 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of co-adsorbed oxygen on crotonaldehyde adsorption over gold nanoclusters: a computational study†
Constantinos D.
Zeinalipour-Yazdi
ab,
David J.
Willock
*b,
Andreia
Machado
b,
Karen
Wilson
c and
Adam F.
Lee
*ad
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: A.F.Lee@warwick.ac.uk
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK. E-mail: willockjd@cardiff.ac.uk
cEuropean Bioenergy Research Institute, Aston University, Birmingham B4 7ET, UK
dSchool of Chemistry, Monash University, Victoria 3800, Australia
First published on 25th November 2013
Abstract
Crotonaldehyde (2-butenal) adsorption over gold sub-nanometer particles, and the influence of co-adsorbed oxygen, has been systematically investigated by computational methods. Using density functional theory, the adsorption energetics of crotonaldehyde on bare and oxidised gold clusters (Au13, d = 0.8 nm) were determined as a function of oxygen coverage and coordination geometry. At low oxygen coverage, sites are available for which crotonaldehyde adsorption is enhanced relative to bare Au clusters by 10 kJ mol−1. At higher oxygen coverage, crotonaldehyde is forced to adsorb in close proximity to oxygen weakening adsorption by up to 60 kJ mol−1 relative to bare Au. Bonding geometries, density of states plots and Bader analysis, are used to elucidate crotonaldehyde bonding to gold nanoparticles in terms of partial electron transfer from Au to crotonaldehyde, and note that donation to gold from crotonaldehyde also becomes significant following metal oxidation. At high oxygen coverage we find that all molecular adsorption sites have a neighbouring, destabilising, oxygen adatom so that despite enhanced donation, crotonaldehyde adsorption is always weakened by steric interactions. For a larger cluster (Au38, d = 1.1 nm) crotonaldehyde adsorption is destabilized in this way even at a low oxygen coverage. These findings provide a quantitative framework to underpin the experimentally observed influence of oxygen on the selective oxidation of crotyl alcohol to crotonaldehyde over gold and gold–palladium alloys.
I. Introduction
Platinum group metal (PGMs) and gold nanoparticles (NPs) are promising heterogeneous catalysts for the selective aerobic oxidation (selox) of alcohols to aldehydes, providing routes that obviate the use of environmentally harmful inorganic oxidants (e.g. CrVI salts1 or permanganates2) and their associated toxic waste,3 handling of hazardous peroxides, or the recovery of expensive organometallic soluble catalysts. Recent research has highlighted Au,4 Pd5,6 and bimetallic nanoparticles thereof7–10 as atom-efficient selox catalysts, able to operate under mild conditions (e.g. reaction temperatures between 60–160 °C and employing ambient air as an oxidant) in non-halogenated solvents, aqueous solutions11 or even solventless.7 The active site in oxide supported Pd NPs for crotyl alcohol selox has been extensively investigated by in situ and operando X-ray spectroscopies.12–15 These reveal that oxygen is critical in weakening the adsorption of the reactively-formed crotonaldehyde product thereby reducing decarbonylation pathways. We have made similar observations over Pd(111)16 and Au/Pd(111)9,17 model single-crystal catalysts, wherein in situ XPS and TPRS measurements show that pre-adsorbed oxygen weakens crotonaldehyde adsorption over pure palladium and Au/Pd surface alloys, thus suppressing undesired decarbonylation pathways. This contrasts with epoxidation wherein oxygen promotes ethene adsorption over silver.18 Enhanced activity of Au/Pd core shell nanoparticles in crotyl alcohol selox has been rationalized by d-charge depletion observed via XANES measurements.10Chin et al. recently explored the influence of co-adsorbed oxygen on methane combustion over Pd and Pt NPs,19,20 while ethanol selective oxidation to ethanal was studied over Au NPs via DFT calculations by Boronat and Corma,21 who found that oxygen lowered the activation barriers for O–H and C–H scission. Delbecq and Sautet also employed DFT in combination with HREELS22 vibrational measurements to study crotonaldehyde adsorption over Pt(111)23 and Pt2Sn/Pt(111)24 alloys, and thereby understand the influence of Sn additives on catalyst selectivity towards the hydrogenation of crotonaldehyde and other α-β-unsaturated aldehydes. DFT has also been applied in conjunction with metastable de-excitation spectroscopy to elucidate the molecular adsorption modes and reaction pathways during crotyl alcohol selox over bare Pd(111).25
In this contribution, the effect of co-adsorbed oxygen on the adsorption of crotonaldehyde is explored for the first time via DFT calculations. In particular, we provide fundamental insight into the impact of co-adsorbed oxygen upon crotonaldehyde adsorption over Au NPs, and discuss the resulting implications for crotyl alcohol selox. We present a systematic computational study of crotonaldehyde adsorption over gold NPs (diameter, d < 1 nm) as a function of co-adsorbed oxygen surface coverage and proximity. The configurational space for adsorbed crotonaldehyde on bare Au13 and Au38 NPs and their oxidised analogues (Au13On, n = 2, 4, 6 and 8 and Au38O2) at effective oxygen coverages between 0–1 monolayers has been explored, and associated adsorption energies calculated. For low oxygen coverage on Au13 particles we find that crotonaldehyde adsorption can be enhanced relative to the bare Au cluster. However, strong destabilisation of crotonaldehyde by co-adsorbed oxygen is found at full oxygen coverage because adsorbed oxygen adatoms occupy positions close to the molecular adsorption site. The concepts discussed are likely extendable to other PGM NPs, and suggest that controlling oxygen surface coverage in situ may aid process optimization during the catalytic selox of allylic alcohols.
II. Methods
Γ point26 DFT calculations were performed with the VASP 5.2 code.27,28 Exchange and correlation effects are considered within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation (XC) functional,29 with the projector augmented-wave (PAW) method30,31 used to represent core states, 1s for C and O, and 1s to 4f for Au. The cut-off energy for the plane-wave basis was set to 400 eV. The nanoparticle/adsorbate models were centered within a 25 × 25 × 25 Å periodic box to ensure a vacuum gap of 15–20 Å around the clusters, with most calculated results obtained at the latter gap. Geometry optimizations of all the atoms in the cluster–adsorbate system were performed with a residual force threshold of 0.015 eV Å−1 using the conjugate-gradient algorithm. The convergence criterion for electronic relaxation was 10−4 eV. The initial charge density was obtained by superposition of atomic charges, and the projection operators were evaluated in reciprocal space. Spin polarization has been tested in several models but found to be unimportant, with calculated adsorption energies agreeing to within 0.5 kJ mol−1 for calculations with and without spin polarisation.Molecular gas phase optimizations were also performed with the Gaussian09 code (rev C.01)32 to allow comparison of results with the Becke's three-parameter, hybrid exchange functional33 combined with the Lee–Yang–Parr non-local correlation functional,34 abbreviated as B3LYP, using the aug-cc-pVTZ35 basis set. Linear dependencies of the basis functions were removed by using the spherical version (5d, 7f) of the aug-cc-pVTZ basis set.
Adsorption energies (Eads) were calculated by subtracting the total energy of the fully-relaxed, isolated adsorbate and nanoparticle from the total energy of the adsorbate-nanoparticle system.
The degree of adsorption-induced charge transfer was estimated through the atomic charges calculated by Bader's atoms-in-molecules method using the numerical grid based approach developed by Henkelman and co-workers.36,37 These authors have noted that the convergence of Bader charges requires the charge density to be produced on a fine grid so that the spacing between grid points is of the order of 0.02 Å.37 VASP works with two grids to represent the charge density, and outputs the finer grid for analysis. The memory requirement for such calculations on the 25 Å cube used to isolate the clusters within the periodic boundary conditions of the simulation was prohibitive on our resources. Accordingly, charge analysis was carried out for each system within a 16 Å cubic simulation cell using the structures optimized within the larger cell. This allowed us to produce a grid with 700 points on a side corresponding to a grid spacing of 0.023 Å. As a check on the numerical accuracy of the analysis we carried out additional calculations for ethene adsorbed in a di-σ fashion to the Au13 cluster. In this case Bader analysis gave C atom charges of −0.1664 |e| and −0.1640 |e|. Since the carbon atoms for ethene should be equivalent by symmetry we infer a numerical accuracy of 0.005 |e|. Accordingly, Bader charges are quoted to two decimal places throughout the manuscript. These ethene calculations help inform on C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond interactions with the various clusters studied, and provide an additional reference point for discussing the crotonaldehyde results.
C double bond interactions with the various clusters studied, and provide an additional reference point for discussing the crotonaldehyde results.
III. Results and discussion
The rotational isomers of free crotonaldehyde were first optimized to determine the lowest energy gas phase conformers. We found four local minima on the potential energy surface, denoted as E-(s)-trans, E-(s)-cis, Z-(s)-trans and Z-(s)-cis as depicted in Fig. 1a–d. The heavy atom molecular framework of all rotamers was found to be planar, indicative of π-conjugation of the CC and CO moieties. As expected for rotational isomers, there is a negligible change of the corresponding bond lengths between the various isomers. The ordering of the relative energies for the four rotamers was found to be E-(s)-trans (−16.0 kJ mol−1) < E-(s)-cis (−6.6 kJ mol−1) < Z-(s)-trans (−4.2 kJ mol−1) < Z-(s)-cis (0.0 kJ mol−1). Of the four rotational isomers identified, E-(s)-trans-crotonaldehyde was the most stable in the gas phase, in agreement with microwave spectroscopy experiments38 on trans-crotonaldehyde. It is noteworthy that this energy trend is found for both plane-wave and atom-centered basis (i.e. B3LYP/aug-cc-pVTZ) methodologies. Subsequently we have used E-(s)-trans to evaluate the effect of co-adsorbed oxygen on the adsorption strength of the desirable product in the selective oxidation of crotyl alcohol, although the general conclusions that follow have also been validated for the second lowest energy conformer (E-(s)-cis).
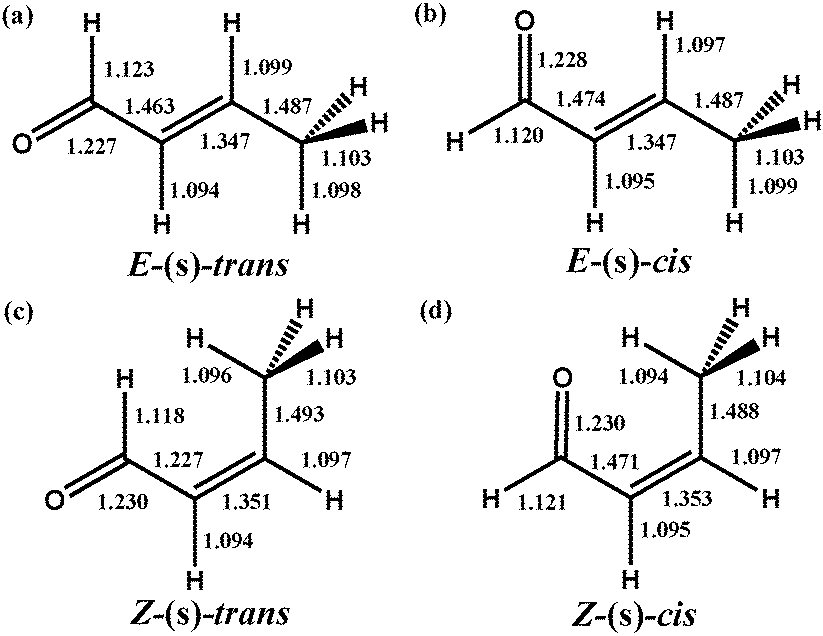 | ||
| Fig. 1 Structure and nomenclature of rotational isomers of crotonaldehyde obtained at Ecut = 400 eV in a 25 × 25 × 25 Å periodic cubic cell. All bond lengths given in Å. | ||
For the gold nanoparticle two particle sizes have been used, Au13 and Au38. In each case the structures were taken from bulk gold (α = β = γ = 90°, a = b = c = 407.82 pm)39 and fully optimized to their nearest energetic minima. Both Au13 and Au38 NPs expose (100)-like and (111)-like facets which are also found on gold NPs of different geometries. Au38 has previously been described as an efficient catalyst for molecular oxygen dissociation with DFT calculation results being confirmed experimentally.40,41 After adsorption of molecular oxygen on a (100) facet a low barrier was calculated to produce O-adatoms in 3-fold hollow sites (h-O) on adjacent (111) sections of the particle. In Fig. 2 we consider the energetics of h-O adsorption on an Au13 cluster by plotting the relative energy of the oxidised cluster compared to Au13 and the corresponding number of gas phase O2 molecules as a function of oxygen coverage. Successive addition of oxygen adatoms thermodynamically stabilizes this system due to the formation of linear O–Au–O structures, which have been previously reported during O2 dissociation on gold clusters42 and employed as an indicator of gold oxide formation.43 In particular, we find the relative energy calculated for oxidised Au13 is linearly dependent on the oxygen coverage. This suggests that in an oxidising atmosphere the Au nanoparticles considered here will become surface oxidised.
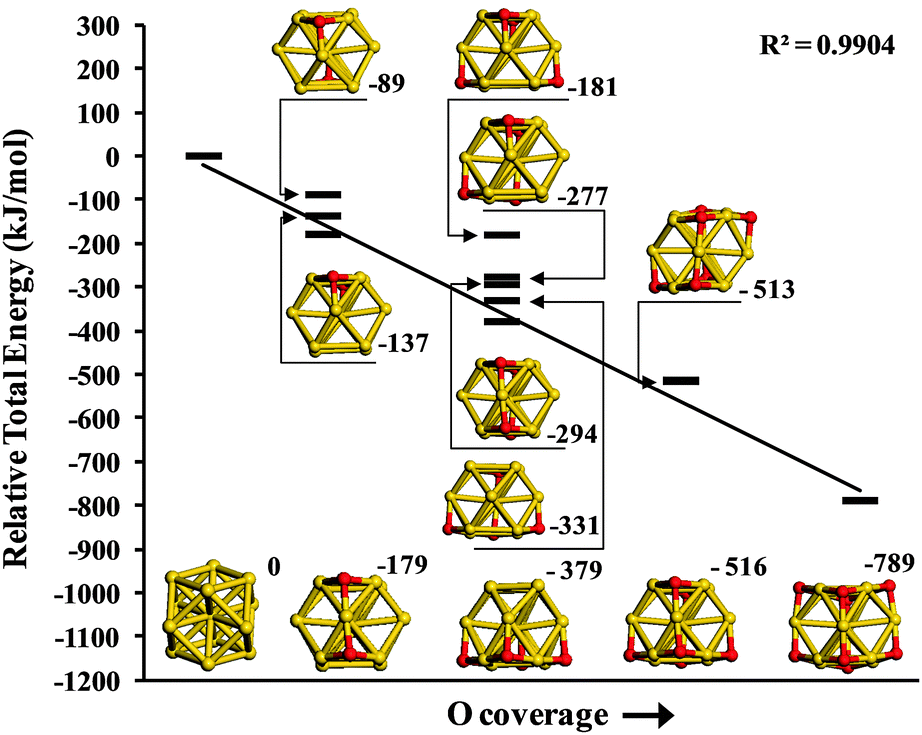 | ||
| Fig. 2 Relative total energy of Au13On, where n = 0, 2, 4, 6 and 8 for the various possible permutations of three-fold hollow oxygen. The least squares line is drawn for minimum energy structure at each oxygen coverage. | ||
We note that DFT in general suggests that small isolated clusters of bare gold (e.g. Au4,44 Au1245) preferentially adopt a planar structure, whereas larger Aun clusters (n > 13) favour 3D morphologies that introduce low-coordination edge and vertex sites alongside (100) facets. However, for surface supported nanoparticles, three-dimensional morphologies46,47 are observed even for small particles. In the present context we note that oxidation of nanoparticles leads to linear O–Au–O structures42 as Au atoms are oxidized,43 and that these will for thermodynamic reasons more easily be accommodated within three dimensional structures.48 Accordingly, our use of bulk like structures allows the comparison of crotonaldehyde adsorption as a function of cluster size and oxygen coverage using a consistent cluster geometry which is also in line with the shapes of the supported Au nano-particles used in selox experimentally.
In the subsequent section we explore the various adsorbed configurations of E-(s)-trans-crotonaldehyde on nanometer scale Au particles and examine the effect of co-adsorbed oxygen and cluster size on the adsorption energetics. The four lowest energy bound structures for each cluster studied are given in Fig. 3. For the bare Au13 cluster (Fig. 3a) the most favourable adsorption geometry is πCC which is some 15 kJ mol−1 lower in energy than the atop and di-σCC modes. The di-σCC structures are akin to ethylene adsorption on neutral, anionic and cationic gold clusters.49 For E-(s)-trans crotonaldehyde adsorption to the clusters we distinguish two di-σCC geometries; in di-σCC-1 the aldehyde group is toward an Au–Au bridge site while for di-σCC-2 it is orientated away from the cluster. For Au13 and Au38 (Fig. 3c) these two alternatives are practically isoenergetic. However, for the oxidized clusters (Au13(h-O)8, Fig. 3b and Au38(h-O)2, Fig. 3d), locating the aldehyde over the bridge site places this group near an h-O atom, which is more destabilizing than locating the methyl group in the same position such that di-σCC-2 becomes notably lower in energy that di-σCC-1.
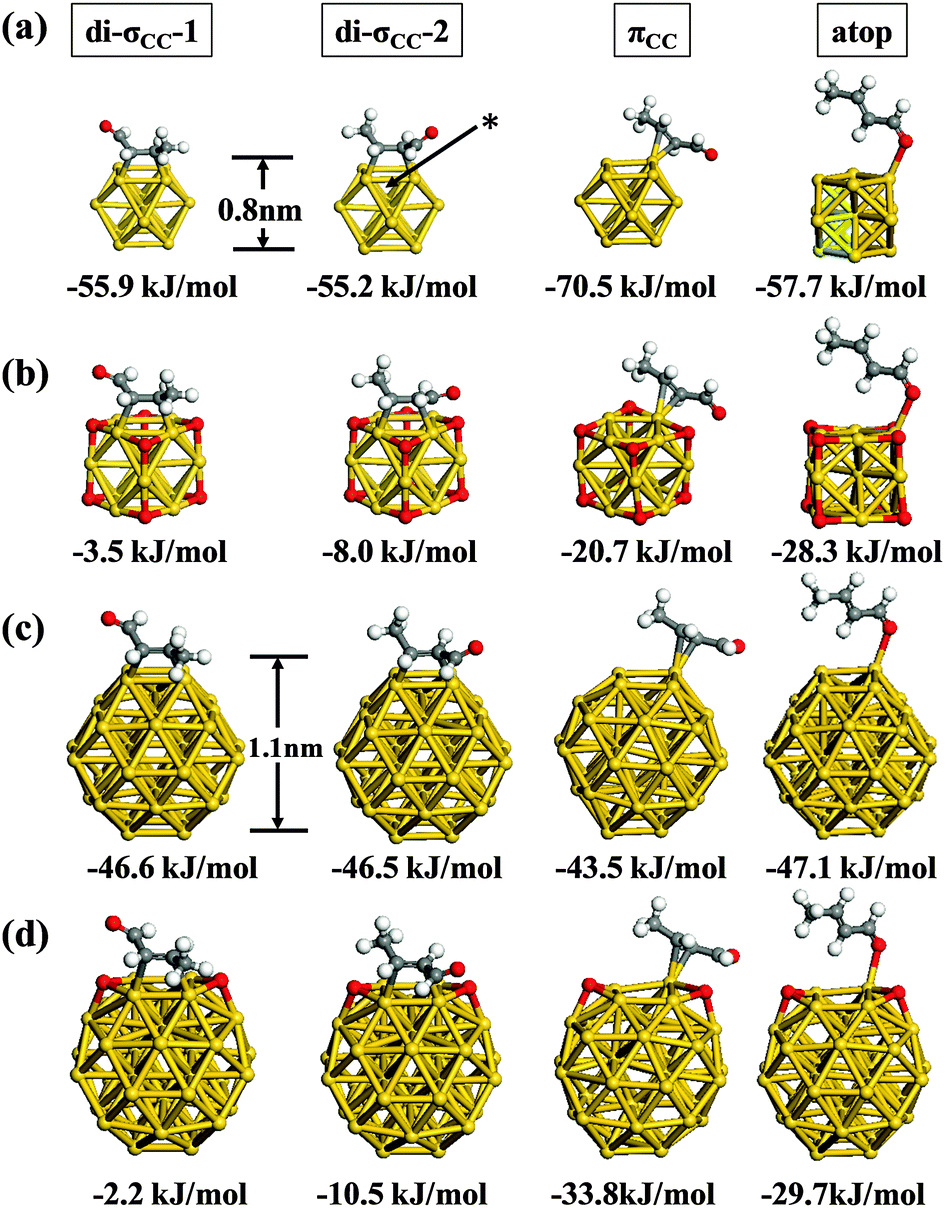 | ||
| Fig. 3 Adsorption energies of di-σCC, πCC and atop-bonded conformations of E-(s)-trans-crotonaldehyde on neutral (a) Au13, (b) Au13(h-O)8, (c) Au38 and (d) Au38(h-O)2. | ||
Delbecq and Sautet have provided a survey of adsorption configurations for acrolein (propenal) and crotonaldehyde on Pt(111),23,24 along with calculated adsorption energies using the PW91 functional. For the E-(s)-trans isomer they considered five adsorption modes. Two of these adsorb via the CC bond; di-σCC for which CC interacts with two surface Pt atoms and πCC which involves only a single Pt atom. Adsorption via the CO moiety gives a further two possibilities; di-σCO and atop with the molecule adsorbed in an end-on fashion through a Pt⋯OC interaction. Finally, a η4 configuration was examined involving both CC and CO bonding to separate Pt atoms. For crotonaldehyde this latter η4 adsorption geometry gave the lowest calculated adsorption energy (−84.5 kJ mol−1).
The adsorption configurations we have located are in general agreement with the earlier work on Pt. Interestingly, the η4 structure for adsorbed E-(s)-trans crotonaldehyde previously shown to have the potential to undergo decarbonylation over Pd(111)25 was not found on Au13O8. This implies that oxidised sub-nanometer Au NPs may afford higher crotonaldehyde selectivity rather than decarbonylation, during crotyl alcohol to crotonaldehyde selox. For the remaining configurations, we find that adsorption through the CC bond and CO moieties are possible on both Au13 and Au38 clusters. The NP structure gives rise to edge and corner sites where the facets meet and these are found to be preferred over adsorption on the facets themselves. For Au13 structures bound through the CC moiety generally adsorb more strongly than those via oxygen bound atop (Fig. 3a). However, in the presence of co-adsorbed oxygen we find that atop adsorption actually becomes more favourable (Fig. 3b). Side reactions that could potentially lead to attack at the allylic bond in the CC bound configurations di-σCC-1, di-σCC-2 and πCC are thus less probable for oxidized sub-nanometer Au NPs because atop adsorption through the aldehyde oxygen atom is energetically favoured. For Au38 this preference is less clear as the different adsorption modes shown in Fig. 3c fall within a narrow range (3.6 kJ mol−1). We have also examined the adsorption energy of di-σ and π-bound E-(s)-trans-crotonaldehyde on Au(111) (see ESI,† Fig. S1) and found that the adsorption energy is essentially zero (1.1 kJ mol−1) indicative that on extended gold surfaces crotonaldehyde can only weakly interact. Comparing the lowest adsorption energies for Pt(111),22,23 Au NPs and Au(111) yields the following trend for crotonaldehyde: Pt(111) (η4: −85 kJ mol−1) > Au13 (πCC: −71 kJ mol−1) > Au38 (atop, di-σCC-1 or di-σCC-2: −47 kJ mol−1) > Au38(h-O)2 (πCC: −34 kJ mol−1) > Au13(h-O)8 (atop: −28 kJ mol−1) > Au(111) (1.1 kJ mol−1).
It is remarkable that the smaller Au cluster gives an adsorption energy just 14 kJ mol−1 less negative than the Pt(111) value. In contrast, the Au38 value is 38 kJ mol−1 less favourable than Pt(111) whereas on Au(111) there is practically no interaction at the PBE-level, revealing a strong particle size dependence of the adsorption energy. This weakened adsorption would imply a pronounced particle size-sensitivity for allylic alcohol selox over gold NPs, with the desired crotonaldehyde product preferentially destabilized over larger particles or small NPs that are oxidised (i.e. Au13O8). It is also apparent from Fig. 3 that the energy differences between adsorption to the different sites on a particle fall into a much narrower range for Au38 (3.1 kJ mol−1) than for Au13 (15.3 kJ mol−1).
Co-adsorbed oxygen exerts an even greater influence on crotonaldehyde adsorption energetics than does particle size. Specifically, we observe a 94%, 86%, 71% and 51% decrease in the magnitude of adsorption energies for E-(s)-trans in the di-σCC-1, di-σCC-2, πCC and atop geometries respectively when Au13 NPs are pre-saturated with oxygen adatoms; this equates to an average decrease of 45 kJ mol−1 upon nanoparticle oxidation.
In later sections we will show that for Au13 the weakening of adsorption observed for the di-σCC geometries on the fully oxidised cluster, Au13(h-O)8, occurs due to the proximity of a h-O atom at the position closest to the bridge site where the molecule is placed (see asterisk in Fig. 3a). A similar arrangement was constructed for the partially oxidized Au38(h-O)2 cluster, however, attempts to locate an oxygen atom in the 3-fold hollow site neighboring the CC bond destabilized E-(s)-trans-crotonaldehyde to such an extent that no optimized adsorption geometry could be found. Interestingly we find that if we place an oxygen at the neighbouring 3-fold hollow and keep di-σ-bound crotonaldehyde at a fixed position, then oxygen diffusion to the adjacent (111)-like facet is observed. However, for Au38(h-O)2, the presence of oxygen adatoms at other adjacent sites also decreased the magnitude of crotonaldehyde adsorption by between 10–45 kJ mol−1 (compare Fig. 3c and d). Such findings provide evidence for a strong oxygen-induced destabilization of allylic aldehyde adsorption over gold nanoparticles. The origin of this dramatic destabilization, and its oxygen coverage dependence, reflects an intriguing and complex bonding mechanism that we explore in the following sections.
Boronat and Corma have shown that molecular O2 adsorption on the (100) facets of Au38 clusters can yield h-O adatoms on neighbouring (111) facets. We have used the idea of oxygen dissociation on (100) leading to h-O on opposing (111) facets to build-up the oxygen adatom population on an Au13 cluster and examined the effect upon E-(s)-trans crotonaldehyde adsorption in the di-σCC-2 and πCC configurations. The dependence of crotonaldehyde adsorption over Au13 on co-adsorbed oxygen coverage is shown in Fig. 4. In Fig. 4, each structure has an O atom placed in the hollow site closest to the adsorbate; this clearly has a dramatic effect on the calculated adsorption energy even at low coverage. Indeed for the di-σCC configuration, dissociation of a single oxygen molecule into such an arrangement leads to 53.1 kJ mol−1 fall in the crotonaldehyde adsorption energy; for intermediate coverages, progressive addition of further oxygen adatoms slightly enhances adsorption with calculated energies increasing from −2.1 (Au13(h-O)2) to −16.5 kJ mol−1 (Au13(h-O)6).
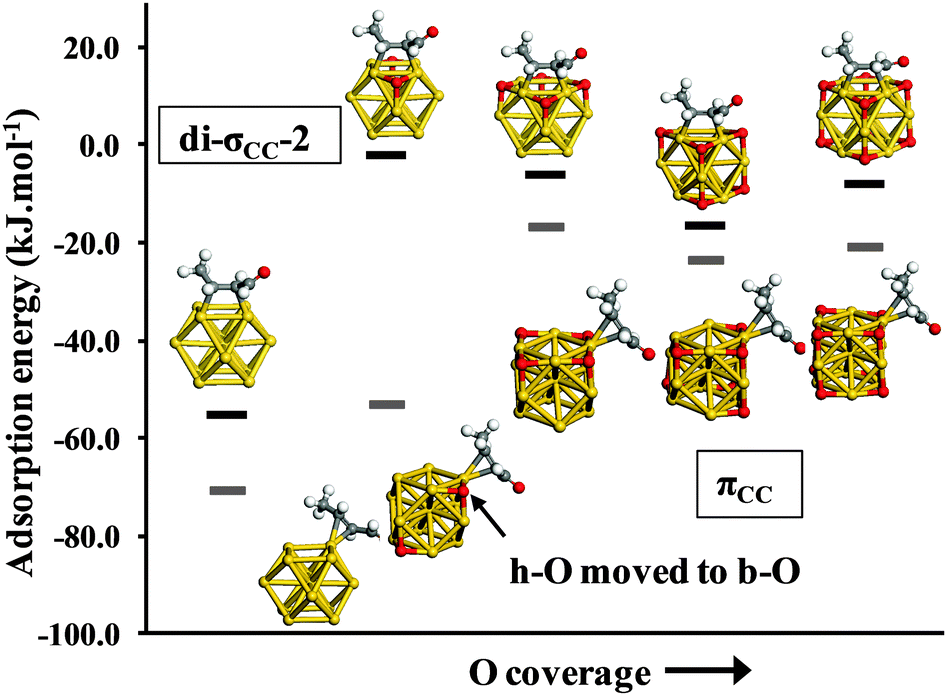 | ||
| Fig. 4 Adsorption energies of di-σCC-2 (black lines) and πCC (grey lines) bound E-(s)-trans-crotonaldehyde as a function of oxygen coverage on neutral Au13, with an O adatom positioned in the hollow site proximate to the molecule. | ||
At full coverage (Au13(h-O)8) adsorption weakens again, being destabilized by 47.2 kJ mol−1 with respect to the bare Au13 cluster. A very similar trend is also observed for the πCC configuration, where on average the adsorption energies are 11 kJ mol−1 stronger than for the di-σCC configuration (excluding Au13O2). For this crotonaldehyde adsorption geometry over Au13O2, the adjacent h-O shifts away from the aldehyde to a two-fold bridged oxygen (b-O) position, relieving the steric interaction.
Fig. 4 highlights the impact of h-O oxygen atoms located in the (111)-like adsorption site neighboring an adsorbed crotonaldehyde molecule. Such proximate adsorption to the CC bond dramatically reduces the strength of crotonaldehyde adsorption. However, there are clearly other sites available for all intermediate oxygen coverage levels, which are only lost at the point of hollow site saturation (Au13(h-O)8). Fig. 5 explores the effect of h-O oxygen atom placement on the adsorption of E-(s)-trans crotonaldehyde in the di-σCC-2 geometry over Au13(h-O)6. The results fall into two broad categories. When the proximal h-O site is occupied, crotonaldehyde adsorption is destabilized (as reported in Fig. 4). Fig. 5 reveals that the oxygen adatom arrangement in Fig. 4 actually represents one of the more strongly bound examples of such structures, with crotonaldehyde adsorption varying between +0.8 and −16.5 kJ mol−1. Locating an oxygen adatom in the (111)-like hollow site nearest to the adsorbed molecule weakens crotonaldehyde binding by an average of 59 kJ mol−1. By comparison, when the proximal h-O site is unoccupied, crotonaldehyde adsorption is enhanced by between 5.5 and 13.6 kJ mol−1 relative to the bare Au13 cluster. This finding has clear implications for practical catalysis, since occupancy of the proximal h-O site can only be guaranteed at saturation oxygen adatom surface coverages, with high binding energy sites for strong crotonaldehyde adsorption available on partially oxidized gold NPs. High oxygen partial pressures/dissolved concentrations should thus eliminate reaction pathways involving crotonaldehyde adsorption at strongly adsorbing sites which likely favour decarbonylation versus desired competing desorption pathways. Fully oxidized gold NP catalysts are anticipated to disfavour the adsorption of related allylic aldehydes with respect to their naked gold counterparts, and may also interact more weakly with other unsaturated organic molecules that typically coordinate through CC functions.
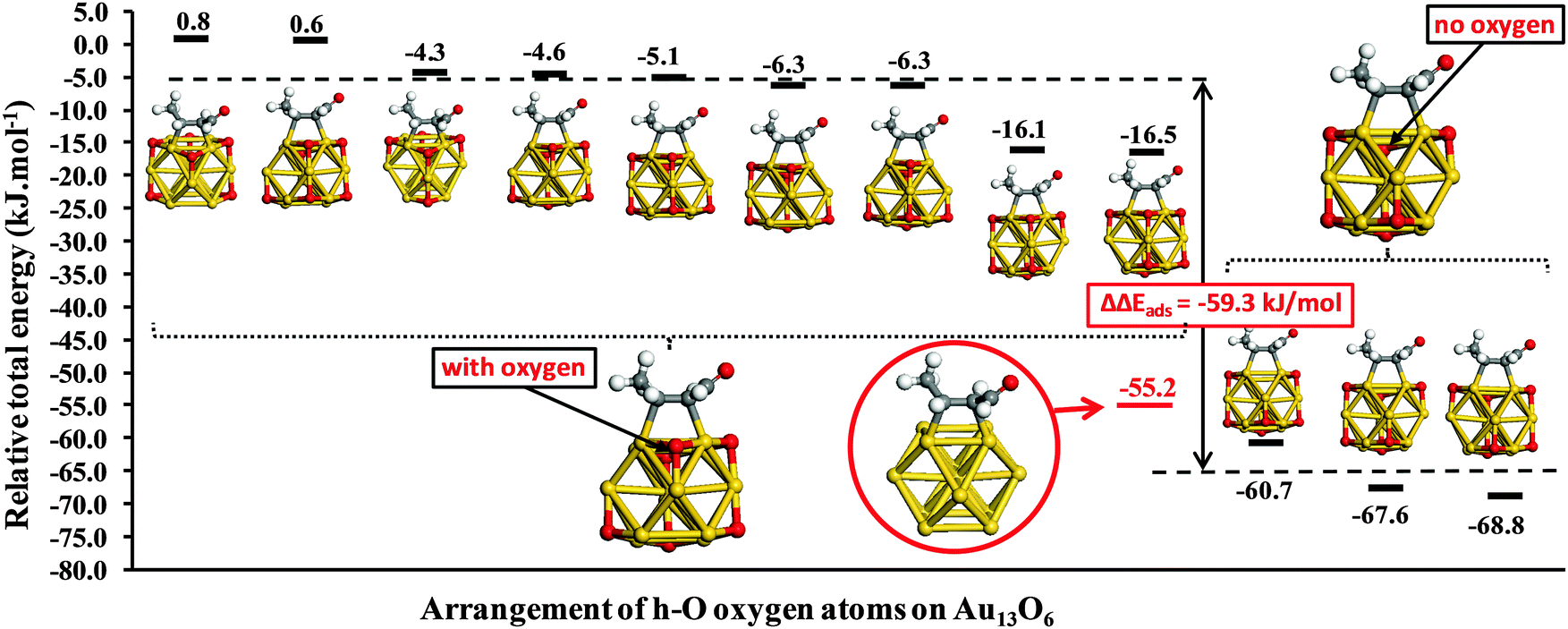 | ||
| Fig. 5 Adsorption energies of E-(s)-trans-crotonaldehyde as di-σCC-2 for different arrangements of h-O oxygen atoms on Au13(h-O)6. | ||
Of the two clusters explored in the present work, Au13 is unique in that each (111) like facets contains only a single three-fold hollow site. Larger clusters possess extended facets, so that even on Au38 each (111) face contains six hollow sites. Occupancy of all such (111) hollow sites by oxygen adatoms is unlikely since this would place h-O atoms close to one another. Boronat and Corma21 suggest that a Au38O16 cluster, in which only a third of the available h-O sites are occupied, represents a saturated oxide monolayer. In this latter scenario, crotonaldehyde adsorption could away from a neighboring h-O atom. It is noteworthy from Fig. 3 that such open adsorption sites on larger clusters would still be destabilized with respect to the bare cluster by 9.7 kJ mol−1 (πCC) and 44.4 kJ mol−1 (di-σCC-1). Hence, nanometre gold nanoparticles partially oxygen covered remain able to adsorb crotonaldehyde with moderate energies (−20 to −40 kJ mol−1) at sites that are not directly adjacent to oxygen adatoms.
Fig. 5 reveals that oxygen adsorption over for Au13 can either enhance or destabilize crotonaldehyde adsorption, and we now advance a bonding model to explain these observations. The di-σCC mode of bonding can be viewed in terms of donation from the CC π-density to empty metal orbitals, and back-donation from the filled metal d-states into the anti-bonding π*-orbitals of the allylic bond. These combined effects change the structures of CC double bonds as the carbon atoms alter from sp2 toward sp3 geometries. The structural analyses presented in Fig. 6 for crotonaldehyde over bare and oxidized Au13 NPs reveal qualitative trends in line with this model. We find generally shorter di-σCC bond lengths (rAu–C) for the more strongly adsorbed configurations (Fig. 6a) accompanied by a decreased dihedral angle φH–C2–C3–H for crotonaldehyde, indicative of sp2 to sp3 rehybridization of the two C atoms in the olefinic bond. For metals with a filled d-shell such as Au, we would also expect the interaction with the bare cluster to be dominated by metal to adsorbate back donation.
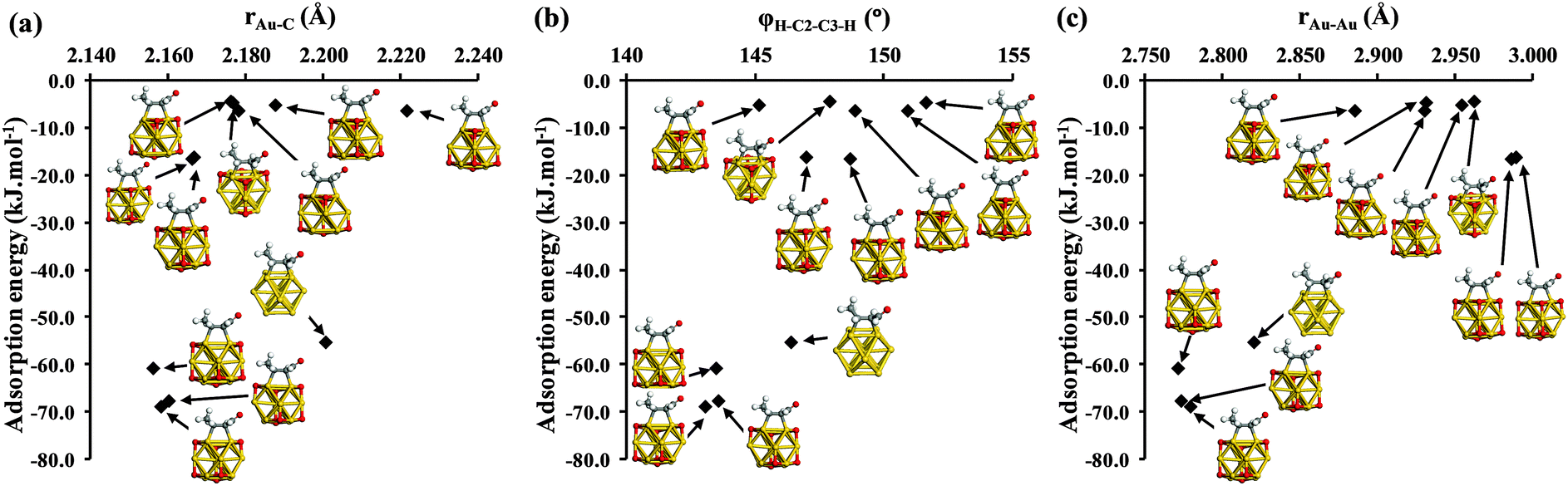 | ||
| Fig. 6 Relationship between crotonaldehyde adsorption energy ΔEads and (a) average gold–carbon bond length rAu–C between crotonaldehyde and Au13 cluster; (b) dihedral bond of crotonaldehyde φH–C2–C3–H; and (c) the gold–gold bond length rAu–Au following di-σCC adsorption of E-(s)-trans crotonaldehyde. Analysis of geometric parameters was performed on the bare and oxidised Au13 NPs shown in Fig. 5. Note the two cluster–adsorbate systems with slightly positive adsorption energies in were omitted for clarity. | ||
The sub-set of structures lacking an h-O atom adjacent to crotonaldehyde exhibit stronger di-σCC-2 adsorption than occurs on the bare Au13 cluster (Fig. 5). These structures have correspondingly shorter rAu–C bonds by ∼0.043 Å, and smaller φH–C2–C3–H dihedral angles (142° versus 147°) than for the bare cluster, consistent with strong rehybridization of the two C atoms in the CC moiety in the presence of h-O adatoms. This likely reflects oxidation of the Au atoms to which the crotonaldehyde is bound; cationic gold, having a smaller effective radius than the neutral atom species, permits closer approach of the adsorbate, while the associated depopulation of d-states facilitates adsorbate to cluster charge donation in concert with the metal to adsorbate back donation that also operates on the bare cluster. The two Au atoms coordinating to crotonaldehyde are also 0.046 Å closer in this set of oxidized clusters than they are for the parent Au13 cluster, probably reflecting distortion of the former arising from the stronger CC binding.
A similar picture emerges for the sub-set of structures in which the three-fold hollow site closest to crotonaldehyde is occupied by oxygen, for which aldehyde adsorption is significantly weaker than on bare Au13. These configurations have generally longer rAu–C bonds (spanning 2.156 Å to 2.222 Å) and φH–C2–C3–H dihedral angles closer to the 180° value expected for the planar CC double bond in the unbound aldehyde. Although the two coordinating Au atoms in this sub-set are again expected to be electron deficient due to adsorbed oxygen, the attractive interactions from molecule-to-metal electron donation have to be balanced against the steric repulsion between crotonaldehyde and the proximate h-O oxygen atom. Fig. 6c shows that oxygen adatoms placed near crotonaldehyde significantly increase the separation of Au atoms to which the CC bond coordinates, resulting in an inverse correlation between aldehyde adsorption and Au–Au bond distance. A simple trigonometric calculation shows that, for sp3 hybridised C atoms, an Au–Au spacing of around 2.80 Å is optimal for Au–C distances of 2.16 Å, i.e. the most strongly bound clusters have geometries consistent with this bonding geometry at the C atoms.
Fig. 7 shows a comparison of the Au 5d partial density of states (PDOS) for the Au13 cluster and associated Au13O6 structures. The bare Au13 states range from around −7 to −1 eV relative to the Fermi level, consistent with the filled d-shell of atomic Au, whereas the Au13O6 PDOS plots show a broader spread of energies, with unoccupied states present above the Fermi level reflecting the formation of linear O–Au–O bonds and concomitant gold oxidation. Formation of empty metal d-states upon cluster oxidation permits localized adsorbate → metal donation as discussed above.
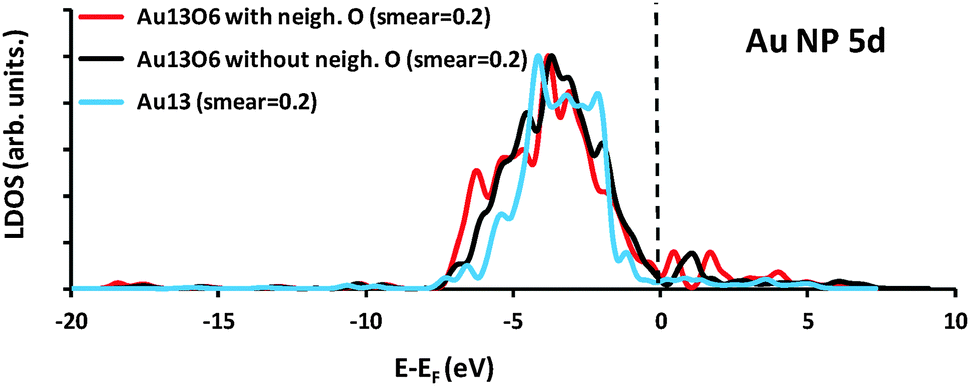 | ||
| Fig. 7 Comparison of partial density of states (PDOS) for the Au 5d states of Au13 (blue), Au13O6 with h-O at adsorption site (red) and Au13O6 without h-O at adsorption site (black). The Fermi level, EF, is indicated by the vertical dashed line. A smearing parameter of 0.2 eV was used in the VASP calculations with no additional smearing applied to the resulting PDOS data. | ||
Further insight into the charge distribution of selected configurations was obtained using Bader analysis (Table 1). Fig. 7 shows that the Au13 cluster has filled d-orbitals and so we would expect a net electron donation from the d-orbitals of the bare Au13 cluster to adsorbed crotonaldehyde with negligible back-donation, however only minimal charge transfer (Δq = 0.04) is actually calculated. It is worth recalling that gold NPs with odd numbers of electrons, such as the neutral Au13 cluster, exhibit high electron affinities50,51 due to the unpaired electron which formally occupies a delocalised state arising from the overlap of Au sp-atomic orbitals. This high electron affinity may allow electron transfer from crotonaldehyde to the cluster and Table 1 suggests that (for Au13) these opposing effects self-compensate. Oxygen addition to Au13 to form Au13O6 results in a net charge transfer of 4.39 e from the cluster to oxygen adatoms, with the six Au atoms doubly coordinated to Oa carrying an average charge of 0.49 e, significantly higher than the quoted average across all 13 Au atoms. Crotonaldehyde adsorption to the Au13O6 cluster lacking an h-O adatom neighbouring the adsorption site shows significant net electron donation from the aldehyde to the cluster (Δq = −0.13), due to the available empty d-orbitals associated with charge withdrawal by the co-adsorbed oxygen, thereby enhancing crotonaldehyde adsorption relative to Au13 (−68.8 kJ mol−1versus −55.2 kJ mol−1 respectively). Bader charge analysis for the Au13O6 where a neighbouring h-O atom is present, reveals a virtually identical charge distribution to that of oxidized cluster with this site vacant. This implies that the electronic structure of crotonaldehyde binding to Au13O6 cluster is similar with or without the proximate h-O. Hence the large energy difference of 52.3 kJ mol−1 between these configurations and attendant molecular destabilization associated with the former can only be attributed to steric interactions between the neighbouring adsorbed oxygen and the aldehyde. Parallel calculations employing ethene as a model allylic adsorbate offer a similar picture; oxidation of the Au13 cluster promotes adsorption unless the proximal h-O site is occupied. For ethene, the additional stabilization due to the availability of empty d-orbitals upon cluster oxidation is greater than for crotonaldehyde (18.2 versus 13.6 kJ mol−1). Conversely, ethene destabilization over Au13O6 by neighbouring h-O is lower (30.9 kJ mol−1) than that observed for crotonaldehyde. The charge distributions for adsorbed ethene with and without neighbouring h-O are also essentially identical, confirming that for adsorbates possessing CC bonds, it is repulsive steric interactions that regulate their adsorption over gold nanoparticles, with ethene less sensitive to such factors than the bulkier allylic aldehyde. We note here that the influence of surface oxygen atoms on the adsorption of ethene is considered for comparison with our crotonaldehyde results. The formation of a oxametallacycle akin to that discussed by Linic and Barteau for ethene epoxidation over Ag52 has not been considered.
| Structure | q Au13 /|e| | q Oads /|e| | 〈qAu〉b/|e| | 〈qΟads〉b/|e| | Δqc/|e| | q C1 /|e| | q C2 /|e| | q O1 /|e| | ΔEads/kJ mol−1 | |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Sum of Bader charges on indicated atom type.
b Average Bader charge on indicated atom type.
c Charge transfer, negative values indicate e transfer from adsorbate to nanoparticle.
d Bader charge on carbon atoms of CC bonded to the cluster, in CrCHO C1 has a CH3 and C2 a CHO substituent.
e Bader charge on aldehyde oxygen atom.
|
||||||||||
| Au13 |

|
0.00 | — | 0.00 | — | — | — | — | — | — |
| Au13O6 |
|
4.39 | −4.39 | 0.34 | −0.73 | — | — | — | — | — |
| CrCHO |

|
— | — | — | — | — | −0.05 | −0.09 | −1.10 | — |
| di-σCC-2-CrCHO-Au13 |

|
0.04 | — | 0.00 | — | 0.04 | −0.09 | −0.15 | −1.09 | −55.2 |
| di-σCC-2-CrCHO -Au13O6 no neighboring O |
|
4.29 | −4.43 | 0.33 | −0.74 | −0.13 | −0.07 | −0.13 | −1.07 | −68.8 |
| di-σCC-2-CrCHO -Au13O6 with neighboring O |
|
4.25 | −4.43 | 0.33 | −0.74 | −0.17 | −0.07 | −0.12 | −1.07 | −16.5 |
| di-σ-Et-Au13 |
|
−0.06 | — | 0.00 | — | −0.06 | −0.17 | −0.16 | — | −84.0 |
| di-σ-Et-Au13O6 no neighboring O |
|
4.26 | −4.44 | 0.33 | −0.74 | −0.18 | −0.15 | −0.14 | — | −102.2 |
| di-σ-Et -Au13O6 with neighboring O |
|
4.22 | −4.43 | 0.32 | −0.74 | −0.21 | −0.14 | −0.13 | — | −53.1 |
The electronic effect of the substituents on the crotonaldehyde CC bond can also be seen; even for the gas phase molecule the two C atoms have significantly different Bader charges. On adsorption both C atoms become slightly more negatively charged but this effect is in-sensitive to the state of the cluster. Further the charge on the aldehyde oxygen atom is practically unchanged from the gas phase for each adsorption mode suggesting that electronic rearrangement within the molecule upon adsorption plays a relatively minor role.
IV. Conclusions
In this study we find that oxidation of well-defined Au nanoparticles strongly influences crotonaldehyde and ethene adsorption. For the 0.8 nm diameter Au13 cluster discussed here, intermediate oxygen coverages enhance crotonaldehyde and ethene adsorption through the CC group by 14–18 kJ mol−1. This effect can be understood in terms of the genesis of empty Au d-states in the oxidised clusters able to receive electrons from these allylic adsorbates. At higher oxygen coverages this bonding picture is still valid, however steric interactions between oxygen adatoms neighbouring the crotonaldehyde (ethene) adsorption site destabilize the molecule by up to 60 kJ mol−1 with respect to bare Au13. We also find that oxidation of these sub-nanometer gold NPs is likely to be favoured in an oxidising atmosphere, so that under normal selox conditions oxygen will be present on the surface.
Increasing the gold nanoparticle size to 1.1 nm (Au38) lowers the crotonaldehyde adsorption energy by 20–40% compared to the bare Au13 cluster. Oxidation to Au38O2, wherein the O adatoms are located close to co-adsorbed crotonaldehyde, weakens the allylic aldehyde adsorption by as much as 90%.
These calculations highlight the importance of both the surface coverage, and location of adsorbed oxygen upon the stability of co-adsorbed crotonaldehyde, the desired product of crotyl alcohol selox. Oxygen bound in a three-fold hollow site adjacent to adsorbed crotonaldehyde dramatically destabilizes the latter due to strong steric repulsion. This finding implies that crotyl alcohol selox catalysed over gold nanoparticles should be conducted under high oxygen partial pressures, in order to desorb the reactively-formed aldehyde product and prevent side-reactions such as crotonaldehyde decarbonylation to propene and CO. Conversely, low oxygen partial pressures, and concomitant low surface oxygen coverages may hinder crotonaldehyde desorption due to enhanced adsorbate → cluster electron transfer relative to unoxidised gold nanoparticles.
Acknowledgements
We thank the EPSRC (EP/E046754/1; EP/G007594/3) for financial support and the award of a Leadership Fellowship (AFL) and studentship (AM). Computational resources for this project were partially provided by UK's National high-performance computing service, HECToR (EP/F067496) through the materials consortium, ARCCA and HPC-Wales supercomputer facilities. We thank Dr Adam Thetford and Soon Wen Hoh for discussions on the charge analysis carried out as part of this work.Notes and references
- D. G. Lee and U. A. Spitzer, J. Org. Chem., 1970, 35, 3589 CrossRef CAS.
- P. V. Prabhakaran, S. Venkatachalam and K. N. Ninian, Eur. Polym. J., 1999, 35, 1743 CrossRef CAS.
- J. Muzart, Tetrahedron, 2003, 59, 5789 CrossRef CAS.
- C. Della Pina, E. Faletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- K. Mori, K. Yamaguchi, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2002, 124, 11572 CrossRef CAS PubMed.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593 CrossRef CAS PubMed.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Wantanabe, C. J. Kiely, D. W. Knight and G. Hutchings, Science, 2006, 311, 362 CrossRef CAS PubMed.
- D. I. Enache, D. W. Knight and G. Hutchings, Catal. Lett., 2005, 103, 43 CrossRef CAS.
- A. F. Lee, S. F. J. Hackett, G. J. Hutchings, S. Lizzit, J. Naugthon and K. Wilson, Catal. Today, 2009, 145, 251 CrossRef CAS PubMed.
- T. Balcha, J. R. Strobl, C. Fowler, P. Dash and R. W. J. Scott, ACS Catal., 2011, 1, 425 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374 CrossRef CAS PubMed.
- A. F. Lee, C. V. Ellis, J. N. Naughton, M. A. Newton, C. M. A. Parlett and K. Wilson, J. Am. Chem. Soc., 2011, 133, 5724 CrossRef CAS PubMed.
- C. V. Gaskell, C. M. A. Parlett, M. A. Newton, K. Wilson and A. F. Lee, ACS Catal., 2012, 2, 2242 CrossRef CAS.
- A. F. Lee, J. N. Naughton, Z. Liu and K. Wilson, ACS Catal., 2012, 2, 2235 CrossRef CAS.
- C. M. A. Parlett, C. V. Gaskell, J. N. Naughton, M. A. Newton, K. Wilson and A. F. Lee, Catal. Today, 2013, 205, 76 CrossRef CAS PubMed.
- A. F. Lee, Z. Chang, P. Ellis, S. F. J. Hackett and K. Wilson, J. Phys. Chem. C, 2007, 111, 18844 CAS.
- J. Naughton, A. F. Lee, S. Thompson, C. P. Vinod and K. Wilson, Phys. Chem. Chem. Phys., 2010, 12, 2670 RSC.
- C. Stegelmann, N. C. Schiodt, C. T. Campbell and P. Stoltze, J. Catal., 2004, 221, 630 CrossRef CAS PubMed.
- Y.-H. Chin and E. Iglesia, J. Phys. Chem. C, 2011, 115, 17845 CAS.
- Y.-H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 15958 CrossRef CAS PubMed.
- M. Boronat and A. Corma, J. Catal., 2011, 284, 138 CrossRef CAS PubMed.
- J. Haubrich, D. Loffreda, F. Delbecq, Y. Jugnet, P. Sautet, A. Krupski, C. Becker and K. Wandelt, Chem. Phys. Lett., 2006, 433, 188 CrossRef CAS PubMed.
- F. Delbecq and P. Sautet, J. Catal., 2002, 211, 398 CAS.
- F. Delbecq and P. Sautet, J. Catal., 2003, 220, 115 CrossRef CAS.
- J. Naughton, A. Pratt, C. W. Woffinden, C. Eames, S. P. Tear, S. M. Thompson, A. F. Lee and K. Wilson, J. Phys. Chem. C, 2011, 115, 25290 CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Kresse and D. Joubert, Phys Rev. B, 1999, 59, 1758 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, H. Nakatsuji, G. A. Petersson, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, T. Vreven, H. Nakai, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, V. Bakken, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, K. Morokuma, R. L. Martin, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, R. C. Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- K. A. Peterson, D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1994, 100, 7410 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules - A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- M. Suzuki and K. Kozima, Bull. Chem. Soc. Jpn., 1969, 42, 2183 CrossRef CAS.
- A. Maeland and T. B. Flanagan, Can. J. Phys., 1964, 42, 2364 CrossRef CAS.
- A. Roldán, S. González, J. M. Ricart and F. Illas, ChemPhysChem, 2009, 10, 348 CrossRef PubMed.
- L. Alves, B. Ballesteros, M. Boronat, J. R. Cabrero-Antonino, P. Concepción, A. Corma, M. A. Correa-Duarte and E. Mendoza, J. Am. Chem. Soc., 2011, 133, 10251 CrossRef CAS PubMed.
- A. Franceschetti, S. J. Pennycook and S. T. Pantelides, Chem. Phys. Lett., 2003, 374, 471 CrossRef CAS.
- K. L. Howard and D. J. Willock, Faraday Discuss., 2011, 152, 135 RSC.
- C. D. Zeinalipour-Yazdi, A. L. Cooksy and A. M. Efstathiou, Surf. Sci., 2008, 602, 1858 CrossRef CAS PubMed.
- D. F. Mukhamedzyanova, N. K. Ratmanova, D. A. Pichugina and N. E. Kuz'menko, J. Phys. Chem. C, 2012, 116, 11507 CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331 CrossRef CAS PubMed.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252 CrossRef CAS PubMed.
- M. Amft, B. Johansson and N. V. Skorodumova, J. Chem. Phys., 2012, 136, 024312 CrossRef PubMed.
- A. Lyalin and T. Taketsugu, J. Phys. Chem. C, 2009, 114, 2484 Search PubMed.
- K. J. Taylor, C. L. Pettiette-Hall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 96, 3319 CrossRef CAS PubMed.
- H. Häkkinan and U. Landman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R2287 CrossRef.
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2002, 124, 310 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The methodology and unit cell for crotonaldehyde adsorption on Au(111), the optimized structures of all nanoparticle/adsorbate models and LDOS plots are given. See DOI: 10.1039/c3cp53691b |
| This journal is © the Owner Societies 2014 |