 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structure and dynamics of carbon dioxide and water containing ices investigated via THz and mid-IR spectroscopy
Marco A.
Allodi
*a,
Sergio
Ioppolo
bc,
Matthew J.
Kelley†
a,
Brett A.
McGuire
a and
Geoffrey A.
Blake
*ab
aDivision of Chemistry and Chemical Engineering, California Institute of Technology, 1200 E California Blvd., Pasadena, CA 91125, USA. E-mail: mallodi@caltech.edu; gab@gps.caltech.edu
bDivision of Geological and Planetary Science, California Institute of Technology, 1200 E California Blvd., Pasadena, CA 91125, USA
cFaculty of Science, Radboud University Nijmegen, IMM, P.O. Box 9010, NL 6500 GL Nijmegen, The Netherlands
First published on 16th December 2013
Abstract
Icy dust grains play a key role in the chemistry of the interstellar medium. The cumulative outcome of recent observations, laboratory studies, and astrochemical models indicates that solid-phase reaction mechanisms may dominate the formation of complex organic molecules such as amino acids and sugars in space. Consequently, the composition and structure of the icy grain mantle may significantly influence solid-phase reaction pathways. In this work, we present a new experimental setup capable of studying astrochemical ice analogs in both the TeraHertz (THz), or far-Infrared (far-IR), region (0.3–7.5 THz; 10–250 cm−1) and the mid-IR (400–4000 cm−1). The instruments are capable of performing a variety of spectroscopic studies that can provide especially relevant laboratory data to support astronomical observations from telescopes such as Herschel, SOFIA, and ALMA. Experimental spectra of astrochemical ice analogs of water and carbon dioxide in pure, mixed, and layered ices were collected at different temperatures under high vacuum conditions with the goal of investigating the structure of the ice. We tentatively observe a new feature in both amorphous solid water and crystalline water at 33 cm−1 (1 THz). In addition, our studies of mixed and layered ices show how it is possible to identify the location of carbon dioxide as it segregates within the ice by observing its effect on the THz spectrum of water ice. The THz spectra of mixed and layered ices are further analyzed by fitting their spectral features to those of pure amorphous solid water and crystalline water ice to quantify the effects of temperature changes on structure. From the results of this work, it appears that THz spectroscopy is potentially well suited to study thermal transformations within the ice.
1 Introduction
Dust grains play a central role in the chemistry of the interstellar medium (ISM). Hydrogen atoms only react efficiently to form molecular hydrogen with a dust grain acting as a third body to stabilize the new molecular species.1–4 In low-mass star-forming regions, analogous to the cloud that formed our own solar system, there exist cores with low enough local temperatures, and high enough density for gas to condense onto dust grains. Simple, gas-phase atoms and molecules accrete onto a dust grain to form an icy outer mantle, and new reaction pathways become available.5–7The diversity of chemistry possible on the ice surface greatly exceeds the possibilities in the gas phase. In an ice, energetic processing, by UV photons or cosmic rays, excites or breaks chemical bonds, leading to chemical reactions of species within the ice.8–13 Thermal processing can occur if different molecules present in the ice initially do not have enough energy to react, and then later gain sufficient energy to overcome a reaction barrier.14–16 In addition, atoms and light molecules freeze out onto the grains and diffuse rapidly over the ice surface inducing chemical reactions.17–24 The ice facilitates the interaction between different species and also serves as a third body to stabilize a new molecule if it forms.
Consequently, these solid-phase reaction mechanisms can potentially lead to the formation of more complex molecules such as sugars and amino acids. Given that recent work has ruled out a gas-phase mechanism in glycine formation,25 and yet glycine has been detected in comets sampled by the STARDUST mission,26 solid-phase reaction mechanisms on ice grains may provide the main pathway for the formation of complex organic molecules (COMs) in the ISM.27 In addition, molecular ices on dust grains likely play a fundamental role in the chemistry of planetary ices, which have been observed on different bodies within our solar system.28,29
The role that the structure of the ice mantle plays in chemical reactions on icy grain surfaces remains an incompletely explored question in understanding the effect of dust grains on the chemistry of the ISM. Given the icy dust grains' ability to facilitate the interaction between different species within the ice as discussed above, it would be useful to understand, specifically, how the ice structure can impact diffusion barriers within the ice. Since COMs form as smaller molecules diffuse through an ice, accurate models of diffusion barriers will improve COM formation models.27,30
Mid-infrared (mid-IR) spectroscopy (450–4000 cm−1) has been fundamental in improving our understanding of ice and dust in the ISM.31–33 As a result, the most abundant simple molecules have been detected in the solid phase via mid-IR astronomical observations.34 To begin investigating structural effects in ices, many laboratory investigations have been carried out on ice mixtures composed of H2O and CO2, the principal focus of this work. Early studies by Bernstein et al. reported how the intermolecular interactions can break the symmetry of different molecules, and thus cause significant changes in the IR spectra, including the presence of peaks not observed in a pure CO2 ice.35 Galvez et al. and Maté et al. characterized extensively co-deposited and sequentially-deposited H2O and CO2 ice.36–38 Their work detected structural differences between mixed ices and identified the differences between CO2 molecules that formed a crystalline layer on top of H2O, and the CO2 molecules that diffused into the H2O sample using Fourier-transform IR (FTIR) spectroscopy and temperature-programmed desorption (TPD) experiments. Öberg et al. investigated segregation within mixed CO2 and H2O ice and found that molecules will form segregated groups as long as it remains more favorable to interact with like molecules as opposed to different species.39 In addition, they also showed that, in contrast with what was previously thought, ice dynamics were thickness dependent. Clearly, IR spectroscopy offers valuable insight into the structure of the ice. However, transitions at IR frequencies correspond to intramolecular vibrational modes of the molecules in the ice, and, thus, IR spectroscopy only detects local changes in structure.
In contrast, TeraHertz (THz), or far-IR, spectroscopy, covering the region from 0.1 to 10 THz (3–330 cm−1), directly probes lower frequency vibrations that correspond to low energy intra- and, especially, intermolecular modes. By virtue of probing intermolecular modes, THz spectroscopy is particularly sensitive to the long range interactions between molecules.40 Since any change to the structure of the ice would affect these long range interactions, THz spectroscopy allows for direct measurements of large-scale structural change. While there has been previous spectroscopic work done in this region,41–45 none of the previous experiments focused on understanding the structure of mixed and layered ices.
There has been some theoretical work focused on mixed and layered H2O and CO2 ices.46,47 Both works focus on the suitability of the mid- and far-IR for studying structural changes in astrophysically-relevant ices. Lee et al. find a wealth of spectroscopic features in the THz region and conclude that the features of H2O ice dominate the spectrum in this region. In addition, they find ice layers at different distances from an H2O–CO2 interface can have different spectral features which are particularly well pronounced at THz wavelengths.47 Their results encourage work in the THz region to investigate these effects experimentally. As observational facilities such as Herschel,48 SOFIA,49 and ALMA50 continue to produce high-quality sub-millimeter and THz data, complementary laboratory data can provide important structural information about the ice and, indeed, give the scientific community the tools needed to interpret observational spectra and understand ice chemistry in the ISM and exoplanetary systems.
In the present study, we employ THz and mid-IR spectroscopy to characterize astrochemical ice analogs of H2O and CO2. We first describe our state-of-the-art setup, which includes a detailed discussion of the time-domain (TD) THz spectrometer built in our lab. To investigate the ice structure, we prepared ices of pure H2O or CO2. We then prepared ices composed of distinct molecular layers, as well as mixtures that were co-deposited, and acquired THz and mid-IR spectra after ice deposition and at different temperatures. Our results provide insight into how different layers of ice interact with each other, and how the segregation of more volatile molecules within an ice can affect the its short- and long-range structure, thus demonstrating the importance of THz spectroscopy in astrochemical ice studies. These laboratory investigations have potential implications for future astronomical work.
2 Experimental methods
The new experimental setup used in this work consists of a high vacuum chamber; a commercially available FTIR spectrometer to cover the mid-IR; and a TD THz spectrometer, constructed in-house, to cover the THz, or far-IR, region. The complete instrument can be seen in Fig. 1. We will discuss each component in detail in the sections below.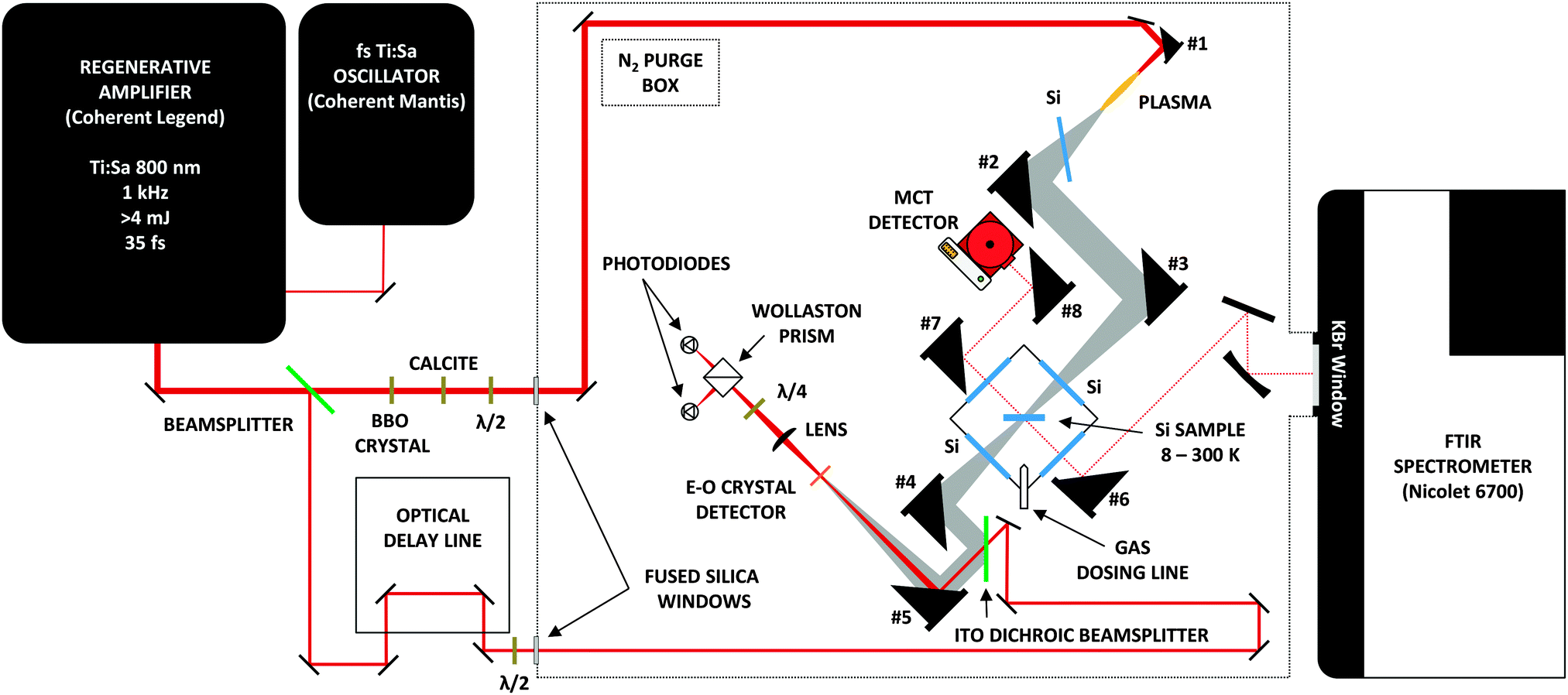 | ||
| Fig. 1 The layout of the Caltech astrochemical ice analog experimental setup, including the home-built time-domain THz spectrometer (not to scale). The mirrors used to steer the generation beam before the BBO are low dispersion, dielectric, high-reflecting mirrors (Newport 10B20UF.20). The 400 and 800 nm beams for generation enter the purge box via a dual-band AR coated 1′′ thick fused silica window (Eksma). The 800 nm detection beam enters the purgebox via 800 nm AR coated 1′′ thick fused silica window (Eksma). The mirrors used to steer the detection beam are protected silver mirrors (Thorlabs PF10-03-P01). Two beam samplers (Newport 10B20-01NC.2) are used in place of 2 mirrors along the detection path to attenuate the beam power, so as not to saturate the photodiodes. The gold coated, 1′′ diameter, 8′′ focal length (f) OAPM (Edmunds) labeled #1 above is used to generate the air plasma. OAPMs #2-4 used to steer the THz beam are 2′′ diameter protected gold mirrors with f = 6′′ (Edmunds Optics). To ensure a tight focus onto the crystal, OAPM #5 has a 2′′ focal length. The OAPM #6 (f = 4′′) steers the mid-IR beam onto the sample, and the beam is collected by OAPM #7 (f = 4′′), and finally, tightly focused by OAPM #8 (f = 2′′) onto the MCT detector. | ||
2.1 Vacuum chamber and IR spectrometer
Studying astrochemically-relevant ice analogues requires high vacuum and low temperatures. To achieve these conditions, we employ a high vacuum chamber (R. C. Janis) that encloses the cold finger of a closed-cycle He cryostat (CTI Cryogenics) capable of cooling a 1′′, high-resistivity, intrinsic Si substrate (University Wafers) to ∼8 K. A heating element, and thermocouple connected to the cryostat allows for precise control of the substrate temperature between ∼8 K and 300 K. As seen in Fig. 2, the vacuum chamber is pumped through a 1.5′′ diameter, flexible, stainless steel pipe connected to a turbomolecular pump (Turbo V 250 Varian inc.). The configuration allows for the facile vertical translation of the main vacuum chamber when necessary to align the spectrometers. A liquid nitrogen reservoir above the TMP can be filled for cryo-pumping. The room temperature base pressure in the vacuum chamber reaches ∼5 × 10−6 Torr, yet when the cryostat is operating, the pressure in the vacuum chamber reaches as low as ∼10−8 Torr.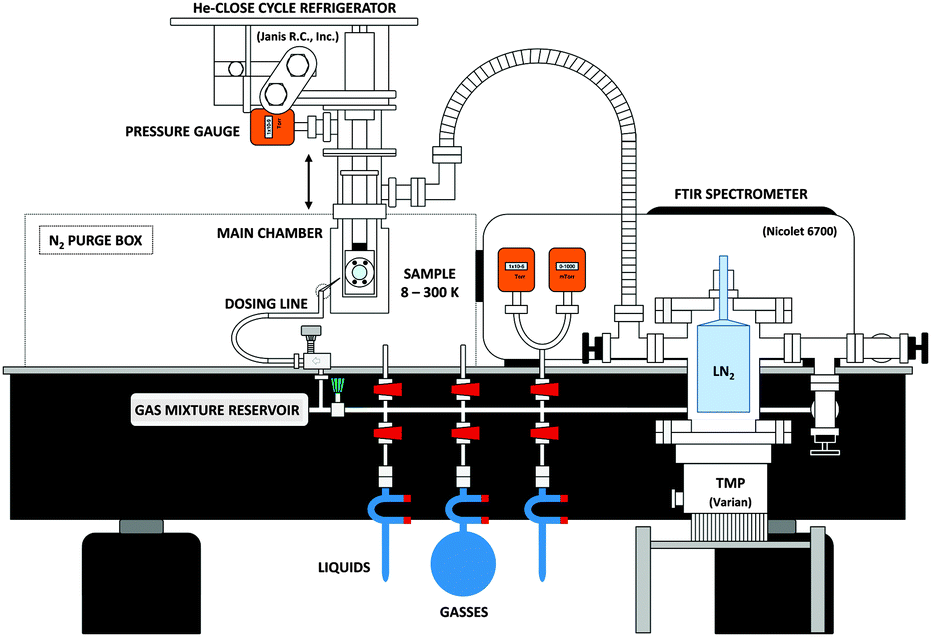 | ||
| Fig. 2 A side view of the experimental setup. The THz spectrometer is not shown here for purposes of clarity. | ||
Samples are prepared for deposition in a stainless-steel dosing line as seen at the bottom of Fig. 2. The line implements a Pirani gauge (Lesker KJL275800) in the range of 10−4 to 1000 Torr, and an active capacitance transmitter (MKS Baratron) in the range between 0.01 to 1100 Torr. The latter allows for mass independent monitoring of the pressure of the gas sample in the line. An all-metal leak valve (Lesker) separates the chamber from the portion of the dosing line where samples enter or are mixed. A 1/8′′ diameter stainless steel pipe connects the leak valve to the vacuum chamber. The end of the pipe is capped with a metal mesh with a 38 μm hole size (McMaster-Carr 85385T117) to ensure a uniform ice deposition, and the end is positioned ∼1′′ away from the substrate.
To deposit a pure ice, vapor fills the evacuated dosing line to a fixed pressure. Then, the leak valve leading into the chamber is opened to allow for a slow deposition of ice onto the Si substrate. The leak valve is closed once the ice on the substrate reaches the desired thickness. To create a layered sample, the dosing line is evacuated and filled with another molecule to a fixed pressure and then deposited via the leak valve.
Sample mixtures are prepared by first filling the dosing line with a sample of one molecule and then closing the valve separating the gas mixture reservoir from the rest of the dosing line. The remaining molecules of the first sample are evacuated using a mechanical pump and the dosing line is refilled with the second molecule. After filling the dosing line with the second molecule, the valve closing the gas mixture reservoir is opened and the two gasses mix in the full volume of the dosing line. The mixture can then be deposited onto the substrate in the sample chamber by opening the leak valve. For the experiments discussed below, water vapor was prepared in the dosing line from a sample of deionized liquid H2O, which was degassed by several freeze–pump–thaw cycles. Gaseous CO2 was used as received from Air Liquide (99.99%).
Our spectrometer operates in two distinct regions of the electromagnetic spectrum. Therefore, two different sources of radiation are used, each entering the chamber along separate paths but at 45° angles to the substrate surface to allow for both beams to be incident on the same sample, as seen in Fig. 1. High-resistivity, 3.5 mm thick, 2′′ diameter, intrinsic Si wafers (University Wafers) act as both windows along the THz path, and one of the two windows along the IR path. A 2′′ diameter Si wafer (Edmunds #68-532) serves as the second IR window.
Mid-IR spectra with a resolution of 1 cm−1 are collected with a Nicolet 6700 FTIR spectrometer using OMNIC software (version 7.4.127, Thermo Scientific). Running in external sample mode, light leaves the spectrometer through a 2′′ diameter KBr window and enters a box purged with N2. A pair of spherical mirrors collimates the light out of the spectrometer, then an off-axis parabolic mirror (OAPM, Edmund Optics) focuses the light onto the sample to a spot size of ∼3/4′′ in diameter. OAPMs collect the light transmitted by the sample and steer the beam onto a mercury cadmium telluride (MCT) detector (Thermo Scientific 840-070300). All of the mid-IR spectra of ice samples shown below are averages of 512 co-added scans.
2.2 THz spectrometer
As seen in Fig. 1, the Caltech TD THz spectrometer relies upon an ultrafast laser system to generate and detect THz pulses. A Ti:Sapphire oscillator (Coherent Mantis) delivers sub 100 fs duration seed pulses containing 80 nm of bandwidth centered around 800 nm, to a chirped pulse regenerative amplifier (Coherent Legend Elite USP) at an 80 MHz repetition rate. The amplifier produces 4 mJ, 37 fs transform-limited pulses centered around 800 nm at a 1 kHz repetition rate. A 99% reflective dielectric mirror acts as a 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 beam splitter to divide the amplifier's output into a THz generation beam, and a THz detection beam.
:1 beam splitter to divide the amplifier's output into a THz generation beam, and a THz detection beam.
The 99% split of the laser's output provides the spectrometer's THz light via a two-color plasma. To make this plasma, a beta-barium borate (BBO) crystal (Eksma Optics), cut for Type 1 second harmonic generation (SHG) at 800 nm, frequency doubles some of the 800 nm light to 400 nm. A birefringent calcite plate (Eksma Optics) then corrects for the normal group velocity dispersion between 800 and 400 nm. Both colors pass through a zero-order dual-wavelength wave plate (Eksma Optics #465-4211) to align the polarizations of the 800 and 400 nm light. Finally, a gold-coated OAPM focuses both colors of light co-linearly in an environment purged with N2(g). The resulting oscillating plasma produces a broad-bandwidth, intense THz pulse, with the same polarization as the optical light fields.51–54 Another OAPM focuses the THz pulse down onto the ice sample in the vacuum chamber. An optical chopper placed before the BBO crystal modulates the THz generation beam at 100 Hz for lock-in detection.
We employ free-space, electro-optic sampling to detect the THz light transmitted through the ice sample.55,56 Optics route the remaining 1% of the 800 nm light from the original 4 mJ pulse down an opto-mechanical delay line (Newport ISL 150 with ESP300 controller) to change the relative delay between the THz generation and detection beams. The detection beam then passes through a half-wave plate (Newport 10RP02-46) which can be rotated to ensure that the linear polarization of the detection beam, and of the THz beam are matched at the detector crystal for maximum signal. An indium tin oxide (ITO) coated glass then serves to recombine the detection beam with the THz beam by reflecting the THz light and transmitting the 800 nm detection light so that the two beams co-axially propagate. An OAPM focuses the recombined beams onto a GaP(110) detector crystal (Incrys, 10 mm × 10 mm × 0.4 mm). The electric field of the THz light interacts with the crystal to produce a DC-bias, as the THz field is effectively DC when compared to optical frequencies. When 800 nm light co-propagates with THz light in the crystal, the THz electric field rotates the polarization of the 800 nm light via the Pockels effect. The magnitude of this change in polarization is proportional to the magnitude of the THz electric field applied. After passing through the detector crystal, a lens refocuses the 800 nm beam to ensure that the photodiodes detect all the light. It then propagates through a quarter-wave plate (Newport 10RP04-46) to create a circularly polarized beam that is split into orthogonal polarizations by a Wollaston Prism (Thorlabs WP20). The signal is measured as a difference signal generated by a pair of balanced photodiodes (Thorlabs PDA36A). When no THz light is incident on the GaP crystal, the photodiodes read the same voltage, but when THz light is present, the photodiodes are no longer balanced, and the resulting difference signal is proportional to the THz electric field.
To measure the entire THz waveform in the time domain, the delay line moves to increase the detection path length so that the detection pulse interacts with the THz field at a different point in time. A digital lock-in amplifier (Stanford Research Systems SR830) records the spectrum as a function of delay. An example time domain trace collected with no sample on the substrate can be seen in Fig. 3. The spectrum is an average of 32 co-added scans, with a 100 ms time constant on the lock-in amplifier. The peak-to-peak signal-to-noise ratio (SNR) of a single scan collected with the lock-in amplifier in the time domain is 325:1.
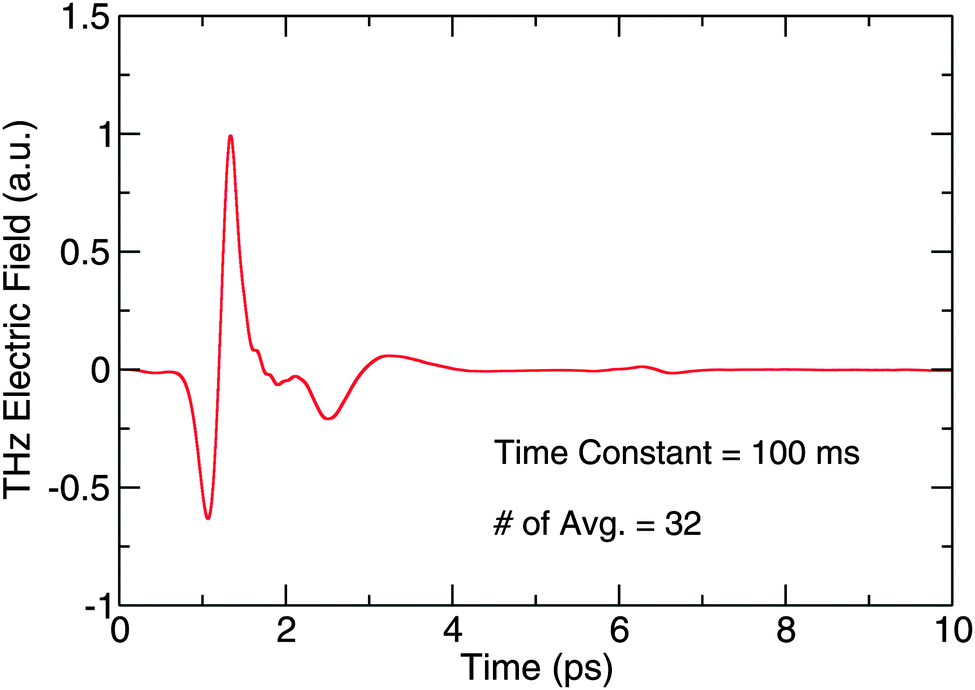 | ||
| Fig. 3 A THz trace of the blank substrate collected by the TD THz spectrometer. | ||
Using a plasma source and electro-optic (EO) sampling with a GaP detector crystal, the Caltech TD THz spectrometer detects a pulsed THz electric field with frequency content from 0.3 THz to 7.5 THz. The bandwidth can be seen in Fig. 4, which is a numerical fast-Fourier transform (FFT) of the data in Fig. 3, and the figure also shows the overlap of our spectrometer with the capabilities of some of the aforementioned sub-millimeter and far-IR telescopes. In addition, Fig. 4 shows the SNR in the frequency domain. By comparing the magnitude of signal on the ordinate to the noise level, which corresponds to the structure beyond 7.5 THz, one can roughly see how the SNR breaks down over the bandwidth of the spectrometer. In the 0.5 to 1.5 THz region, we report a SNR of 1800:1. From 1.5 to 4.5 THz, the SNR decreases by a factor of roughly 3 per 100 GHz starting at 1800:1 until it reaches 30:1, and the SNR remains flat at that value from 4.5 to 6.5 THz. The SNR then falls off again from 6.5 to 7.5 THz, where the signal meets the noise level.
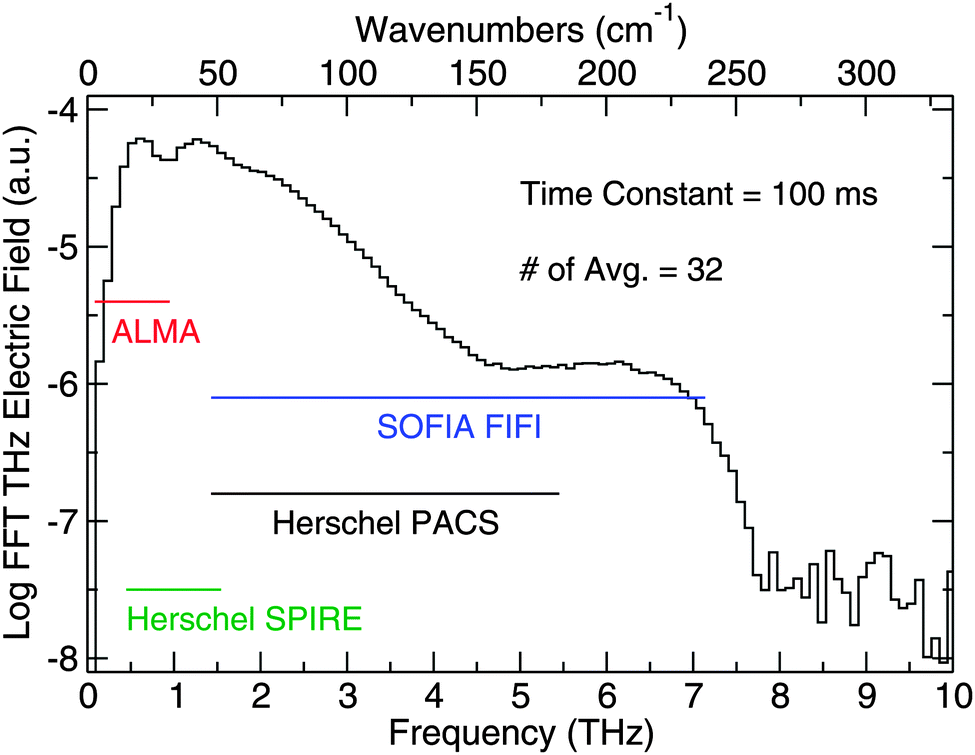 | ||
| Fig. 4 The bandwidth of the THz spectrometer. The ordinate provides information on the relative strength of the THz electric field within the THz pulse at different frequencies. The horizontal lines indicate the spectral coverage of observational facilities in the THz region. | ||
The length of the scan in the time domain limits the resolution of the data in the frequency domain. While in theory only the length of the delay line limits the frequency-domain resolution, etalons produced in the EO detector crystal, and other optics through which the THz light passes, can add structure to the data. Small amounts of drift in the frequency position of the etalons over the course of an experimental run can be caused by environmental fluctuations and can change the etalon positions in the sample relative to the background scan. The etalons will then not divide out during background subtraction because of the time required to collect a spectrum. While there exist ways to push etalon peaks further away from the main THz pulse or remove them entirely, alternatively, one can scan a smaller window in the time domain to avoid these recurrences of the THz pulse. In this work, we co-added 32 spectra in the time domain to create an average which we subsequently used to calculate a FFT using boxcar apodization and zero-padding the length to the next power of 2. The spectra were collected over a 10 ps window in the time domain, chosen to avoid etalon features, which corresponds to a frequency domain resolution of 94 GHz (3 cm−1). The sample spectra were then divided by a reference scan of the bare Si substrate to provide the absorption spectra shown in the following figures.
The spectral coverage of the combined THz and FTIR capabilities can be seen in Fig. 5. The spectrometer coverage extends from 10–4000 cm−1 with a gap from 250–400 cm−1 and a small gap between 580–630 cm−1 because the signal falls below the noise in those ranges. Using this broad spectral coverage, we are able to draw conclusions about the ices from both the THz and mid-IR regions.
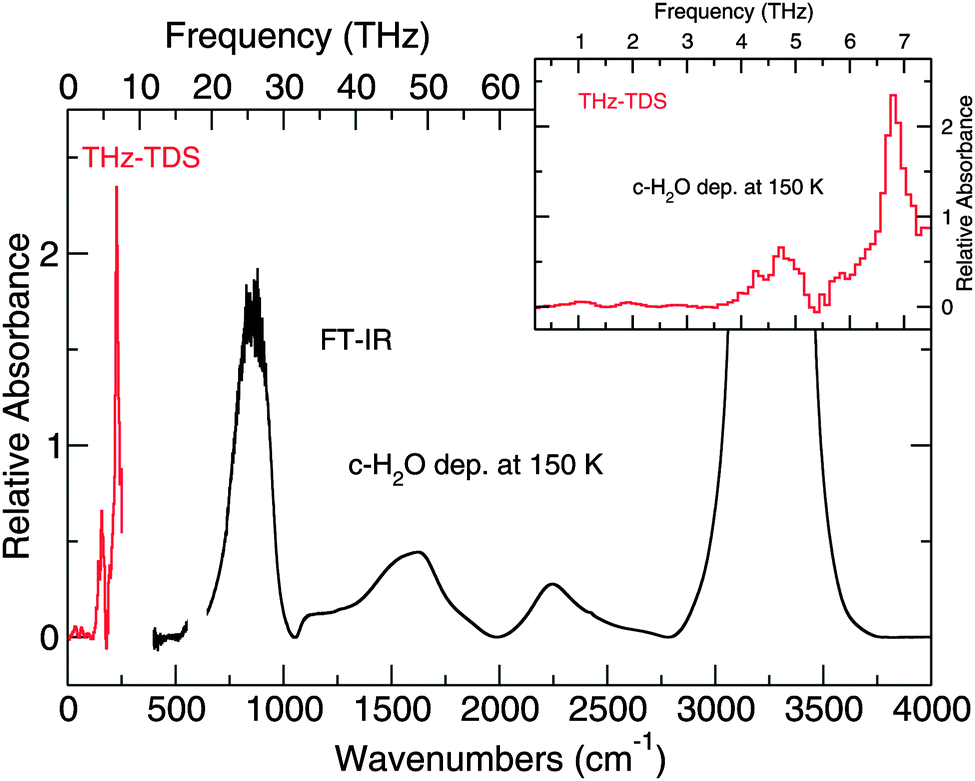 | ||
| Fig. 5 The spectrum of crystalline H2O ice deposited at 150 K and collected at 150 K. This spectrum demonstrates the complete spectral window covered by the spectrometer. The inset shows a zoom of the region collected with the THz-TDS. | ||
3 Results and discussion
We present the results in three subsections. THz and mid-IR results for pure ices of H2O and CO2 are discussed first. Identifying the features and structure of pure ices allows us to understand how such features change when molecules are mixed or layered. Second, results from experiments of H2O:CO2 mixed ices that are co-deposited (CD) are discussed. Finally, data collected from layered ice samples are presented. The layered experiments performed are crystalline sequential (CS) deposition, amorphous sequential (AS) deposition, and inverse sequential (IS) deposition, where we have used the nomenclature established in the literature.36Table 1 summarizes the experiments discussed below.| Experiment | Method | Ratio | R dep (μm min−1) | Thickness (μm) | T dep (K) | Thermal history (K) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Deposition of single species (dep), co-deposition (co-dep), and sequential deposition (CS, AS, and IS) are performed under different laboratory conditions; Rdep is the deposition rate of a selected molecule expressed in μm min−1; thickness is expressed in μm; Tdep is the substrate temperature during deposition; thermal history is the temperatures that the ice have been sequentially exposed to. The heating ramp rate is 20 K min−1, in all cases. | ||||||||||
| Pure ice | ||||||||||
| H2O | dep | — | 0.08 | 0.3, 2.6, 5.3 | 150 | 150 | ||||
| H2O | dep | — | 0.08 | 0.4, 1.6, 3.2 | 10 | 10, 150 | ||||
| H2O | dep | — | 0.06 | 4.8 | 150 | 150, 75, 10, 75, 150 | ||||
| H2O | dep | — | 0.12 | 2.8 | 10 | 10, 75, 150, 75, 10, 150 | ||||
| CO2 | dep | — | 0.13 | 0.1, 0.3, 0.6, 1.4 | 75 | 75, 60, 75, 60, 75, 60, 75, 60 | ||||
| CO2 | dep | — | 0.13 | 0.2 | 60 | 60, 10, 75 | ||||
| Mixed ice | ||||||||||
| H2O:CO2 |
co-dep | 1:5 |
0.22 | 2.1 | 60 | 60, 175, 75, 10, 150 | ||||
| H2O:CO2 |
co-dep | 1:10 |
0.8 | 2.3 | 60 | 60, 10, 60, 100, 60, 125, 60, 150, 60, 175, 60 | ||||
| Layered ice | H2O | CO2 | H2O | CO2 | H2O | CO2 | H2O | H2O and CO2 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| H2O and CO2 on top | CS | — | 0.4 | 0.2 | 2 | 0.3 | 60 | 75 | 60, 175, 75, 10, 150 | 75, 60, 10 |
| H2O and CO2 on top | AS | — | 0.7 | 0.6 | 2 | 0.3 | 60 | 60 | 60 | 60, 10, 60, 100, 60, 150, 175, 150 |
| CO2 | H2O | CO2 | H2O | CO2 | H2O | CO2 | CO2 and H2O | |||
|---|---|---|---|---|---|---|---|---|---|---|
| CO2 and H2O on top | IS | — | 0.8 | 0.9 | 0.2 | 2 | 60 | 60 | 60 | 60, 10, 60, 100, 60, 150 |
All ices deposited in this study fall into the “thick” regime (>100 monolayers) and are approximately 0.5–5 μm thick for H2O ice.‡ To help ensure consistent thickness, we used the height of the OH librational band between 700–900 cm−1 to asses when to stop deposition of water. The following figures show that the relative thickness of the different ices is comparable. These experiments and others in the cryostat60 have shown that the ice is stable for at least 24 h as long as the temperature is kept below the sublimation temperature of a given ice. Moreover, the residual gasses in the vacuum chamber consist mainly of H2O, accreting at a rate of roughly 1 × 1012 molecules cm−2 s−1 (0.001 μm h−1) when the substrate is kept at 10 K. Thus, relative to the ice thickness used in our study, the background water deposition can be considered negligible over the course of the experiment.
It must be noted here that in all of the following THz spectra, there is noise at 3.2 THz that is only the width of one frequency bin. This noise spike appears to be some form of laser noise that we were unable to filter out while conducting these experiments. As such, the point corresponding to this noise feature is not plotted in most of the following spectra.
3.1 Spectral fitting of the THz data
To quantify the changes in the structure of mixed ices relative to the structure of pure ices samples, a spectral fitting procedure was used. The procedure makes use of a singular value decomposition to compare how the THz spectra of the mixtures differ from those of the pure ices. A set of four pure H2O ice spectra forms the basis of components used to create a fit (Fig. 6). The spectrum of ASW was collected at 10 K from an ice that was deposited at 10 K. For the spectra of crystalline H2O, a pure H2O ice was deposited at 150 K and then cooled to 75 K and 10 K, with spectra collected at each of these temperatures. The coefficients resulting from the singular value decomposition reflect the weight of a given spectrum in the fit. The fits can be seen plotted in the figures that follow of mixed and layered ices.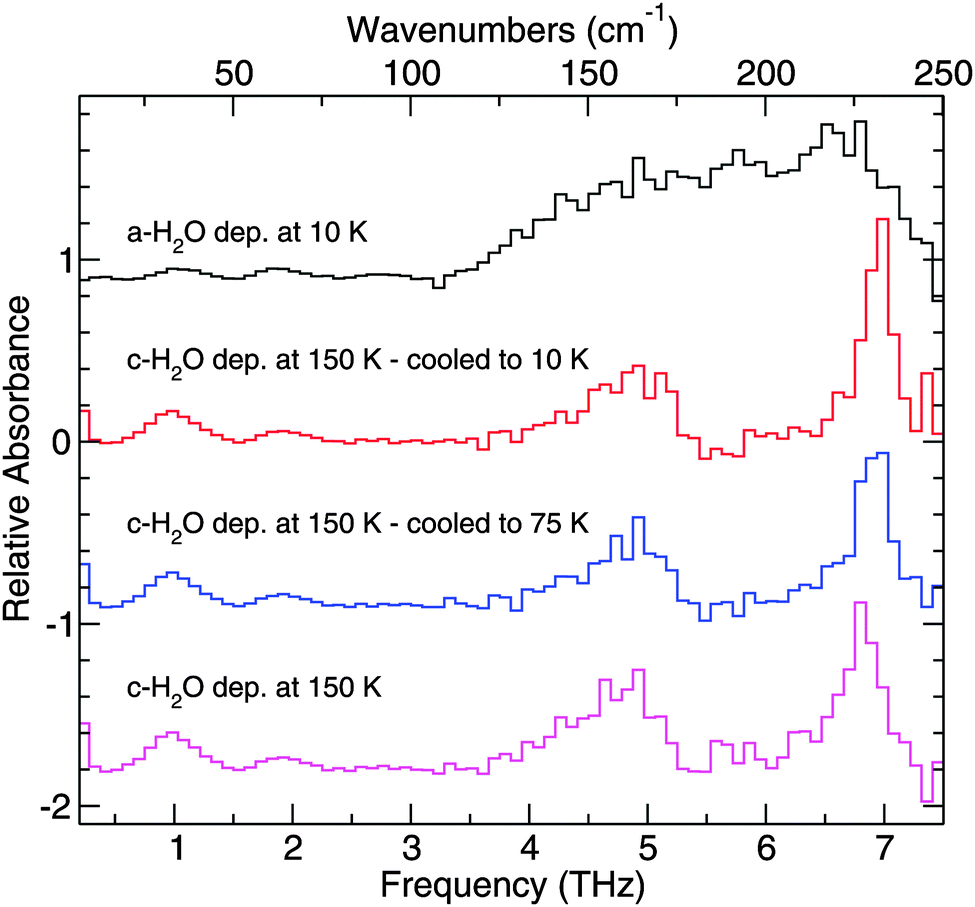 | ||
| Fig. 6 Component spectra used to create the fits to the THz data. The top spectrum corresponds to the THz spectra of amorphous H2O ice, while the bottom three spectra correspond to crystalline H2O ice. The spectra are labeled with their deposition temperature and temperature at which the spectra were collected. For the top and bottom traces, the spectra were collected at the deposition temperature. | ||
To normalize the fit coefficients for experiment-to-experiment differences in the ices, the coefficients for a given component are multiplied by the integrated area of the mid-IR H2O bending mode at ∼1600 cm−1 in the pure ice spectrum for that component. The integrated area of the bending mode of the ice mixture being fit defines the value for 100% area. The coefficients of the fit components are reported as a percentage of the total area of the H2O bending mode of a given ice mixture being analyzed.
The degree to which the sum of the normalized coefficient percentages approaches 100% indicates the ability of the four component basis to reproduce the spectra of the mixtures being analyzed. In cases where the structure of the mixed or layered ice is not expected to differ from the pure ices structure, such as in the CS deposition, the sum of the fit coefficient percentages are close to 100%. In some cases, however, the sum of the fit coefficient percentages does not approach 100%. This does not signify that the fit procedure works poorly, since the fit still qualitatively reproduces the spectral data – instead, it shows that the structure of that ice differs somehow from the structure of the pure ice components. When the fit components of a given ice sum to a percentage much less than 100%, that ice cannot be described by a basis consisting only of pure H2O ice components. Thus, a poor fit actually provides useful information about what the structure of the ice does not look like. In a few specific cases, such as in the CD 60 K/125 K and the CD 60 K/175 K, the procedure was incapable of qualitatively fitting the data accurately and thus shows that the structure of the ice being fit differs significantly from the structure of a pure ice. However, since the fit was unsuccessful, there is no value in trying to quantify the difference from the structure of a pure ice, and so fit percentages are not reported for these data. The specific fits are discussed in more detail in the context of the specific experiments below.
3.2 Pure ices
Pure H2O has been deposited in two different forms, amorphous solid water (ASW) and crystalline H2O.61 Cooling the substrate down below 100 K before deposition ensures deposition of porous ASW, which lacks a well-defined long-range crystal structure. Crystalline H2O can be formed in two ways. If the substrate is kept above 140 K, crystalline H2O ice will form upon deposition. Another way to form crystalline H2O is to deposit ASW at temperatures less than 100 K, which is then annealed. Above 140 K, ASW undergoes a non-equilibrium transition to the more thermodynamically stable, crystalline H2O form.Fig. 6 highlights the THz spectroscopic differences between ice structures formed at different deposition temperatures. Both crystalline and amorphous H2O have an absorption feature at 1.9 THz. ASW resulting from deposition at 10 K has one broad absorption feature starting at approximately 3.1 THz and extending out past 7.5 THz. In the corresponding region for crystalline H2O, deposited at 150 K, distinct peaks with the following assignments40,62 are seen: 6.8 THz (hydrogen-bonded bilayer stretch), 5.7 THz (out-of-phase vibration within the bilayer), 4.7 THz (proton disordered motion), 4.2 THz, and 1.9 THz (corresponding to an O–O–O bend) which are in good agreement with theory and previous experiments.43,63–65 The peak positions of the THz features for the pure ices studied are summarized in Table 2.
| H2O | |||
|---|---|---|---|
| Amorphous | Crystalline | ||
| THz | cm−1 | THz | cm−1 |
| The transitions of H2O ice can be assigned as follows:a O–O–O bend.b Proton disordered vibration.c Out-of-phase vibration of molecules within a bilayer.d Hydrogen bond stretch between bilayers.e The feature at 4.2 THz is blended with the feature at 4.7 THz. | |||
| 1.0 | 33 | 1.0 | 33 |
| 1.9 | 63 | 1.9a | 63 |
| 5.5 | 181 | 4.2e | 139 |
| 4.7b | 155 | ||
| 5.7c | 188 | ||
| 6.8d | 224 | ||
In addition to the features described above, a feature appears in these spectra at 1 THz that has not been previously observed in far-IR experiments, and so we tentatively report a new feature at 1 THz. This feature has likely not been seen in previous work because a TD THz spectrometer, like the one used in this study, has a higher signal-to-noise around this band than a comparable far-IR FTIR study. Over the 0.1 to 3 THz region, a plasma emitter produces more photons than the incoherent source in an FTIR, and so features which would have been below the noise level of the previous work can be detected with our TD THz spectrometer. As such, previous experiments did not extend below 50 cm−1 and were thus unable to see this feature.
The results of the other method of preparing crystalline H2O are shown in Fig. 7. In contrast with Fig. 6, where crystalline water ice was prepared by deposition at 150 K, the crystalline H2O ice presented in Fig. 7 was deposited as ASW at 60 K, then annealed to 175 K and subsequently cooled. In these experiments, H2O transitions from compact amorphous to crystalline ice start at ∼140 K, in good agreement with the literature. Annealing the ice to 175 K should ensure that the amorphous to crystalline transition is fully completed.66 There will be some mass lost from the ice when annealing at such high temperatures. However, because of the thickness of the ice, such loss is negligible. Under these vacuum conditions, H2O molecules start to desorb from the ice at ∼170 K.
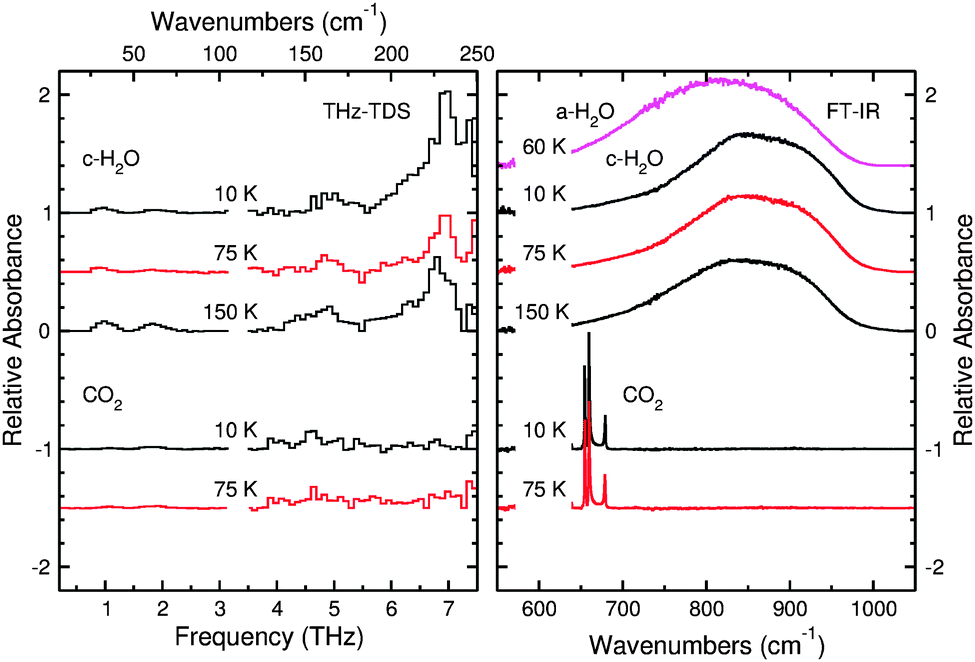 | ||
| Fig. 7 THz spectra (left panel) and mid-IR spectra (right panel) of pure crystalline H2O and pure CO2 at different temperatures. Both molecules were deposited at 60 K and then the ices were annealed. The H2O was annealed at 175 K, while the CO2 was annealed at 75 K. | ||
The H2O spectra in both Fig. 6 and 7 illustrate the spectral changes as a function of temperature. As the cryostat cools the ice sample, the THz feature that peaks at 6.8 THz becomes sharper and more intense at 10 K relative to the same feature at 150 K because cooling the sample will reduce the thermal population of hot-band states of the vibration. The same is true for the shoulder at 5.7 THz. These changes in shape may be useful for determining the temperature of an ice detected in space from its THz spectrum.
Fig. 7 shows the absorption spectrum for pure CO2 ice. Although Moore and Hudson reported that CO2 should have one narrow feature at 3.3 THz,43 we observe no unambiguous features in our spectra. This is likely caused by a combination of sensitivity and, especially, resolution, owing to the proximity of this feature to the noise spike at 3.2 THz in our experiments. Molecules of CO2 begin to desorb at ∼90 K from the ice, and an ice of pure CO2 will be fully desorbed at 100 K under these experimental conditions. In the following sections, we will show that the desorption temperature of CO2 varies with respect to the ice composition.
In conjunction with the THz spectra, the right panel of Fig. 7 also shows the mid-IR spectra collected with the FTIR spectrometer from the same samples. This portion of the spectrum shows the librational mode of water ice and is a useful region of the spectrum to consider because resonances corresponding to the bending mode of CO2 also appear in this same spectral window. A spectrum of ASW is presented first for comparison, and one can easily see the differences in shape between the amorphous and crystalline ice. The ASW peak is more symmetric than that of crystalline H2O, and this asymmetry becomes more pronounced as the crystalline ice cools from 150 to 10 K. These FTIR spectra are also useful because CO2 shows clear resonances distinct from those of H2O ice. There are four peaks which appear in the spectrum of CO2. The two main peaks centered around 660 cm−1 are the main features of the CO2 bending mode for S-polarized light. The peak at 680 cm−1 is the signature of the CO2 bending mode for P-polarized light. Given that the angle of incidence of the IR beam is 45° and that there is no polarizer along the beam path, the FTIR spectra in this work will show contributions from both polarizations of light. The final peak centered around 640 cm−1 is much weaker, and it corresponds to the 13CO2 bending mode. Analysis of FTIR data for CO2 allows for an effective determination of whether a mixed ice sample still contains small amounts of CO2 since the THz spectra of this molecule do not provide distinct and strong absorption features.
3.3 Co-deposition of mixed ices
The importance of CO2 in this study stems from how its presence affects the structure of H2O ice. In the ISM, such a mixture could arise from the formation of CO2 within a polar ice.67 Consequently, it is interesting to know if and how the presence of CO2 would affect the structure of the ice in which it exists.:CO2 ratio of ∼5:1.
 | ||
| Fig. 8 THz spectra (left panel) and mid-IR spectra (right panel) of mixed H2O and CO2 ice co-deposited. Data for mixed ices that were annealed directly after deposition are presented in the top three traces of the figure. The spectra were collected after cooling. In the bottom three traces, the spectra collected from a mixed ice that was cooled immediately after deposition and stepwise annealed are presented. The spectra are labeled with the temperature at which they were collected. The dashed lines in the plot correspond to the fits calculated for the data from the spectra of the pure species, which are plotted with a polynomial spline fit for clarity. Note that in the bottom traces, the effect of the CO2 on the H2O ice structure can be seen from the blue shift of the ASW feature, and after annealing, the 4.5 THz transition is missing from the spectrum. The inability of the fits to reproduce the spectra show that the structure of the ices differ greatly from the structure of pure ices. The components of the fits are summarized in Table 3. The superscripts denote the temperature to which the ices were annealed: aannealed to 125 K, bannealed to 175 K. | ||
Annealing at such a high temperature should force the water to become completely crystalline,66 and one can see that the spectra of the mixture exhibit THz features that are quite similar to crystalline H2O at 10 K and 75 K but differing in intensity and structure. Compared with the spectrum of pure crystalline H2O ice at 75 K, the 6.8 THz, 5.7 THz, and 4.7 THz features are not as intense in the spectrum of this ice. Even at 10 K, where the pure ice features are sharpest and most intense, the 6.8 THz feature in Fig. 8 does not appear to change compared to the spectrum at 75 K.
The changes in these transitions results from the presence of CO2 in the ice. One can see in the right panel of Fig. 8 the resonance corresponding to the CO2 bending vibrational mode. The shape of this peak is qualitatively different from that of pure CO2 since now only one peak with a Lorentzian lineshape is observed as opposed to the three peaks observed previously. Fig. 8 shows that, even though the ice was annealed to a temperature above the CO2 sublimation temperature, not all of the CO2 molecules leave the ice. This effect has been previously observed by temperature-programmed desorption (TPD) experiments using a quadrupole mass spectrometer (QMS).68 As a result of the co-deposition of the two molecules, some CO2 molecules remain trapped by the molecules deposited on top of them. We thus observe the “interior” CO2 as discussed previously in the literature.36–38 Clearly, after annealing, the trapped CO2 molecules affect the ability of the H2O molecules to reorient themselves to form long-range order within the solid ice and properly crystallize, but the observed structure does differ from that of pure ASW.
By comparing the mid-IR data given in the top three spectra in Fig. 8 with the top three spectra in Fig. 7 (right panel), it is possible to conclude that the H2O lacks a fully crystalline structure. However, from the THz spectra alone, the lack of crystalline H2O features is also clear. In addition, given that features in the THz region involve the longer range coordinated motion of many molecules, the THz data also provide insight into the location of the CO2 molecules within the ice by observing which spectral features are disrupted.
To quantitatively characterize the structure of the ice, we fitted the THz spectral profile of the features shown in Fig. 8 as discussed in Section 3.1. The fits to these spectra plotted in Fig. 8 agrees well qualitatively with the spectra of the mixed ice. The fits show that the pure H2O bands at 4.7 and 5.7 THz are blueshifted in the spectra of the mixtures while the 6.8 THz feature is always fit well by using a spectrum of pure H2O ice acquired at the same temperature as the mixed ice. In all of these cases, a fit component corresponding to ASW was needed to improve the fit. The specific fit components for each spectrum are summarized in Table 3. Since the percentages listed in the table do not approach 100%, they provide another piece of evidence showing that the structure of these ices differs from the structure of pure crystalline water ice.
| Experiments | Fit component (percentage) | ||||
|---|---|---|---|---|---|
| ASW | c-H2O | c-H2O | c-H2O | Totala (%) | |
| 10 K | 10 K | 75 K | 150 K | ||
| a The percentage does not always equal 100% because the structure of the ice cannot always be described by the components of pure H2O used. b These are the data for the CD experiments which were directly annealed. c These are the data for the CD experiments which were cooled first and then stepwise annealed. d In this table 60 K/100 K is defined as a spectrum collected at 60 K for an ice that was first annealed to 100 K. This convention holds for all lines in the experiments column where two temperature values are listed. e No percentages are reported in this case since the fitting procedure produced a poor fit to the actual spectra. As such, a quantitative analysis is not meaningful. | |||||
| CDb 10 K | 59 | 18 | 77 | ||
| CDb 75 K | 21 | 17 | 38 | ||
| CDb 150 K | 20 | 47 | 67 | ||
| CDc 10 K | 69 | 69 | |||
| CDc 60 K/125 Kd | —e | ||||
| CDc 60 K/175 K | —e | ||||
| CS 10 K | 99 | 99 | |||
| CS 60 K | 99 | 99 | |||
| AS 10 K | 95 | 95 | |||
| AS 60 K | 87 | 87 | |||
| AS 60 K/ 100 K | 73 | 12 | 85 | ||
| AS 150 K | 66 | 21 | 87 | ||
| AS 150 K/175 K | 61 | 22 | 83 | ||
| IS 10 K | 33 | 33 | |||
| IS 60 K | 60 | 60 | |||
| IS 60 K/100 K | 52 | 8 | 60 | ||
:CO2 ratio of ∼10:1. In this set of experiments, the ice was first cooled to 10 K and a spectrum was collected. Then the ice was warmed back up to the deposition temperature of 60 K. The spectra at 10 K and 60 K agree qualitatively with the shape of the spectrum of pure, ASW in Fig. 6, however, the relative absorbance of this mixed ice does not appear as strong as that of the pure H2O. We can see from the mid-IR data in Fig. 8 that both CO2 and H2O are present in this ice. It is worth noting that the shape of the mid-IR CO2 bending mode in these unannealed spectra differs qualitatively from the shape of the bending mode in the annealed spectra at the top of the figure. The peak in the unannealed spectrum appears to have a flat top as opposed to a smooth curve and is redshifted relative to the annealed spectrum. Also, the H2O librational mode appears broader in the unannealed spectra than in the annealed spectra.
This ice was then annealed. First, the ice was annealed to 100 K and cooled down to 60 K. Both the THz and FTIR spectra show no appreciable change compared to the unannealed spectra at 60 K. The ice was then subsequently annealed to 125 K, 150 K, and 175 K, with a cool down cycle returning the ice to 60 K each time after annealing. There is no significant change to the ice after annealing until the ice is annealed up to 175 K. However, the THz spectrum of the ice after annealing to 175 K does not match the spectrum taken of the ice that was directly annealed. As discussed above, the THz spectra of the ices that were directly annealed after deposition show features of crystalline H2O. However, the spectra of the ice that was cooled before being annealed show features that correspond more to the structure of ASW.
A comparison between the bottom set of spectra in Fig. 8 and the spectral fits as discussed above shows that the presence of CO2 in the ice causes a blue shift of the broad band of amorphous water ice at 10 K. Moreover, although the last spectrum annealed to 175 K shows a weak band around 6.8 THz because of crystalline H2O, the 4.7 and 5.7 THz bands are blue shifted and/or weak. In this case, and for all spectra annealed above 100 K, the fit cannot reproduce the data satisfactorily. This shows that the structure of this ice is significantly different from the structure of the pure ices used in the fits.
The mid-IR FTIR data provide a strong basis for interpreting our THz spectra. None of the data in Fig. 8 (right panel) show the characteristic asymmetric feature of crystalline H2O in the H2O librational band (see Fig. 7), thus strengthening the interpretation that CO2 disrupts the crystallization process of the H2O ice. In addition, as previously mentioned, one can observe a difference in the features between the directly annealed spectra and the stepwise annealed spectra. With this information in hand, one can learn more about the structure of the ice from the THz spectra.
In particular, the THz region has the potential to indicate the degree of crystallization, and the history of transformations that an ice has undergone. Generally speaking, spectroscopy is not sensitive to the history of transformations an ice has undergone. However, if the different ices were to start at the same unstable equilibrium point and then undergo different, irreversible non-equilibrium transitions, the different histories would result in different spectra. For the ice that was annealed directly after deposition, we note that it does appear, from the THz spectra, to have some crystalline H2O structure. In this case, the THz spectra provide some insight into the degree of crystallinity. The ice was annealed to 175 K, where one would have expected the crystallization process to be completed (see Fig. 7). The fact that its features are attenuated somewhat relative to the spectrum of pure crystalline H2O deposited at 60 K and annealed to 175 K likely results from the presence of segregated CO2 within the ice. Since it is known that segregation occurs quickly in thick ices,39 this ice most likely contains large regions of crystalline H2O, but the pockets of segregated CO2 prevent a fully uniform crystalline solid from forming, and, thus, the peak strengths and shapes differ from that of pure crystalline H2O.
In the other experiment, by cooling the ice immediately after deposition, the ice was taken below the temperature at which segregation can occur. While some segregation likely took place during the deposition process at 60 K, by cooling first and stopping segregation, the CO2 is left more well mixed with H2O. Then, by restarting the annealing process in steps, the CO2 molecules were forced to segregate more locally into smaller domains. In contrast, when the first ice was annealed directly after deposition at 60 K, the segregation rate increased when the temperature was raised. This likely encouraged the formation of large domains of segregated CO2, and large regions composed entirely of H2O molecules. Consequently, the features of crystalline H2O are more pronounced in the directly annealed spectra when compared with the ice that was cooled immediately after deposition. This mechanism can potentially explain the differences observed in the THz spectra between the two sets of experiments.
The results from the quantitative fits discussed above provide further evidence to support the differences in the structure of the ices in the two different experiments. The spectra of the ice that was directly annealed can be fit quite well using these components as seen in Fig. 8. In contrast, the best fit of the stepwise annealed data still show a peak at 4.5 THz meaning that using the fit components corresponding to pure H2O ice, the fit to the data is unsatisfactory. Consequently, the poor nature of the fit shows that the structure of the stepwise annealed ice differs significantly from the directly annealed ice which can be fit in this multicomponent fashion.
It would also be valuable to know the location of the segregated CO2 in the structure of the H2O ice. The 6.8 THz feature in crystalline H2O corresponds to the stretching of hydrogen-bonded bilayers of H2O molecules.40 Since this feature is weak in the THz spectra of the ice that was cooled after deposition, the CO2 molecules must interfere with the hydrogen bonding between bilayers of crystalline H2O ice. Most likely, the CO2 molecules segregate together between bilayers and disrupt the H-bond stretching that give rise to the feature at 6.8 THz. As discussed by Maté et al.,37 “interior” CO2 must be located within some type of channels or pores within the ice. However, these authors were unable to determine specifically the location of “interior” CO2 within their H2O ice. By using THz spectroscopy and the known assignment of these attenuated features in the crystalline H2O ice, our results provide new insight into the specific location of the segregated CO2 within the H2O ice. The disruption of the feature at 6.8 THz shows that the CO2 molecules segregate between the bilayers of the crystalline H2O ice structure.
We would be remiss if we did not discuss the previous work done in the far-IR on ice mixtures. Moore and Hudson in 1994 also co-deposited a mixture of H2O and CO2 and observed a disruption of the crystalline water features after annealing a co-deposited mixture.43 Our results, however, are an expansion on the previous work in two ways. First, we performed two different CD experiments, one which was directly annealed, and one which was stepwise annealed, and observed different results. Second, given recent theoretical advances in understanding crystalline H2O structure,40 and an improved knowledge of segregation dynamics,39 we can infer the position of the CO2 within the ice and provide new insight into the structure of ice mixtures by determining what types of pores and channels exist for the CO2 to occupy.
It must be noted that the ratio of H2O:CO2 is different in the two ices discussed above, resulting from the experimental realities of deposition. In an attempt to achieve the same ratio in both ices, the mixing ratio in the dosing line was adjusted to try to account for the mass loss (predominantly of CO2) when the ice was annealed directly to 175 K. However, the two ratios used were different by too large an amount, which led to the different mixtures observed in the ice. This results in a difference in the ice mixtures of less than a factor of two. Even considering these differences between the ices, the results of our study remain relevant. One might expect that a higher concentration of CO2 in H2O would disrupt the crystallization to a greater degree than having a lower concentration of CO2. However, our results indicate that crystallization of water occurs more readily in the directly annealed mixture that also has a slightly higher concentration of CO2 with respect to the stepwise annealed mixture. As such, we can conclude that segregation of CO2 still trapped in a H2O ice is influenced by the annealing procedure.
3.4 Layered ices
Layered ices present another interesting system where THz spectroscopy may provide some added insight into the long-range molecular structure.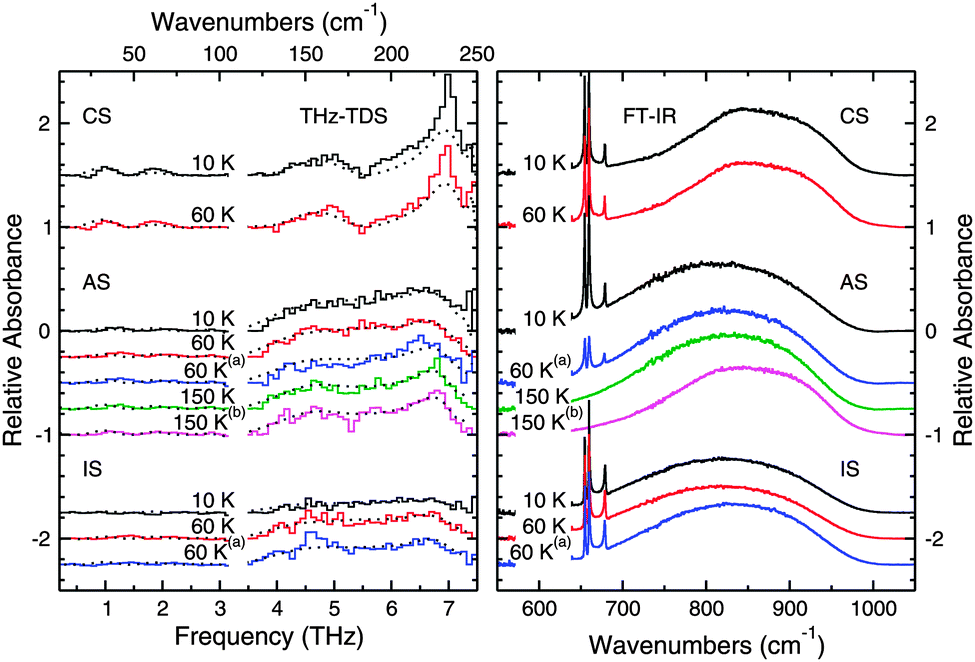 | ||
| Fig. 9 THz spectra (left panel) and mid-IR spectra (right panel) of layered ice samples. The spectra collected from the crystalline sequential (CS) deposition are shown in the top two traces, those from the amorphous sequential (AS) deposition are shown in the middle traces. The spectra collected from the inverse sequential (IS) deposition are shown in the bottom three traces. All spectra are labeled according to the temperature at which they were collected and the conditions under which they were annealed. The dashed lines in the plot correspond to the fits calculated for the data from the spectra of the pure species, which are plotted with a polynomial spline fit for clarity. The superscripts denote thermal processing as follows: (a) annealed to 100 K, (b) annealed to 175 K. | ||
The data from the CS deposition were also fit with the spectral components from pure water ices as discussed above and the fits are plotted along with the data in Fig. 9. In this case, the fits can reproduce qualitatively and quantitatively all the observed features at the selected temperatures, thus confirming that the CO2 is not affecting the structure of the crystalline H2O ice. The 6.8 THz feature is, however, stronger in these CS data than in the fits. This difference is most likely because of the different experimental procedures used to prepared crystalline water ice.
There are, however, differences between the spectra of the CS deposition, seen in the top two spectra of Fig. 9, and the spectra of the AS deposition seen in the middle five spectra in Fig. 9. The CS deposition has a THz spectrum that appears qualitatively identical to the annealed pure ices in Fig. 7, while this is not the case for the AS experiments. The crystalline H2O features present in the AS spectra after annealing to 175 K are not as sharp or strong as the features present in both the pure annealed H2O ice or the ice in the CS deposition. From our quantitative fits of the data, we observe that up to 60 K, the spectra can be fit with just an ASW component, while above 60 K, a crystalline H2O at 150 K component must also be included to accurately fit the data. The percentage of the crystalline H2O component of the fit increases with increasing temperature as shown in Table 3, however since the total percentage does not approach 100%, the structure of this ice, even after annealing, is somehow different from that of a pure crystalline H2O ice. Also, CO2 is still present in the ice at 100 K indicating that some CO2 diffused into the water layers underneath.
As seen in Fig. 9 (right panel), both CO2 and H2O are present in the ice, and by looking at the shape of the CO2 bending mode peaks, we can tell that the ice layers remain mostly separate. The bending mode features show the three sharp peaks seen in pure CO2 as opposed to the one broadened feature seen in the well mixed ices discussed above. One would expect, then, to simply see THz features corresponding to amorphous H2O; however, the THz spectrum is quite flat and nearly featureless. There does appear to be a subtle bump corresponding to the 4–7.5 THz amorphous H2O ice absorption, but it is not especially intense. The CO2 molecules clearly disrupt the THz active motions in what should be an amorphous H2O even though amorphous H2O is clearly present from the FTIR data.
The layered ice was then warmed to 60 K, and the THz features corresponding to ASW become more pronounced. While there is little change in the spectrum collected with the FTIR, it becomes possible to tell from the THz data that there is ASW present in the sample. The ice was then annealed to 100 K and cooled back to 60 K. The spectrum after annealing now has a slightly different shape, but is qualitatively similar to the previous spectrum taken at 60 K. While the mid-IR spectra do show some differences in this case, the differences are not as pronounced as in the THz region.
These spectra can be fit just with an ASW at 10 K component, except for the last spectrum annealed to 100 K, where a crystalline H2O component at 150 K was included to improve the fit. In this case, we note that the proton-disordered vibrational band at 4.5 THz is present in the data and cannot be fit with the components hereby selected because the 6.8 THz band is completely suppressed by the presence of the CO2 diffusing into the water top layers. A wider sample of spectral fitting components and a higher spectra resolution may explain the band at 4.5 THz.
When comparing the AS and CS experiments, the THz spectra provide more information about the structure of the ice when used in conjunction with the mid-IR data. The mid-IR spectra in Fig. 9 show that the water in the AS experiments crystallizes after annealing to 175 K and has significant similarities with the CS spectrum taken at 60 K. However, the THz spectra of these two ices are quite different. As such, the THz spectra provide more information regarding the structure of the ices than just the mid-IR spectra do alone.
The AS deposition shows results that are different from the results obtained in the CS experiments, but that are similar to the results obtained from the different CD experiments reported above. These spectra are qualitatively similar to those in the CD experiment that was annealed directly after deposition, seen in the top half of Fig. 8. Both the spectra in Fig. 8 and 9 and show the 6.8, 5.7, and 4.7 THz features of crystalline water ice that are attenuated and distorted relative to these features in pure crystalline H2O ice, and this can be seen in our fits. Clearly, the CO2 was able to diffuse into the structure of the ASW layer underneath and was trapped in the pores or channels of the ASW.37 The presence of these CO2 domains within the ASW prevented the crystallization process from proceeding as it would in the pure H2O ice. We can thus infer that, while there are large enough domains of water to crystallize, the presence of CO2 prevents these domains from joining to form a well ordered macroscopic crystal. Instead, crystallization is apparent in both the mid-IR and THz spectra, but the THz features are sensitive to the degree of crystallization of the ice and indicate that the ice differs in structure from that of a pure H2O sample.
The deposition in the IS experiment again points to the importance of THz spectroscopy in the study of the molecular structure of the ice. The fact that the feature corresponding to ASW is missing from these spectra is unsurprising from the results present in the literature. Maté et al. suggest that as H2O deposits, CO2 molecules are immediately incorporated into the structure of the ASW formed.37 This disruption of the structure of ASW would explain the flatness of the spectrum at 10 K. Once the sample is heated to 60 K, segregation begins to occur, and as Fig. 9 clearly shows, CO2 is still present in the ice after annealing to 100 K. Consequently, a broad feature corresponding to that of ASW appears in both of the spectra taken at 60 K (before and after annealing to 100 K). Since the mid-IR spectra sense only the local environment in which the molecules interact, the THz spectra provide the opportunity to draw stronger conclusions.
4 Conclusions
We present a new setup consisting of two spectrometers capable of studying astrochemical ice analogs from the THz, or far-IR, region (0.3–7.5 THz; 10–250 cm−1) up through the mid-IR (400–4000 cm−1) with a THz spectral resolution of 3 cm−1 and a mid-IR spectral resolution of 1 cm−1. This combination of spectral regions is powerful because we are sensitive to intramolecular modes in the mid-IR and intermolecular modes in the THz. Also, we can compare our mid-IR spectrum to the large body of literature already available in this spectral region. In addition, the new setup described in this work is capable of making a significant contribution to the body of experimental laboratory data that is needed to interpret astronomical observations from Herschel, SOFIA, and ALMA. Below, we enumerate our conclusions:1. Our THz spectra of pure H2O ice agree well with previous literature studies, and we tentatively report a new feature in both crystalline H2O and amorphous solid water at 1 THz. This feature may be especially useful for astronomical observations given the current coverage of observational facilities.
2. For experiments where we co-deposited H2O and CO2, we observed different structural changes within the H2O ice based on the rate of segregation. From the differences in the THz spectra, we observe that CO2 can segregate to disrupt the hydrogen bonding between bi-layers of crystalline H2O ice and thus infer the location of the segregated CO2.
3. In the experiments of sequentially deposited ices, we distinctly observed the impact of CO2 molecules on the structure of the H2O ice. Although we were unable to observe any features corresponding to pure CO2 ice, its effect on the THz spectrum was clear – CO2 disrupted the interactions between H2O molecules so that the resonances of H2O ice were attenuated, distorted, and/or shifted.
4. Given the differences in the THz spectra of the CO2 and H2O containing ices, THz spectroscopy has the potential to indicate the degree of crystallinity of an ice and the history of thermal transformations within the ice, provided that the history consists of non-equilibrium transitions away from a point of unstable equilibrium.
5. Our work demonstrates the importance of THz spectroscopy in the study of astrochemical ices because features in the THz region are sensitive to long range structural change. In addition, the temperature dependence of the shape of a feature in the THz spectrum of a molecule may provide a useful tool for determining the local temperature of an ice detected in space.
Acknowledgements
The authors thank Daniel B. Holland and P. Brandon Carroll for helpful discussions as well as the referees whose comments helped improve the manuscript. This work was supported by the NSF CRIF:ID and CSDM programs and the NASA Exobiology and Laboratory Astrophysics programs. M.A.A thanks Katie Kirby for proofreading help, and was supported by the Department of Defense (DoD) Air Force Office of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship, 32 CFR 168a. S.I. acknowledges support from a Niels Stensen Fellowship and a Marie Curie Fellowship (FP7-PEOPLE-2011-IOF-300957). B.A.M. gratefully acknowledges funding by an NSF Graduate Research Fellowship.References
- R. Gould and E. Salpeter, Astrophys. J., 1963, 138, 393–407 CAS.
- R. Gould, E. Salpeter and T. Gold, Astrophys. J., 1963, 138, 408 CAS.
- S. Cazaux and A. G. G. M. Tielens, Astrophys. J., 2004, 604, 222 CAS.
- H. B. Perets, O. Biham, G. Manic, V. Pirronello, J. Roser, S. Swords and G. Vidali, Astrophys. J., 2005, 627, 850 CAS.
- L. Allamandola, M. Bernstein, S. Sandford and R. Walker, Space Sci. Rev., 1999, 90, 219–232 CrossRef CAS.
- E. F. van Dishoeck, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12249–12256 CrossRef CAS PubMed.
- E. Herbst and E. F. van Dishoeck, Annu. Rev. Astron. Astrophys., 2009, 47, 427–480 CrossRef CAS PubMed.
- L. Allamandola, S. Sanford and G. Valero, Icarus, 1988, 76, 225–252 CrossRef CAS.
- M. Moore and R. Hudson, Icarus, 2000, 145, 282–288 CrossRef CAS.
- R. Hudson and M. Moore, Icarus, 2000, 145, 661–663 CrossRef CAS.
- G. Strazzulla, G. Baratta and M. Palumbo, Spectrochim. Acta, Part A, 2001, 57, 825–842 CrossRef CAS.
- S. Ioppolo, M. E. Palumbo, G. A. Baratta and V. Mennella, Astron. Astrophys., 2009, 493, 1017–1028 CrossRef CAS.
- U. Raut, D. Fulvio, M. J. Loeffler and R. A. Baragiola, Astrophys. J., 2012, 752, 159 Search PubMed.
- J. A. Noble, F. Dulieu, E. Congiu and H. J. Fraser, Astrophys. J., 2011, 735, 121 Search PubMed.
- P. Theule, F. Duvernay, A. Ilmane, T. Hasegawa, O. Morata, S. Coussan, G. Danger and T. Chiavassa, Astron. Astrophys., 2011, 530, A96 CrossRef PubMed.
- J. A. Noble, P. Theule, F. Borget, G. Danger, M. Chomat, F. Duvernay, F. Mispelaer and T. Chiavassa, Mon. Not. R. Astron. Soc., 2013, 428, 3262–3273 CrossRef CAS PubMed.
- N. Watanabe and A. Kouchi, Astrophys. J., Lett., 2002, 571, L173 CrossRef CAS.
- G. W. Fuchs, H. M. Cuppen, S. Ioppolo, C. Romanzin, S. E. Bisschop, S. Andersson, E. F. van Dishoeck and H. Linnartz, Astron. Astrophys., 2009, 505, 629–639 CrossRef CAS PubMed.
- S. Ioppolo, H. M. Cuppen, C. Romanzin, E. F. van Dishoeck and H. Linnartz, Astrophys. J., 2008, 686, 1474 CAS.
- N. Miyauchi, H. Hidaka, T. Chigai, A. Nagaoka, N. Watanabe and A. Kouchi, Chem. Phys. Lett., 2008, 456, 27–30 CrossRef CAS PubMed.
- E. Matar, E. Congiu, F. Dulieu, A. Momeni and J. L. Lemaire, Astron. Astrophys., 2008, 492, L17–L20 CrossRef CAS.
- U. Raut and R. A. Baragiola, Astrophys. J., Lett., 2011, 737, L14 CrossRef.
- J. Kalvāns and I. Shmeld, Astron. Astrophys., 2010, 521, A37 CrossRef PubMed.
- Q. Chang and E. Herbst, Astrophys. J., 2012, 759, 147 Search PubMed.
- R. L. Pulliam, B. A. McGuire and A. J. Remijan, Astrophys. J., 2012, 751, 1 Search PubMed.
- J. E. Elsila, D. P. Glavin and J. P. Dworkin, Meteorit. Planet. Sci., 2009, 44, 1323–1330 CrossRef CAS.
- R. T. Garrod, Astrophys. J., 2013, 765, 60 Search PubMed.
- W. Grundy, L. Young and E. Young, Icarus, 2003, 162, 222–229 CrossRef CAS.
- B. Buratti, D. Cruikshank, R. Brown, R. Clark, J. Bauer, R. Jaumann, T. McCord, D. Simonelli, C. Hibbitts, G. Hansen, T. Owen, K. Baines, G. Bellucci, J. Bibring, F. Capaccioni, P. Cerroni, A. Coradini, P. Drossart, V. Formisano, Y. Langevin, D. Matson, V. Mennella, R. Nelson, P. Nicholson, B. Sicardy, C. Sotin, T. Roush, K. Soderlund and A. Muradyan, Astrophys. J., 2005, 622, L149–L152 CAS.
- R. T. Garrod, S. L. W. Weaver and E. Herbst, Astrophys. J., 2008, 682, 283–302 CAS.
- J. R. Brucato, G. A. Baratta and G. Strazzulla, Astron. Astrophys., 2006, 455, 395–399 CrossRef CAS.
- S. E. Bisschop, G. W. Fuchs, A. C. A. Boogert, E. F. van Dishoeck and H. Linnartz, Astron. Astrophys., 2007, 470, 749–759 CrossRef CAS.
- K. I. Öberg, H. J. Fraser, A. C. A. Boogert, S. E. Bisschop, G. W. Fuchs, E. F. van Dishoeck and H. Linnartz, Astron. Astrophys., 2007, 462, 1187–1198 CrossRef.
- K. I. Öberg, A. C. A. Boogert, K. M. Pontoppidan, S. van den Broek, E. F. van Dishoeck, S. Bottinelli, G. A. Blake and N. J. Evans, Astrophys. J., 2011, 740, 109 Search PubMed.
- M. Bernstein, D. Cruikshank and S. Sandford, Icarus, 2005, 179, 527–534 CrossRef CAS PubMed.
- O. Gálvez, I. K. Ortega, B. Maté, M. A. Moreno, B. Martn-Llorente, V. J. Herrero, R. Escribano and P. J. Gutiérrez, Astron. Astrophys., 2007, 472, 691–698 CrossRef.
- B. Maté, O. Galvez, B. Martin-Llorente, M. A. Moreno, V. J. Herrero, R. Escribano and E. Artacho, J. Phys. Chem. A, 2008, 112, 457–465 CrossRef PubMed.
- O. Galvez, B. Maté, V. J. Herrero and R. Escribano, Icarus, 2008, 197, 599–605 CrossRef CAS PubMed.
- K. I. Öberg, E. C. Fayolle, H. M. Cuppen, E. F. van Dishoeck and H. Linnartz, Astron. Astrophys., 2009, 505, 183–194 CrossRef.
- G. Profeta and S. Scandolo, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 024103 CrossRef.
- J. E. Bertie, Appl. Spectrosc., 1968, 22, 634–640 CrossRef CAS.
- J. E. Bertie and S. M. Jacobs, J. Chem. Phys., 1977, 67, 2445–2448 CrossRef CAS PubMed.
- M. Moore and R. Hudson, Astron. Astrophys., Suppl. Ser., 1994, 103, 45–56 CAS.
- M. Moore and R. Hudson, Astrophys. J., 1992, 401, 353–360 CAS.
- M. Moore and R. Hudson, Radiat. Phys. Chem., 1995, 45, 779–789 CrossRef.
- N. Plattner, M. W. Lee and M. Meuwly, Faraday Discuss., 2010, 147, 217–230 RSC.
- M. W. Lee, N. Plattner and M. Meuwly, Phys. Chem. Chem. Phys., 2012, 14, 15464–15474 RSC.
- J. D. Green, N. J. Evans II, J. K. Jorgensen, G. J. Herczeg, L. E. Kristensen, J.-E. Lee, O. Dionatos, U. A. Yildiz, C. Salyk, G. Meeus, J. Bouwman, R. Visser, E. A. Bergin, E. F. van Dishoeck, M. R. Rascati, A. Karska, T. A. van Kempen, M. M. Dunham, J. E. Lindberg, D. Fedele and the DIGIT Team, Astrophys. J., 2013, 770, 123 Search PubMed.
- R. Simon, N. Schneider, J. Stutzki, R. Guesten, U. U. Graf, P. Hartogh, X. Guan, J. G. Staguhn and D. J. Benford, Astron. Astrophys., 2012, 542, L12 CrossRef PubMed.
- C. Qi, K. I. Öberg, D. J. Wilner, P. DAlessio, E. Bergin, S. M. Andrews, G. A. Blake, M. R. Hogerheijde and E. F. van Dishoeck, Science, 2013, 341, 630–632 CrossRef CAS PubMed.
- D. Cook and R. Hochstrasser, Opt. Lett., 2000, 25, 1210–1212 CrossRef CAS.
- X. Xie, J. Dai and X.-C. Zhang, Phys. Rev. Lett., 2006, 96, 075005 CrossRef.
- K.-Y. Kim, J. H. Glownia, A. J. Taylor and G. Rodriguez, Opt. Express, 2007, 15, 4577–4584 CrossRef CAS.
- K. Y. Kim, A. J. Taylor, J. H. Glownia and G. Rodriguez, Nat. Photonics, 2008, 2, 605–609 CrossRef CAS.
- D. Auston, A. Johnson, P. Smith and J. Bean, Appl. Phys. Lett., 1980, 37, 371–373 CrossRef CAS PubMed.
- Q. Wu and X. Zhang, Appl. Phys. Lett., 1995, 67, 3523–3525 CrossRef CAS PubMed.
- P. A. Gerakines, W. A. Schutte, J. M. Greenberg and E. F. van Dishoeck, Astron. Astrophys., 1995, 296, 810 CAS.
- M. S. Westley, G. A. Baratta and R. A. Baragiola, J. Chem. Phys., 1998, 108, 3321–3326 CrossRef CAS PubMed.
- M. Satorre, M. Domingo, C. Milln, R. Luna, R. Vilaplana and C. Santonja, Planet. Space Sci., 2008, 56, 1748–1752 CrossRef CAS PubMed.
- L. J. Karssenmeijer, S. Ioppolo, M. C. van Hermert, M. A. Allodi, G. A. Blake and H. M. Cuppen, Astrophys. J. Search PubMed , in press.
- J. B. Bossa, K. Isokoski, M. S. de Valois and H. Linnartz, Astron. Astrophys., 2012, 545, A82 CrossRef PubMed.
- S. Krishnamurthy, R. Bansil and J. Wiafe-Akenten, J. Chem. Phys., 1983, 79, 5863–5870 CrossRef CAS PubMed.
- J. E. Bertie and E. Whalley, J. Chem. Phys., 1967, 46, 1271–1284 CrossRef CAS PubMed.
- D. D. Klug, J. S. Tse and E. Whalley, J. Chem. Phys., 1991, 95, 7011–7012 CrossRef CAS PubMed.
- D. M. Hudgins, S. A. Sandford, L. J. Allamandola and A. G. G. M. Tielens, Astrophys. J., Suppl. Ser., 1993, 86, 713–870 CrossRef CAS.
- P. Jenniskens and D. F. Blake, Astrophys. J., 1996, 473, 1104 CAS.
- S. Ioppolo, G. Fedoseev, T. Lamberts, C. Romanzin and H. Linnartz, Rev. Sci. Instrum., 2013, 84, 073112 CrossRef CAS PubMed.
- E. C. Fayolle, K. I. Öberg, H. M. Cuppen, R. Visser and H. Linnartz, Astron. Astrophys., 2011, 529, A74 CrossRef PubMed.
Footnotes |
| † Present Address: Technology and Applications Center, Newport Corporation, 1791 Deere Avenue, Irvine, CA 92606, USA. |
| ‡ To estimate the ice thickness, we first obtained the column densities for H2O and CO2 from the integrated intensity of their IR bands using a modified Lambert–Beer equation. We selected the O–H bending mode for H2O (1660 cm−1) and the O–C–O bending mode for CO2 (660 cm−1), and used the relevant absorption band strength as reported in Gerakines et al. 1995.57 The ice thickness is subsequently estimated by dividing the column density of each species by a conversion factor that is proportional to the density of the species in the ice. The density of H2O used is 0.9 g cm−3,58 and the density of CO2 used is 1.5 g cm−3.59 |
| This journal is © the Owner Societies 2014 |