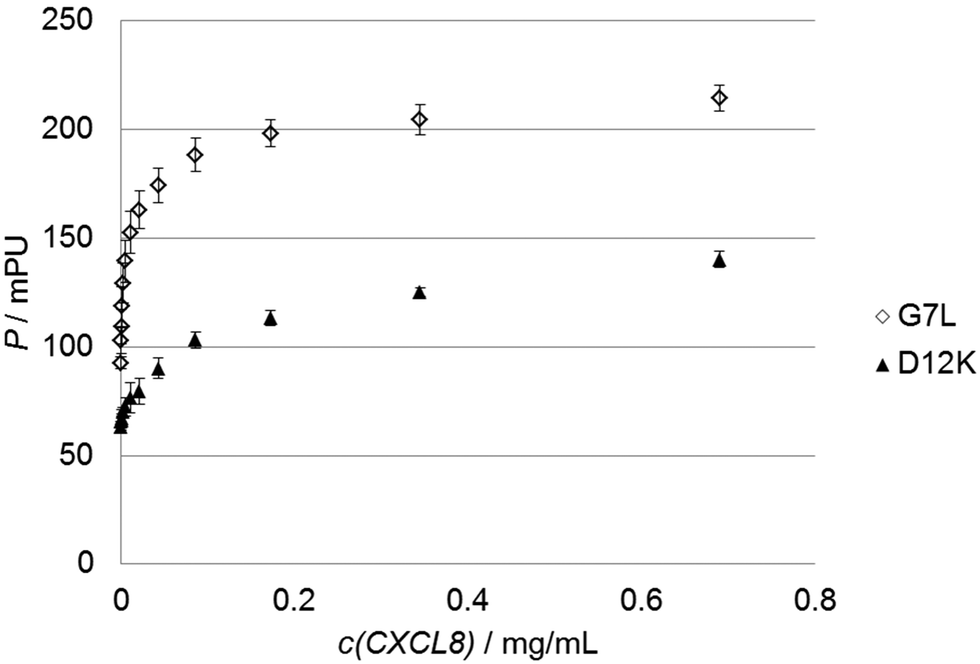 | ||
| Fig. 3 Sample graphs for binding experiments. Binding isotherms of the peptide with the highest (D12K, black triangles) and lowest (G7L, white diamonds) Kd values are shown. | ||
| Mutant | Type | Calculated | Measured |
|---|---|---|---|
| M1A | Ala-Scan | −0.81 | 0.38 |
| W2A | Ala-Scan | −0.32 | 2.71 |
| D3A | Ala-Scan | 2.16 | 2.00 |
| F4A | Ala-Scan | 2.03 | 2.06 |
| D5A | Ala-Scan | 2.96 | 2.74 |
| D6A | Ala-Scan | 1.55 | 2.72 |
| G7A | Ala-Scan | 0.18 | −0.41 |
| M8A | Ala-Scan | 0.99 | 0.45 |
| P9A | Ala-Scan | 0.77 | 2.97 |
| P10A | Ala-Scan | 0.50 | 1.49 |
| A11A wildtype | Ala-Scan | 0.00 | 0.00 |
| D12A | Ala-Scan | 3.32 | 2.20 |
| E13A | Ala-Scan | 2.34 | 1.56 |
| D14A | Ala-Scan | 2.46 | 2.59 |
| Y15A | Ala-Scan | 1.14 | 1.17 |
| S16A | Ala-Scan | 0.28 | −0.26 |
| P17A | Ala-Scan | 0.85 | 2.24 |
| A11D | Decrease in ΔΔG | −2.04 | −0.49 |
| P10F | Decrease in ΔΔG | −1.78 | 1.08 |
| M8D | Decrease in ΔΔG | −1.76 | 0.22 |
| G7I | Decrease in ΔΔG | −1.17 | −0.77 |
| P9F | Neutral on ΔΔG | −0.03 | 1.64 |
| D6M | Neutral on ΔΔG | 0.00 | 1.64 |
| P10L | Neutral on ΔΔG | 0.08 | 1.37 |
| G7L | Neutral on ΔΔG | 0.23 | −1.20 |
| E13H | Increase in ΔΔG | 3.08 | 3.64 |
| D12K | Increase in ΔΔG | 4.84 | 5.03 |
| F4R | Increase in ΔΔG | 4.85 | 3.48 |
| G7W | Increase in ΔΔG | 5.81 | −1.06 |
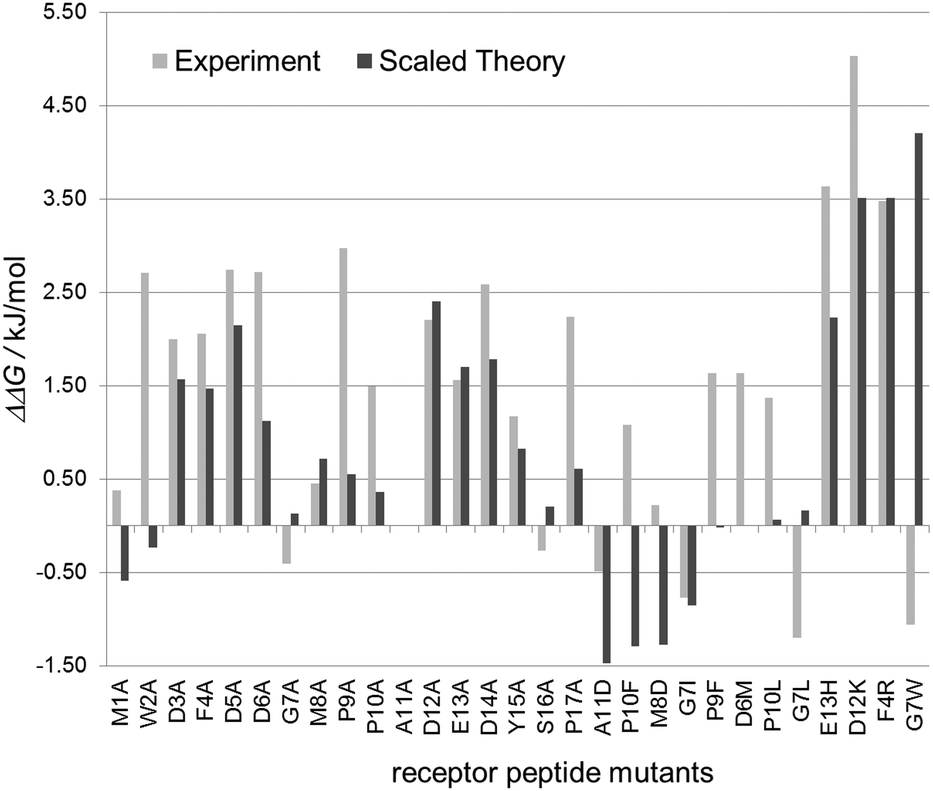 | ||
| Fig. 4 Comparison of theoretical and experimental data. When divided by a factor of three theoretical values (dark grey) correspond reasonably well with experimental values (light gray) for binding energy differences. | ||
These experimental values corresponded reasonably well to the predicted values. The average error in affinity change between experiment and theory was only −0.3 kJ mol−1 or 0.08 kJ mol−1 including or excluding the proline substitutions, respectively.
The large increases in binding energy for an alanine exchange at the positions of the negatively charged aspartate and glutamate residues (D3A, D5A, D6A, D12A, E13A and D14A) were confirmed to within 1 kJ mol−1. The increases in binding energy for the proline residues (P9A, P10A, P17A) were, on average, 1.5 kJ mol−1 higher than the values predicted by the computational model. This difference can be attributed, at least in part, to the expected loss of flexibility of the unbound peptide, which is not accounted for in the computational model.
The predicted increase in binding free enthalpy for the exchange of phenylalanine 4 (F4A) and tyrosine 15 (Y15A) agreed well with that of the computational model. Small predicted gains in the binding enthalpy for the substitution of glycine 7 (G7A) and serine 16 (S16A) were found to be small losses in the binding enthalpy. Alanine substitutions in the N-terminal methionine (M1A) and tryptophan residues (W2A) that were predicted to lead to a decrease in binding energy had an opposite effect in the experiment.
A similar set of observations was made for the selected mutants: increases in binding energy (ΔΔG > 0) were mostly predicted correctly (E13H; D12K, F4R). Mutations predicted to have mainly no effect (ΔΔG ≈ 0) led to a loss in affinity, while the all tested gain-of-affinity (ΔΔG < 0) predictions exhibited changes in either direction in the experiment. The three mutants replacing glycine residues are of particular interest: while the computer model predicts a loss of affinity for G7W, a minor loss for G7L (and G7A) and a gain in affinity for G7I, all three individual mutations however show a slight increase in affinity in the binding experiment. This can be rationalized by the increase in rigidity of the peptide backbone upon the exchange of the flexible glycine that lacks a side chain and can be rotated freely around the NH–Cα and Cα–CO-bonds. In a similar manner, substitution of either of the proline residues for which gains (P10A, P17A), losses (P10F) and negligible effects (P9F) were predicted led to a loss of affinity in the experiments as substitution of proline led to a more flexible peptide backbone. Binding of a more rigid peptide results in smaller entropic costs compared to the binding of a flexible peptide, so that rigid peptides generally exhibit higher affinities. Binding energies in the computer model had been computed against a fixed unbound model.
The various reports in the literature on the binding of receptor derived peptides are difficult to compare to the data obtained in this work due to a variety of sequences and conditions used (summarized Table S1, ESI†). Peptide lengths range from 1722,26 to 40 amino acids19 derived from the human receptor19,26 or its rabbit homolog.23,29 Remarkably, binding assays were conducted at pH values between 5.522 and 8.029 so that the degree of protonation in the binding partners differs in the individual studies. Finally, as different assay formats were used data were reported as Kd for affinity assays and as Ki for competition assays, making a comparison even more difficult.
The clear trends indicated by the experimental data provide valuable clues on how to improve the docking model in order to obtain more reliable predictions for inhibitor peptide design. An overall normalization of the calculated energy differences as suggested by the measured data is sensible, since in general computational models still tend to have difficulties matching the energy scale in experiments. Substitutions of proline and glycine residues lead to changes in backbone flexibility that need to be considered in the computer model. While the replacement of the rigid proline residues leads to a more flexible backbone and a loss of affinity, replacing the flexible glycine residue with other amino acids produces a more rigid backbone and a measurable increase in affinity. As the flexibility of the free peptide determines the change in entropy upon binding to the protein and thereby has an impact on free energy, entropic effects have to be included in the model to make correct predictions in these cases. Similar findings have been reported by Yang et al. in their study of second mitochondria-derived activator of caspase (Smac) to X-linked inhibitor of apoptosis (XIAP) using computational docking based on the MM-GBSA (molecular mechanics and generalized Born surface area) approach and comparison to experimental data reported in the literature.39 They propose to compensate for entropic effects by introducing a ligand reorganization free energy.
Experimental
Computational model
A free-energy based simulation method was applied in which each protein backbone conformation was assigned an “internal free-energy” resulting from integration of the solvent degrees of freedom. In this approach sampling of the backbone conformational space is decoupled from the computation of the relative free energies for each conformation.40 All docking simulations were performed using the all-atom physics based force field PFF0240,41 that considers the Lennard-Jones potential, electrostatic interactions, solvent interactions, hydrogen bonds and a torsional potential to parameterize the internal free energy of a protein microstate. Parameters were assigned as described previously.7The receptor–ligand binding energy was computed from the published structure of a complex of CXCL8 bound to receptor-derived peptide22 (PDB No: 1ILP) in which the non-natural amino acid aminohexanoic acid (Ahx) in position 7 was replaced by glycine for simplification. Relaxation simulation and computation were performed as described previously.7 In brief, the complex was relaxed by keeping the chemokine backbone rigid while allowing the peptide to search its lowest energy conformation. Protein and peptide were separated and the interaction energy was calculated as the energy difference between the CXCL8–peptide complex and the isolated binding partners, ensuring convergence of geometries and energies by performing up to 10 simulations of the wildtype complex. All peptides were expected to assume a similar open-chain conformation in complex with the chemokine, and only the differences in binding energies were considered in the results, in which the peptide reference energies cancel out exactly. Therefore, the reference structure of the free peptides in solution was not computed. The resulting free energy differences therefore only permit a comparison of the relative binding energies of peptides in the bound state, while the absolute interaction energy is overestimated. Since the scale of the energy changes in the force field is known, energy differences were fitted to the experimental data using a linear model with zero offset, resulting in an overall correction factor of 3. The scaled computational data were used to analyze the impact of the mutation as it allows direct comparison with experimental data.
Materials
Salts and culture media for protein expression and purification were purchased from Merck (Darmstadt, Germany) or Carl Roth (Karlsruhe, Germany); hen egg white lysozyme, ampicillin, DNase I, Triton X-100, and Triton X-114 were obtained from Sigma-Aldrich-Fluka (Taufkirchen, Germany). All Fmoc-protected amino acids were commercially obtained (Novabiochem, Darmstadt, Germany, or IRIS Biotech, Marktredwitz, Germany) and used without further purification. Solvents and coupling reagents for peptide synthesis were purchased from Biosolve (Valkenswaard, Netherlands). All columns for protein purification were obtained from GE Healthcare (Chalfont St Giles, UK).Peptides
For assay development the CXCL8 receptor peptide reported by Skelton22 was synthesized on a Syro II multiple peptide synthesizer (Syro II, MultiSyntech, Germany) using standard Fmoc chemistry as previously described.42,43 The identity of the CXCL8 receptor peptide was confirmed by mass spectrometry. The crude peptide was purified over an RP-18 column (Supelco Analytical, Bellefonte, USA) on an ÄKTA purifier HPLC system (GE Healthcare Europe, Freiburg) with a gradient of acetonitrile/water. Fluorescently labeled peptides were commercially obtained from peptides & elephants (Potsdam) and the fluorescein-coupled control peptide Fluo-AVLPALP was purchased from Biomatik (Cambridge, Ontario, Canada). All peptides were used without further purification. The peptides were dissolved in 0.1% NH3 solution and diluted in PBS (PBS in a 1:4 dilution: 35 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.1% Triton X-100, pH 7.5) to a final concentration of 20 nM or 40 nM.
Protein expression
CXCL8 was expressed as described previously.35 In brief, 10 mL of an overnight culture of E. coli BL21 (DE3) RIL cells (Novagen, Merck, Darmstadt, Germany) transformed with the pET22b-IL8 vector was used to inoculate 800 mL of LB-medium with 60 μg mL−1 ampicillin and incubated at 160 rpm and 37 °C. Expression of CXCL8 was induced at OD600 of 0.6–0.8 by addition of isopropylthiogalactoside (IPTG) to a final concentration of 0.1 mM and continued for 2 h at 30 °C. The cells were centrifuged for 45 min at 8 °C and 8555 × g. The pellet was then resuspended in 4 mL buffer A (40 mM sodium phosphate, 90 mM NaCl, pH 7.5) and additionally 1 mM EDTA was added. Lysis was induced by addition of lysozyme (0.2 mg mL−1) and DNase I (0.1 mg mL−1, 3000 U mg−1). One protease inhibitor tablet (Complete Mini®, Roche, Basel, Switzerland) was added additionally. After 1.5 h of incubation on ice an equal volume of 0.5% Triton X-114 was added. Further cell disruption was accomplished by sonication for 3 × 30 s at 50% of maximum power (Sonopuls, Bandelin Electronics, Berlin, Germany). The cell suspension was frozen in liquid nitrogen and thawed at room temperature. In an optimized purification procedure the lysate was heated to 70 °C for 10–15 min according to the report by Cheng et al. to precipitate a large fraction of the E. coli proteins while recovering CXCL8 in solution.34 After incubation with more DNase for 30 min at RT and centrifugation at 4 °C and 4754 × g for 45 min the supernatant was filtered through a 0.2 μm syringe top filter and subjected to cation exchange chromatography on a 5 mL HiTrap SP FF column (GE Healthcare, Munich, Germany) using 2 CV of buffer A for washing and a linear salt gradient over 10 CV from buffer A to 70% buffer B (1.5 M NaCl and 40 mM sodium phosphate, pH 7.4) for elution. Alternatively, after prolonged washing with 10 CV of buffer A, CXCL8 was eluted with 10 CV of 70% of buffer B. This resulted in a sharper CXCL8 peak so that further concentration steps could be omitted. CXCL8 containing fractions were lyophilized, dissolved in a small volume of distilled water and desalted on a desalting column HiPrep 26/10 (GE Healthcare, Munich, Germany) using buffer C (35 mM NaCl, 40 mM phosphate, pH 7.4). Protein concentrations were determined by the bicinchoninic acid (BCA)-assay (Thermo Scientific, Rockford, IL, USA; according to the manufacturer's instructions) against a standard curve of commercial CXCL8 (Genscript, Hong Kong) or lysozyme.35Fluorescence polarization
Fluorescence polarization measurements were performed on a plate reader (Infinite F200; Tecan, Crailsheim, Germany) equipped with a fluorescence polarization module and a filter set for an excitation wavelength of 485 ± 20 nm and emission of 535 ± 20 nm. Peptide solutions were diluted to 40 nM (optimization experiments) or 20 nM (library measurement) in 1/4 PBS and 1:2 dilution series of purified CXCL8 in buffer C were prepared in 96-well plates. 0.1% of Triton X-100 was added to all buffers to prevent non-specific binding of the peptides to the microtiter plates.36 Both solutions were transferred into 384-well, black, flat-bottom, polystyrene microtiter plates (Greiner Bio-One, Frickenhausen, Germany). For transferring large sample numbers a manual high throughput pipetting instrument (Liqudator96, Steinbrenner Laborsysteme GmbH, Wiesenbach, Germany) was used. 20 μL of protein solution was added to 20 μL of peptide solution yielding a final volume of 40 μL per well. The mixture of equal volumes of buffer C from protein purification and 1/4 PBS for peptide dilution resulted in a final concentration of 25 mM phosphate buffer and 35 mM NaCl. In high throughput experiments, all data points were measured in quadruplicate and all measurements were repeated three times. Polarization is reported in millipolarization units (mPU). Data analysis was performed using Excel version 2007 and 2010 (Microsoft, Redmond, USA) and Sigma Plot 10 (Systat Software, Chicago, IL, USA). Dissociation constants (Kd) were determined by non-linear regression using a single-site single-ligand binding model.
Conclusion
In the work presented here we have established a binding assay based on fluorescence polarization to confirm the predictions of a computer model for the interaction of receptor derived peptides with the chemokine ligand CXCL8. Using the all-atom physics based free energy force field PFF02 that was originally designed for the de novo prediction of protein folding points and published structural data of a CXCL8–peptide complex, the changes in binding energy upon amino acid exchanges in the peptide sequence were predicted. A binding assay was established using recombinant CXCL8 and a synthetic receptor-derived type peptide and assay conditions were optimized to obtain a sufficient dynamic range for measurements. This assay turned out to be a straightforward means to determine affinities of protein–peptide complexes and thereby validate predictions on binding energy. According to experimental findings a scaling factor was introduced into the computer model. Some flexibility needs to be introduced into the docking algorithm to better reflect the impact of exchanges involving flexible residues like glycine and rigid residues like proline on the entropic term in free binding enthalpy. Also, salt effects need to be included since both binding partners are highly charged and experimental data indicate a dependence of affinity on salt concentration. To accommodate the fact that IL-8 tends to form dimers,9,31 the assay is currently being extended to include a trapped dimer and a non-dimerizing variant of CXCL8.44 A new round of predictions and polarization experiments are under our active investigation and will be published in due course.Thus, the assay presented here will constitute a useful tool to improve the computational model by iterative cycles of predictions, experimental tests and improvement of the algorithm resulting in a more reliable model and peptides with improved affinity. The presented combination of computer model and fluorescence polarization can be easily applied to other pairs of chemokines and receptor-derived peptides by exploiting homologies in the chemokine family, but also to other protein peptide pairs as far as suitable structural data are available as a starting point.
Acknowledgements
We are grateful for the support from the volunteers of POEM@HOME, where a part of the simulations was carried out. We want to thank Mettler-Toledo for permitting us to work with the Liquidator96™. This work received financial support from the ‘Concept for the Future’ of Karlsruhe Institute of Technology (KIT) within the framework of the German Excellence Initiative (Research Group 26-2) and the Landesstiftung Baden-Württemberg (HPC-5). M.G. received support from the DFG-Center for Functional Nanostructures (subproject E2.3). I.M. was funded by a PhD fellowship from the German Academic Exchange Service (DAAD). T.S. received a grant from the Carl-Zeiss-Stiftung.Notes and references
- T. Kortemme and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14116–14121 CrossRef CAS PubMed.
- T. Herges and W. Wenzel, Biophys. J., 2004, 87, 3100–3109 CrossRef CAS PubMed.
- A. Schug, T. Herges and W. Wenzel, Phys. Rev. Lett., 2003, 91, 158102 CrossRef CAS.
- J. Schymkowitz, J. Borg, F. Stricher, R. Nys, F. Rousseau and L. Serrano, Nucleic Acids Res., 2005, 33, W382–W388 CrossRef CAS PubMed.
- D. Chivian, D. E. Kim, L. Malmstrom, P. Bradley, T. Robertson, P. Murphy, C. E. Strauss, R. Bonneau, C. A. Rohl and D. Baker, Proteins, 2003, 53(suppl 6), 524–533 CrossRef CAS PubMed.
- S. J. Darnell, L. LeGault and J. C. Mitchell, Nucleic Acids Res., 2008, 36, W265–W269 CrossRef CAS PubMed.
- I. Meliciani, K. Klenin, T. Strunk, K. Schmitz and W. Wenzel, J. Chem. Phys., 2009, 131, 034114 CrossRef PubMed.
- M. Baggiolini, A. Walz and S. L. Kunkel, J. Clin. Invest., 1989, 84, 1045–1049 CrossRef CAS PubMed.
- D. Rossi and A. Zlotnik, Annu. Rev. Immunol., 2000, 18, 217–242 CrossRef CAS PubMed.
- B. Moser, B. Dewald, L. Barella, C. Schumacher, M. Baggiolini and I. Clark-Lewis, J. Biol. Chem., 1993, 268, 7125–7128 CAS.
- C. Bizzarri, A. R. Beccari, R. Bertini, M. R. Cavicchia, S. Giorgini and M. Allegretti, Pharmacol. Ther., 2006, 112, 139–149 CrossRef CAS PubMed.
- M. R. Attwood, N. Borkakoti, G. A. Bottomley, E. A. Conway, I. Cowan, A. G. Fallowfield, B. K. Handa, P. S. Jones, E. Keech, S. J. Kirtland, G. Williams and F. X. Wilson, Bioorg. Med. Chem. Lett., 1996, 6, 1869–1874 CrossRef CAS.
- X. D. Yang, J. R. Corvalan, P. Wang, C. M. Roy and C. G. Davis, J. Leukocyte Biol., 1999, 66, 401–410 CAS.
- R. Horuk and H. P. Ng, Med. Res. Rev., 2000, 20, 155–168 CrossRef CAS.
- T. N. Wells, C. A. Power, J. P. Shaw and A. E. Proudfoot, Trends Pharmacol. Sci., 2006, 27, 41–47 CrossRef CAS PubMed.
- G. M. Clore, E. Appella, M. Yamada, K. Matsushima and A. M. Gronenborn, J. Biol. Chem., 1989, 264, 18907–18911 CAS.
- J. Lee, R. Horuk, G. C. Rice, G. L. Bennett, T. Camerato and W. I. Wood, J. Biol. Chem., 1992, 267, 16283–16287 CAS.
- R. B. Gayle 3rd, P. R. Sleath, S. Srinivason, C. W. Birks, K. S. Weerawarna, D. P. Cerretti, C. J. Kozlosky, N. Nelson, T. Vanden Bos and M. P. Beckmann, J. Biol. Chem., 1993, 268, 7283–7289 CAS.
- R. T. Clubb, J. G. Omichinski, G. M. Clore and A. M. Gronenborn, FEBS Lett., 1994, 338, 93–97 CrossRef CAS.
- S. R. Leong, R. C. Kabakoff and C. A. Hebert, J. Biol. Chem., 1994, 269, 19343–19348 CAS.
- M. E. Hammond, V. Shyamala, M. A. Siani, C. A. Gallegos, P. H. Feucht, J. Abbott, G. R. Lapointe, M. Moghadam, H. Khoja, J. Zakel and P. Tekamp-Olson, J. Biol. Chem., 1996, 271, 8228–8235 CrossRef CAS PubMed.
- N. J. Skelton, C. Quan, D. Reilly and H. Lowman, Structure, 1999, 7, 157–168 CrossRef CAS.
- A. Ravindran, P. R. B. Joseph and K. Rajarathnam, Biochemistry, 2009, 48, 8795–8805 CrossRef CAS PubMed.
- S. H. Park, F. Casagrande, L. Cho, L. Albrecht and S. J. Opella, J. Mol. Biol., 2011, 414, 194–203 CrossRef CAS PubMed.
- A. Ravindran, K. V. Sawant, J. Sarmiento, J. Navarro and K. Rajarathnam, J. Biol. Chem., 2013, 288, 12244–12252 CrossRef CAS PubMed.
- M. R. Attwood, E. A. Conway, R. M. Dunsdon, J. R. Greening, B. K. Handa, P. S. Jones, S. C. Jordan, E. Keech and F. X. Wilson, Bioorg. Med. Chem. Lett., 1997, 7, 429–432 CrossRef CAS.
- C. Ezerzer, M. Dolgin, J. Skovorodnikova and N. Harris, Peptides, 2009, 30, 1296–1305 CrossRef CAS PubMed.
- E. F. Barter and M. J. Stone, Biochemistry, 2012, 51, 1322–1331 CrossRef CAS PubMed.
- L. Rajagopalan and K. Rajarathnam, J. Biol. Chem., 2004, 279, 30000–30008 CrossRef CAS PubMed.
- M. Houimel and L. Mazzucchelli, J. Leukocyte Biol., 2009, 85, 728–738 CrossRef CAS PubMed.
- H. B. Lowman, W. J. Fairbrother, P. H. Slagle, R. Kabakoff, J. Liu, S. Shire and C. A. Hebert, Protein Sci., 1997, 6, 598–608 CrossRef CAS PubMed.
- A. Koltermann, W. Boidol, J. Daum, P. Scholz and P. Donner, J. Biotechnol., 1997, 54, 29–42 CrossRef CAS.
- B. Goger, Y. Halden, A. Rek, R. Mosl, D. Pye, J. Gallagher and A. J. Kungl, Biochemistry, 2002, 41, 1640–1646 CrossRef CAS PubMed.
- H. T. Cheng, K. C. Huang, H. Y. Yu, K. J. Gao, X. Zhao, F. Li, J. Town, J. R. Gordon and J. W. Cheng, Protein Expression Purif., 2008, 61, 65–72 CrossRef CAS PubMed.
- D. Wiese and K. Schmitz, J. Immunol. Methods, 2011, 364, 77–82 CrossRef CAS PubMed.
- N. J. Moerke, Curr. Prot. Chem. Biol., John Wiley & Sons, Inc., 2009 Search PubMed.
- S. J. Allen, S. Ribeiro, R. Horuk and T. M. Handel, Protein Expression Purif., 2009, 66, 73–81 CrossRef CAS PubMed.
- A. Datta and M. J. Stone, Protein Sci., 2003, 12, 2482–2491 CrossRef CAS PubMed.
- C. Y. Yang, H. Y. Sun, J. Y. Chen, Z. Nikolovska-Coleska and S. M. Wang, J. Am. Chem. Soc., 2009, 131, 13709–13721 CrossRef CAS PubMed.
- T. Herges and W. Wenzel, Biophys. J., 2004, 87, 3100–3109 CrossRef CAS PubMed.
- A. Verma and W. Wenzel, Biophys. J., 2009, 96, 3483–3494 CrossRef CAS PubMed.
- P. Wadhwani, S. Afonin, M. Ieronimo, J. Buerck and A. S. Ulrich, J. Org. Chem., 2006, 71, 55–61 CrossRef CAS PubMed.
- P. Wadhwani, E. Strandberg, N. Heidenreich, J. Burck, S. Fanghanel and A. S. Ulrich, J. Am. Chem. Soc., 2012, 134, 6512–6515 CrossRef CAS PubMed.
- K. Rajarathnam, G. N. Prado, H. Fernando, I. Clark-Lewis and J. Navarro, Biochemistry, 2006, 45, 7882–7888 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp53850h |
| This journal is © the Owner Societies 2014 |