 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Allosteric control of kinesin's motor domain by tubulin: a molecular dynamics study†
Aliaksei
Krukau
ab,
Volker
Knecht
ac and
Reinhard
Lipowsky
*a
aTheory & Bio-Systems, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: lipowsky@mpikg.mpg.de; Fax: +49 331 5679602; Tel: +49 331 5679600
bLeibniz-Rechenzentrum, Bavarian Academy of Sciences, 85748 Garching, Germany
cBiomolecular Dynamics, University of Freiburg, 79104 Freiburg, Germany
First published on 6th February 2014
Abstract
Molecular motors such as kinesin are essential for many biological processes. These motors have two motor domains, which bind to tubulin filaments, hydrolyze ATP, and transduce the released chemical energy into directed movements. The general principles of this chemomechanical coupling are now well-established but the underlying molecular mechanisms remain elusive because small conformational changes within large proteins are difficult to detect experimentally. Here, we use atomistic molecular dynamics simulations to monitor such changes within a single motor domain of KIF1A, which belongs to the kinesin-3 motor family. The nucleotide binding pocket of this domain can be empty or occupied by ATP or ADP. For these three nucleotide states, we determine the mobility of the backbone of the protein, both in solution and attached to tubulin. Only one subdomain of the motor domain is found to exhibit a strongly increased mobility upon binding to tubulin: the neck linker that presumably acts as a mechanical transmitter to the other motor domain in dimeric kinesin-3 motors. Furthermore, upon binding to tubulin, the neck linker mobility becomes sensitive to the bound nucleotide and is highly increased after phosphate release, which implies undocking of this linker from the core of the motor domain. These simulation results are consistent with experimental data from EPR spectroscopy, FRET, and cryo-electron microscopy. A detailed analysis of our simulation data also reveals that the undocking of the neck linker in the ADP-kinesin-tubulin state arises from allosteric interactions between the nucleotide and tubulin and that the β-sheet core undergoes a twist both during phosphate release and ATP binding. The computational approach used here can be applied to other motor domains and mechanoenzymes in order to identify allosteric interactions between the subdomains of these proteins.
1 Introduction
Molecular motors such as kinesin that interact with cytoskeletal filaments are essential for many biological processes including intracellular cargo transport, cell motility, and cell division. These cytoskeletal motors are intriguing because they involve a wide range of scales, from the cleavage of a single covalent bond during ATP hydrolysis up to the transport over mesoscopic or even macroscopic scales. In order to elucidate the connection between these different scales, a wide variety of different experimental methods has been applied. The molecular structure of kinesin motors has been studied using X-ray crystallography1–5 and cryo-electron microscopy (Cryo-EM).4,6,7 The conformational changes within these motor proteins have been probed by electron paramagnetic resonance (EPR) spectroscopy,6,8,9 Förster or fluorescence resonance energy transfer (FRET),6 mutagenesis,9,10 and biochemical methods.11 The mechanical stepping of single motors can be controlled by laser traps and resolved by optical microscopy.12 The latter methods are also useful in order to determine how multiple motors work together in the environment of the cell.13From the theoretical point of view, the transport properties of single motors14 and small teams of motors15,16 can be understood in terms of stochastic processes that describe the motors as mechanoenzymes, which undergo both chemical transitions and mechanical steps. These theories have clarified the general principles that govern the chemomechanical coupling of the motors as well as the dependence of their transport properties on intrinsic motor parameters and external control parameters. However, these stochastic theories cannot determine the underlying molecular mechanisms, which are related to relatively small conformational changes within the large motor proteins. Here, we address these molecular mechanisms via atomistic molecular dynamics (MD) simulations using the motor domain of KIF1A mouse kinesin, which belongs to the kinesin-3 family, as an instructive example. In contrast to previous simulation studies,17–23 we will explicitly study the allosteric role of tubulin by simulating three nucleotide states of the motor domain both in solution and attached to tubulin. A quantitative comparison between these six different states reveals that, when the motor domain is bound to tubulin, the phosphate release from the nucleotide binding pocket leads to a strong increase in the mobility of the neck linker. The associated undocking of this linker from the core of the motor domain, which is essential for the motility of kinesin-1 and related constructs,6,7,8 may also play an important role for the motility of both monomeric24,25 and dimeric kinesin-3.26–28 To the best of our knowledge, we report here the first simulation study that provides evidence for allosteric interactions between kinesin and tubulin or, more generally, between a molecular motor and the associated filament.
Many different kinesin motors have been identified that form a large superfamily of proteins.27,29 All members of this superfamily possess a motor domain. Here, we study the motor domain of KIF1A mouse kinesin as an example for a kinesin-3 motor. The latter motors can exist as monomers or homodimers.27 In fact, both monomeric24,25 and dimeric26–28 KIF1A can move progressively along microtubules.
High-resolution X-ray crystal structures of kinesin's motor domain are now available for several members of the kinesin superfamily.1–5 The high-resolution structure of the kinesin-3 motor KIF1A is displayed in Fig. 1, based on X-ray data from ref. 5. As shown in this figure, the motor domain contains several important subdomains: the nucleotide binding pocket, the microtubule binding site, and the neck linker together with the α6-helix.
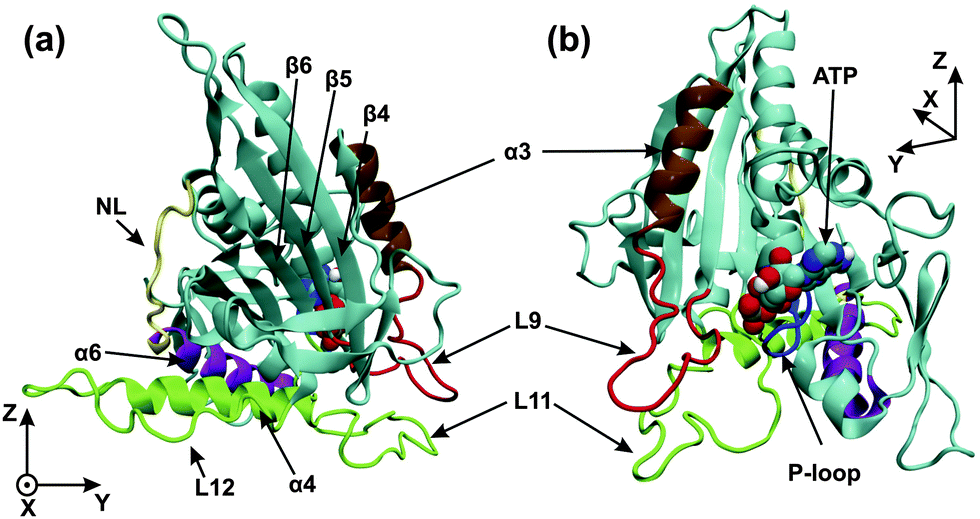 | ||
| Fig. 1 (a) Front and (b) back view of the motor domain in the ATP state. The image is generated from a structural model based on X-ray data (PDB 1vfw) for KIF1A mouse kinesin,5 a kinesin-3 motor, with a docked neck linker using VMD.30 The microtubule binding site (green) consists of the L11-loop, the L12-loop, and the α4-helix. The nucleotide binding pocket consists of the P-loop (blue) and the L9-loop (red), which is directly connected to the α3-helix (ochre). The last subdomain at the C-terminal end of the motor domain is the neck linker (NL, yellow), which is preceded by the α6-helix (purple). The C-terminus of the α6-helix is in close proximity to the C-terminus of the α4-helix. The coordinates x, y, and z are used to describe the conformational changes of the motor domain. | ||
The nucleotide binding pocket can be empty or occupied by ATP or ADP. As shown in Fig. 1, this pocket consists of several regions: the P-loop, which interacts with the phosphoryl groups of the bound nucleotide, as well as the L9-loop, which is directly connected to the α3-helix; the latter two structural elements form the so-called switch-I region, which is located close to the γ-phosphate and is involved in the cleavage of this phosphate from ATP.11
The microtubule binding site or switch-II region is responsible for the interactions of the motor domain with the filament, and changes its affinity to tubulin upon release of phosphate or ADP from the nucleotide binding pocket. The microtubule binding site contains the L11- and L12-loops, as well as the α4-helix, see Fig. 1. The L11-loop (or switch-II-loop) is located very close to the L9-loop (or switch-I-loop) of the nucleotide binding pocket, and these two loops are believed to be crucial for the cross-talk between the nucleotide binding pocket and the microtubule binding site.5
The neck linker is a short peptide segment of about 10 residues, see NL in Fig. 1, and provides a connection, together with the α6-helix, between the motor domain and the neck region of dimeric kinesin-3. Mutagenesis and biochemical experiments indicate that the neck-linker determines the direction of movement of the kinesin motors along tubulin filaments.10 The conformation and mobility of the neck linker as probed by EPR spectroscopy, FRET, and Cryo-EM were found to be sensitive to the nucleotide state when kinesin-1 and related constructs were attached to tubulin but not for these motors in solution.6,8,9 Because of its nucleotide-dependent mobility, the neck linker should be essential for force generation6,9 even though the molecular displacements are expected to be small.12
Some atomistic molecular dynamics (MD) simulations have been performed previously in order to address the conformational changes of kinesin's motor domain in solution17 and attached to tubulin.19 The latter simulations were restricted to very short runs of only 1 ns. More recently, atomistic MD simulations were performed applying secondary structure restraints as deduced from Cryo-EM maps.22 The corresponding constrained MD trajectories had an overall run time of 42 ns. Atomistic20,21 and coarse-grained18 MD simulations were used to study force generation by the neck linker. Coarse-grained MD was also applied recently to the diffusion of kinesin along filaments.23
In our MD study, both the motor domain and the tubulin dimer were modelled with atomic resolution and fully immersed in about 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 45000 water molecules, see Methods section below. We distinguished three different nucleotide states, denoted by ATP, ADP or Empty, of the motor domain. Our ATP structures are based on AMP-PNP structures from the protein database. We first validated our simulation code by calculating the crystal structure of the motor domain in the ATP state and demonstrating that this computed structure is in good agreement with the crystal structure as obtained from X-ray diffraction. We then studied the different nucleotide states of the motor domain both in solution and attached to tubulin. Each of these six states was simulated for 300 ns. For the ATP state of the motor domain, the initial configuration of our simulations was provided by the corresponding X-ray structure, see Fig. 1. For the ADP and Empty states, we modified this initial configuration by removing the γ-phosphate and the ATP molecule, respectively, from the motor domain. Comparing the conformations of the Empty state with those of the ATP state, we can determine the conformational changes during ATP binding. Likewise, a comparison of the ATP state with the ADP state provides insight into the conformational changes associated with phosphate release.
000 and 45000 water molecules, see Methods section below. We distinguished three different nucleotide states, denoted by ATP, ADP or Empty, of the motor domain. Our ATP structures are based on AMP-PNP structures from the protein database. We first validated our simulation code by calculating the crystal structure of the motor domain in the ATP state and demonstrating that this computed structure is in good agreement with the crystal structure as obtained from X-ray diffraction. We then studied the different nucleotide states of the motor domain both in solution and attached to tubulin. Each of these six states was simulated for 300 ns. For the ATP state of the motor domain, the initial configuration of our simulations was provided by the corresponding X-ray structure, see Fig. 1. For the ADP and Empty states, we modified this initial configuration by removing the γ-phosphate and the ATP molecule, respectively, from the motor domain. Comparing the conformations of the Empty state with those of the ATP state, we can determine the conformational changes during ATP binding. Likewise, a comparison of the ATP state with the ADP state provides insight into the conformational changes associated with phosphate release.
Using this MD set-up, we first studied the mobility of the 351 Cα-atoms of KIF1A's motor domain by calculating the positional fluctuations of these atoms. Essentially, all of these fluctuations are found to remain unchanged or become reduced when the motor domain binds to tubulin, with one remarkable exception: the neck linker, which exhibits an essentially constant mobility both in solution and in the Empty and ATP states of the kinesin-3–tubulin complex but a strongly enhanced mobility in the ADP/kinesin-3/tubulin state. This enhanced mobility of the neck linker implies an undocking of this linker from the core of the motor domain, in agreement with experimental data from EPR spectroscopy, FRET, and Cryo-EM.6–8 Furthermore, a detailed analysis of our simulation data also shows that the enhanced mobility of the neck linker arises from rotational movements of the α4-helix (or switch-II-helix), which belongs to the microtubule binding site, and the α6-helix, which is directly connected to the neck linker. These helix rotations are caused by L11–L9 loop interactions, which are modified because the L9-loop (or switch-I-loop) moves away from the nucleotide during phosphate release. In this way, our simulations provide direct evidence for an allosteric coupling mechanism that leads from phosphate release to neck linker undocking via a sequence of conformational changes involving several subdomains, namely the L9 loop, the L11 loop, the α4-helix, and the α6-helix.
Our paper is organized as follows. In the “Results” section, we first describe the mobility of the different subdomains for the three nucleotide states of the motor domain, both in solution and attached to tubulin. We show that the neck linker of ADP–kinesin-3 exhibits a strongly enhanced mobility upon binding to tubulin. Next, we determine the rotations of the α6- and α4-helices in the six different states of the motor domain and show that these rotations are quite different for the motor domain in solution and attached to tubulin. We also find a tilt of the strand orientations within the β-sheet core, both during phosphate release and ATP binding. Finally, we study the interactions between the L9- and L11-loops for the kinesin-3–tubulin complex and find that the L9-loop moves away from the nucleotide during phosphate release. In the “Discussion” section, we compare our results with available experimental data and argue that our simulation results provide direct evidence for tubulin's allosteric control of the chemomechanical coupling between phosphate release and neck linker undocking. The Methods section at the end contains a detailed description of our computational approach.
2 Results
2.1 Subdomain mobilities of kinesin-3 in solution and attached to tubulin
The motor domain of KIF1A mouse kinesin (hereafter called ‘kinesin-3’) consists of 361 residues and, thus, of 361 Cα-atoms that define the protein's backbone. In this subsection, we will first discuss the mobility of these Cα-atoms by calculating the standard deviations or root-mean-square-fluctuations (rmsf) of their coordinates. These fluctuations are displayed in Fig. 2(a) and (b) for kinesin-3 in solution and attached to tubulin, respectively. Inspection of this figure reveals that the overall fluctuations of the backbone of kinesin-3 are strongly reduced upon binding to tubulin. The largest decrease is observed for the microtubule binding site, which is located between the Cα-numbers 250 and 310. The mobility of the L11- and L12-loops of kinesin-3 attached to tubulin is about half of that in solution, and the mobility of the α4-helix drops by 25% from 0.2 to 0.15 nm upon attachment to tubulin.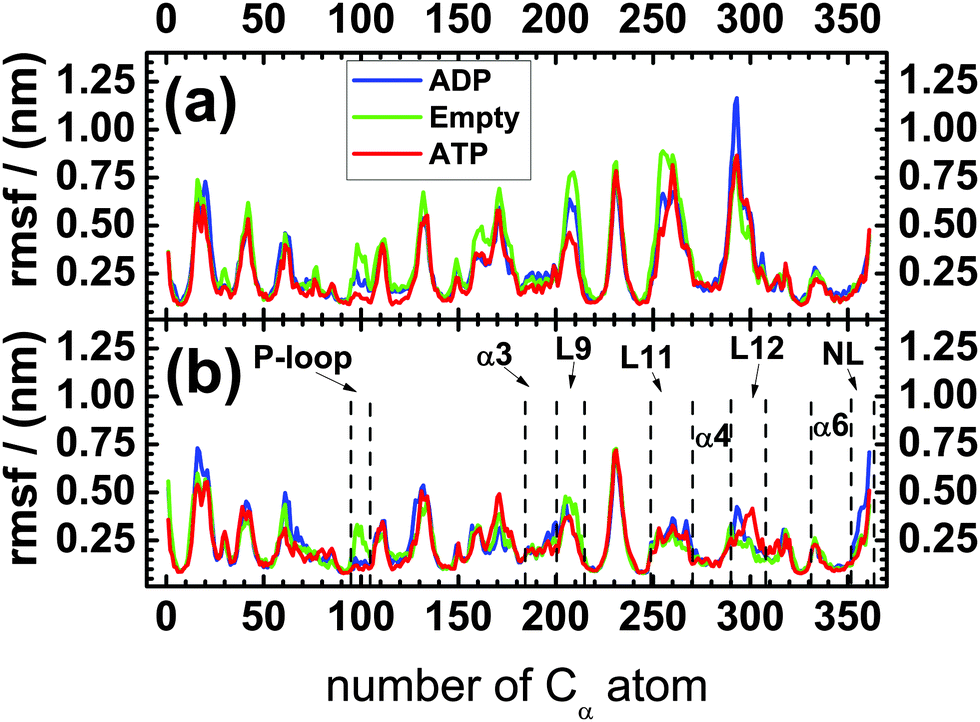 | ||
| Fig. 2 Standard deviations or root-mean-square-fluctuations (rmsf) of the 361 Cα-atoms of the kinesin's backbone (a) in solution and (b) attached to a tubulin dimer for the three nucleotide states ADP (blue), Empty (green), and ATP (red). The x-axis is labeled by the numbers of the Cα-atoms: the first Cα-atom with number 1 corresponds to the N-terminus of the motor domain, the last Cα-atom with number 361 represents the C-terminus of this domain and of the neck linker. The different loops (P-loop, L9, L11, L12) and helices (α3, α4, α6) as well as the neck linker (NL) have been introduced in Fig. 1. | ||
In order to corroborate this peculiar behavior of the neck linker, we also performed a principal component analysis of its mobility. The outcome of this analysis for the Cα-atoms of the neck linker is shown in Fig. 3. In solution, we found small differences in the first eigenvalue, but these differences are smaller than the statistical error, see Fig. 3(a). Therefore the mobility of the neck linker in solution is essentially independent of the nucleotide state. In contrast, the fluctuations of the neck linker depend strongly on the nucleotide state when the motor domain is attached to tubulin as shown in Fig. 3(b). The first eigenvalue for the ADP state is now 2.3 and 3.7 times larger than for the Empty and ATP nucleotide states, respectively. In fact, all first four eigenvalues are 2–3 times larger for the ADP compared to the ATP or Empty states. Upon binding to tubulin, the first eigenvalue is increased by a factor 2.1 and 1.4 for the ADP and the Empty states and decreased by a factor 1.4 for the ATP state, as follows from a comparison of the values in Fig. 3(a) and (b).
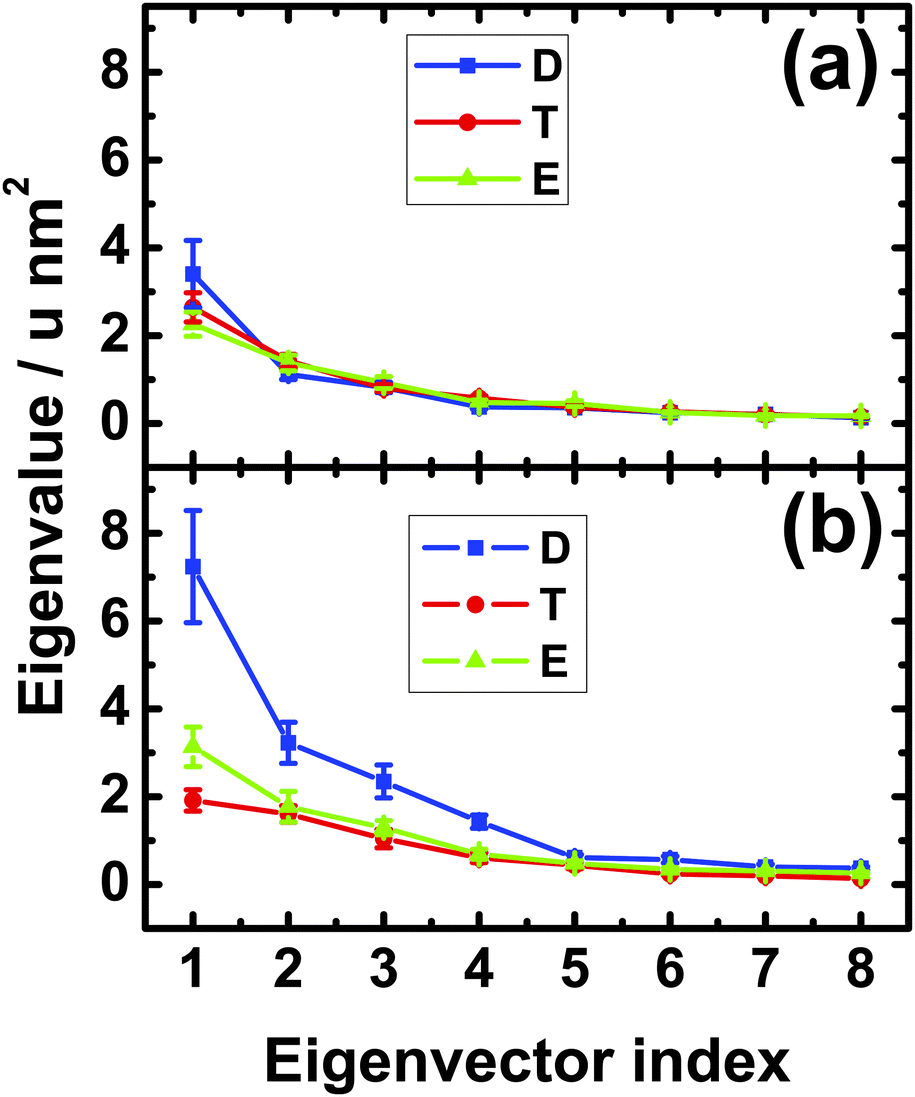 | ||
| Fig. 3 Eigenvalues of the covariance matrix for the Cα atoms of the neck linker region, residues 351–361, for the motor domain (a) in solution and (b) attached to a tubulin dimer. The eigenvalues for the ADP, ATP, and Empty nucleotide states are displayed in blue (D), red (T), and green (E), respectively. The parameter u denotes the atomic mass unit. | ||
Therefore, for the motor domain bound to tubulin, both the root-mean-square-fluctuations in Fig. 2(b) and the eigenvalues of the principal component analysis in Fig. 3(b) imply that the mobility of the neck linker is hardly changed during ATP binding whereas this mobility is strongly increased during phosphate release. The latter increase corresponds to an undocking of the neck linker from the core of the motor domain.
For the motor domains of kinesin-120,21 and kinesin-5,31 the neck linker interacts with the N-terminal cover strand, located between the Cα-numbers 0 and 9, and forms a cover neck bundle upon ATP binding. We did not analyze this structural feature in the present study but, for kinesin-3 attached to tubulin, the mobility of the N-terminal cover strand is strongly reduced for the ATP compared to the Empty state; see Fig. 2(b), as one would expect if a cover neck bundle is formed during ATP binding.
2.2 Rotations of α4- and α6-helices in solution and attached to tubulin
As shown in Fig. 1, the last subdomain of kinesin-3 that precedes the neck linker is the α6-helix which is in close contact with the α4-helix, the latter being part of the microtubule binding site. Therefore, we have also analyzed the conformation of the α4- and the α6-helices to study possible interactions4,5 between the microtubule binding site and the neck linker.In order to visualize the dependence of the molecular structure of kinesin-3 on its nucleotide state, we have compared the dominant conformations of the ATP and ADP states as well as those of the ATP and Empty states. For a given pair of states, this comparison was performed by superimposing their β-sheet cores and identifying the rotations of the α4- and α6-helices. In order to describe these rotations, we used the Cartesian coordinate system (x, y, z) as shown in Fig. 1 and described in more detail in the Appendix and Fig. S1 and S2, see the ESI.†
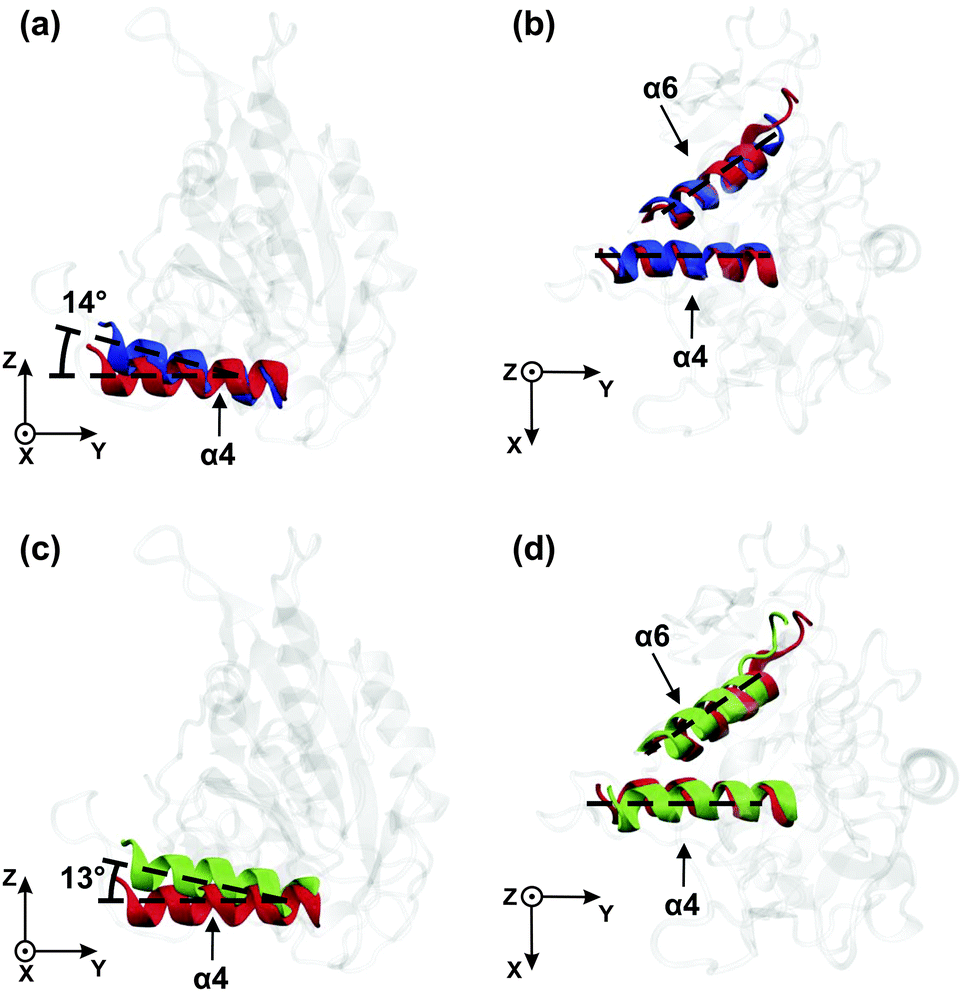 | ||
| Fig. 4 Kinesin-3 in solution: dominant orientations of the α4- and α6-helices for the three nucleotide states ATP (red), ADP (blue), and Empty (green) when projected onto the yz- and the xy-plane: (a and b) ATP cleavage and phosphate release (red → blue) and (c and d) ATP binding (green → red). In both processes, the helices undergo pronounced rotations parallel to the yz-plane as depicted in (a) and (c). | ||
Relative to its orientation in the ATP state (red), the α4-helix in the ADP state (blue) is rotated by 14° around the x-axis as shown in Fig. 4(a). Thus, the latter rotation is observed within the yz-plane. In contrast to the α4-helix, the α6-helix does not undergo any significant rotation in the yz-plane during the transition from the ATP to the ADP state. Furthermore, when projected onto the xy-plane, neither the α4- nor the α6-helices change their orientation, see Fig. 4(b).
In Fig. 4(c) and (d), the orientations of the α4- and α6-helices for the Empty state (in green) are shown together with those for the ATP state (in red). When the motor domain binds ATP, the α4-helix rotates by about 13° within the yz-plane, see Fig. 4(c), whereas both the α4- and the α6-helices remain essentially unchanged when projected onto the xy-plane, see Fig. 4(d). Furthermore, inspection of all four subfigures Fig. 4(a)–(d) shows that the helix orientations in the Empty state closely resemble those in the ADP state.
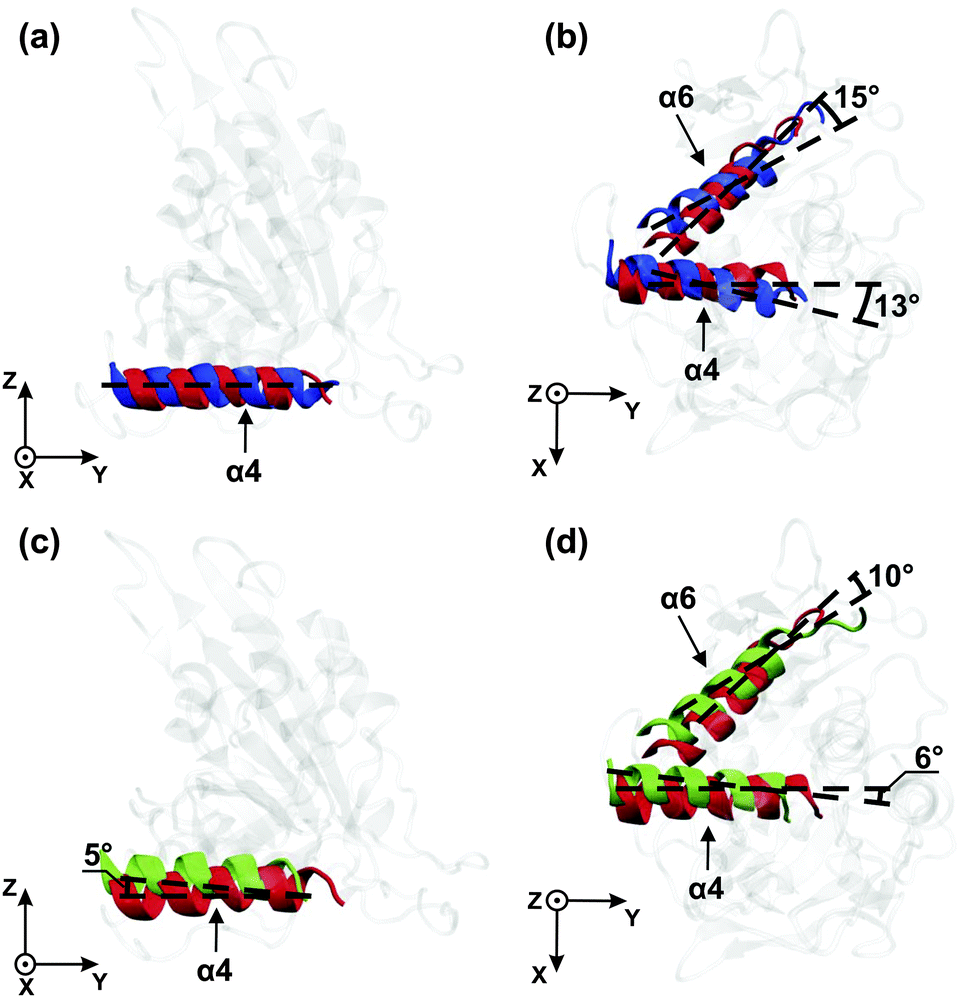 | ||
| Fig. 5 Kinesin-3 attached to tubulin: dominant orientations of the α4- and α6-helices for the three nucleotide states ATP (red), ADP (blue), and Empty (green) when projected onto the yz- and xy-planes: (a and b) ATP cleavage and phosphate release (red → blue) and (c and d) ATP binding (green → red). In both cases, the helices undergo pronounced rotations parallel to the xy-plane as depicted in (b) and (d). | ||
In Fig. 5(c) and (d), the orientations of the helices are compared for the Empty state (green) with those for the ATP state (red). Upon binding of ATP, the α4-helix undergoes relatively small rotations in the yz-plane and in the xy-plane, respectively, while the α6-helix undergoes no appreciable rotation in the yz-plane and a rotation by 10° in the xy-plane. In order to directly determine the effect of these helix rotations onto the neck linker, we have also measured the neck linker's average displacement and its root-mean-square-fluctuations in the different nucleotide states, see Table S1 in the ESI.†
Very recently, it has been proposed32,33 that the β-sheet core of kinesin-1 may undergo a twist during nucleotide release, similar to myosin motors.34 In order to find out whether such twists may occur in kinesin-3, we have studied the relative orientations of neighboring β-strands within the β-sheet core of this motor, as explained in the Methods section. For kinesin-3 attached to tubulin, we find that the β4-strand is rotated by about 10° relative to its neigboring strands β3 and β5, both during phosphate release and during ATP binding, see Table 1.
| State | ϕ 34 (°) | ϕ 45 (°) | ϕ 56 (°) |
|---|---|---|---|
| ATP | 20 ± 1.0 | 10 ± 0.7 | 16 ± 1.3 |
| ADP | 8.6 ± 0.5 | 18 ± 1.0 | 17 ± 1.2 |
| Empty | 9.3 ± 2.0 | 19 ± 1.8 | 15 ± 1.8 |
2.3 Interactions of L9- and L11-loops in kinesin-3 attached to tubulin
One possible pathway for the mechanical transmission of conformational changes from the nucleotide binding pocket to the microtubule binding site is provided by the interactions between the L9-loop, which is located in the vicinity of the γ-phosphate of the ATP, and the L11-loop, which is attached to the N-terminus of the α4-helix, the latter being part of the microtubule binding site, see Fig. 1. The dominant conformations of the L9- and L11-loop in the ATP and the ADP nucleotide state are displayed in Fig. 6(a) and (b), respectively. In the ATP nucleotide state, essentially the complete L9-loop is found to be close to the γ-phosphate and probably participates in the ATP hydrolysis. In addition, the central part of the L9-loop has an α-helical conformation and interacts with the N-terminal part of the L11-loop. At the same time, the C-terminal part of the L9-loop interacts with the N-terminal part of the α4-helix, see Fig. 6(a). In the ADP nucleotide state, on the other hand, the L9-loop changes its conformation, moving away from the nucleotide, and the central part of this loop starts to interact with the central part of the L11-loop, which induces the rotation of the α4-helix in the xy-plane, see Fig. 6(b).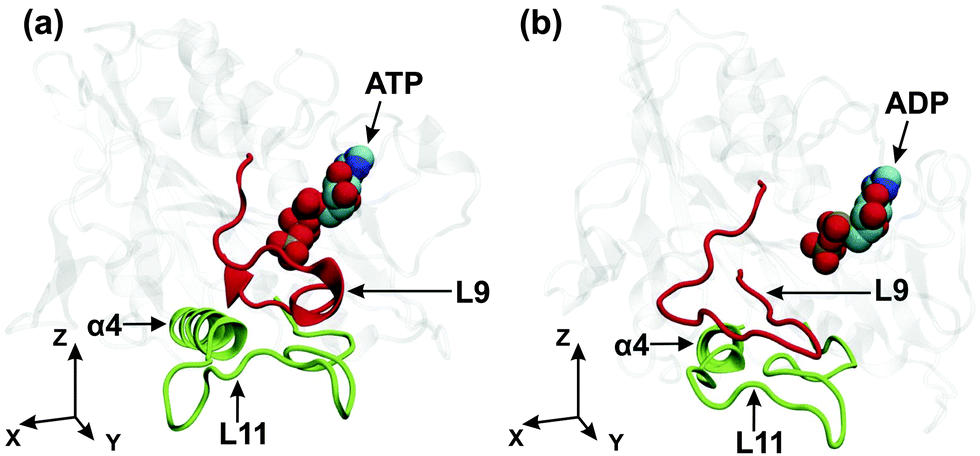 | ||
| Fig. 6 Kinesin-3 attached to tubulin: dominant conformations of the L9-loop (red) and the L11-loop (green) for (a) the ATP and (b) the ADP states of the motor domain. As before, the (x,y)-plane is tangential to the tubulin surface and the y-axis is parallel to the long principal axis of the α4-helix in the ATP state. Comparison of the two conformations reveals that the γ-phosphate release from the nucleotide binding pocket causes the L9-loop to move away from the nucleotide, to change its interaction pattern with the L11-loop, see the corresponding atomic separations as described in Fig. 7 below, and to induce a rotation of the α4-helix within the (x,y)-plane as in Fig. 5(b). | ||
To describe the observed conformational changes between the ATP and the ADP state in a more quantitative manner, we calculated the separations of different atoms within these two regions of kinesin-3 attached to tubulin, see Fig. 7. In the ATP nucleotide state, contacts are found between the peptide segments Ala206–Arg216 of L9 and Ala255–Lys261 of L11, between the segments Thr213–Arg216 of L9 and Ala248–Ala255 of L11, as well as between the segments Ser215–Arg216 of L9 and Asn272–Thr276 of the α4-helix, as indicated by the purple circles in Fig. 7(a). In addition, several very stable hydrogen bonds were observed between L9-Thr213 and L11-Ala255 (existence probability 0.861), L9-Ser214 and L11-Glu253(0.962), as well as L9-Arg216 and L11-Asn272(0.721). The last hydrogen bond connects the C-terminus of the L9-loop to the N-terminus of the α4-helix, and thus may stabilize the orientation of the helix. When ATP is replaced by ADP in the binding pocket, almost all contacts present in the ATP state disappear, see purple circles in Fig. 7(b). Furthermore, all hydrogen bonds that were formed in the ATP state are destroyed as well. However, a region of stable contact is observed between the peptide segments Ala207–Thr213 of L9 and Arg264–Ala269 of L11, see red circle in Fig. 7(b). In addition, two stable hydrogen bonds are found between L9-Asn209 and L11-Glu267(0.95) as well as L9-Met210 and L11-Glu267(0.883).
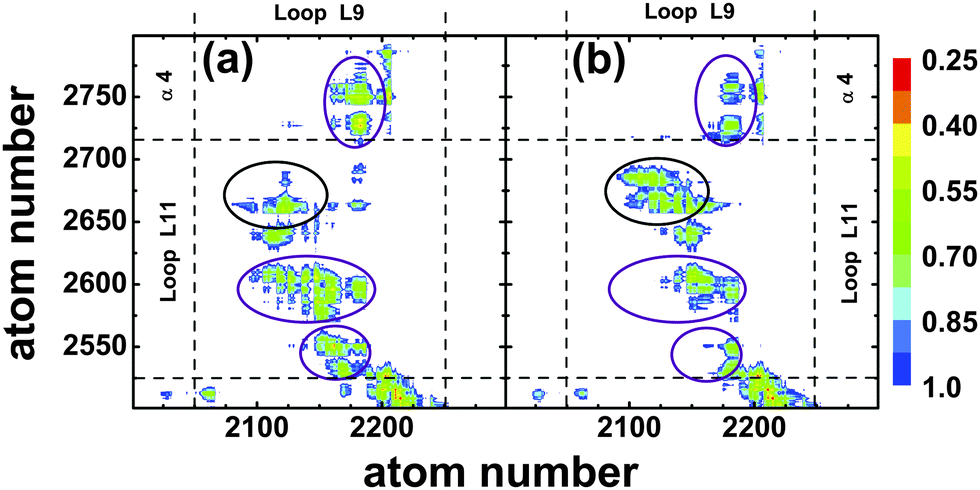 | ||
| Fig. 7 Kinesin-3 attached to tubulin: distance between the atoms of the L9-loop, which belongs to the nucleotide binding pocket, and the L11-loop, which is part of the microtubule binding site, for (a) the ATP and (b) the ADP nucleotide states. The color code represents the average distance between two atoms in nm. In the ATP state, the central part of the L9-loop interacts with the N-terminal part of the L11-loop (lower purple ellipses) and the C-terminal part of the L9-loop interacts with the N-terminal part of the α4-helix (upper purple ellipses). All of these interactions are strongly reduced in the ADP state, which exhibits, however, enhanced interactions between the inner part of the L9-loop and the C-terminal part of the L11-loop (black ellipses). | ||
Thus, after the release of the γ-phosphate, conformational changes in the L9-loop induce the breaking of several hydrogen bonds between the loops, destabilizing the ATP state orientation of the α4-helix. At the same time, the appearance of contacts between the central segments of the loops facilitate rotation of the α4-helix, and hydrogen bonds between residues in these segments stabilize the new orientation of this helix. The complete removal of the ATP from the binding pocket leads to a three-fold decrease in the stability of the hydrogen bonds between the termini of the L9- and L11-loops. However, for this Empty state, the appearance of new contacts between any segments of the loops is not observed. Therefore, the destabilization of hydrogen bonds between the termini of the L9- and L11-loops in the Empty state, without the creation of contacts between their central parts, leads to a rotation of the α4-helix with a magnitude only half of that in the ADP state.
3 Conclusions
In the molecular dynamics simulations reported here, we studied the motor domain of KIF1A mouse kinesin and focussed on those subdomains that should play an important role for free energy transduction and motility of kinesin-3 motors: the nucleotide binding pocket, the microtubule binding site, and the neck linker. First, we determined the mobility of these subdomains as described by the positional fluctuations of the Cα-atoms of the protein's backbone.For the motor domain in solution, we do not observe significant differences in the mobility of the neck linker for the three nucleotide states, see Fig. 2(a) and Fig. 3(a). This independence of the mobility on the nucleotide state agrees with the results of EPR6 and FRET measurements.6,8 Comparing our simulation data on the mobility of the neck linker in solution and attached to tubulin, see the data for NL in Fig. 2(a) and (b), we find that fluctuations of the neck linker in the ADP nucleotide state almost double in amplitude upon binding to tubulin. In contrast, the fluctuations of the neck linker in the ATP state are slightly reduced when the motor domain is attached to tubulin. The increase in the mobility of the neck linker in the ADP relative to the ATP state, when the motor domain is attached to tubulin, again agrees with Cryo-EM and EPR data.6,9 In fact, in our simulations, the neck linker is found to be the only region within the motor domain, for which tubulin binding leads to an increased mobility, see Fig. 2.
Furthermore, when the motor domain is attached to tubulin, the mobility of the neck linker is slightly higher in the Empty state compared to the ATP nucleotide state, see Fig. 3(b). This result correlates well with Cryo-EM experiments,7 which show that the neck linker was docked onto the motor head in the ATP state, but did not have any pronounced structure in the Empty state.
One important result of our simulation study is the remarkable difference between the conformational changes of the motor domain in solution and attached to tubulin. In solution, the α4-helix is found to rotate in the (y, z)-plane during γ-phosphate release, i.e., during the transition from the ATP to the ADP state, see Fig. 4(a), which agrees with experiment as previously mentioned. However, inspection of Fig. 4(b) and (d) also shows that we do not observe any rotation of the α6-helix. Therefore, in solution, we find no evidence for a direct coupling between the α4-helix, which belongs to the microtubule binding site, and the α6-helix, which is directly connected to the neck linker.
However, when the motor domain is attached to tubulin, the helices are found to behave in a completely different way. First, during phosphate release, the α4-helix no longer rotates in the yz-plane, but rather in the xy-plane, which is tangential to the tubulin surface. Most likely, the rotation of the α4-helix within the yz-plane is suppressed by the steric hindrance provided by tubulin. Second, and even more importantly, we now observe a large rotation of the α6-helix during phosphate release, see Fig. 5(b). In this way, we observe direct crosstalk between the microtubule binding site and the neck linker when the motor domain is attached to tubulin.
We also analyzed the distance between atoms of the L9-loop, which belongs to the nucleotide binding pocket, and those of the L11-loop, which is part of the microtubule binding site. When the motor domain is attached to tubulin, the interactions between these loops are found to depend on the nucleotide within the binding pocket.
In the ATP state, the central and the C-terminal parts of the L9-loop are in contact with the N-terminal part of the L11-loop and the α4-helix, respectively, as shown in Fig. 6(a). The loops are connected by several stable hydrogen bonds. However, the release of the γ-phosphate from the nucleotide binding pocket induces a change in the conformation of L9, the disappearance of the contacts between the L9-loop and the termini of the L11-loop and the α4-helix, as well as the destruction of the previously existing hydrogen bonds between the loops, see Fig. 6(b). These changes lead to an increased mobility of the L11-loop and the α4-helix. At the same time, the appearance of contacts between the central regions of the L9- and L11-loops, as described in Fig. 7(b), facilitates the large rotation of the α4-helix, as observed in the (xy)-plane tangential to the tubulin surface.
The release of the complete ATP molecule from the binding pocket, on the other hand, leads only to a destabilization of the hydrogen bonds between the termini of the two loops and to a small rotation of the α4-helix in the Empty state, see Fig. 5(c) and (d). This rotation increases the mobility of the neck linker only slightly, compared to the ADP state, and does not lead to its complete undocking from the core of the motor domain.
The conformational changes of kinesin-3 during phosphate release as deduced from our simulations of the ATP and ADP state are consistent with the scheme shown in Fig. 8. First, phosphate release leads to a conformational change of the L9-loop, which moves further away from the bound nucleotide, see Fig. 6, and modifies the interactions of this loop with the L11-loop. The latter loop is directly connected to the α4-helix and then acts to rotate this helix. Furthermore, because the C-terminus of the α4-helix is in close proximity to the C-terminus of the α6-helix, the rotation of α4 leads to a rotation of α6. The latter rotation generates a force on the N-terminus of the neck linker, which is then pulled away from its docked position, see the measured displacements in Table S1 (ESI†) and increases the neck linker's mobility, see Fig. 2.
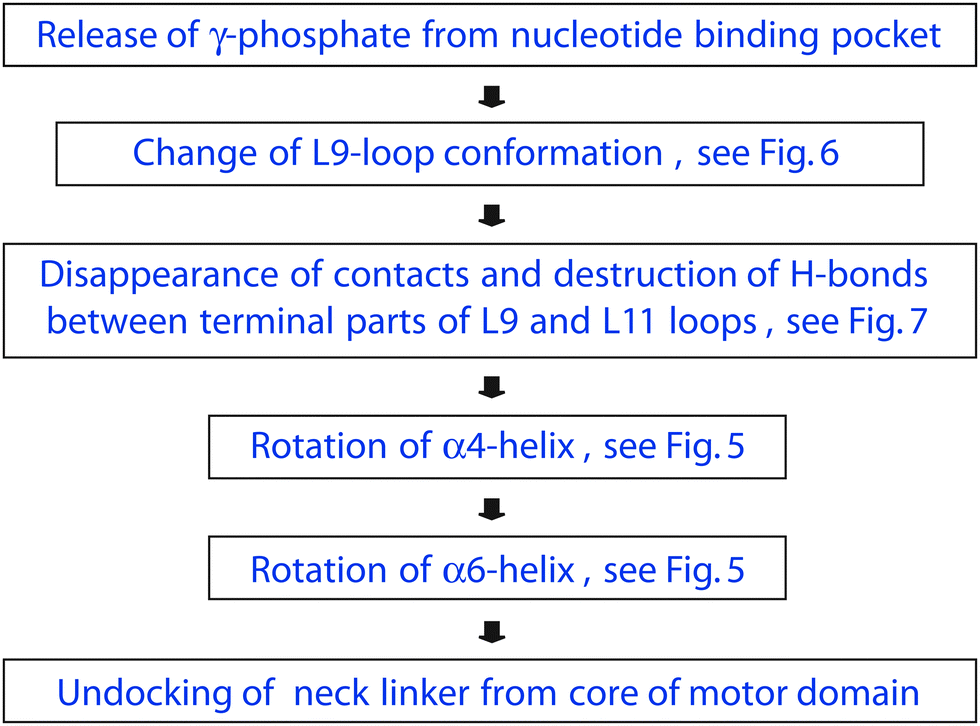 | ||
| Fig. 8 Chemomechanical coupling between phosphate release and neck linker undocking for kinesin-3 attached to tubulin as mediated by allosteric interactions between the motor's subdomains. First, phosphate release affects the L9-loop conformation, which moves away from the nucleotide and loses many contacts with the L11-loop. These reduced interactions between the two loops rotate the α4-helix and, in this way, modify the molecular interactions between the α4- and the α6-helices. As a result, the α6-helix undergoes a substantial rotation as well and undocks the neck linker from the motor domain's core. These allosteric interactions are absent for the motor domain in solution, see Fig. 4. | ||
It is interesting to note that our chemomechanical coupling scheme as summarized in Fig. 8 predicts that the rotation of the α4 helix precedes the undocking of the neck linker. The rotation of the α4 helix, which is part of the microtubule binding site, may act to release the motor domain from its strongly bound ATP state. Such a sequence of conformational transitions seems to be consistent with the motility model for monomeric KIF1A as reviewed in ref. 25. In the latter model, the KIF1A motor becomes loosely bound to the microtubule before it undergoes a conformational change that biases its diffusion towards the plus end of the filament.
We also determined the strand orientations within the β-sheet core for the different nucleotide states. For kinesin-3 attached to tubulin, we found a twist within this core, both during phosphate release and during ATP binding, see Table 1.
Since the members of the kinesin-3 family have motor domains that are close structural homologs, the allosteric coupling mechanism found here for KIF1A mouse kinesin should apply to other kinesin-3 motors as well. In addition, the recent observation26–28 that KIF1A motors can form dimers in vivo indicates that a similar allosteric coupling mechanism may even apply to other members of the kinesin superfamily but this remains to be studied. Apart from such an extension to different kinesins, our computational approach can be applied to other motor domains and mechanoenzymes as well because it is based on a relatively simple strategy: consider the mechanoenzyme in different states that are distinguished by their ligands or binding partners and determine, for these different states, the mobility of important subdomains within the protein. Ligand-induced changes in the mobilities will then reveal allosteric interactions between these subdomains and the ligands. Other possible targets for future applications of our computational approach include the rotary motor F1-ATPase35 and the actin-based stepping motor myosin V.36
4 Methods
4.1 Crystal structure of KIF1A's motor domain and code validation
The motor domain of KIF1A mouse kinesin consisting of 361 residues was studied in explicit water using MD simulations with atomic resolution. In order to validate our computational method, we first simulated the crystal structure of this protein in the ATP state and compared it with the corresponding X-ray crystal structure,5 protein data bank id 1VFW. The missing loops, L11 and L12, of KIF1A corresponding to residues 252–266 and 287–300, respectively, were modeled using the ModLoop server.37,38 The complete unit cell of the crystal, which contains four copies of the motor domain and 5139 water molecules, see Fig. 9, was constructed using the Computational Crystallography Toolbox (cctbx) plug-in for PyMOL.39 Using the X-ray crystal structure as the initial configuration, we performed 10 simulation runs with a 4 fs time step in an NPT ensemble at temperature T = 350 K and pressure P = 1 bar. Each run took 30 ns, which implies a total simulation time of 300 ns. | ||
| Fig. 9 Crystal structure of KIF1A mouse kinesin, protein data bank id 1VFW. The simulation box (black frame) is identical with the unit cell, which is a rectangular cuboid and contains four copies of the protein. The unit cell has the dimensions 4.26 nm × 5.55 nm × 15.67 nm. All protein molecules are displayed in ribbon representation. In addition, the four copies within the simulation box are depicted in transparent spacefilling representation and distinguished by four different colors. The blue background is provided by water molecules. | ||
In order to estimate the overall accuracy of our simulations, we saved the simulated configurations of the crystal every 2 ps which leads to a total number of 15 × 104 configurations. Using these configurations, we calculated the root-mean-square deviation (RMSD) of the simulated crystal from the X-ray crystal structure, averaging over all Cα atoms of the protein. This RMSD value was found to be 0.25 ± 0.02 nm, where the standard deviation was calculated using the 40 protein copies used in the 10 simulation runs. Thus, within the accuracy of this computation, the RMSD value of the simulated crystal is compatible with the spatial resolution of 0.23 nm as estimated for the X-ray crystal structure.5
For the motor domain in aqueous solution and attached to tubulin, we found characteristic rotations of the α4 and α6 helices. In order to estimate the accuracy of these simulation results, we also determined the RMSD values for the ends of these helices from the crystal structure simulations. The N- and C-termini of the α4-helix were found to have the RMSD values of 0.26 ± 0.07 nm and 0.32 ± 0.1 nm, respectively. Likewise, the RMSD values for the N- and C-termini of the α6-helix were found to be 0.19 ± 0.04 nm and 0.16 ± 0.03 nm. Thus, the RMSD values of the ends of these two helices are also compatible with the spatial resolution of the X-ray crystal structure.
We also analyzed the orientations of the α4- and α6-helices in our crystal simulations. We found that the α4- and α6-helices of the simulated crystal structure were rotated by 4° ± 1.6° and 4.6° ± 1.5°, respectively, compared to the X-ray crystal structure. Such a rotation angle is again compatible with the spatial resolution of 0.23 nm for the X-ray structure. Indeed, if we rotate the α4-helix, which has a length of 2.15 nm, around one of its ends by 4°, the other end of the helix is displaced by 0.15 nm. The same consideration applies to the α6-helix with a length of 2.18 nm.
4.2 Motor domain in solution
In solution, we studied the ATP, ADP, and Empty states of the motor domain, corresponding to bound ATP, bound ADP, and no bound nucleotide. As an initial configuration for the ATP state, we used again the X-ray structure of KIF1A mouse kinesin in the ATP state,5 protein data bank id 1VFW. As in the crystal structure simulations, the missing loops, L11 and L12, of the motor domain corresponding to residues 252–266 and 287–300, respectively, were modeled using the ModLoop server.37,38 The protein was placed in a dodecahedral box of initial edge length 9 nm containing 15263 simple point charge (SPC) water molecules40 and 4 Cl− ions to ensure electroneutrality of the system. The simulations were performed using periodic boundary conditions. The structure of the missing loops was energy minimized for 106 steps via steepest descent and equilibrated for 5 ns with a 1 fs time step in an NPT ensemble at temperature T = 300 K and pressure P = 1 bar. Afterwards the system was simulated at T = 350 K for 60 ns and a structure cluster analysis based on the Daura algorithm41 was performed to find the dominant conformation of the missing loops.
The ADP and Empty nucleotide states of the motor head were created from its ATP state by removing the γ-phosphate and ATP, respectively, from the nucelotide binding pocket. Both ATP and ADP were complexed with Mg2+ ions. Depending on the nucleotide state, three to five Cl− ions were placed in the boxes to ensure electroneutrality of the systems. These systems were energy minimized for 106 steps via steepest descent and then equilibrated for 10 ns with a 1 fs time step in an NPT ensemble at T = 300 K and P = 1 bar. To achieve better sampling of conformational space, each system was simulated 10 times using different initial velocities. Every production run continued 30 ns with a 4 fs time step and configurations were saved every 10 ps. So each system was simulated for a total time of 300 ns and 3 × 104 configurations were available for analysis. The production runs were conducted in an NPT ensemble at T = 350 K and P = 1 bar. The GROMOS43a1 force field was used for the motor domains and the ions. The GROMOS43a1 parameter set was also used to model ATP and ADP, apart from the partial charges which were taken from.42 A spherical cutoff of 1 nm was used for the short-range Lennard-Jones and electrostatic interactions; long range electrostatic interactions were taken into account by particle mesh Ewald summation.43 The Berendsen scheme for temperature and pressure control44 with the relaxation times of 0.1 ps and 1 ps, respectively, was used, and all simulations were performed using the GROMACS software package.45
4.3 Motor domain attached to tubulin
One motor domain of KIF1A mouse kinesin in the ATP state attached to a tubulin dimer,4 protein data bank id 2hxf, was taken as the initial configuration for the simulations. The missing loops L11 and L12 of the motor domain, and the missing regions of the E-hook of tubulins, corresponding to residues 440–451 and 438–455 for α- and β-tubulin, respectively, were modeled using the scheme described above. One example for the resulting configuration of the kinesin-3–tubulin complex is shown in Fig. S1 in the ESI.† The Empty and ADP states of the motor domain were created as in solution. The protein complexes were placed in dodecahedral boxes of initial edge length 13 nm with 45732 SPC water molecules in every box. Depending on the nucleotide state 48–50 Na+ ions were added to obtain electrically neutral systems, which were energy minimized, equilibrated and simulated using the protocol described above. Tubulin, GTP, and GDP were modeled using the GROMOS43a1 force field.
4.4 Analysis methods
Dominant conformations were obtained from a structure cluster analysis using the algorithm by Daura et al.41 The largest clusters were considered to provide the dominant contributions and were represented by the corresponding central configurations. Angles between regions of the motor domain in the different states were calculated using VMD.30 A principal component analysis was performed for the Cα atoms of the region of interest using the analysis tools of GROMACS.Two residues were considered to be hydrogen bonded when the distance between the donor X1H and the acceptor X2 did not exceed 0.35 nm and the angle X1HX2 exceeded 130°; here X1 represents a nitrogen, oxygen or sulfur atom, and X2 represents a nitrogen or oxygen atom.
4.5 Strand orientations within the β-sheet core
The β-sheet core of kinesin-3 consists of eight strands, β1 to β8, corresponding to the residues 54–57 for β1, 7–12 for β2, 324–330 for β3, 91–97 for β4, 237–248 for β5, 218–230 for β6, 138–150 for β7, and 153–156 for β8, in the spatial order of the strands and according to PDB ID 1VFW.For the β-sheet core of myosin, a twist has been found between neighboring β-strands.34 In order to investigate the possibility that analogous twists occur in the β-sheet core of kinesin-3, as recently proposed for kinesin-1 in ref. 32 and 33 the orientation of the strand βi extending from residue ni to residue ni + li was characterized by the unit vector êi, that points from the Cα-atom of residue ni + 1 to the Cα-atom of residue ni + li − 1.
The average tilt angle ϕij between two neighboring strands βi and βj was evaluated via ϕij = min (〈ψij〉, 180° − 〈ψij〉) with 〈ψij〉 ≡ 〈arccos(êi·êj)〉. For each state, the angle 〈ψ〉 represents an average over all configurations. The corresponding standard error was evaluated by averaging over the individual trajectories. As a result, the tilt angle ϕ34 between the strands β3 and β4 as well as the tilt angle ϕ45 between the strands β4 and β5 are found to change by about 10°, both during phosphate release and during ATP binding, whereas the tilt angle ϕ56 is found to be essentially independent of the nucleotide state, see Table 1 in the main text.
Acknowledgements
We thank an anonymous reviewer for useful comments about the processivity of monomeric KIF1A and possible twists within the β-sheet core.References
- F. J. Kull, E. P. Sablin, R. Lau, R. J. Fletterick and R. D. Vale, Nature, 1996, 380, 550–555 CrossRef CAS PubMed.
- F. Kozielski, S. Sack, A. Marx, M. Thormählen, E. Schönbrunn, V. Biou, A. Thompson, E.-M. Mandelkow and E. Mandelkow, Cell, 1997, 91, 985–994 CrossRef CAS.
- Y.-H. Song, A. Marx, J. Müller, G. Woehlke, M. Schliwa, A. Krebs, A. Hoenger and E. Mandelkow, EMBO J., 2001, 20, 6213–6225 CrossRef CAS PubMed.
- M. Kikkawa, E. Sablin, Y. Okada, H. Yajima, R. Fletterick and N. Hirokawa, Nature, 2001, 411, 439–445 CrossRef CAS PubMed.
- R. Nitta, M. Kikkawa, Y. Okada and N. Hirokawa, Science, 2004, 305, 678–683 CrossRef CAS PubMed.
- S. Rice, A. Lin, D. Safer, C. Hart, N. Naber, B. Carragher, S. Cain, E. Pechatnikova, E. Wilson-Kubalek, M. Whittaker, E. Pate, R. Cooke, E. Taylor, R. Milligan and R. Vale, Nature, 1999, 402, 778–784 CrossRef CAS PubMed.
- G. Skiniotis, T. Surrey, S. Altmann, H. Gross, Y.-H. Song, E. Mandelkow and A. Hoenger, EMBO J., 2003, 22, 1518–1528 CrossRef CAS PubMed.
- C. V. Sindelar, M. J. Budney, S. Rice, N. Naber, R. Fletterick and R. Cooke, Nat. Struct. Biol., 2002, 9, 844–848 CAS.
- S. Rice, Y. Cui, C. Sindelar, N. Naber, M. Matuska, R. Vale and R. Cooke, Biophys. J., 2003, 84, 1844–1854 CrossRef CAS.
- R. B. Case, D. W. Pierce, N. Hom-Booher, C. L. Hart and R. D. Vale, Cell, 1997, 90, 959–966 CrossRef CAS.
- D. D. Hackney, The Enzymes: Energy Couling and Molecular Motors, Academic Press, 2003, vol. XXIII, pp. 88–142 Search PubMed.
- S. M. Block, Biophys.J., 2007, 92, 2986–2995 CrossRef CAS PubMed.
- M. W. Elting and J. A. Spudich, Dev. Cell, 2012, 23, 1084–1091 CrossRef CAS PubMed.
- S. Liepelt and R. Lipowsky, Phys. Rev. Lett., 2007, 98, 258102 CrossRef.
- M. J. I. Müller, S. Klumpp and R. Lipowsky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4609–4614 CrossRef PubMed.
- F. Berger, C. Keller, S. Klumpp and R. Lipowsky, Phys. Rev. Lett., 2012, 108, 208101 CrossRef.
- W. Wriggers and K. Schulten, Biophys. J., 1998, 75, 646–661 CrossRef CAS.
- C. Hyeon and J. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17382–17387 CrossRef CAS PubMed.
- I. Aprodu, M. Soncini and A. Redaelli, J. Biomech., 2008, 41, 3196–3201 CrossRef PubMed.
- W. Hwang, M. Lang and M. Karplus, Structure, 2008, 16, 62–71 CrossRef CAS PubMed.
- A. Khalil, D. Appleyard, A. Labno, A. Georges, M. Karplus, A. Belcher, W. Hwang and M. Lang, PNAS, 2008, 105, 19247–19252 CrossRef CAS PubMed.
- M. Li and W. Zheng, Biochemistry, 2012, 51, 5022–5032 CrossRef CAS PubMed.
- R. Kanada, T. Kuwata, H. Kenzaki and S. Takada, PLoS Comput. Biol., 2013, 9, e1002907 CAS.
- Y. Okada, H. Yamazaki and Y. Sekineaizawa, Cell, 1995, 81, 769–780 CrossRef CAS.
- M. Hirokawa, R. Nitta and Y. Okada, Nat. Rev. Mol. Cell Biol., 2009, 10, 877–884 CrossRef PubMed.
- J. W. Hammond, D. Cai, T. L. Blasius, Z. Li, Y. Jiang, G. T. Jih, E. Meyhofer and K. J. Verhey, PLoS Biol., 2009, 7, e1000072 Search PubMed.
- K. J. Verhey and J. W. Hammond, Nat. Rev. Mol. Cell Biol., 2009, 10, 765–777 CrossRef CAS PubMed.
- Y. Yue, Y. Sheng, H.-N. Zhang, Y. Yu, L. Huo, W. Feng and T. Xu, Biochem. Biophys. Res. Commun., 2013, 435, 441–446 CrossRef CAS PubMed.
- N. Hirokawa, Y. Noda, Y. Tanaka and S. Niwa, Nat. Rev. Mol. Cell Biol., 2009, 10, 682–696 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- A. Goulet, W. M. Behnke-Parks, C. V. Sindelar, J. Major, S. S. Rosenfeld and C. A. Moores, J. Biol. Chem., 2012, 287, 44654–44666 CrossRef CAS PubMed.
- B. Gigant, W. Wang, B. Dreier, Q. Jiang, L. Pecqueur, A. Plückthun, C. Wang and M. Knossow, Nat. Struct. Mol. Biol., 2013, 20, 1001–1008 CAS.
- J. C. Cochran and F. J. Kull, Nat. Struct. Mol. Biol., 2013, 20, 920–921 CAS.
- P.-D. Coureux, A. L. Wells, J. Menetrey, C. M. Yengo, C. A. Morris, H. L. Sweeney and A. Houdusse, Nature, 2003, 425, 419–423 CrossRef CAS PubMed.
- T. Yoshidome, Y. Ito, M. Ikeguchi and M. Kinoshita, J. Am. Chem. Soc., 2011, 133, 4030–4039 CrossRef CAS PubMed.
- D. Trivedi, C. David, D. J. Jacobs and C. M. Yengo, Biophys. J., 2012, 102, 2545–2555 CrossRef CAS PubMed.
- A. Fiser, R. K. G. Do and A. Sali, Protein Sci., 2000, 9, 1753–1773 CrossRef CAS PubMed.
- A. Fiser and A. Sali, Bioinformatics, 2003, 19, 2500–2501 CrossRef CAS PubMed.
- P. D. Adams, P. v. Afonide, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- W. Wriggers and K. Schulten, Biophys. J., 1997, 73, 624–639 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden and H. Lee, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS PubMed.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Appendix on the choice of the Cartesian coordinate system with two Fig. S1 and S2 as well as Table S1 with neck linker displacements. See DOI: 10.1039/c3cp53367k |
| This journal is © the Owner Societies 2014 |