DOI:
10.1039/C4CE01618A
(Paper)
CrystEngComm, 2014,
16, 11064-11077
1,2,4,5-Benzenetetrasulfonic acid and 1,4-benzenedisulfonic acid as sulfo analogues of pyromellitic and terephthalic acids for building coordination polymers of manganese†
Received
4th August 2014
, Accepted 15th October 2014
First published on 16th October 2014
Abstract
The new polysulfonic acids H4B4S (1,2,4,5-benzenetetrasulfonic acid) and H2BDS (1,4-benzenedisulfonic acid) were used for the preparation of five manganese coordination polymers. The reactions of H4B4S and MnCO3 were performed in dimethylformamide (DMF) or N-methylpyrrolidone (NMP) as solvent in sealed glass ampoules at elevated temperatures. Two benzenetetrasulfonates could be obtained, [NH2(CH3)2]2{Mn(B4S)(DMF)2} (I) (P21/c, Z = 2, a = 942.02(5) pm, b = 1684.86(6) pm, c = 918.45(5) pm, β = 97.746(6)°, R1; wR2 (Io > 2σ(Io)) = 0.0357; 0.0771) and [HNMP]2{Mn(B4S)(NMP)2} (II) (P21/c, Z = 2, a = 931.7(1) pm, b = 1049.2(1) pm, c = 1944.9(1) pm, β = 113.529(3)°, R1; wR2 (Io > 2σ(Io)) = 0.0470; 0.0952). Both compounds exhibit anionic chains according to ∞1{Mn(B4S)2/2(L)2}2− (L = DMF, NMP). The Mn2+ ions are in octahedral coordination of oxygen atoms. The charge of the anionic chains is either compensated by dimethylammonium cations, [NH2(CH3)2+], or by protonated NMP molecules, HNMP+. Solvothermal reactions of H2BDS and MnCO3 in the solvents DMF, NMP and dimethylacetamide (DMA), respectively, yielded the disulfonates Mn(BDS)(DMF)2 (III) (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 1, a = 514.3(1) pm, b = 926.2(1) pm, c = 940.2(1) pm, α = 93.552(8)°, β = 99.993(7)°, γ = 99.237(7)°, R1; wR2 (Io > 2σ(Io)) = 0.0290; 0.0637), Mn(BDS)(DMA)2 (IV) (P, Z = 2, a = 936.00(4) pm, b = 984.94(4) pm, c = 1034.75(5) pm, α = 81.606(2)°, β = 88.941(2)°, γ = 82.364(2)°, R1; wR2 (Io > 2σ(Io)) = 0.0387; 0.1046) and Mn(BDS)(NMP)2 (V) (P, Z = 2, a = 962.97(3) pm, b = 976.76(3) pm, c = 1034.23(3) pm, α = 89.371(1)°, β = 78.839(1)°, γ = 87.604(2)°, R1; wR2 (Io > 2σ(Io)) = 0.0243; 0.0713). In the crystal structures of the three compounds, the octahedrally coordinated Mn2+ ions are linked by the disulfonate ions to layers according to ∞2{Mn(BDS)4/4(L)2} (L = DMF, DMA, NMP). The thermal analysis of all compounds shows that they can be desolvated completely and that the remaining solvent-free sulfonates exhibit decomposition temperatures up to 500 °C that is exceptionally high when compared to the respective carboxylates. Temperature-dependent magnetic susceptibility measurements of [NH2(CH3)2]2{Mn(B4S)(DMF)2} show paramagnetic behavior of the Mn2+ ions.
, Z = 1, a = 514.3(1) pm, b = 926.2(1) pm, c = 940.2(1) pm, α = 93.552(8)°, β = 99.993(7)°, γ = 99.237(7)°, R1; wR2 (Io > 2σ(Io)) = 0.0290; 0.0637), Mn(BDS)(DMA)2 (IV) (P, Z = 2, a = 936.00(4) pm, b = 984.94(4) pm, c = 1034.75(5) pm, α = 81.606(2)°, β = 88.941(2)°, γ = 82.364(2)°, R1; wR2 (Io > 2σ(Io)) = 0.0387; 0.1046) and Mn(BDS)(NMP)2 (V) (P, Z = 2, a = 962.97(3) pm, b = 976.76(3) pm, c = 1034.23(3) pm, α = 89.371(1)°, β = 78.839(1)°, γ = 87.604(2)°, R1; wR2 (Io > 2σ(Io)) = 0.0243; 0.0713). In the crystal structures of the three compounds, the octahedrally coordinated Mn2+ ions are linked by the disulfonate ions to layers according to ∞2{Mn(BDS)4/4(L)2} (L = DMF, DMA, NMP). The thermal analysis of all compounds shows that they can be desolvated completely and that the remaining solvent-free sulfonates exhibit decomposition temperatures up to 500 °C that is exceptionally high when compared to the respective carboxylates. Temperature-dependent magnetic susceptibility measurements of [NH2(CH3)2]2{Mn(B4S)(DMF)2} show paramagnetic behavior of the Mn2+ ions.
Introduction
The connection of metal ions or metal oxo-clusters by polydentate ligands leads to frameworks of various dimensionalities with respect to the linkage. Chain-type compounds (1D), layer structures (2D), and complex networks (3D) have been reported.1,2 They are usually named coordination polymers (CPs) and those compounds with frameworks exhibiting large open pores are referred to as metal–organic frameworks (MOFs).3 By the choice of the metal that is used for building CPs and MOFs, specific properties can be implemented. For example, rare earth metals may lead to interesting luminescence properties,4,5 while transition metals like Mn, Fe, Co and Ni can cause paramagnetism or even cooperative magnetic phenomena.5–7 The dimensionality and the porosity of CPs and MOFs can be tuned by the choice of the polydentate ligands, the so-called linkers.8–10 The majority of used linkers are polycarboxylate anions and neutral nitrogen-containing heterocycles. One of the reasons for the extensive use of polycarboxylates is that the respective polycarboxylic acids are commercially available in a great variety or that they can at least be prepared very easily. Unfortunately, this is not the case for the sulfo analogues of these acids, the so-called polysulfonic acids. Only a limited number of these acids are available and most of the compounds are even lacking reliable synthesis protocols. On the other hand, it has been shown that polysulfonate anions are interesting linkers because they show different coordination chemistry compared to carboxylates. For example, polysulfonates are weaker ligands making them good candidates for the formation of very flexible host structures. This has been intensively studied for those polysulfonic acids which are available up to now.11,12 However, it remains a problem that a lot of potentially interesting polysulfonic acids were not accessible. This was especially true for the analogues of very simple and widely used polycarboxylic acids like 1,4-benzenedicarboxylic acid (“terephthalic acid”), 1,3,5-benzenetricarboxylic acid (“trimesic acid”), 1,2,4,5-benzenetetrasulfonic acid (“pyromellitic acid”), and 1,2,3,4,5,6-benzenehexasulfonic acid (“mellitic acid”). Thus, some years ago, we initiated a research program aiming at the development of simple and scalable preparation routes for polysulfonic acids and their use for the preparation of new CPs and MOFs. In the course of these investigations, we were also able to prepare the sulfo analogues of the abovementioned carboxylic acids (Fig. 1).13–15 It turned out that the metal salts of these acids usually show low-dimensional structures (1D and 2D) with the metals additionally coordinated by solvent molecules. In all cases, however, the solvent can be removed by heating without decomposition of the organic linker. In some cases, the desolvated compounds are thermally stable up to 600 °C that is remarkably high, especially when compared to the respective carboxylates. This is of special interest if temperature sensible functions of the desolvated compounds like gas storage are envisioned. As a further result of our ongoing research, we here present the new manganese compounds of 1,4-benzenedisulfonic acid (H2BDS, 1) and 1,2,4,5-benzenetetrasulfonic acid (H4B4S, 2).
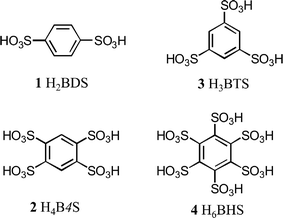 |
| | Fig. 1 Selected polysulfonic acids which have been prepared in our groups as sulfo analogues of well-known polycarboxylic acids. The manganese salts of 1 and 2 are presented in this work. | |
Experimental section
Preparation of compounds
General.
The reactions were carried out in thick-walled glass ampoules (length: 300 mm, diameter: 16 mm). The ampoules were loaded with the reactants, torch-sealed under vacuum, and placed in a resistance furnace. The ampoules are heated up to 105 °C within 6 h, held for 24 h and cooled down to room temperature within 96 h. The products were obtained as colorless crystals which were separated from the supernatant by decantation. [NH2(CH3)2]2{Mn(B4S)(DMF)2}: 20 mg (0.043 mmol) of H4B4S, 14 mg (0.122 mmol) of MnCO3, and 3 ml of DMF. [HNMP]2{Mn(B4S)(NMP)2}: 20 mg (0.043 mmol) of H4B4S, 5 mg (0.043 mmol) of MnCO3, and 3 ml of NMP. Mn(BDS)(DMF)2: 20 mg (0.073 mmol) of H2BDS, 17 mg (0.148 mmol) of MnCO3, and 3 ml of DMF. Mn(BDS)(DMA)2: 20 mg (0.073 mmol) of H2BDS, 8 mg (0.070 mmol) of MnCO3, and 3 ml of DMA. Mn(BDS)(NMP)2: 20 mg (0.073 mmol) of H2BDS, 17 mg (0.148 mmol) of MnCO3, and 3 ml of NMP. The acids H2BDS and H4B4S were prepared as previously described.13–15 MnCO3 turned out to be the best starting product, while the use of manganese acetate or nitrate usually leads to undesired by-products (solvates of the salts). However, even under different reaction conditions, the product usually contains traces of MnCO3 that is not seen in XRD but in magnetochemistry.
IR spectroscopy
IR data were collected with a Bruker Tensor 27 spectrometer using the attenuated total reflection (ATR) method on some dried single crystals. The specimen was prepared and placed onto the detector head in a glove box and transferred to the spectrometer under inert atmosphere. The IR data were processed with the OPUS 6.5 program.16
[NH2(CH3)2]2{Mn(B4S)(DMF)2}.
IR(ATR): 3471 (w), 3118 (m), 2807 (w), 2451 (w), 1651 (s), 1614 (m), 1465 (m), 1441 (m), 1376 (m), 1309 (w), 1274 (m), 1239 (s), 1183 (s), 1136 (s), 1109 (s), 1041 (s), 937 (m), 892 (m), 856 (m), 828 (m), 686 (s), 669 (m), 644 (s), 573 (m), 546 (s) cm−1. [HNMP]2{M(B4S)(NMP)2}: 3548 (m), 3461 (w), 3368 (m), 3095 (m), 1656 (s), 1518 (m), 1476 (m), 1459 (m), 1410 (m), 1311 (m), 1240 (s), 1177 (s), 1107 (s), 1045 (s), 1014 (s), 980 (m), 932 (m), 685 (s), 665 (s), 642 (s), 566 (m) cm−1. Mn(BDS)(DMF)2: 2992 (w), 2938 (w), 2898 (w), 1652 (s), 1501 (w), 1440 (w), 1415 (m), 1389 (m), 1237 (s), 1184 (s), 1139 (s), 1109 (s), 1050 (s), 1011 (s), 870 (w), 846 (m), 685 (m), 664 (s), 579 (s), 570 (s), 528 (w) cm−1. Mn(BDS)(DMA)2: 3542 (m), 3347 (m), 3221 (m), 3097 (w), 1633 (s), 1470 (w), 1393 (m), 1167 (s), 1128 (s), 1109 (s), 1035 (s), 998 (s), 833 (s), 663 (s), 564 (s), 531 (w) cm−1. Mn(BDS)(NMP)2: 3551 (m), 3345 (m), 3225 (m), 3097 (w), 1633 (m), 1394 (m), 1169 (s), 1131 (s), 1110 (s), 1042 (s), 1002 (s), 834 (s), 673 (s), 565 (s), 535 (m) cm−1.
X-ray structure determination
Single crystals of the compounds [HNMP]2{Mn(B4S)(NMP)2}, Mn(BDS)(DMF)2, Mn(BDS)(DMA)2, and Mn(BDS)(NMP)2 were selected under protecting oil and transferred into the cold nitrogen stream (120 K) of a single-crystal diffractometer (BRUKER APEX II). The crystals of [NH2(CH3)2]2{Mn(B4S)(DMF)2} are measured at 296 K on a single-crystal diffractometer (Stoe IPDS I). The collection of intensity data was performed at the abovementioned temperature for the respective best specimen. The structure solutions were successful by applying direct methods, and the structures were expanded by Fourier techniques. Refinement of the structures with introduction of anisotropic displacement parameters for all non-hydrogen atoms was performed after the data have been corrected for absorption effects. All calculations were performed with the SHELX program, and the data were processed with the programs X-Red and X-Shape.17 After the refinement of the structure [HNMP]2{Mn(B4S)(NMP)2}, a large electron density remained which could be attributed to the disorder of the [HNMP]+ cation and the SO3 groups of the benzenetetrasulfonate anions. The disorder was well resolved using split positions for the respective molecules leading to an occupancy ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 for the SO3 groups of the B4S4− anions and 7:3 for the [HNMP]+ cation and gave an R1 = 0.0387. Also in the compound Mn(BDS)(DMA)2, a disorder of the solvent molecule occurs. The DMA molecule with the numbering O2/C21/C22/N2/C23/C24 has two arrangements. The disorder was well resolved using split positions for the DMA molecules leading to an occupancy ratio of 7.5:2.5 and gave an R1 = 0.0348. Tables 4 and 5 give details of the data collection and the obtained crystallographic data. In Tables 6 and 7, important distances are summarized.
:4 for the SO3 groups of the B4S4− anions and 7:3 for the [HNMP]+ cation and gave an R1 = 0.0387. Also in the compound Mn(BDS)(DMA)2, a disorder of the solvent molecule occurs. The DMA molecule with the numbering O2/C21/C22/N2/C23/C24 has two arrangements. The disorder was well resolved using split positions for the DMA molecules leading to an occupancy ratio of 7.5:2.5 and gave an R1 = 0.0348. Tables 4 and 5 give details of the data collection and the obtained crystallographic data. In Tables 6 and 7, important distances are summarized.
Thermal analysis
The investigation of the thermal behavior was performed using a TGA/DSC apparatus (Mettler-Toledo GmbH, Schwerzenbach, Switzerland). In a flow of dry oxygen, about 5 mg of the respective compound were placed in a corundum crucible and heated at a rate of 10 K min−1 up to 1050 °C. The collected data were processed using the software of the analyzer (Mettler-Toledo STARe V9.3).18 Thermal decomposition data for [NH2(CH3)2]2{Mn(B4S)(DMF)2}, [HNMP]2{Mn(B4S)(NMP)2}, Mn(BDS)(DMF)2, Mn(BDS)(DMA)2 and Mn(BDS)(NMP)2 are presented in Tables 2 and 3 and Fig. 8 and 10.
Powder diffraction
XRD measurements were measured with the help of the powder diffractometer STADIP using Cu-Kα radiation (λ = 154.06 pm). The compounds under investigation were prepared on a flat sample holder. The diffraction data were processed with the WinXPow 2007 program package.19
Magnetic property measurements
The magnetic property measurements were carried out using a Quantum Design Physical Property Measurement System using a vibrating sample magnetometer. The measurement was performed in the temperature range of 3–300 K with flux densities up to 80 kOe. 15.569 mg of the polycrystalline sample was packed in a polypropylene capsule and fixed to a brass sample holder rod.
Results and discussion
Crystal structures
Benzenetetrasulfonates.
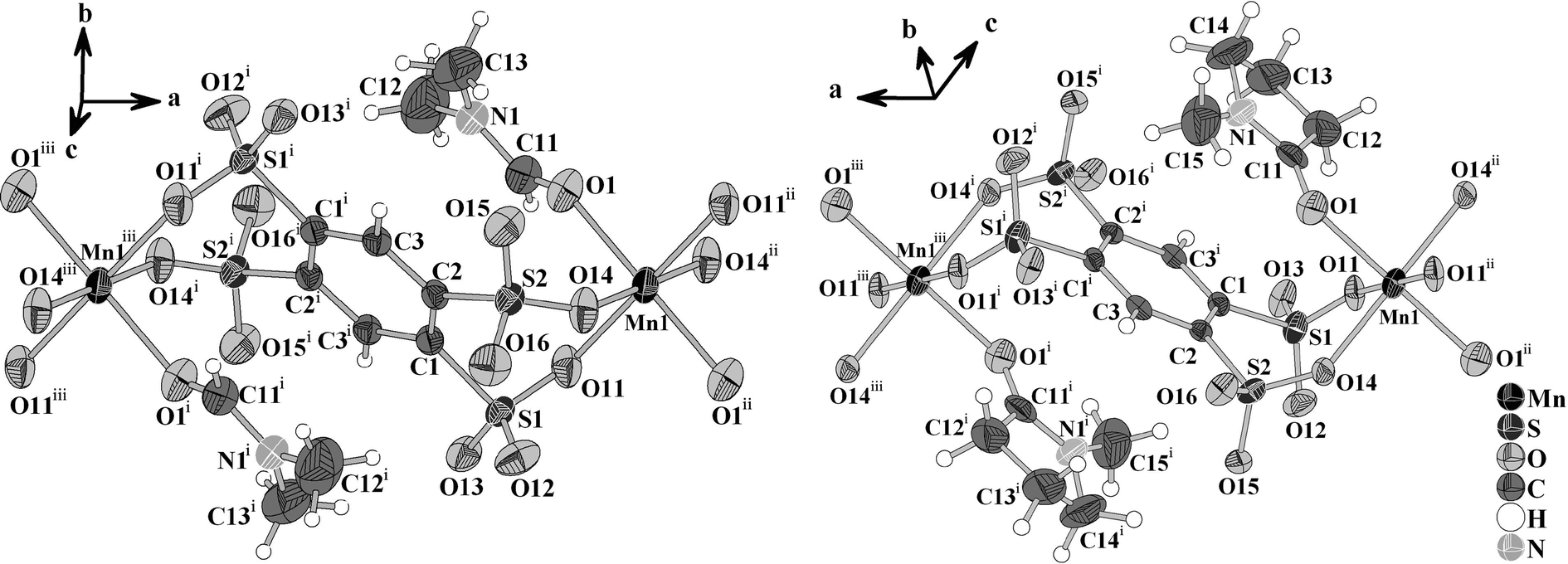
The crystal structures of [NH2(CH3)2]2{Mn(B4S)(DMF)2} (I) and [HNMP]2{Mn(B4S)(NMP)2} (II) exhibit anionic chains according to ∞1{Mn(B4S)2/2(L)2}2− (L = DMF, NMP). Within the chains, both the Mn2+ ions and the centroids of the benzene rings of the anions are located on special crystallographic positions with −1 site symmetry (Wyckoff sites 2b and 2a). The Mn2+ ions are coordinated by two bidentate B4S4− ligands and two solvent molecules (Fig. 2). The resulting [MnO6] octahedra are quite regular and display distances Mn–O of 217.1(2), 215.4(2) and 216.4(2) pm for compound I. The longest bond is found for the coordinating DMF molecules, and the shorter ones are found for the B4S4− ligands. In II, the situation is opposite and the shortest distance Mn–O of 215.7(2) pm is that to the NMP molecule, while the sulfonate ligands shows slightly larger values of 218.3(3) and 219.1(4) pm, respectively. Another significant difference in the anionic chains of both compounds is the orientation of the [SO3] moieties of the tetrasulfonates with respect to each other. When viewed along the S⋯S direction of neighboring [SO3] groups, it is obvious that the oxygen atoms have a staggered conformation in I and an eclipsed one in II (Fig. 3).
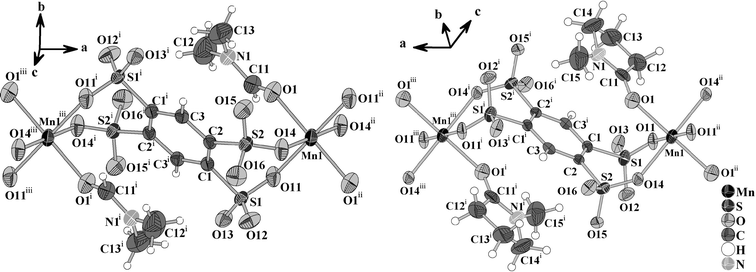 |
| | Fig. 2 Sections of the anionic chains {Mn(B4S)(L)2}2− (L = DMF, NMP) in I (left) and II (right). The Mn atoms and the centroids of the benzene ring are located on special crystallographic sites bearing inversion symmetry. The displacement ellipsoids are drawn at a 50% probability level. Symmetry codes: I: (i) = −x, −y, −z; (ii) = 1 − x, −y, −z; (iii) = −1 + x, y, z; II: (i) = −x, −1 − y, −1 − z; (ii) = −1 − x, −1 − y, −1 − z; (iii) = 1 + x, y, z. | |
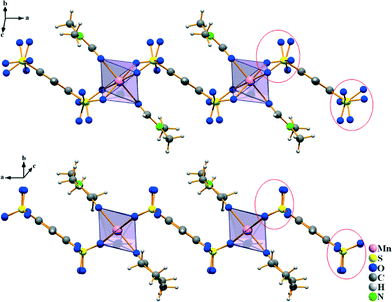 |
| | Fig. 3 A closer look on the anionic chains in I (top) and II (bottom) shows the differences between the compounds. When viewed along the S⋯S direction of neighboring [SO3] groups, the oxygen atoms are either in a staggered (I) or an eclipsed (II) conformation (as emphasized by the red circles). | |
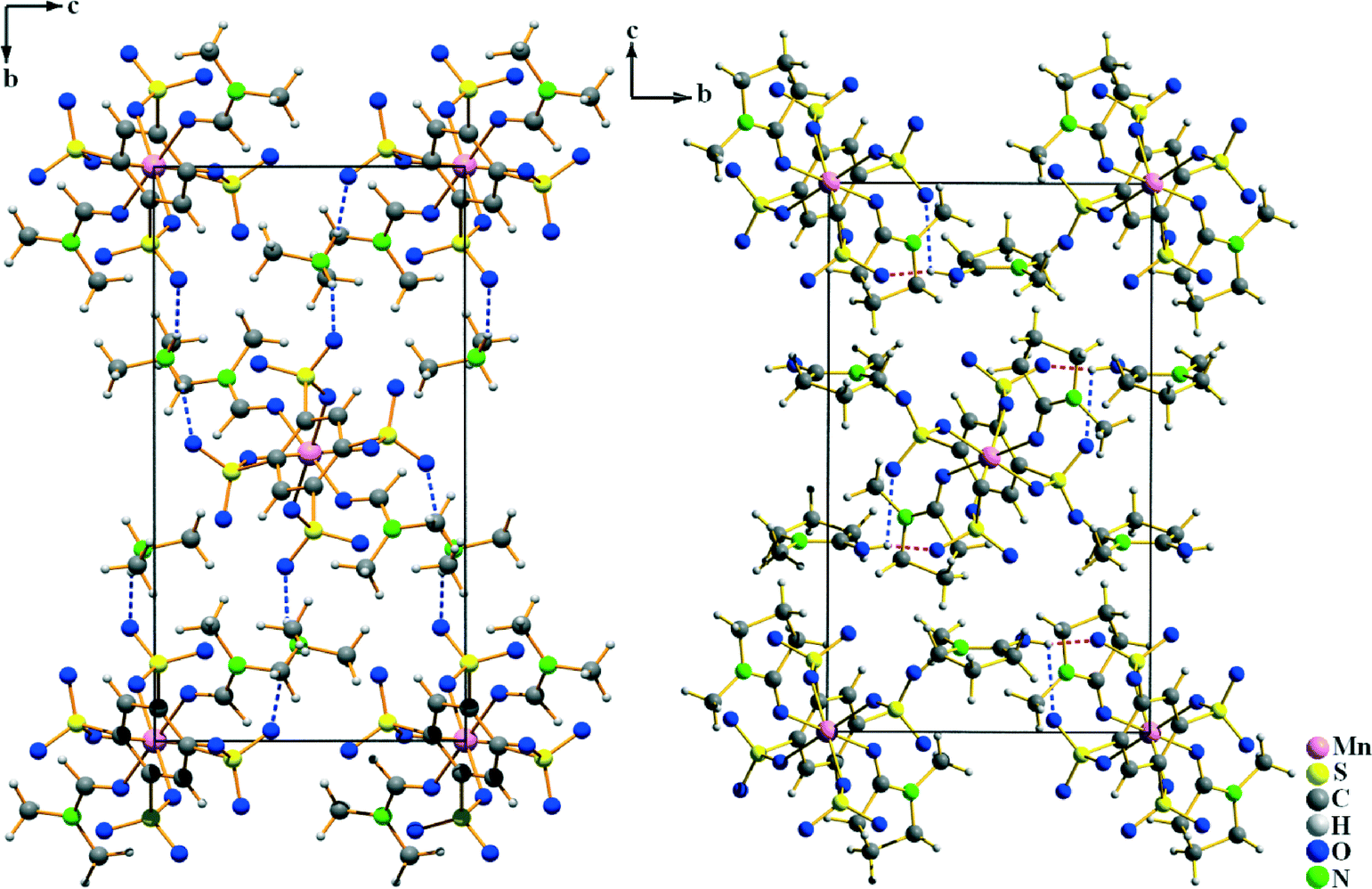
The ∞1{Mn(B4S)(L)2}2− chains in both compounds are aligned along the [100] direction (Fig. 4). They are arranged in densest rod packing fashion and their charge is compensated by dimethylammonium cations (in I) and by protonated NMP molecules (in II), respectively. The [NH2(CH3)2]+ originate obviously from the partial decomposition of the DMF solvent. The protonation of the NMP molecule can be easily identified by inspection of the C–O distance. It is found at 128.5(6) pm for the [HNMP]+ ion, while the respective distance is 123.7(3) pm in the manganese coordinated NMP molecule. Furthermore, the proton can be found in the Fourier map during the crystal structure refinement, even if it was refined by a constrained model. For both of the different cations, hydrogen bonds to the non-coordinating oxygen atoms of the [SO3] group could be identified. In I, the donor–acceptor distances D–A are found at 283.1(3) and 291.5(3) pm and the angles ∠DHA are 155(3)° and 160(3)°, hinting at medium–strong hydrogen bonds.20 In compound II, the hydrogen bonds with donor–acceptor distances of 243.3(4) and 283.8(5) pm with the respective angles ∠DHA being 136(5)° and 115(4)° (cf.Table 1) are quite strong.20 The observed hydrogen bonds are emphasized in Fig. 4.
 |
| | Fig. 4 Crystal structures of I (left) and II (right) viewed along the [100] direction of the monoclinic unit cells. In this direction, the anionic chains shown in Fig. 3 are oriented. The charge compensation is achieved by [NH2(CH3)2]+ and [HNMP]+ cations which also show hydrogen bonding to neighboring oxygen atoms of [SO3] groups. These are emphasized as dashed lines in red (strong H-bonds) and blue (medium strong H-bonds). | |
Table 1 Hydrogen bonds in [NH2(CH3)2]2{Mn(B4S)(DMF)2} (I) and [HNMP]2{Mn(B4S)(NMP)2} (II) with d(H⋯A) < r(A) + 200 pm and ∠DHA > 110 deg. The strong hydrogen bonds are marked in bolda
|
|
D–H |
d(D–H)/pm |
d(H⋯A)/pm |
∠DHA/° |
d(D⋯A)/pm |
A |
|
Symmetry codes: (i) = x, −1/2 − y, 1/2 + z; (ii) = 1 + x, −3/2 − y, 1/2 + z.
|
|
I
|
N2–H21 |
93(4) |
202(4) |
160(3) |
291.5(3) |
O13i |
| N2–H22 |
100(4) |
190(4) |
155(3) |
283.1(3) |
O16 |
|
II
|
O2–H1 |
85.0(1)
|
175(4)
|
136(5)
|
243.3(4)
|
O15
ii
|
| O2–H1 |
85.0(1) |
237(5) |
115(4) |
283.8(5) |
O12ii |
Benzenedisulfonates.
The manganese benzenedisulfonates Mn(BDS)(DMF)2 (III), Mn(BDS)(DMA)2 (IV), and Mn(BDS)(NMP)2 (V) crystallize with triclinic symmetry. The structures are similar in the way that the Mn2+ ions are octahedrally coordinated by oxygen atoms that belong to four disulfonate ligands and two solvent molecules (Fig. 5). The solvent molecules are in trans orientation with respect to each other at the apices of the [MnO6] octahedra. In III, the [MnO6] octahedron bears inversion symmetry, i.e. the Mn2+ ion is located on a special site of the triclinic unit cell (Wyckoff position 1g). In IV and V, the manganese atoms are situated on general sites (Wyckoff position 2i). In all of the three compounds, the distances Mn–O lie in a narrow range between 213 and 219 pm (Table 7).
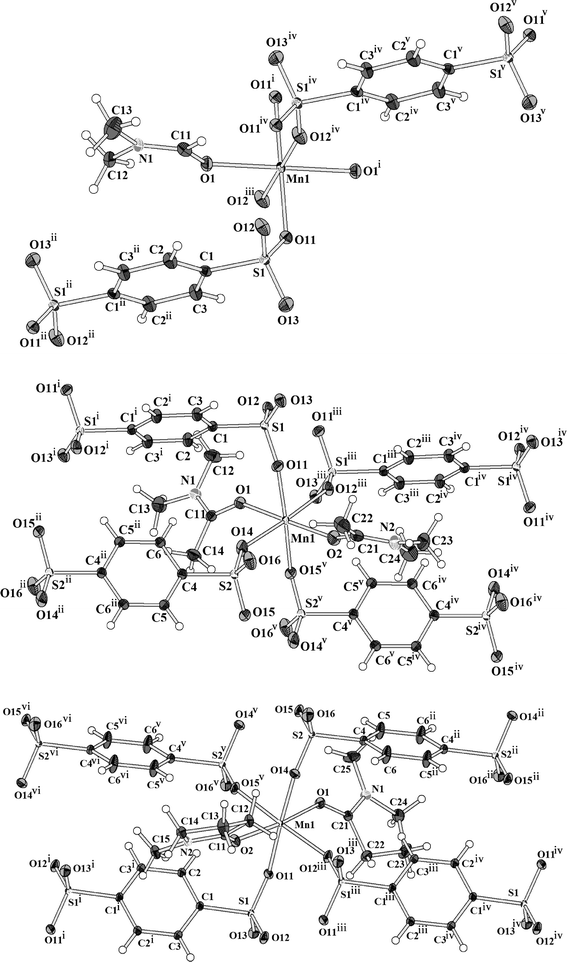 |
| | Fig. 5 Coordination of the Mn2+ ions in Mn(BDS)(DMF)2 (III, top), Mn(BDS)(DMA)2 (IV, middle), and Mn(BDS)(NMP)2 (V, bottom). Note that in the structure of III the manganese site show inversion symmetry. The displacement ellipsoids are drawn at a 50% probability level. For compounds IV and V, the asymmetric unit has two half BDS2− moieties, each lying at independent inversion centres. Symmetry codes: III: (i) = −x, 1 − y, 1 − z; (ii) = 1 − x, 2 − y, 1 − z; (iii) = −1 + x, y, z; (iv) = 1 − x, 1 − y, 1 − z; (v) = x, −1 + y, z; IV: (i) = 1 − x, 1 − y, 1 − z; (ii) = 1 − x, 1 − y, 2 − z; (iii) = 1 − x, −y, 1 − z; (iv) = x, −1 + y, z; (v) = 1 − x, −y, 2 − z; V: (i) = −x, −y, 2 − z; (ii) = 2 − x, −y, 1 − z; (iii) = 1 − x, −y, 2 − z; (iv) = 1 + x, y, z; (v) = 1 − x, −y, 1 − z; (vi) = −1 + x, y, z. | |
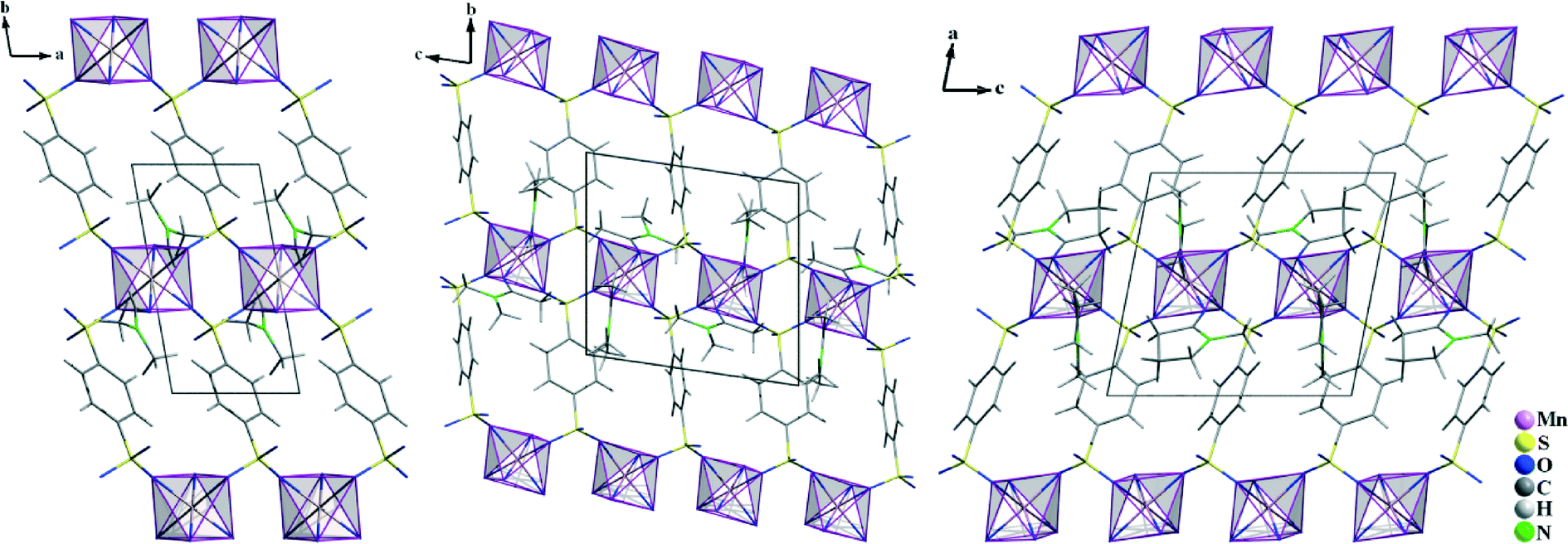
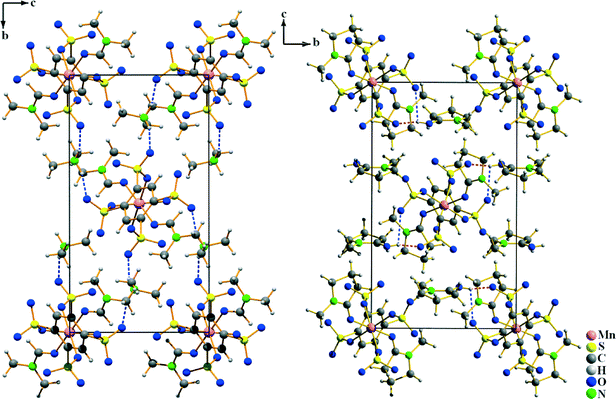
In all of the three compounds, the Mn2+ ions are linked by the benzenedisulfonate ligands to layers according to ∞2{Mn(BDS)4/2(L)2/1} (L = DMF, DMA, NMP) (Fig. 6). The structural differences arise from the arrangement of the BDS2− anions with respect to each other in the individual structures. In III, the benzenedisulfonate anions have the same orientation, while in IV and V, the single BDS2− anions are twisted with respect to each other (Fig. 6). The solvent molecules L point into the interlayer spacing, and weak interactions to the non-coordinating oxygen atoms of the [SO3] moieties of adjacent layers can be assumed.
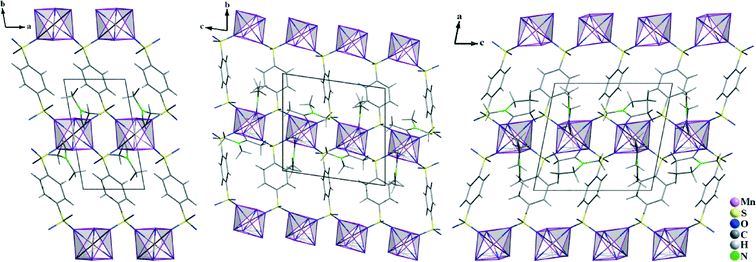 |
| | Fig. 6 Crystal structures of Mn(BDS)(DMF)2 (III, left), Mn(BDS)(DMA)2 (IV, middle) and Mn(BDS)(NMP)2 (V, right). For all structures, the view is onto the ∞2{Mn(BDS)4/2(L)2/1} layers. The structures differ due to the different orientation of the BDS2− anions with respect to each other. | |
Thermal behaviour
Thermal decomposition of I and II.
The thermal decomposition of [NH2(CH3)2]2{Mn(B4S)(DMF)2} (I) and [HNMP]2{Mn(B4S)(NMP)2} (II) was monitored by means of DSC/TG measurements in a flow of dry oxygen (Fig. 7 and Table 2). Both compounds decompose in multi-step processes; however, these steps are not clearly resolved in any case. Nevertheless, it seems clear that the first endothermic steps can be attributed to the loss of the two solvent molecules and the decomposition of the cations under release of NHMe2 for compound I and NMP for compound II, respectively. This is in good accordance with the observed mass loss of 33% (calcd. 33%) for I and 47% (calcd. 46%) for II (cf.Table 2). In both cases, this should lead formally to the formation of the dihydrogentetrasulfonate “Mn(H2B4S)”, which subsequently decomposes in the course of three exothermic steps. According to XRD investigations at 650 °C (compound I) and 600 °C (compound II), respectively, the decomposition leads to a mixture of MnSO4 and Mn3O4 (Fig. 8).21,22 For I, small amounts of MnO2 are also observed in the diffraction pattern.23 In a final step, the manganese(II)sulfate decomposes to Mn2O3 according to the XRD pattern of the final residue.24
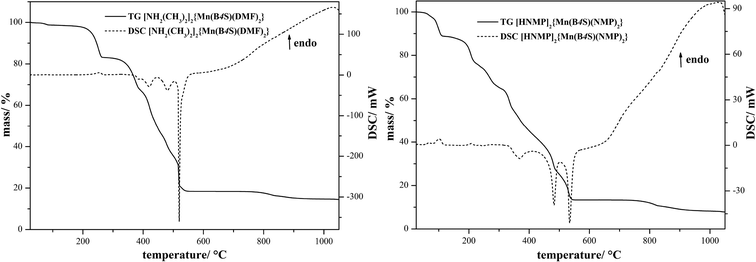 |
| | Fig. 7 DSC/TG diagrams of the thermal decompositions of [NH2(CH3)2]2{Mn(B4S)(DMF)2} (I) and [HNMP]2{Mn(B4S)(NMP)2} (II). The decompositions are composed of endothermic steps which can be attributed to the loss of solvent molecules and decompositions of the cations [NH2(CH3)2]+ and [HNMP]+, respectively. Subsequently, the intermediate product, most probably Mn(H2B4S), decomposes in various exothermic steps, leading finally to Mn2O3 and Mn3O4. | |
Table 2 Thermal decomposition data of the anionic chain compounds I and II
| [NH2(CH3)2]2{Mn(B4S)(DMF)2} (I) |
| Stage |
T
onset/°C |
T
end/°C |
T
max/°C |
Mass loss obsd./% |
Mass loss calcd./% |
Elimination/decomposition |
| 1 |
40 |
85 |
79 |
2 |
33 |
Loss of two equiv. of NH(CH3)2 and two equiv. of DMF |
| 2 |
214 |
270 |
260 |
15 |
| 3 |
326 |
392 |
375 |
16 |
| 4 |
392 |
442 |
420 |
21 |
— |
Decomposition to Mn3O4 and MnSO4 and MnO2 |
| 5 |
455 |
512 |
480 |
10 |
| 6 |
512 |
550 |
520 |
18 |
| 7 |
770 |
845 |
830 |
4 |
— |
Decomposition to Mn2O3 and 4Mn3O4 (calcd. 11%) |
| ∑ |
|
|
|
86 |
89 |
|
|
|
| [HNMP]2{Mn(B4S)(NMP)2} (II) |
| 1 |
51 |
108 |
65; 100 |
11 |
23 |
Loss of two equiv. of NMP |
| 2 |
185 |
225 |
208 |
13 |
| 3 |
225 |
300 |
235; 268 |
12 |
23 |
Loss of two equiv. of NMP |
| 4 |
308 |
352 |
335 |
11 |
| 5 |
152 |
400 |
370 |
11 |
— |
Decomposition to Mn3O4 and MnSO4 |
| 6 |
466 |
500 |
484 |
17 |
| 7 |
506 |
550 |
535 |
12 |
| 8 |
720 |
830 |
826 |
5 |
— |
Decomposition to Mn2O3 and Mn3O4 (calcd. 9%) |
| ∑ |
|
|
|
92 |
91 |
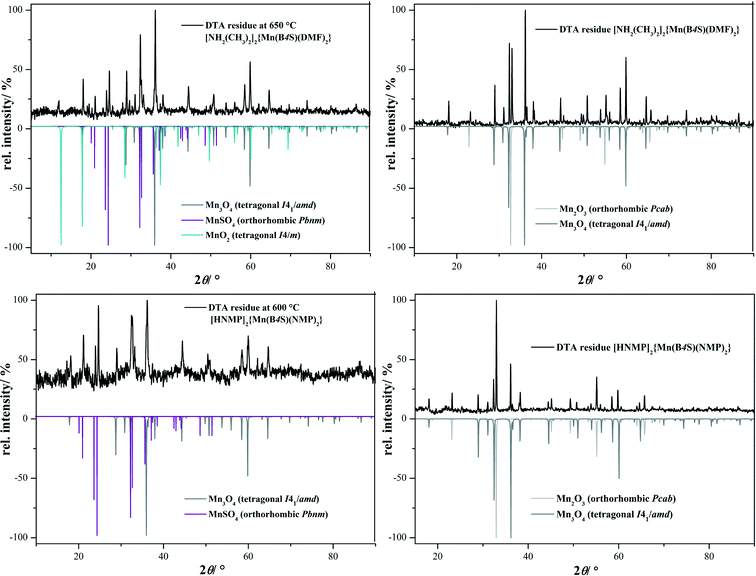 |
| | Fig. 8 Powder patterns of the intermediates (left) and final decomposition products (right) of I (top) and II (bottom). | |
Thermal decomposition of III, IV, and V.
The thermal decomposition of compounds III, IV, and V was investigated by DSC/TG measurements in a flow of dry oxygen (Fig. 9 and Table 3). All of the compounds decompose in three-step processes. The first steps are endothermic and can be attributed to the loss of the solvent molecules. The solvent-free manganese benzenedisulfonates Mn(BDS) show high thermal stabilities, and the decompositions start clearly above 500 °C. These decompositions lead in a first step to MnSO4 (for IV, a small amount of Mn3O4 is also seen, cf.Fig. 10).22,23,26 The anhydrous manganese sulfate occurs in two different modifications, as can be seen from XRD measurements of the decomposition products obtained at 650 °C (Fig. 10). As a result of the preparation of the sample on a flat sample holder, reflections of the hydrate Mn(SO4)·2H2O might also occur.27 In any case, the manganese sulfate decomposes further leading essentially to Mn3O4, although in the case of compound II Mn2O3 is also found as a minor decomposition product (Fig. 10).23,25 The quantification of the crystalline phases contained in powder samples of the final product was estimated with respect to the reflection intensities as determined by Rietveld refinements.28
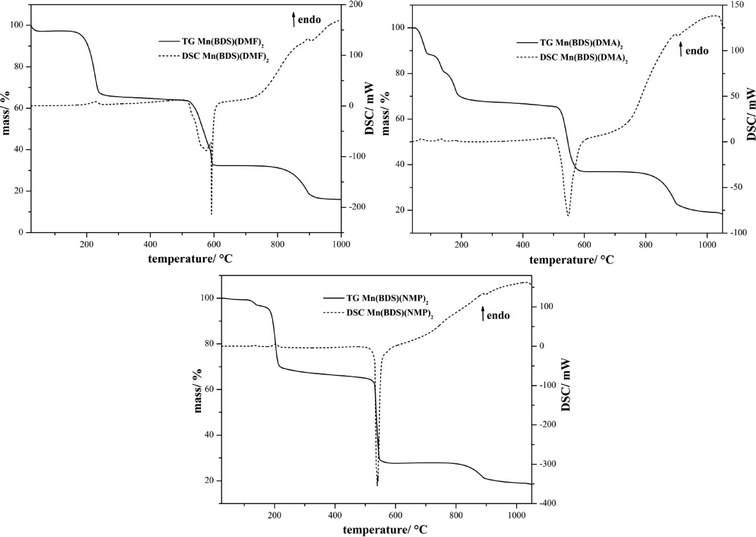 |
| | Fig. 9 TG/DSC diagram of the thermal decomposition of Mn(BDS)(DMF)2 (III), Mn(BDS)(DMA)2 (IV), and Mn(BDS)(NMP)2 (V). The decomposition of all compounds starts with the loss of the solvent molecules. Finally, the solvent-free compounds decompose exothermally at temperatures above 500 °C. | |
Table 3 Thermal decomposition data of the benzenedisulfonates III, IV, and V
| Mn(BDS)(DMF)2 (III) |
| Stage |
T
onset/°C |
T
end/°C |
T
max/°C |
Mass loss obsd./% |
Mass loss calcd./% |
Elimination/decomposition |
| I |
190 |
245 |
230 |
34 |
33 |
Loss of two equiv. of DMF |
| II |
516 |
607 |
577; 592 |
33 |
32 |
Decomposition to MnSO4 (calcd. 34%) |
| III |
860 |
906 |
895 |
17 |
17 |
Decomposition to Mn3O4 (calcd. 17%) |
| ∑ |
|
|
|
84 |
82 |
|
|
|
| Mn(BDS)(NMP)2 (IV) |
| I |
110 |
220 |
135; 204 |
34 |
40 |
Loss of two equiv. of NMP |
| II |
525 |
560 |
540 |
39 |
40 |
Decomposition to MnSO4 (calcd. 31%) |
| III |
775 |
896 |
890 |
9 |
— |
Decomposition to Mn2O3 and 9Mn3O4 (calcd. 16%) |
| ∑ |
|
|
|
82 |
84 |
|
|
|
| Mn(BDS)(DMA)2 (V) |
| I |
50 |
195 |
68; 135; 180 |
34 |
37 |
Loss of two equiv. of DMA |
| II |
508 |
593 |
547 |
31 |
— |
Decomposition to Mn3O4 and MnSO4 |
| III |
868 |
904 |
895 |
20 |
— |
Decomposition to Mn3O4 (calcd. 16%) |
| ∑ |
|
|
|
85 |
84 |
|
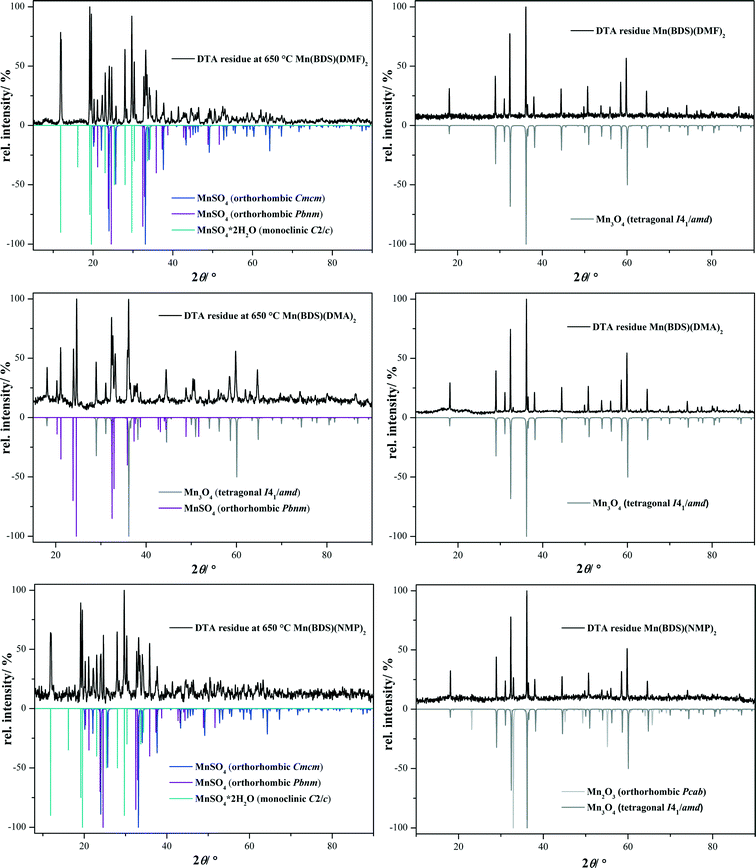 |
| | Fig. 10 Powder patterns of the intermediate (left) and final product (right) of III (top), IV (middle) and V (bottom). | |
Magnetic properties of [NH2(CH3)2]2{Mn(B4S)(DMF)2}
The temperature-dependent magnetic susceptibility and inverse magnetic susceptibility data (χ and χ−1) are depicted in Fig. 11 (top). The inverse magnetic susceptibility shows linear variation with temperature, indicating Curie–Weiss behavior [χ = C/(T − θP)] over the whole temperature range. A fit of the data set resulted in an effective magnetic moment of 5.19(1) μB/Mn atom and a Weiss constant of θP = −1.9(5) K, indicating minute antiferromagnetic interactions in the paramagnetic domain. The effective magnetic moment is in between the values for a Mn2+(h.s.) ion (μtheo = 5.92 μB) and a Mn3+(h.s.) ion (μtheo = 4.90 μB).29 From crystal chemical considerations, a Mn2+ ion in high spin configuration is to be expected.
 |
| | Fig. 11 Magnetic behavior of [NH2(CH3)2]2{Mn(B4S)(DMF)2}. The temperature-dependent magnetic susceptibility and inverse magnetic susceptibility (upper part) reveal the paramagnetic behaviour of the compound. In zero-field-cooled/field-cooled measurement mode (middle), a small amount of ferromagnetic impurity (MnCO3) can be identified. The magnetization isotherms (bottom) gave no hint of a magnetic ordering phenomenon. | |
Table 4 Crystallographic data of [NH2(CH3)2]2{Mn(B4S)(DMF)2} and [HNMP]2{Mn(B4S)(NMP)2} and their determination
|
|
[NH2(CH3)2]2{Mn(B4S)(DMF)2} |
[HNMP]2{Mn(B4S)(NMP)2} |
| Lattice parameters |
a = 942.02(5) pm |
a = 931.70(7) pm |
|
b = 1684.86(6) pm |
b = 1049.19(7) pm |
|
c = 918.45(5) pm |
c = 1944.87(12) pm |
|
β = 97.746(6)° |
β = 113.529(3)° |
| Density (calculated g cm−3) |
1.58 |
1.62 |
| Cell volume |
1444.4(1) Å3 |
1743.1(2) Å3 |
| No. of formula units |
2 |
2 |
| Cryst. syst. |
Monoclinic |
Monoclinic |
| Space group |
P21/c (no. 14) |
P21/c (no. 14) |
| Measuring device |
Stoe IPDS I |
Bruker APEX II |
| Radiation |
Mo-Kα, λ = 71.07 pm |
Mo-Kα, λ = 71.07 pm |
| Temp. |
296 K |
120 K |
| Absorption correction |
Numerical |
Numerical |
|
μ
|
8.2 cm−1 |
7.0 cm−1 |
| Measured reflections |
22 160 |
77 638 |
| Unique reflections |
3560 |
5757 |
| With Io > 2σ(Io) |
2330 |
4016 |
|
R
int; Rσ |
0.0668; 0.0514 |
0.0512; 0.0384 |
| GOF (all data) |
0.870 |
1.029 |
|
R
1; wR2 (Io > 2σ(Io)) |
0.0357; 0.0771 |
0.0501; 0.1310 |
|
R
1; wR2 (all data) |
0.0643; 0.0837 |
0.0818; 0.1484 |
| Max./min. electron density |
0.62/−0.26 e/Å3 |
1.10/−0.47 e/Å3 |
| CCDC number |
951310
|
959360
|
Table 5 Crystallographic data of Mn(BDS)(DMF)2, Mn(BDS)(DMA)2, and Mn(BDS)(NMP)2
|
|
Mn(BDS)(DMF)2 |
Mn(BDS)(DMA)2 |
Mn(BDS)(NMP)2 |
| Lattice parameters |
a = 514.30(7) pm |
a = 936.00(4) pm |
a = 962.97(3) pm |
|
b = 926.20(12) pm |
b = 984.94(4) pm |
b = 976.76(3) pm |
|
c = 940.20(13) pm |
c = 1034.75(5) pm |
c = 1034.23(3) pm |
|
α = 93.552(8)° |
α = 81.606(2)° |
α = 89.3710(10)° |
|
β = 99.993(7)° |
β = 88.941(2)° |
β = 78.8390(10)° |
|
γ = 99.237(7)° |
γ = 82.364(2)° |
γ = 87.604(2)° |
| Density (calculated g cm−3) |
1.68 |
1.65 |
1.71 |
| Cell volume |
433.48(10) Å3 |
935.35(7) Å3 |
953.55(5) Å3 |
| No. of formula units |
1 |
2 |
2 |
| Cryst. syst. |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P (no. 2) |
P (no. 2) |
P (no. 2) |
| Measuring device |
Bruker APEX II |
Bruker APEX II |
Bruker APEX II |
| Radiation |
Mo-Kα, λ = 71.07 pm |
Mo-Kα, λ = 71.07 pm |
Mo-Kα, λ = 71.07 pm |
| Temp. |
120 K |
120 K |
120 K |
| Absorption correction |
Numerical |
Numerical |
Numerical |
|
μ
|
10.5 cm−1 |
9.8 cm−1 |
9.6 cm−1 |
| Measured reflections |
9686 |
25 447 |
46 913 |
| Unique reflections |
3443 |
8169 |
11 970 |
| With Io > 2σ(Io) |
2587 |
6324 |
9790 |
|
R
int; Rσ |
0.0393; 0.0520 |
0.0726; 0.0502 |
0.0315; 0.0237 |
| GOF (all data) |
0.956 |
1.045 |
1.021 |
|
R
1; wR2 (Io > 2σ(Io)) |
0.0290; 0.0637 |
0.0348; 0.0939 |
0.0243; 0.0713 |
|
R
1; wR2 (all data) |
0.0462; 0.0670 |
0.0504; 0.1002 |
0.0314; 0.0733 |
| Max./min. electron density |
0.46/−0.40 e/Å3 |
1.09/−0.59 e/Å3 |
0.59/−0.47 e/Å3 |
| CCDC number |
951313
|
951315
|
951316
|
Table 6 Selected distances (pm) and angles (deg.) for [NH2(CH3)2]2{Mn(B4S)(DMF)2} and [HNMP]2{Mn(B4S)(NMP)2}
|
|
[NH2(CH3)2]2{Mn(B4S)(DMF)2} |
[HNMP]2{Mn(B4S)(NMP)2} |
| [MnO6] |
Mn1 |
–O1(2×) |
217.1(2) |
Mn1 |
–O1(2×) |
215.7(2) |
| –O11(2×) |
216.4(2) |
–O11(2×) |
219.1(4) |
| –O14(2×) |
215.4(2) |
–O14(2×) |
218.3(3) |
|
|
| O1– |
Mn1 |
–O11 |
90.47(7) |
O1– |
Mn1 |
–O11 |
89.4(1) |
| O1– |
Mn1 |
–O14 |
89.79(7) |
O1– |
Mn1 |
–O14 |
88.4(1) |
| O11– |
Mn1 |
–O14 |
86.11(6) |
O11– |
Mn1 |
–O14 |
96.3(1) |
|
|
| [SO3] |
S1 |
–O11 |
146.3(2) |
S1 |
–O11 |
146.1(4) |
| –O12 |
143.4(2) |
–O12 |
144.2(4) |
| –O13 |
144.8(2) |
–O13 |
144.2(4) |
| –C1 |
179.4(2) |
–C1 |
180.2(2) |
|
|
| S2 |
–O14 |
145.6(2) |
S2 |
–O14 |
141.9(3) |
| –O15 |
143.0(2) |
–O15 |
149.3(3) |
| –O16 |
144.3(2) |
–O16 |
143.3(4) |
| –C2 |
181.0(2) |
–C2 |
179.5(2) |
|
|
| O11– |
S1 |
–O12 |
113.8(1) |
O11– |
S1 |
–O12 |
112.9(2) |
| O11– |
S1 |
–O13 |
110.4(1) |
O11– |
S1 |
–O13 |
111.6(2) |
| O12– |
S1 |
–O13 |
114.1(1) |
O12– |
S1 |
–O13 |
114.6(2) |
| O11– |
S1 |
–C1 |
104.9(1) |
O11– |
S1 |
–C1 |
105.4(2) |
| O12– |
S1 |
–C1 |
107.1(1) |
O12– |
S1 |
–C1 |
106.8(2) |
| O13– |
S1 |
–C1 |
105.7(1) |
O13– |
S1 |
–C1 |
104.7(2) |
|
|
| O14– |
S2 |
–O15 |
113.2(1) |
O14– |
S2 |
–O15 |
111.2(2) |
| O14– |
S2 |
–O16 |
111.3(1) |
O14– |
S2 |
–O16 |
115.4(2) |
| O15– |
S2 |
–O16 |
113.2(1) |
O15– |
S2 |
–O16 |
110.1(2) |
| O14– |
S2 |
–C2 |
107.5(1) |
O14– |
S2 |
–C2 |
107.9(1) |
| O15– |
S2 |
–C2 |
105.1(1) |
O15– |
S2 |
–C2 |
105.3(1) |
| O16– |
S2 |
–C2 |
105.9(1) |
O16– |
S2 |
–C2 |
106.2(2) |
Table 7 Selected distances (pm) and angles (deg.) for Mn(BDS)(DMF)2, Mn(BDS)(DMA)2, and Mn(BDS)(NMP)2
|
|
Mn(BDS)(DMF)2 |
Mn(BDS)(DMA)2 |
Mn(BDS)(NMP)2 |
| [MnO6] |
Mn1 |
–O1 (2×) |
215.09(9) |
Mn1 |
–O1 |
213.4(1) |
Mn1 |
–O1 |
213.07(5) |
|
|
|
–O11 (2×) |
218.41(9) |
|
–O2 |
211.9(1) |
|
–O2 |
213.51(5) |
|
|
|
–O12 (2×) |
214.49(9) |
|
–O11 |
219.6(1) |
|
–O11 |
219.19(5) |
|
|
|
|
|
|
–O12 |
219.0(1) |
|
–O12 |
219.12(4) |
|
|
|
|
|
|
–O14 |
217.7(1) |
|
–O14 |
217.25(5) |
|
|
|
|
|
|
–O15 |
219.4(1) |
|
–O15 |
218.89(5) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O1– |
Mn1 |
–O11 |
91.86(4) |
O1– |
Mn1 |
–O2 |
174.94(5) |
O1– |
Mn1 |
–O2 |
172.86(2) |
|
|
O1– |
Mn1 |
–O12 |
93.09(4) |
O1– |
Mn1 |
–O11 |
91.07(4) |
O1– |
Mn1 |
–O11 |
87.52(2) |
|
|
O11– |
Mn1 |
–O12 |
90.16(4) |
O1– |
Mn1 |
–O12 |
89.16(4) |
O1– |
Mn1 |
–O12 |
90.10(2) |
|
|
|
|
|
|
O1– |
Mn1 |
–O14 |
90.04(5) |
O1– |
Mn1 |
–O14 |
91.12(2) |
|
|
|
|
|
|
O1– |
Mn1 |
–O15 |
85.67(5) |
O1– |
Mn1 |
–O15 |
87.32(2) |
|
|
|
|
|
|
O2– |
Mn1 |
–O11 |
92.06(5) |
O2– |
Mn1 |
–O11 |
91.18(2) |
|
|
|
|
|
|
O2– |
Mn1 |
–O12 |
86.94(5) |
O2– |
Mn1 |
–O12 |
96.91(2) |
|
|
|
|
|
|
O2– |
Mn1 |
–O14 |
94.04(5) |
O2– |
Mn1 |
–O14 |
90.43(2) |
|
|
|
|
|
|
O2– |
Mn1 |
–O15 |
91.21(5) |
O2– |
Mn1 |
–O15 |
85.70(2) |
|
|
|
|
|
|
O11– |
Mn1 |
–O12 |
88.75(4) |
O11– |
Mn1 |
–O12 |
89.37(2) |
|
|
|
|
|
|
O11– |
Mn1 |
–O14 |
87.99(5) |
O11– |
Mn1 |
–O14 |
177.52(2) |
|
|
|
|
|
|
O11– |
Mn1 |
–O15 |
176.73(4) |
O11– |
Mn1 |
–O15 |
91.74(2) |
|
|
|
|
|
|
O12– |
Mn1 |
–O14 |
176.63(5) |
O12– |
Mn1 |
–O14 |
88.56(2) |
|
|
|
|
|
|
O12– |
Mn1 |
–O15 |
91.36(5) |
O12– |
Mn1 |
–O15 |
177.15(2) |
|
|
|
|
|
|
O14– |
Mn1 |
–O15 |
91.84(5) |
O14– |
Mn1 |
–O15 |
90.26(2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| [SO3] |
S1 |
–O11 |
1.46.2(1) |
S1 |
–O11 |
146.2(1) |
S1 |
–O11 |
146.19(5) |
|
|
|
–O12 |
1.45.1(1) |
|
–O12 |
145.9(1) |
|
–O12 |
146.13(5) |
|
|
|
–O13 |
1.44.2(1) |
|
–O13 |
144.4(1) |
|
–O13 |
144.65(5) |
|
|
|
–C1 |
1.76.8(1) |
|
–C1 |
177.8(1) |
|
–C1 |
177.21(6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
S2 |
–O14 |
145.4(1) |
S2 |
–O14 |
146.02(5) |
|
|
|
|
|
|
–O15 |
145.0(1) |
|
–O15 |
145.89(5) |
|
|
|
|
|
|
–O16 |
144.1(1) |
|
–O16 |
144.25(6) |
|
|
|
|
|
|
–C4 |
177.5(1) |
|
–C4 |
177.94(6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O11– |
S1 |
–O12 |
112.07(5) |
O11– |
S1 |
–O12 |
112.51(7) |
O11– |
S1 |
–O12 |
112.85(3) |
|
|
O11– |
S1 |
–O13 |
112.01(6) |
O11– |
S1 |
–O13 |
112.61(7) |
O11– |
S1 |
–O13 |
112.33(3) |
|
|
O12– |
S1 |
–O13 |
114.01(6) |
O12– |
S1 |
–O13 |
114.65(7) |
O12– |
S1 |
–O13 |
113.89(3) |
|
|
O11– |
S1 |
–C1 |
106.41(6) |
O11– |
S1 |
–C1 |
105.84(6) |
O11– |
S1 |
–C1 |
106.18(3) |
|
|
O12– |
S1 |
–C1 |
105.02(6) |
O12– |
S1 |
–C1 |
104.07(6) |
O12– |
S1 |
–C1 |
105.07(3) |
|
|
O13– |
S1 |
–C1 |
106.61(6) |
O13– |
S1 |
–C1 |
106.15(7) |
O13– |
S1 |
–C1 |
105.64(3) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O14– |
S2 |
–O15 |
113.02(8) |
O14– |
S2 |
–O15 |
112.84(3) |
|
|
|
|
|
|
O14– |
S2 |
–O16 |
113.84(8) |
O14– |
S2 |
–O16 |
111.95(3) |
|
|
|
|
|
|
O15– |
S2 |
–O16 |
112.37(8) |
O15– |
S2 |
–O16 |
114.61(3) |
|
|
|
|
|
|
O14– |
S2 |
–C4 |
104.89(7) |
O14– |
S2 |
–C4 |
106.48(3) |
|
|
|
|
|
|
O15– |
S2 |
–C4 |
105.98(7) |
O15– |
S2 |
–C4 |
104.19(3) |
|
|
|
|
|
|
O16– |
S2 |
–C4 |
105.84(7) |
O16– |
S2 |
–C4 |
105.85(3) |
The 100 Oe measurement in zero-field-cooled/field-cooled mode (inset of Fig. 11, center) reveals a trace amount of ferromagnetic impurity at TC = 32.5(5) K. This is consistent with the ferromagnetic ordering temperature of MnCO3 (TC = 32.2 K).30 The ZFC/FC measurement was repeated with a magnetic flux density of 500 Oe in order to saturate the ferromagnetic MnCO3 impurity (Fig. 11, center) to show the extrinsic nature of this anomaly. The MnCO3 impurity obviously remains from the synthesis which starts from MnCO3 as the manganese source.
Fig. 11 (bottom) shows the magnetization isotherms of the [NH2(CH3)2]2{Mn(B4S)(DMF)2} sample measured at 5, 10, 25 and 50 K with magnetic flux densities up to 80 kOe. The 5 K isotherm exhibits pronounced curvature without showing any indication of a magnetic ordering phenomenon, the curvature of the 10 K isotherm is less pronounced and the 25 and 50 K isotherms show linear dependence of temperature as is expected for a paramagnetic material.
Taking the trace amount of MnCO3 into account, one can assume that the compound contains Mn2+ ions in high spin configuration. No magnetic ordering phenomenon was evident within the investigated temperature range.
Conclusion
In the course of our investigations on the preparation of novel polysulfonic acids and their use in the synthesis of polysulfonates, we have obtained five manganese polysulfonates based on the acids H4B4S (1,2,4,5-benzenetetrasulfonic acid) and H2BDS (1,4-benzenedisulfonic acid). The compounds were prepared by solvothermal reactions at elevated temperatures. While the tetrasulfonates exhibit unique anionic chains according to ∞1{Mn(B4S)2/2(L)2}2− (L = DMF, NMP), the disulfonates show layer-type structures. For both kinds of compound, thermoanalytical measurements show that the sulfonates have exceptionally high decomposition temperatures, especially when compared to related polycarboxylates.31 Further investigations shall now elucidate the structures of the solvent-free compounds in order to investigate if they are porous MOF-type materials. The final decomposition products are Mn3O4 and Mn2O3. For the chain-type tetrasulfonate [NH2(CH3)2]2{Mn(B4S)(DMF)2}, magneto chemical investigations have also been performed, proving the ferromagnetic behavior of the compound. Magnetic coupling, however, is not observed. The results presented show that polysulfonates are a highly interesting class of compounds that need further investigation. The fundament for these investigations is the development of preparative routes for polysulfonic acids, and thus, the strength of this project is the combination of expertise from organic chemistry and that from inorganic chemistry.
Acknowledgements
The authors thank Dipl.-Chem. Wolfgang Saak and Dr. Marc Schmidtmann for the collection of the X-ray data and Florian Behler for the measurements of the IR spectra. Financial support by the Deutsche Forschungsgemeinschaft is also gratefully acknowledged.
References
-
(a) C. Janiak, Dalton Trans., 2003, 2781–2804 RSC;
(b) A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
-
(a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS PubMed;
(b) U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC;
(c) S. Bauer and N. Stock, Chem. Unserer Zeit, 2008, 42, 12–19 CrossRef CAS.
-
(a) J.-M. Shi, W. Xu, Q.-Y. Liu, F.-L. Liu, Z.-L. Huang, H. Lei, W.-T. Yu and Q. Fang, Chem. Commun., 2002, 756–757 RSC;
(b) G.-F. Liu, Z.-P. Qiao, H.-Z. Wang, X.-M. Chen and G. Yang, New J. Chem., 2002, 26, 791–795 RSC;
(c) T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651–1657 CrossRef CAS;
(d) Y. Zhu, Z. Sun, Y. Zhao, J. Zhang, X. Lu, N. Zhang, L. Liu and F. Tong, New J. Chem., 2009, 33, 119–124 RSC;
(e) N. Kerbellec, L. Catala, C. Daiguebonne, A. Gloter, O. Stephan, J.-C. Bünzli, O. Guillou and T. Mallah, New J. Chem., 2008, 32, 584–587 RSC;
(f) X. Yang, J. H. Rivers, W. J. McCarty, M. Wiester and R. A. Jones, New J. Chem., 2008, 32, 790–793 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
-
(a) J. A. Real, E. Andres, M. C. Munoz, M. Julve, T. Granier, A. Bousseksou and F. Varret, Science, 1995, 268, 265–267 CAS;
(b) L. G. Beauvais, M. P. Shores and J. R. Long, J. Am. Chem. Soc., 2000, 122, 2763–2772 CrossRef CAS.
-
(a) H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 1734–1744 RSC;
(b) D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770–818 RSC;
(c) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC;
(d) J. Larionova, Y. Guari, C. Sangregorio and C. Guérin, New J. Chem., 2009, 33, 1177–1190 RSC.
-
(a) L. J. Murray, M. Dincǎ and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC;
(b) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS PubMed;
(c) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC;
(d) C.-J. Li, Z.-J. Lin, M.-X. Peng, J.-D. Leng, M.-M. Yang and M.-L. Tong, Chem. Commun., 2008, 6348–6350 RSC;
(e) S. S. Iremonger, P. D. Southon and C. J. Kepert, Dalton Trans., 2008, 6103–6105 RSC;
(f) S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS PubMed;
(g) S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC.
- Z. Hulvey, E. H. L. Falcao, J. Echert and A. K. Cheetham, J. Mater. Chem., 2009, 19, 4307–4309 RSC.
-
(a) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., 2004, 116, 2388–2430 CrossRef;
(b) G. Férey, Chem. Mater., 2001, 13, 3084–3098 CrossRef;
(c) J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS PubMed.
-
(a) B. D. Chandler, G. D. Enright, K. A. Udachin, S. Pawsey, J. A. Ripmeester, D. T. Cramb and G. K. H. Shimizu, Nat. Mater., 2008, 7, 229–235 CrossRef CAS PubMed;
(b) G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430–1449 RSC;
(c) G. B. Deacon, A. Gitlits, G. Zelesny, D. Stellfeldt and G. Meyer, Z. Anorg. Allg. Chem., 1999, 625, 764–772 CrossRef CAS;
(d) G. B. Deacon, R. Harika, P. C. Junk, B. W. Skelton and A. H. White, New J. Chem., 2007, 31, 634–645 RSC;
(e) D. J. Hoffart, S. A. Dalrymple and G. K. H. Shimizu, Inorg. Chem., 2005, 44, 8868–8875 CrossRef CAS PubMed;
(f) V. Videnova-Adrabinska, Coord. Chem. Rev., 2007, 251, 1987–2016 CrossRef CAS PubMed.
-
(a) K. T. Holman, A. M. Pivovar, J. A. Swift and M. D. Ward, Acc. Chem. Res., 2001, 34, 107–118 CrossRef CAS PubMed;
(b) K. T. Holman, S. M. Martin, D. P. Parker and M. D. Ward, J. Am. Chem. Soc., 2001, 123, 4421–4431 CrossRef CAS PubMed;
(c) M. J. Horner, K. T. Holman and M. D. Ward, J. Am. Chem. Soc., 2007, 129, 14640–14660 CrossRef CAS PubMed;
(d) V. A. Russell, C. C. Evans, W. Li and M. D. Ward, Science, 1997, 276, 575–579 CrossRef CAS;
(e) J. A. Swift, A. M. Pivovar and A. M. Reynolds, J. Am. Chem. Soc., 1998, 24, 5887–5894 CrossRef;
(f) J. A. Swift, A. M. Reynolds and M. D. Ward, Chem. Mater., 1998, 10, 4159–4168 CrossRef CAS.
- A. Mietrach, T. W. T. Muesmann, J. Christoffers and M. S. Wickleder, Eur. J. Inorg. Chem., 2009, 5328–5334 CrossRef CAS.
- T. W. T. Muesmann, A. Mietrach, J. Christoffers and M. S. Wickleder, Z. Anorg. Allg. Chem., 2010, 636, 1307–1312 CrossRef CAS.
-
(a) A. Mietrach, T. W. T. Muesmann, C. Zilinski, J. Christoffers and M. S. Wickleder, Z. Anorg. Allg. Chem., 2011, 637, 195–200 CAS;
(b) T. W. T. Muesmann, C. Zitzer, A. Mietrach, T. Klüner, J. Christoffers and M. S. Wickleder, Dalton Trans., 2011, 40, 3128–3141 RSC;
(c) T. W. T. Muesmann, C. Zitzer, M. S. Wickleder and J. Christoffers, Inorg. Chim. Acta, 2011, 369, 45–48 CrossRef CAS PubMed.
-
BrukerOptik GmbH, OPUS 6.5, Germany, 2009 Search PubMed.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed;
(b)
X-RED 1.22, Stoe & Cie, Darmstadt, Germany, 2001 Search PubMed;
(c)
X-SHAPE 1.06f, Stoe & Cie, Darmstadt, Germany, 1999 Search PubMed.
-
Stare V 9.3, Mettler-Toledo GmbH, Schwerzenbach, Switzerland, 2009 Search PubMed.
-
WinXPOW 2007, Stoe & Cie, Darmstadt, Germany, 2006 Search PubMed.
-
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Kirfel and G. Will, High Temp. – High Pressures, 1974, 6, 525–527 CAS.
- G. Aminoff, Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem., 1927, 64, 475–490 CAS.
-
Y. D. Kondrashev, A. I. Zaslavskii, Golden Book of Phase Transitions, Wroclaw, 2002, vol. 1, 1–123 Search PubMed.
- S. Geller, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 821–828 CrossRef.
- G. Will, Acta Crystallogr., 1965, 19, 854–857 CrossRef CAS.
- F. Golinska, A. Lodzinska and F. Rozploch, Pol. J. Chem., 1984, 58, 31–39 CAS.
-
High Score Plus V 3.0d, PANalytical B. V., Almelo, Netherlands, 2011 Search PubMed.
-
H. Lueken, Magnetochemie, B.G. Teubner, Stuttgart, 1999 Search PubMed.
-
(a) I. Maartense, Phys. Rev., 1969, 188, 924–930 CrossRef CAS;
(b) A. Kosterov, T. Frederichs and T. von Dobeneck, Phys. Earth Planet. Inter., 2006, 154, 234–242 CrossRef CAS PubMed.
-
(a) L.-F. Song, C.-H. Jiang, C.-L. Jiao, J. Zhang, L.-X. Sun, F. Xu, Q.-Z. Jiao, Y.-H. Xing, Y. Du, Z. Cao and F.-L. Huang, J. Therm. Anal. Calorim., 2010, 102, 1161–1166 CrossRef CAS;
(b) R.-Q. Zhong, R.-Q. Zou, M. Du, T. Yamada, G. Maruta, S. Takeda, J. Li and Q. Xu, CrystEngComm, 2010, 12, 677–681 RSC;
(c) Y. Liu, H. Li, Y. Han, X. Lv, H. Hou and Y. Fan, Cryst. Growth Des., 2012, 12, 3505–3513 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence