
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Greasy tails switch 1D-coordination [{Zn2(OAc)4(4′-(4-ROC6H4)-4,2′:6′,4′′-tpy)}n] polymers to discrete [Zn2(OAc)4(4′-(4-ROC6H4)-4,2′:6′,4′′-tpy)2] complexes†
Y. Maximilian
Klein
a,
Edwin C.
Constable
a,
Catherine E.
Housecroft
*a,
Jennifer A.
Zampese
a and
Aurélien
Crochet
b
aDepartment of Chemistry, University of Basel, Spitalstrasse 51, CH4056 Basel, Switzerland. E-mail: catherine.housecroft@unibas.ch; Fax: +41 61 267 1018; Tel: +41 61 267 1008
bDepartment of Physics, University of Fribourg, Chemin du Musée 3, CH-1700 Fribourg, Switzerland. E-mail: aurelien.crochet@unifr.ch
First published on 22nd September 2014
Abstract
The homologous series of 4′-(4-ROC6H4)-4,2′:6′,4′′-tpy ligands with R = Me, Et, nPr, nBu, npentyl, nhexyl, nheptyl, noctyl, nnonyl and ndecyl (1–10, respectively) are reported, including single crystal structures of 6 and 7. Reactions of zinc(II) acetate with 1–10 have been investigated using room temperature crystallization methods (diffusion or layering). For ligands with the shortest alkoxy substituents, 1-dimensional coordination polymers [{Zn2(OAc)4(L)}n] (L = 1, 2 or 3) are formed. In each polymer, the 4′-(4-ROC6H4)-4,2′:6′,4′′-tpy ligands bind zinc through the two outer pyridine donors. The polymer structures are similar with the n-propyl chain adopting a folded conformation in [{Zn2(OAc)4(3)}n] which allows it to fit in the cavity occupied by methyl or ethyl groups in [{Zn2(OAc)4(1)}n] and [{Zn2(OAc)4(2)}n]. Reaction between 5 and Zn(OAc)2·2H2O gives both the coordination polymer [{2Zn2(OAc)4(5)·2H2O}n] and the discrete complex [Zn2(OAc)4(5)2]. Although the zig-zag form of the polymer chain in [{2Zn2(OAc)4(5)·2H2O}n] mimics those in [{Zn2(OAc)4(L)}n] (L = 1, 2 or 3), packing interactions differ and the wider separation of the chains in a sheet results in the incorporation of water molecules in the lattice. π-Stacking between pyridine rings in [{Zn2(OAc)4(L)}n] (L = 1, 2 or 3) produces infinite assemblies in contrast to isolated tetradecker π-stacks in [{2Zn2(OAc)4(5)·2H2O}n]. This assembly is replicated in [{4Zn2(OAc)4(7)·3H2O}n] (n-heptoxy substituents). In contrast, the n-hexoxy-containing coordination polymer crystallizes with acetic acid in the lattice; [{Zn2(OAc)4(6)·MeCO2H}n] consists of zig-zag polymer chains which π-stack in a manner which is unique among the other polymers. Further lengthening of the alkoxy chain favours the formation of [Zn2(OAc)4(L)2] (L = 8, 9 or 10) which are analogues of [Zn2(OAc)4(5)2]. In each, the 4′-(4-ROC6H4)-4,2′:6′,4′′-tpy ligand is monodentate. The alkoxy chains are in extended (or close to extended) conformations and pack into planar sheets with interdigitated chains. Pockets in the sheets are occupied by methyl groups of {Zn2(OAc)4} units in the adjacent sheet in a ball-and-socket assembly motif. The study shows that coordination polymers [{Zn2(OAc)4(L)}n] in which π-stacking are the dominant interactions are favoured for small alkoxy substituents (ligands 1–3); for ligands 8–10, discrete complexes [Zn2(OAc)4(L)2] in which van der Waals interactions dominate are observed. In the intermediate range (ligands 5–7), the preference between the two structure types appears to be marginal.
Introduction
4,2′:6′,4′′-Terpyridine (4,2′:6′,4′′-tpy) ligands are readily accessible building blocks for the assembly of coordination polymers and networks.1 In contrast to the bis(chelating) mode of 2,2′:6′,2′′-terpyridine (the most commonly encountered terpyridine isomer), 4,2′:6′,4′′-tpy offers a V-shaped building block with a fixed internal angle of 120°. Despite possessing three N-donors, 4,2′:6′,4′′-tpy typically coordinates through only the outer pyridine domains. This preference was initially demonstrated in the 1-dimensional coordination polymer [{ZnCl2(4,2′:6′,4′′-tpy)}n],2 and has now been observed in a wide range of [{ZnX2(4′-R-4,2′:6′,4′′-tpy)}n] (X = Cl, Br) and [{Zn2(OAc)4(4′-R-4,2′:6′,4′′-tpy)}n] complexes.1 Recently, there has been interest in the incorporation of coordinatively non-innocent carboxylate groups (e.g. 4-carboxyphenyl, 2-carboxyphenyl) in the 4′-position of the 4,2′:6′,4′′-tpy domain.3–7 The introduction of heterocyclic substituents (pyridine,8–17 imidazole18 and pyrimidine19) in the 4′-position of 4,2′:6′,4′′-tpy is a simple means of increasing structural diversity and enhancing the dimensionality. Ditopic bis(4,2′:6′,4′′-terpyridine) ligands remain underexplored,20,21 and open further routes to developing 2- and 3-dimensional assemblies containing 4,2′:6′,4′′-tpy metal-binding domains.In 2002, Cave and Raston reported the synthesis and structure of the helical coordination polymer [{ZnCl2L}n] in which L is 4′-(4-octoxyphenyl)-4,2′:6′,4′′-terpyridine.22 Despite incorporating long alkyl chains, the polymer is structurally similar to others in the [{ZnX2(4′-R-4,2′:6′,4′′-tpy)}n] family.1 In contrast, we have observed that the reaction of 4′-(4-dodecoxyphenyl)-4,2′:6′,4′′-terpyridine with zinc(II) acetate leads to a discrete molecule containing a {Zn2(OAc)4} paddle wheel unit with monodentate 4′-(4-dodecoxyphenyl)-4,2′:6′,4′′-tpy ligands in the axial sites.23 This breaks the common pattern of reactions of Zn(OAc)2·2H2O with 4′-X-4,2′:6′,4′′-tpy ligands leading to zigzag chains with the tpy domain bridging between {Zn2(OAc)4} units.1 We now present a systematic study of the reactions of Zn(OAc)2·2H2O with a homologous series of 4′-(4-ROC6H4)-4,2′:6′,4′′-tpy ligands (1–10, Scheme 1) in which R is an alkyl substituent, and we illustrate the competition between π-stacking interactions of aromatic domains and van der Waals interactions between alkoxy chains as a function of chain length in determining the structure type.
 | ||
| Scheme 1 Structures of the ligands and numbering scheme for NMR spectroscopic assignments. | ||
Experimental
Electrospray mass spectra were measured on a Bruker Esquire 3000 instrument using MeCN solutions of samples. Absorption spectra were recorded on Varian Carry 5000 spectrometer, and IR spectra on a Shimadzu FTIR-8400S or Perkin Elmer UATR Two spectrophotometer. 1H and 13C NMR spectra were recorded using a Bruker Avance III-500 or 400 NMR spectrometers at 295 K; chemical shifts were referenced to residual solvent peaks with respect to δ(TMS) = 0 ppm.Commercial sources or synthetic and characterization details for precursors, as well as IR spectroscopic data for the ligands are given in the ESI.†
4′-(4-Methoxyphenyl)-4,2′:6′,4′′-terpyridine (1)
p-Anisaldehyde (1.21 mL, 1.36 g, 10.0 mmol) was dissolved in EtOH (60 mL). 4-Acetylpyridine (2.27 mL, 2.47 g, 20.0 mmol) was added, followed by crushed KOH (1.12 g, 20.0 mmol). The mixture changed from colourless to yellow, to red. Aqueous NH3 (25%, 49.3 mL, 44.9 g, 320 mmol) was added dropwise and the mixture was stirred at room temperature overnight. The white precipitate that formed was collected by filtration, washed with water (3 × 10 mL) and EtOH (3 × 10 mL) and was recrystallized from EtOH. Compound 1 was obtained as a white solid (1.61 g, 4.73 mmol, 47.3%). M.p. 203.5 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.78 (m, 4H, HA2), 8.07 (m, 4H, HA3), 8.00 (s, 2H, HB3), 7.71 (m, 2H, HC2), 7.08 (m, 2H, HC3), 3.91 (s, 3H, HMe); 13C{1H}NMR (126 MHz, CDCl3) δ/ppm 161.1 (CC4), 155.3 (CA4), 150.8 (CB4), 150.7 (CA2), 146.3 (CB2), 130.2 (CC1), 128.5 (CC2), 121.3 (CA3), 118.6 (CB3), 114.9 (CC3), 55.6 (CMe). UV-VIS (MeCN, 2.5 × 10−5 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1) 269 (36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600). ESI-MS m/z 340.3 [M + H]+ (calc. 340.1). Found C 77.19, H 5.08, N 12.36; required for C22H17N3O C 77.86, H 5.05, N 12.38.
600). ESI-MS m/z 340.3 [M + H]+ (calc. 340.1). Found C 77.19, H 5.08, N 12.36; required for C22H17N3O C 77.86, H 5.05, N 12.38.
4′-(4-Ethoxyphenyl)-4,2′:6′,4′′-terpyridine (2)
4-Ethoxybenzaldehyde (1.50 g, 10.0 mmol) was dissolved in EtOH (60 mL), then 4-acetylpyridine (2.27 mL, 2.47 g, 20.0 mmol) was added, followed by crushed KOH (1.12 g, 20.0 mmol). Colour changes, addition of aqueous NH3 (25%, 49.3 mL, 44.9 g, 320 mmol), work up and recrystallization were as for the preparation of 1. Compound 2 was obtained as a white solid (2.23 g, 6.31 mmol, 63.1%). M.p. 211.7 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.78 (m, 4H, HA2), 8.07 (m, 4H, HA3), 8.00 (s, 2H, HB3), 7.70 (m Hz, 2H, HC2), 7.06 (m, 2H, HC3), 4.13 (q, J = 7.0 Hz, 2H, HOCH2), 1.48 (t, J = 7.0 Hz, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.5 (CC4), 155.3 (CA4), 150.8 (CB4), 150.6 (CA2), 146.3 (CB2), 130.0 (CC1), 128.5 (CC2), 121.34 (CA3), 118.5 (CB3), 115.4 (CC3), 63.9 (COCH2), 14.9 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 269 (38300). ESI-MS m/z 354.3 [M + H]+ (calc. 354.2). Found C 76.59, H 5.64, N 11.35; required for C23H19N3O·0.5H2O C 76.22, H 5.56, N 11.59.
4′-(4-nPropoxyphenyl)-4,2′:6′,4′′-terpyridine (3)
4-Propoxybenzaldehyde (1.63 mL, 1.69 g, 10.0 mmol) was dissolved in EtOH (60 mL); 4-acetylpyridine (2.27 mL, 2.47 g, 20.0 mmol) was added followed by crushed KOH (1.12 g, 20.0 mmol). Colour changes, addition of aqueous NH3 (25%, 49.3 mL, 44.9 g, 320 mmol), work up and recrystallization were as for the preparation of 1. Compound 3 was obtained as a white solid (2.21 g, 6.01 mmol, 60.1%). M.p. 195.8 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.77 (m, 4H, HA2), 8.05 (m, 4H, HA3), 7.98 (s, 2H, HB3), 7.68 (m, 2H, HC2), 7.06 (m, 2H, HC3), 4.00 (t, J = 6.5 Hz, 2H, HOCH2), 1.86 (m, 2H, HCH2Me), 1.08 (t, J = 7.4 Hz, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.7 (CC4), 155.2 (CA4), 150.7 (CB4), 150.6 (CA2), 146.3 (CB2), 129.87 (CC1), 128.4 (CC2), 121.3 (CA3), 118.4 (CB3), 115.4 (CC3), 69.9 (COCH2), 22.7 (CCH2Me), 10.7 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (38700). ESI-MS m/z 368.3 [M + H]+ (calc. 368.2). Found C 77.82, H 5.79, N 11.60; required for C24H21N3O C 78.45, H 5.76, N 11.44.
4′-(4-nButoxyphenyl)-4,2′:6′,4′′-terpyridine (4)
Compound 4 has previously been prepared by a grinding method,24 but we find the following more convenient; NMR spectra were not previously assigned.4-Butoxybenzaldehyde (1.76 mL, 1.82 g, 10.0 mmol) was dissolved in EtOH (60 mL), then 4-acetylpyridine (2.27 mL, 2.47 g, 20.0 mmol) was added to the solution and crushed KOH (1.12 g, 20.0 mmol) was added in one portion. Colour changes, addition of aqueous NH3 (25%, 49.3 mL, 44.9 g, 320 mmol), work up and recrystallization were as for the preparation of 1. Compound 4 was obtained as a white solid (2.15 g, 5.63 mmol, 56.3%). M.p. 188.9 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.78 (m, 4H, HA2), 8.06 (m, 4H, HA3), 7.99 (s, 2H, HB3), 7.69 (m, 2H, HC2), 7.13–7.02 (m, 2H, HC3), 4.06 (t, J = 6.5 Hz, 2H, HOCH2), 1.82 (m, 2H, HOCH2CH2), 1.53 (m, 2H, HCH2Me), 1.01 (t, J = 7.4 Hz, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.7 (CC4), 155.3 (CA4), 150.8 (CB4), 150.6 (CA2), 146.3 (CB2), 129.9 (CC1), 128.4 (CC2), 121.3 (CA3), 118.5 (CB3), 115.4 (CC3), 68.1 (COCH2), 31.4 (COCH2CH2), 19.4 (CCH2Me), 14.0 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (38400). ESI-MS m/z 382.3 [M + H]+ (calc. 382.2). Found C 78.12, H 6.18, N 11.17; required for C25H23N3O C 78.71, H 6.08, N 11.02.
4′-(4-nPentoxyphenyl)-4,2′:6′,4′′-terpyridine (5)
4-Pentoxybenzaldehyde (1.40 g, 7.29 mmol) was dissolved in EtOH (30 mL), then 4-acetylpyridine (1.70 mL, 1.85 g, 15.0 mmol) was added to the solution and crushed KOH (0.84 g, 15.0 mmol) was added in one portion. Colour changes, addition of aqueous NH3 (25%, 49.3 mL, 32.4 mL, 29.4 g, 210 mmol), work up and recrystallization were as for the preparation of 1. Compound 5 was obtained as a white solid (1.74 g, 4.40 mmol, 60.3%). M.p. 172.3 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.79 (m, 4H, HA2), 8.12 (m, 4H, HA3), 8.02 (s, 2H, HB3), 7.70 (m, 2H, HC2), 7.07 (m, 2H, HC3), 4.05 (t, J = 6.6 Hz, 2H, HOCH2), 1.84 (m, 2H, HOCH2CH2), 1.48 (m, 2H, HCH2CH2Me), 1.41 (m, 2H, HCH2Me), 0.96 (t, J = 7.2 Hz, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.8 (CC4), 155.1 (CA4), 150.9 (CB4), 150.0 (CA2), 146.9 (CB2), 129.8 (CC1), 128.5 (CC2), 121.5 (CA3), 118.7 (CB3), 115.5 (CC3), 68.4 (COCH2), 29.0 (COCH2CH2), 28.3 (CCH2CH2Me), 22.6 (CCH2Me), 14.2 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (35500). ESI-MS m/z 396.4 [M + H]+ (calc. 396.2). Found C 78.12, H 6.44, N 10.43; required for C26H25N3O C 78.96, H 6.37, N 10.62.
4′-(4-nHexoxyphenyl)-4,2′:6′,4′′-terpyridine (6)
4-Hexoxybenzaldehyde (0.62 g, 2.99 mmol) was dissolved in EtOH (20 mL), then 4-acetylpyridine (0.79 mL, 0.87 g, 7.00 mmol) was added to the solution and crushed KOH (0.39 g, 7.00 mmol) was added in one portion. Colour changes, addition of aqueous NH3 (25%, 27.7 mL, 25.2 g, 180 mmol), work up and recrystallization were as for the preparation of 1. Compound 6 was obtained as a white solid (0.76 g, 1.86 mmol, 62.1%). M.p. 183.4 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.79 (m, 4H, HA2), 8.12 (m, 4H, HA3), 8.03 (s, 2H, HB3), 7.70 (m, 2H, HC2), 7.07 (m, 2H, HC3), 4.05 (t, J = 6.6 Hz, 2H, HOCH2), 1.83 (m, 2H, HOCH2CH2), 1.50 (m, 2H, HOCH2CH2CH2), 1.37 (m, 4H, HCH2), 0.92 (m, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.8 (CC4), 155.07 (CA4), 150.9 (CB4), 150.0 (CA2), 146.9 (CB2), 129.8 (CC1), 128.5 (CC2), 121.5 (CA3), 118.8 (CB3), 115.5 (CC3), 68.4 (COCH2), 31.7 (CCH2CH2Me), 29.3 (COCH2CH2), 25.9 (COCH2CH2CH2), 22.7 (CCH2Me), 14.2 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (37100). ESI-MS m/z 410.4 [M + H]+ (calc. 410.2). Found C 78.60, H 6.63, N 10.25; required for C27H27N3O C 79.19, H 6.65, N 10.26.
4′-(4-nHeptoxyphenyl)-4,2′:6′,4′′-terpyridine (7)
4-Heptoxybenzaldehyde (0.95 mL, 0.94 g, 4.27 mmol) was dissolved in EtOH (40 mL), then 4-acetylpyridine (0.97 mL, 1.06 g, 8.54 mmol) was added to the solution and crushed KOH (0.48 g, 8.54 mmol) was added in one portion. Colour changes, addition of aqueous NH3 (25%, 21.1 mL, 19.2 g, 137 mmol), work up (but using 5 mL H2O and EtOH instead of 10 mL) and recrystallization were as for the preparation of 1. Compound 7 was obtained as a white solid (1.15 g, 2.72 mmol, 63.8%). M.p. 188.1 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.79 (m, 4H, HA2), 8.07 (m, 4H, HA3), 8.00 (s, 2H, HB3), 7.70 (m, 2H, HC2), 7.06 (m, 2H, HC3), 4.04 (t, J = 6.6 Hz, 2H, HOCH2), 1.84 (m, 2H, HOCH2CH2), 1.49 (m, 2H, HOCH2CH2CH2), 1.38 (m, 2H, HOCH2CH2CH2CH2), 1.33 (m, 4H, HCH2), 0.91 (m, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.7 (CC4), 155.3 (CA4), 150.8 (CB4), 150.7 (CA2), 146.3 (CB2), 129.8 (CC1), 128.5 (CC2), 121.3 (CA3), 118.5 (CB3), 115.4 (CC3), 68.4 (COCH2), 31.9 (CCH2CH2Me), 29.4 (COCH2CH2), 29.2 (COCH2CH2CH2CH2), 26.2 (COCH2CH2CH2), 22.8 (CCH2Me), 14.3 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (38500). ESI-MS m/z 424.5 [M + H]+ (calc. 424.2). Found C 78.82, H 6.96, N 10.00; required for C28H29N3O C 79.40, H 6.90, N 9.92.
4′-(4-nOctoxyphenyl)-4,2′:6′,4′′-terpyridine (8)
Compound 8 has previously been prepared by a grinding method,24 but we find the following more convenient; NMR spectra were not previously assigned.4-Octoxybenzaldehyde (1.41 g, 6 mmol) was dissolved in EtOH (50 mL), then 4-acetylpyridine (1.36 mL, 1.48 g, 12 mmol) was added to the solution and crushed KOH (0.67 g, 12 mmol) was added in one portion. Colour changes, addition of aqueous NH3 (25%, 30.8 mL, 28 g, 200 mmol), work up (but using 5 mL H2O and EtOH instead of 10 mL) and recrystallization were as for the preparation of 1. Compound 8 was obtained as a white solid (1.54 g, 3.52 mmol, 58.7%). M.p. 189.8 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.80 (m, 4H, HA2), 8.14 (m, 4H, HA3), 8.04 (s, 2H, HB3), 7.70 (m, 2H, HC2), 7.07 (m, 2H, HC3), 4.05 (t, J = 6.6 Hz, 2H, HOCH2), 1.83 (m, 2H, HOCH2CH2), 1.49 (m, 2H, HOCH2CH2CH2), 1.33 (overlapping m, 8H, HCH2), 0.90 (m, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.8 (CC4), 155.0 (CA4), 151.0 (CB4), 149.7 (CA2), 147.1 (CB2), 129.7 (CC1), 128.5 (CC2), 121.6 (CA3), 118.9 (CB3), 115.5 (CC3), 68.4 (COCH2), 32.0 (CCH2CH2Me), 29.5 (CCH2), 29.4 (CCH2), 29.3 (COCH2CH2), 26.2 (COCH2CH2CH2), 22.8 (CCH2Me), 14.3 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (35800). ESI-MS m/z 438.5 [M + H]+ (calc. 438.3). Found C 79.14, H 7.11, N 9.59; required for C29H31N3O C 79.60, H 7.14, N 9.60.
4′-(4-nNonoxyphenyl)-4,2′:6′,4′′-terpyridine (9)
4-Nonoxybenzaldehyde (1.62 g, 6.52 mmol) was dissolved in EtOH (50 mL), then 4-acetylpyridine (1.81 mL, 1.98 g, 16 mmol) was added to the solution and crushed KOH (0.90 g, 16 mmol) was added in one portion. Colour changes, addition of aqueous NH3 (25%, 38.5 mL, 35.1 g, 300 mmol), work up (but using 7 mL H2O and EtOH instead of 10 mL) and recrystallization were as for the preparation of 1. Compound 9 was obtained as a white solid (1.83 g, 4.05 mmol, 62.1%). M.p. 167.7 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.79 (m, 4H, HA2), 8.10 (m, 4H, HA3), 8.01 (s, 2H, HB3), 7.70 (m, 2H, HC2), 7.06 (m, 2H, HC3), 4.04 (t, J = 6.6 Hz, 2H, HOCH2), 1.83 (m, 2H, HOCH2CH2), 1.48 (m, 2H, HOCH2CH2CH2), 1.37 (m, 2H, HOCH2CH2CH2CH2), 1.35–1.23 (overlapping m, 8H, HCH2), 0.88 (m, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.8 (CC4), 155.1 (CA4), 150.9 (CB4), 150.2 (CA2), 146.7 (CB2), 129.8 (CC1), 128.5 (CC2), 121.5 (CA3), 118.7 (CB3), 115.4 (CC3), 68.4 (COCH2), 32.0 (CCH2CH2Me), 29.7 (CCH2CH2CH2CH2Me), 29.5 (COCH2CH2CH2CH2), 29.4 (CCH2CH2CH2Me), 29.3 (HOCH2CH2), 26.2 (COCH2CH2CH2), 22.8 (CCH2Me), 14.3 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (37600). ESI-MS m/z 452.5 [M + H]+ (calc. 452.3). Found C 80.09, H 7.33, N 9.37; required for C30H33N3O C 79.79, H 7.37, N 9.30.
4′-(4-nDecoxyphenyl)-4,2′:6′,4′′-terpyridine (10)
4-([4,2′:6′,4′′-Terpyridin]-4′-yl)phenol (see ESI†) (0.35 g, 1.00 mmol) was dissolved in acetone (40 mL), K2CO3 (0.27 g, 2.00 mmol) was added to the solution and the reaction mixture was refluxed for 15 min. 1-Bromodecane was added and the reaction mixture was refluxed overnight. The brown precipitate was filtered, dissolved in chloroform and washed with aq. K2CO3 solution. The organic layers were dried over MgSO4 and the solvent removed. The off-white solid so obtained was purified by column chromatography (SiO2, ethylacetate:cyclohexane 1:1, Rf = 0.6). Compound 10 was obtained as a white solid (0.11 g, 0.24 mmol, 23.6%). M.p. 153.8 °C. 1H NMR (500 MHz, CDCl3) δ/ppm 8.80 (m, 4H, HA2), 8.13 (m, 4H, HA3), 8.03 (s, 2H, HB3), 7.70 (m, 2H, HC2), 7.07 (m, 2H, HC3), 4.05 (t, J = 6.6 Hz, 2H, HOCH2), 1.83 (m, 2H, HOCH2CH2), 1.49 (m, 2H, HOCH2CH2CH2), 1.42–1.12 (overlapping m, 12H, HCH2), 0.88 (m, 3H, HMe); 13C{1H} NMR (126 MHz, CDCl3) δ/ppm 160.8 (CC4), 155.0 (CA4), 151.0 (CB4), 149.8 (CA2), 147.1 (CB2), 129.7 (CC1), 128.5 (CC2), 121.6 (CA3), 118.8 (CB3), 115.5 (CC3), 68.4 (COCH2), 32.0 (CCH2), 29.7 (CCH2), 29.71 (CCH2), 29.5 (CCH2), 29.5 (CCH2), 29.3 (COCH2CH2), 26.2 (CCH2), 22.8 (COCH2CH2CH2), 14.3 (CMe). UV-VIS (MeCN, 2.5 × 10−5 M) λ/nm (ε/dm3 mol−1 cm−1) 270 (40132). ESI-MS m/z 466.6 [M + H]+ (calc. 466.3). Found C 80.04, H 7.54, N 9.29; required for C31H35N3O C 79.96, H 7.58, N 9.02.
[{Zn2(OAc)4(1)}n]
An MeOH solution of 1 (17.0 mg, 0.05 mmol) was placed in a vial inside a vial containing an MeOH solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol); the total volume of solvent was 20 mL. Colourless crystals of [{Zn2(OAc)4(1)}n] (5.6 mg, 0.008 mmol, 16%) grew within 2 weeks. IR (solid, ν/cm−1) 3074 (w), 2986 (w), 2930 (w), 2833 (w), 2051 (w), 1635 (s), 1617 (s), 1598 (s), 1539 (m), 1519 (m), 1423 (s), 1347 (m), 1327 (m), 1297 (m), 1259 (m), 1243 (m), 1222 (m), 1180 (m), 1120 (w), 1067 (m), 1051 (w), 1036 (m), 1025 (m), 993 (m), 936 (w), 897 (w), 850 (m), 832 (s), 813 (m), 774 (w), 747 (w), 733 (w), 685 (m), 666 (s), 652 (m), 641 (m), 619 (s), 568 (m), 526 (m), 497 (m). Found C 47.51, H 4.02, N 5.78; required for C30H29N3O9Zn2·3H2O C 47.39, H 4.64, N 5.53.[{Zn2(OAc)4(2)}n]
A solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol) in MeOH (8 mL) was layered over a solution of 2 (17.7 mg, 0.05 mmol) in CHCl3 (5 mL). Colourless crystals of [{Zn2(OAc)4(2)}n] (3.1 mg, 0.008 mmol, 9%) were obtained after 2 weeks. IR (solid, ν/cm−1) 2972 (w), 1637 (s), 1617 (s), 1604 (s), 1539 (m), 1520 (m), 1506 (m), 1469 (m), 1422 (s), 1326 (m), 1297 (m), 1270 (m), 1245 (s), 1221 (m), 1184 (m), 1124 (w), 1068 (m), 1047 (w), 1029 (m), 981 (m), 935 (w), 897 (w), 851 (m), 835 (s), 817 (m), 751 (w), 732 (w), 688 (w), 666 (s), 652 (s), 642 (m), 620 (s), 604 (m), 529 (m), 497 (w). Found C 51.31, H 4.55, N 5.63; required for C31H31N3O9Zn2C 51.69, H 4.34, N 5.83.[{Zn2(OAc)4(3)}n]
A solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol) in MeOH (8 mL) was layered over a solution of 3 (18.4 mg, 0.05 mmol) in CHCl3 (5 mL). Colourless crystals of [{Zn2(OAc)4(3)}n] (19.7 mg, 0.027 mmol, 54%) grew within 2 weeks. IR (solid, ν/cm−1) 2975 (w), 1635 (s), 1617 (s), 1604 (s), 1539 (m), 1520 (m), 1506 (m), 1469 (m), 1424 (s), 1345 (m), 1326 (m), 1298 (m), 1271 (m), 1245 (s), 1220 (m), 1184 (m), 1124 (w), 1068 (m), 1047 (w), 1029 (m), 981 (m), 935 (w), 897 (w), 851 (m), 835 (s), 817 (m), 749 (w), 733 (w), 688 (m), 666 (s), 653 (s), 642 (m), 620 (s), 605 (m), 529 (s), 497 (m). Found C 53.03, H 4.64, N 6.17; required for C32H33N3O9Zn2 C 52.34, H 4.53, N 5.72.[{Zn2(OAc)4(4)}n]
A solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol) in MeOH (8 mL) was layered over a solution of 4 (19.1 mg, 0.05 mmol) in CHCl3 (5 mL). Colourless crystals of [{Zn2(OAc)4(4)}n] (16.36 mg, 0.014 mmol, 28%) grew within 2 weeks, but were not of X-ray quality. For the bulk sample: found C 51.74, H 4.87, N 5.57; required for C33H35N3O9Zn2·H2O C 51.72, H 4.87, N 5.48.[{2Zn2(OAc)4(5)·2H2O}n] and [Zn2(OAc)4(5)2]
A solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol) in MeOH (8 mL) was layered over a solution of 5 (19.8 mg, 0.05 mmol) in CHCl3 (5 mL). Colourless blocks of [Zn2(OAc)4(5)2] mixed with crystals of [{2Zn2(OAc)4(5)·2H2O}n] were obtained. The bulk sample was characterized by powder X-ray diffraction (see text).[{Zn2(OAc)4(6)·MeCO2H}n]
A solution of Zn(OAc)2·2H2O (44.0 mg, 0.20 mmol) in MeOH (8 mL) was layered over a solution of 6 (41.0 mg, 0.1 mmol) in CHCl3 (5 mL) as solvents. Colourless crystals of [{Zn2(OAc)4(6)·MeCO2H}n] (30 mg, 0.136 mmol, 68%) were obtained. IR (solid, ν/cm−1) 2929 (w), 1636 (s), 1616 (s), 1519 (m), 1425 (s), 1293 (w), 1240 (m), 1182 (m), 1068 (w), 1031 (m), 850 (m), 834 (s), 731 (w), 666 (s), 653 (s), 621 (s), 603 (m), 528 (m). Found C 53.02, H 5.31, N 5.42; required for C35H39N3O9Zn2·H2O·MeCO2H C 52.91, H 5.20, N 5.29.[{4Zn2(OAc)4(7)·3H2O}n]
A solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol) in MeOH (8 mL) was layered over a solution of 7 (21.2 mg, 0.05 mmol) in CHCl3 (5 mL). After 2 weeks, colourless crystals of {2[Zn2(OAc)4(7)]·3H2O} (4.8 mg, 0.004 mmol, 6%) had grown. Found C 53.48, H 5.50, N 5.26; required for C144H158N12O36Zn8·3H2O C 53.88, H 5.15, N 5.24.[Zn2(OAc)4(8)2]
A solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol) in MeOH (8 mL) was layered over a solution of 8 (21.9 mg, 0.05 mmol) in CHCl3 (5 mL). Colourless crystals of [Zn2(OAc)4(8)2] (19.4 mg, 0.016 mmol, 31%) were obtained after 2 weeks. IR (solid, ν/cm−1) 2923 (w), 1639 (s), 1597 (s), 1516 (m), 1422 (s), 1296 (w), 1243 (m), 1179 (m), 1065 (w), 1029 (w), 997 (w), 849 (w), 828 (s), 804 (w), 680 (w), 664 (m), 637 (m), 620 (m), 604 (m), 523 (m). Satisfactory analysis for the bulk sample could not be obtained.[Zn2(OAc)4(9)2]
A solution of Zn(OAc)2·2H2O (44.0 mg, 0.20 mmol) in MeOH (8 mL) was layered over a solution of 9 (22.6 mg, 0.05 mmol) in CHCl3 (5 mL). After 2 weeks, colourless crystals of [Zn2(OAc)4(9)2] (58 mg, 0.046 mmol, 91%) had grown. IR (solid, ν/cm−1) 2951 (w), 2924 (w), 2851 (w), 1639 (s), 1619 (s), 1595 (s), 1539 (s), 1519 (s), 1481 (m), 1463 (m), 1424 (s), 1399 (s), 1298 (m), 1242 (m), 1178 (m), 1118 (w), 1073 (w), 1064 (m), 1030 (m), 1011 (m), 998 (m), 956 (w), 895 (w), 852 (m), 831 (s), 807 (m), 725 (w), 692 (w), 681 (m), 665 (s), 644 (m), 637 (m), 620 (s), 604 (s), 524 (m), 497 (w). Found C 62.33, H 6.08, N 6.46; required for C68H78N6O10Zn2·2H2O C 62.53, H 6.33, N 6.43.[Zn2(OAc)4(10)2]
A solution of Zn(OAc)2·2H2O (21.9 mg, 0.100 mmol) in MeOH (8 mL) was layered over a solution of 10 (23.3 mg, 0.05 mmol) in CHCl3 (5 mL). After 2 weeks, colourless crystals of [Zn2(OAc)4(10)2] (13.5 mg, 0.010 mmol, 21%) had grown. IR (solid, ν/cm−1) 2923 (m), 2851 (w), 1638 (s), 1619 (s), 1597 (s), 1560 (m), 1539 (m), 1519 (s), 1462 (w), 1424 (s), 1399 (s), 1343 (w), 1297 (m), 1242 (s), 1179 (m), 1118 (w), 1073 (m), 1064 (m), 1030 (m), 1017 (m), 997 (m), 897 (w), 849 (m), 830 (s), 808 (m), 735 (w), 724 (w), 680 (m), 664 (s), 644 (m), 637 (s), 620 (s), 603 (s), 524 (s). Found C 64.49, H 6.40, N 6.56; required for C70H82N6O10Zn2 C 64.76, H 6.37, N 6.47.Crystallography
Single crystal data were collected on a Bruker APEX-II diffractometer with data reduction, solution and refinement using the programs APEX25 and SHELXL97 or SHELX-13.26 ORTEP diagrams and structure analysis used Mercury v. 3.0.1 or 3.3.27,28 Crystallographic data for all structures are given in Table 1. Powder diffractograms were measured on a STOE STADI P diffractometer equipped with a Cu Kα1 radiation (λ = 1.540598 Å) and a Mythen1K detector.| Compound | 6 | 7 | [{Zn2(OAc)4(1)}n] | [{Zn2(OAc)4(2)}n] |
|---|---|---|---|---|
| Formula | C27H27N3O | C28H29N3O | C30H29N3O9Zn2 | C31H31N3O9Zn2 |
| Formula weight | 409.52 | 423.54 | 706.34 | 720.37 |
| Crystal colour and habit | Colourless block | Colourless plate | Colourless block | Colourless plate |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Cc | P21/c | P21/c |
| a, b, c/Å | 10.0691(8), 10.7273(9), 11.9802(9) | 19.3470(19), 11.0840(13), 10.8074(10) | 7.9572(6), 15.2139(11), 24.9697(17) | 7.9713(10), 15.4036(16), 25.046(3) |
| α, β, γ/° | 93.200(3), 106.786(3), 117.792(3) | 90, 92.930(7), 90 | 90, 90.666(5), 90 | 90, 90.562(10), 90 |
| U/Å3 | 1067.91(15) | 2314.5(4) | 3022.6(4) | 3075.1(6) |
| D c/Mg m−3 | 1.274 | 1.215 | 1.552 | 1.556 |
| Z | 2 | 4 | 4 | 4 |
| μ(Cu-Kα)/mm−1 | 0.612 | 0.581 | 2.455 | 2.425 |
| Refln. collected (Rint) | 20505 (0.0288) |
8976 (0.0456) | 25658 (0.0579) |
27516 (0.0831) |
| Unique refln. | 3824 | 2852 | 5359 | 5485 |
| Refln. for refinement | 3549 | 2562 | 4536 | 4159 |
| Parameters | 281 | 291 | 402 | 411 |
| R 1 (R1 all data) | 0.0334 (0.0357) | 0.0349 (0.0417) | 0.0528 (0.0620) | 0.0574 (0.0782) |
| wR2 (wR2 all data) | 0.0916 (0.0937) | 0.0855 (0.0893) | 0.1428 (0.1487) | 0.1438 (0.1550) |
| Goodness of fit | 1.044 | 1.035 | 1.097 | 1.073 |
| CCDC deposition | 1002439 | 1002432 | 1002429 | 1002430 |
| Compound | [{Zn2(OAc)4(3)}n] | [{2Zn2(OAc)4(5)·2H2O}n] | [Zn2(OAc)4(5)2] | [{Zn2(OAc)4(6)·MeCO2H}n] |
|---|---|---|---|---|
| Formula | C32H33N3O9Zn2 | C68H78N6O20Zn2 | C60H62N6O10Zn2 | C37H43N3O11Zn2 |
| Formula weight | 734.39 | 1560.94 | 1157.94 | 836.52 |
| Crystal colour and habit | Colourless block | Colourless block | Colourless block | Colourless block |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P21/c |
P |
P |
P21/c |
| a, b, c/Å | 8.1511(6), 16.0769(12), 24.7374(18) | 15.1794(6), 17.1547(7), 17.2922(7) | 10.8959(6), 11.2529(7), 12.1372(7) | 8.0908(9), 29.735(3), 16.6368(18) |
| α, β, γ/° | 90, 90.796(3), 90 | 115.890(2), 100.999(2), 108.227(2) | 109.804(2), 102.546(2), 91.431(2) | 90, 99.076(9), 90 |
| U/Å3 | 3241.4(4) | 3554.1(3) | 1358.81(14) | 3952.3(7) |
| D c/Mg m−3 | 1.505 | 1.455 | 1.415 | 1.406 |
| Z | 4 | 2 | 1 | 4 |
| μ(Cu-Kα)/mm−1 | 2.312 | 2.163 | 1.626 | 2.004 |
| Refln. collected (Rint) | 77184 (0.0382) |
65543 (0.0533) |
21259 (0.0274) |
25130 (0.1318) |
| Unique refln. | 5861 | 12842 |
4875 | 7029 |
| Refln. for refinement | 5531 | 10005 |
4568 | 4459 |
| Parameters | 420 | 910 | 355 | 485 |
| R 1 (R1 all data) | 0.0331 (0.0348) | 0.0516 (0.0685) | 0.0324 (0.0344) | 0.1061 (0.1554) |
| wR2 (wR2 all data) | 0.0981 (0.1007) | 0.1364 (0.1490) | 0.0845 (0.0863) | 0.2826 (0.3108) |
| Goodness of fit | 1.062 | 1.043 | 1.051 | 1.136 |
| CCDC deposition | 1002431 | 1002437 | 1002436 | 1002440 |
| Compound | [{4Zn2(OAc)4(7)·3H2O}n] | [Zn2(OAc)4(8)2] | [Zn2(OAc)4(9)2] | [Zn2(OAc)4(10)2] |
|---|---|---|---|---|
| Formula | C144H164N12O39Zn8 | C66H74N6O10Zn2 | C68H78N6O10Zn2 | C70H82N6O10Zn2 |
| Formula weight | 3216.06 | 1242.10 | 1270.14 | 1298.20 |
| Crystal colour and habit | Colourless block | Colourless block | Colourless block | Colourless plate |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P |
P |
P |
P |
| a, b, c/Å | 15.1693(8), 17.4457(10), 17.6212(16) | 14.2398(7), 14.7602(7), 15.7924(8) | 10.9315(7), 11.2063(8), 13.3817(9) | 10.7384(5), 11.1810(6), 13.7983(7) |
| α, β, γ/° | 104.114(4), 112.607(4), 109.132(3) | 89.058(2), 66.110(2), 87.738(2) | 96.615(3), 102.460(3), 99.944(3) | 91.221(3), 103.627(3), 94.827(3) |
| U/Å3 | 3683.4(5) | 3032.5(3) | 1556.69(18) | 1602.91(14) |
| D c/Mg m−3 | 1.447 | 1.493 | 1.355 | 1.345 |
| Z | 1 | 2 | 1 | 1 |
| μ(Cu-Kα)/mm−1 | 2.099 | 1.493 | 1.466 | 1.435 |
| Refln. collected (Rint) | 43849 (0.0505) |
42825 (0.0365) |
28581 (0.0345) |
30683 (0.0438) |
| Unique refln. | 12927 |
10870 |
5584 | 5791 |
| Refln. for refinement | 10051 |
9374 | 5065 | 4752 |
| Parameters | 938 | 763 | 391 | 518 |
| R 1 (R1 all data) | 0.0594 (0.0772) | 0.0443 (0.0515) | 0.0348 (0.0390) | 0.0444 (0.0567) |
| wR2 (wR2 all data) | 0.1704 (0.1876) | 0.1122 (0.1179) | 0.0877 (0.0911) | 0.1151 (0.1241) |
| Goodness of fit | 1.035 | 1.049 | 1.031 | 1.023 |
| CCDC deposition | 1002433 | 1002435 | 1002438 | 1002434 |
Results and discussion
Ligand synthesis and characterization
Compounds 1–9 were prepared using the one pot method of Wang and Hanan29 summarized in Scheme 2, and were isolated in yields ranging from 47.3 to 63.1%. A convenient route to compound 10 proved to be the reaction of nucleophilic 4-([4,2′:6′,4′′-terpyridin]-4′-yl)phenol (formed by deprotection of 1 with pyridinium chloride) with 1-bromodecane (Scheme 3); this strategy is analogous to that used for the preparation of polyethyleneoxy-functionalized tpy derivatives.30 The electrospray mass spectrum of each compound exhibited a base peak corresponding to [M + H]+. The 1H and 13C{1H} NMR spectra of 1–10 were assigned using COSY, NOESY, HMQC and HMBC methods, and representative 1H NMR spectra are shown in Fig. S1.† The NOESY spectrum of 1 is shown in Fig. 1 and illustrates that crosspeaks between protons HA3/HB3, HB3/HC2 and HC3/HOMe (see Scheme 1 for labelling) allow unambiguous assignments of the aromatic protons in the A and C rings. The appearance of a triplet or multiplet around ∂ 4 ppm in the 1H NMR spectrum of each compound along with an HMQC associated signal in the 13C NMR spectrum at ∂ 55.6 ppm in 1 or in the range ∂ 64–69 ppm for 2–10 was consistent with the presence of the alkoxy substituent. Fig. S2† shows the solution absorption spectra of the ten 4'-(4-ROC6H4)-4,2′:6′,4′′-tpy ligands. Each exhibits an intense absorption band around 270 nm with a shoulder at 305 nm. Dominant contributions to these bands come from π* ← π and π* ← n transitions. | ||
| Scheme 2 Synthetic approach to compounds 1–9. | ||
 | ||
| Scheme 3 Synthetic route to 10. | ||
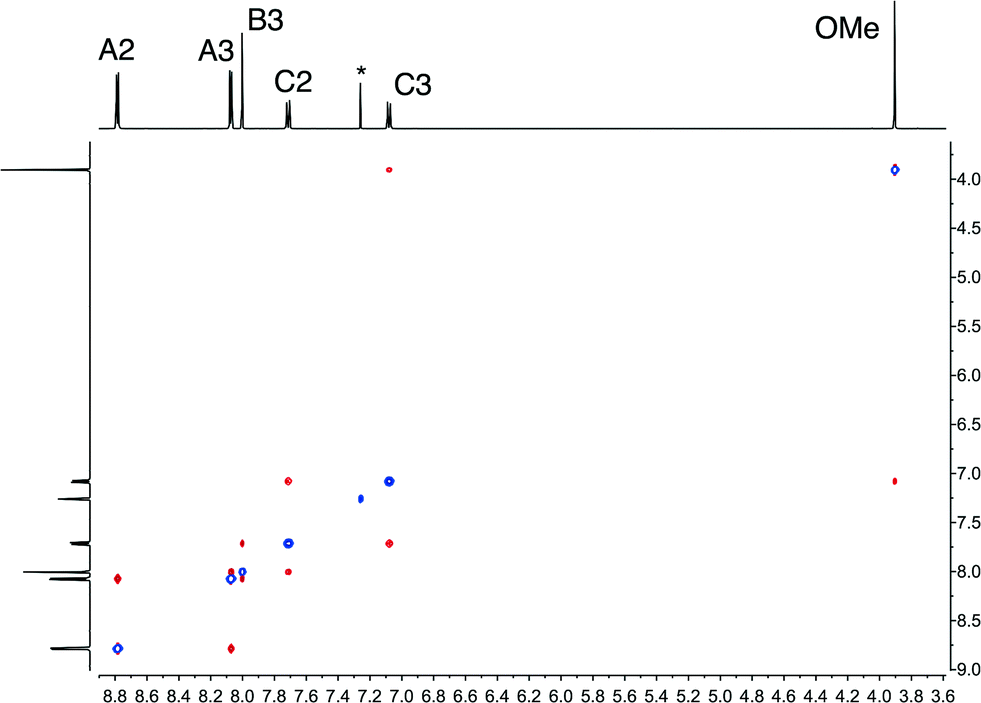 | ||
| Fig. 1 500 MHz NOESY spectrum of compound 1 in CDCl3 (295 K). The signal marked * is residual CHCl3. | ||
Single crystals of 6 and 7 suitable for X-ray analysis were serendipitously obtained from layer diffusion of solutions of 6 in CHCl3 and Zn(OAc)2·2H2O in MeOH, or by slow evaporation of the solvent from a MeOH solution of 7, respectively. Fig. 2 and S3† show the structures of these compounds and important bond parameters are given in the figure captions. All bond lengths and angles are as expected. At the molecular level, the two structures are similar with the alkyl chain in an extended conformation and the phenyltpy domain twisted. The angles between the rings containing atoms N1/N2, N2/N3 and N2/C16 are 17.6, 22.7 and 14.3° in 6, and 16.9, 27.9 and 20.0° in 7. Compounds 6 and 7 crystallize in the triclinic P and monoclinic Cc space groups, respectively, and the packing of molecules of 6 and 7 necessarily differs. In both structures, the primary assembly motif is a ribbon built up by translation (Fig. 3a) with CH⋯N short contacts (in 6, C14H14a⋯N1i = 2.57 Å, symmetry code i = x, 1 + y, z; in 7, C2H2a⋯N3i = 2.67 Å, symmetry code i = x, y, 2 + z). In 6, adjacent ribbons are related by inversion leading to interdigitation of hexoxy chains and the assembly of planar sheets (Fig. 3b). The packing, dictated by P symmetry, resembles that observed in 4′-(4-nbutoxyphenyl)-4,2′:6′,4′′-terpyridine (CSD31 code ACUKAK),32 4′-(4-dodecoxyphenyl)-4,2′:6′,4′′-terpyridine (QATQEJ),23 and 4′-(4-noctadecoxyphenyl)-2,2′:6′,2′′-terpyridine (JERNUQ)33 and 4′-(4-noctoxyphenyl)-2,2′:6′,2′′-terpyridine (JERPAY).33 A similar layer arrangement is also observed in 4′-(4-octoxyphenyl)-4,2′:6′,4′′-terpyridine34 (XAYPOC); each sheet is built up from centrosymmetric pairs of molecules but in this case, additional symmetry in the space group C2/c, affects the relationship between molecules in adjacent sheets. In 7, neighbouring ribbons are related by a glide plane giving rise to an off-axis interdigitation of heptoxy chains, all of which point in the same direction (Fig. 3c). The overall packing in 7 (Fig. 3d) involves a combination of π-stacking of aromatic rings, CH⋯π contacts and inter-chain van der Waals interactions, with no one type apparently predominant. It is likely that the energy difference between the two different molecular arrangements in the lattices of 6 and 7 is small, and that environmental effects influence the observed packing.
 | ||
| Fig. 2 Structure of compound 6 with ellipsoids plotted at 40% probability level. Selected bond metrics: N1–C1 = 1.3375(14), N1–C5 = 1.3393(14), N2–C10 = 1.3449(14), N2–C6 = 1.3428(13), N3–C11 = 1.3397(15), N3–C15 = 1.3379(15), C19–O1 = 1.3601(13), O1–C22 = 1.4361(13) Å; C1–N1–C5 = 115.75(9), C10–N2–C6 = 117.28(9), C11–N3–C15 = 116.00(9), C19–O1–C22 = 117.75(8)°. | ||
 | ||
| Fig. 3 (a) Assembly of molecules of 6 into ribbons; molecules are related by translation and an analogous motif occurs in 7. (b) Part of one planar sheet in 6 showing interdigitation of chains. (c) Oblique interdigitation of heptoxy chains in adjacent ribbons of molecules of 7; magenta ribbon is related to the pale grey one by a glide plane. (d) Packing of molecules of 7 with heptoxy chains oriented in the same direction. | ||
Coordination polymers formed between Zn(OAc)2·2H2O and ligands 1–3
Layer diffusion of solutions of 1–3 with Zn(OAc)2·2H2O yielded single crystals suitable for X-ray diffraction. Structure determination confirmed the formation of the one-dimensional coordination polymers [{Zn2(OAc)4(1)}n], [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n]. Elemental analytical data and solid state IR spectra were recorded for the bulk materials. All three compounds crystallize in the space group P21/c with unit cell dimensions that are similar (Table 1) and related to the cell parameters of a family of [{Zn2(OAc)4(4′-X-4,2′:6′,4′′-tpy)}n] polymers1 where X = Ph,35 4-BrC6H4,36 4-MeSC6H4,36 biphenyl-4-yl,37 pentafluorobiphenyl-4-yl37 and tBu.38 Because of the similarity in the structures of [{Zn2(OAc)4(1)}n], [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n], we discuss only [{Zn2(OAc)4(1)}n] in detail, and then present a comparison of the three compounds. Fig. 4 shows the repeat unit in [{Zn2(OAc)4(1)}n]. The polymer chain comprises paddle-wheel {Zn2(OAc)4} units interconnected by 1 which coordinates through the two outer N-donors. The bond parameters of the {Zn2(OAc)4} entity (Fig. 4 caption) are typical. The 4,2′:6′,4′′-tpy is virtually planar (angles between the rings containing atoms N1/N2 and N2/N3 = 4.6 and 4.0°, respectively) and the phenyl substituent is twisted 28.6° out of the plane of the pyridine ring to which it is bonded. The methoxy group lies in the plane of the phenyl ring. This, along with the C19–O1–C22 bond angle of 117.2(4)° and the difference in C19–O1 (1.370(5) Å) and O1–C22 (1.432(6) Å) bond lengths, is consistent with extension of the π-delocalization from the phenyl ring to atom O1.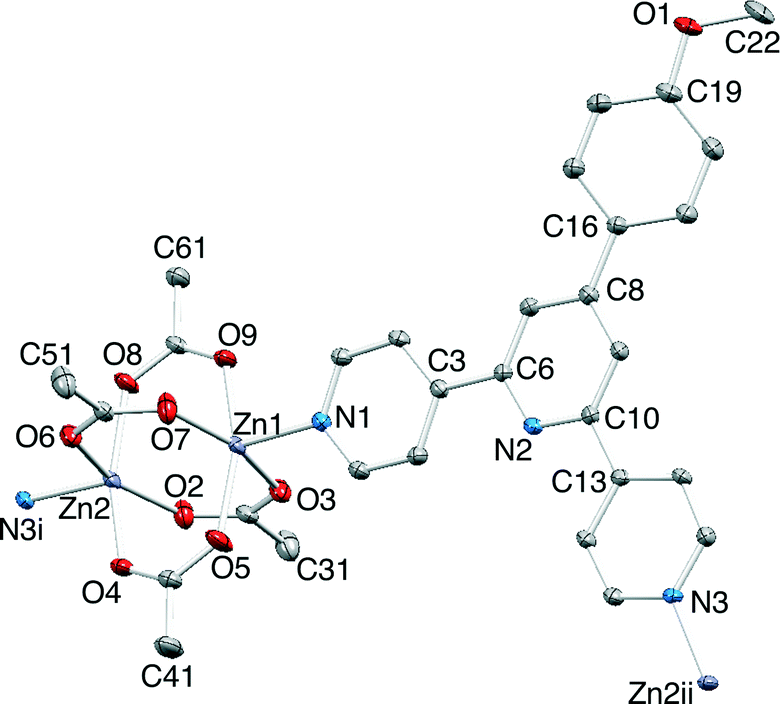 | ||
| Fig. 4 Repeat unit in [{Zn2(OAc)4(1)}n] with ellipsoids plotted at 40% probability level. Symmetry codes: i = 1 + x, 1/2 − y, −1/2 + z; ii = −1 + x, 1/2 − y, 1/2 + z. Selected bond distances: Zn1–N1 = 2.028(3), Zn2–N3i = 2.034(3), Zn1–O5 = 2.032(3), Zn1–O7 = 2.037(3), Zn1–O9 = 2.039(3), Zn1–O3 = 2.061(3), Zn2–O4 = 2.032(3), Zn2–O2 = 2.038(3), Zn2–O8 = 2.046(3), Zn2–O6 = 2.069(3), Zn1⋯Zn2 = 2.9058(7) Å. | ||
Each polymer chain in [{Zn2(OAc)4(1)}n] has a zigzag form and chains assemble into sheets with the 4-methoxyphenyl group pointing into a V-shaped cavity in an adjacent chain (red or blue chains in Fig. 5a). This assembly replicates that observed in related complexes, and detailed discussion here is unnecessary.1 Of relevance to the present study is the packing of the sheets. Fig. 5a shows that the chains in adjacent sheets are arranged so that the 4,2′:6′,4′′-tpy domains are offset from one another, facilitating face-to-face π-interactions between the pyridine rings containing N2 and N3 and those with N2i and N3i (symmetry code i = −1 − x, 1 − y, 1 − z). The interplane and centroid⋯centroid distances are 3.32 and 3.68 Å, respectively. As Fig. 5b shows, the π-interactions extend to form a tetradecker stack involving symmetry related pyridine rings containing atoms N1ii and N1iii (symmetry codes ii = − x, 1 − y, 1 − z; iii = − 1 + x, y, z); the distance from the centroid of the N1-containing ring to the least squares planes through the N2/N3 bipyridine domain is 3.35 Å. The tetradecker π-stack is the repeat assembly motif which results in efficient interactions between slipped 4,2′:6′,4′′-tpy units of adjacent sheets to give infinite π-stacked oblique columns running through the lattice.
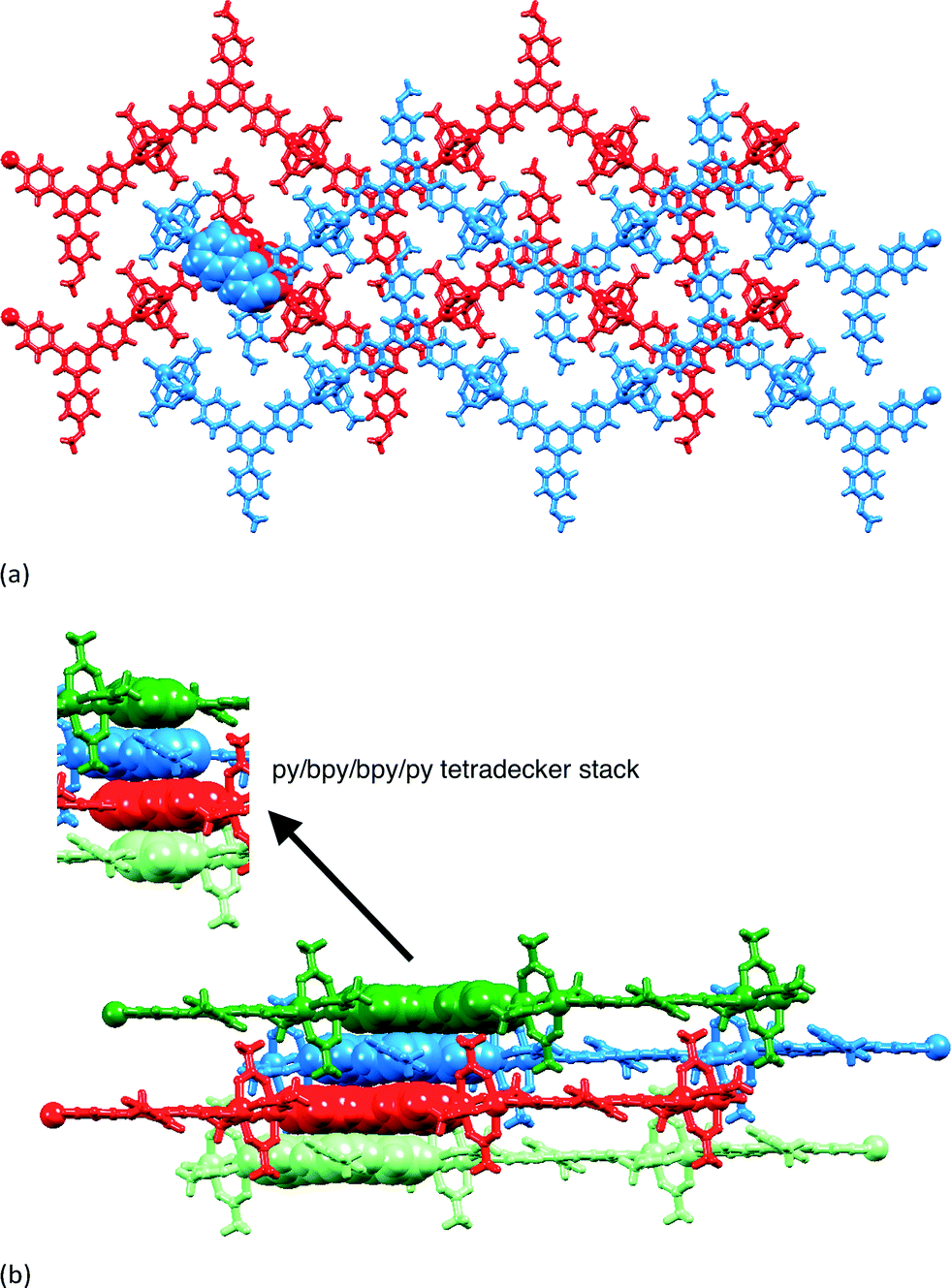 | ||
| Fig. 5 (a) Packing of zigzag chains in [{Zn2(OAc)4(1)}n]; chains coloured blue lie in one sheet, interacting through π-stacking of tpy domains with chains in the adjacent sheet (red); a representative π-interaction is shown in space-filling representation. (b) Infinite column of face-to-face interactions between slipped tpy domains can be described in terms of repeating tetradecker π-stacked py/bpy/bpy/py assembly motifs (top left). | ||
The repeat units in the polymers of [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n] (Fig. S4 and S5†) are structurally analogous to that in [{Zn2(OAc)4(1)}n], and bond parameters (see respective figure captions) are comparable. As in [{Zn2(OAc)4(1)}n], the 4,2′:6′,4′′-tpy units in [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n] are close to planar (angles between the rings containing N1/N2 and N2/N3 are both 6.4° in [{Zn2(OAc)4(2)}n], and are 6.2 and 7.6° in [{Zn2(OAc)4(3)}n]). The phenyl substituent is twisted through 27.3° with respect to the plane of the central pyridine ring in [{Zn2(OAc)4(2)}n] compared to 28.2° in [{Zn2(OAc)4(3)}n], and 28.6° in [{Zn2(OAc)4(1)}n]. The polymer chains in [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n] adopt the same zigzag configuration as in [{Zn2(OAc)4(1)}n], and the packing of the chains into sheets and stacking interactions between the sheets follow the same assembly principles in all three compounds. In [{Zn2(OAc)4(2)}n], the parameters that define the tetradecker π-stack assembly motif (see the description of [{Zn2(OAc)4(1)}n] and Fig. 5b) are interplane and centroid⋯centroid distances of 3.32 and 3.70 Å for the central bpy/bpy domain and a centroid-to-plane distance of 3.38 Å for the py/bpy stacking interaction. These distances are close to the corresponding values of 3.32, 3.68 and 3.35 Å in [{Zn2(OAc)4(1)}n], but increase to 3.39, 3.75 and 3.41 Å on going to [{Zn2(OAc)4(3)}n]. An associated parameter that increases is the distance between adjacent chains in one sheet, and this is assessed from the distance d defined in Fig. 6a. This dimension has been used in an overview of related [{Zn2(OAc)4(4′-X-4,2′:6′,4′′-tpy)}n] structures in which d varies from 12.088 Å (X = tBu) to 13.347 Å (X = biphenyl-4-yl).1 In [{Zn2(OAc)4(1)}n], [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n], d = 12.417(6), 12.601(6) and 13.277(3) Å, respectively, and the trend is consistent with the accommodation of the increasingly bulkier alkyl group in the V-shaped cavity of an adjacent chain (Fig. 6b–d). It is significant that the n-propyl chain is not in an extended conformation (Fig. S5†). Folding of the chain allows accommodation in the pocket (Fig. 6d) without the need for the chains to move further apart. Such lateral movement would be detrimental to the π-stacking interactions between chains in adjacent sheets.
 | ||
| Fig. 6 (a) Distance d measures the separation of central pyridine rings of 4,2′:6′,4′′-tpy units in adjacent chains in the same sheet. Accommodation of the alkoxy substituent in the V-shaped pocket of an 4,2′:6′,4′′-tpy unit in an adjacent chain: (b) methoxy, (c) ethoxy, (d) n-propoxy. | ||
Reaction between Zn(OAc)2·2H2O and 4 (the n-butoxy-tailed ligand) under the same layering conditions as for reactions between Zn(OAc)2·2H2O and 2 or 3 led to the formation of crystals, but none was suitable for structural determination. However, elemental analysis of the bulk sample was consistent with the formulation [{Zn2(OAc)4(4)}n·H2O] and the inclusion of H2O in the lattice is consistent with that observed in [{2Zn2(OAc)4(5)·2H2O}n] (see below) as the alkoxy chain lengthens.
A frustrated system: [{2Zn2(OAc)4(5)·2H2O}n] and [Zn2(OAc)4(5)2]
Room temperature layering of an MeOH solution of Zn(OAc)2·2H2O over a CHCl3 solution of 5 with a molar ratio Zn(OAc)2 to 5 of 2:1 resulted in the formation of colourless blocks. Elemental analysis of the bulk sample was not definitive in terms of the product, and single crystal X-ray diffraction confirmed the presence of two materials: the 1D-coordination polymer [{2Zn2(OAc)4(5)·2H2O}n] and the discrete complex [Zn2(OAc)4(5)2].
The compound [{2Zn2(OAc)4(5)·2H2O}n] crystallizes in the triclinic space group P, with two independent {Zn2(OAc)4} and two independent ligands 5 in the asymmetric unit (Fig. 7). This contrasts with [{Zn2(OAc)4(1)}n], [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n] (monoclinic, P21/c and one independent [{Zn2(OAc)4}(L)] unit). The second feature that distinguishes the polymer containing the pentoxy-tailed ligand 5 from those of containing the methoxy, ethoxy and n-propoxy-substituted ligands is the presence of solvent in the lattice (see later). As Fig. 7 shows, the 1D-chain in [{2Zn2(OAc)4(5)·2H2O}n] mimics that in the earlier polymers in the series and the bond parameters for the {Zn2(OAc)4} units are unexceptional. The tpy domains of the two independent ligands are more slightly twisted than in the complexes with shorter alkoxy chains; in [{2Zn2(OAc)4(5)·2H2O}n] the angles between the planes of the rings containing N1/N2 and N2/N3 are 4.5 and 9.1°, and between those with N4/N5 and N5/N6 are 5.5 and 10.4°. The phenyl rings in the two 4,2′:6′,4′′-tpy ligands are twisted 26.3 and 27.4° with respect to each central pyridine ring. The most noticeable difference between the two independent ligands is the conformation of the pentoxy chain. The chain containing atom O2 is in an approximately extended conformation, and the maximum deviation of an alkyl C atom from the plane through the phenyl ring to which the chain is attached is 1.17 Å for C50; the terminal atom C52 lies 0.77 Å out of this plane. In contrast, the chain containing O1 is twisted out of the plane of the phenyl ring containing C16 (Fig. 7); the terminal atom C26 lies 3.02 Å away from the plane of the phenyl ring containing C16.
 | ||
| Fig. 7 Repeat unit in [{2Zn2(OAc)4(5)·2H2O}n] with ellipsoids plotted at 40% probability level; water molecules are omitted. Symmetry codes: i = −2 + x, −1 + y, z; ii = 2 + x, 1 + y, z. Selected bond distances: Zn1–O70 = 2.027(3), Zn1–N1 = 2.029(3), Zn1–O90 = 2.043(3), Zn1–O60 = 2.046(3), Zn1–O80 = 2.050(3), Zn2–O81 = 2.011(3), Zn2–O71 = 2.032(3), Zn2–O61 = 2.034(4), Zn2–N6ii = 2.036(3), Zn2–O91 = 2.050(3), Zn3–O100 = 2.024(3), Zn3–N3 = 2.030(3), Zn3–O130 = 2.035(3), Zn3–O120 = 2.042(3), Zn3–O110 = 2.047(3), Zn3–Zn4 = 2.8967(6), Zn4–O111 = 2.015(3), Zn4–O131 = 2.032(3), Zn4–N4 = 2.045(3), Zn4–O121 = 2.054(3), Zn4–O101 = 2.066(3), Zn1⋯Zn2 = 2.9048(6) Å. | ||
At first glance, the packing of the chains in [{2Zn2(OAc)4(5)·2H2O}n] appears the same as in [{Zn2(OAc)4(1)}n] (compare Fig. 8a with Fig. 5a). However, adjacent chains in the same sheet (e.g. blue in Fig. 8a) are not equally separated. The distance d defined in Fig. 6a has alternating values of 12.521(6) and 15.719(6) Å within a sheet in [{2Zn2(OAc)4(5)·2H2O}n]. The smaller and larger separations correspond, respectively, to accommodation of folded or extended pentoxy chains (Fig. 8a). The slippage of the chains affects the π-stacking interactions. In contrast to the infinite π-stacked assemblies in [{Zn2(OAc)4(1)}n], [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n] (Fig. 5b), 4,2′:6′,4′′-tpy domains in [{2Zn2(OAc)4(5)·2H2O}n] assemble in discrete tetradecker π-stacks (Fig. 8b) made up of three pairs of bpy/bpy interactions. For the central bpy/bpy domain of the stack (yellow space-filling representation in Fig. 8b), the interplane and centroid⋯centroid distances are 3.31 and 3.65 Å; for the outer bpy/bpy stacking interaction (green to yellow in Fig. 8b), the angle between the least squares planes through the bpy units is 6.3° and the centroid⋯plane separation is 3.34 Å.
 | ||
| Fig. 8 (a) Packing of zigzag chains in [{2Zn2(OAc)4(5)·2H2O}n]; chains coloured blue and red lie in adjacent sheets. (b) Discrete tetradecker π-stack; pyridine rings coloured green or yellow (space-filling representation) are, respectively, attached to n-pentoxyphenyl domains with extended or folded chains. | ||
In contrast to [{Zn2(OAc)4(1)}n], [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n], the change in packing associated with the longer alkyl chains in [{Zn2(OAc)4(5)}n] gives rise to cavities in the lattice that are occupied by H2O molecules (modelled as one full, one half and two quarter occupancies). The partial occupancies mean that detailed discussion of interactions involving these molecules is not meaningful. Solvent inclusion indicates that the packing of the 1-dimensional polymer chains is less efficient with n-pentoxy-substituted ligands than with the methoxy, ethoxy and n-propoxy-substituted ligands. Note that the elemental analytical data for the bulk material containing the n-butoxy-substituted ligand 4 are also consistent with the presence of water in the lattice.
The colourless blocks of the polymer [{2Zn2(OAc)4(5)·2H2O}n] were accompanied in the same crystallization tube by colourless crystals of the discrete complex [Zn2(OAc)4(5)2]. The Zn: ligand ratio of 1:1 in [Zn2(OAc)4(5)2] differs from the 2:1 ratio observed in [{2Zn2(OAc)4(5)·2H2O}n], and the discrete molecular structure is described in detail in a later section along with the structures of [Zn2(OAc)4(8)2], [Zn2(OAc)4(9)2] and [Zn2(OAc)4(10)2].
The bulk sample containing ligand 5 was characterized by powder X-ray diffraction and the data in Fig. S6† confirm the presence of both components.
[{Zn2(OAc)4(6)·MeCO2H}n] and [{4Zn2(OAc)4(7)·3H2O}n]
Crystallization experiments combining zinc(II) acetate with either ligand 6 or 7 resulted in colourless blocks, single crystal X-ray structure determinations of which confirmed 1-dimensional coordination polymers [{Zn2(OAc)4(6)·MeCO2H}n] and [{4Zn2(OAc)4(7)·3H2O}n]. Elemental analysis for the bulk samples was consistent with formulations of [{Zn2(OAc)4(6)·H2O·MeCO2H}n] and [{4Zn2(OAc)4(7)·3H2O}n]. The coordination modes of ligands 6 and 7 mimic those of 1–3 and 5 in the respective polymers. Fig. S7 and S8† show the structures of the repeat units in [{Zn2(OAc)4(6)·MeCO2H}n] and [{4Zn2(OAc)4(7)·3H2O}n], the asymmetric units in which contain one and two independent ligands, respectively. In [{Zn2(OAc)4(6)·MeCO2H}n], the 4,2′:6′,4′′-tpy domain is slightly twisted (angles between the least squares planes of the rings containing N1/N2 and N2/N3 = 3.5 and 17.6°) and the phenyl ring deviates 15.4° from the plane of the pyridine ring containing N2 (see Fig. S7† for atom numbering). In [{4Zn2(OAc)4(7)·3H2O}n], the corresponding angles are 9.4, 11.6 and 29.7° for one independent ligand, and 0.5, 11.8 and 26.1° for the second.In [{Zn2(OAc)4(6)·MeCO2H}n], the n-hexoxy chain is in an approximately extended conformation and adjacent chains pack into sheets in an analogous manner to those in [{Zn2(OAc)4(1)}n], [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n]. However, comparison of Fig. 9a with Fig. 6b–d illustrates that the increased chain length forces the chains much further apart, while retaining a similar overall packing regime; the distance d (defined in Fig. 6a) is 16.78(1) Å. The voids between adjacent chains in a sheet (Fig. 9a) are partially occupied by acetate groups from {Zn2(OAc)4} in adjacent sheet and the remaining cavities are filled by acetic acid molecules (Fig. 9b). Supporting evidence for [{Zn2(OAc)4(6)·MeCO2H}n] rather than a salt containing protonated 6 and acetate ions is two-fold. First, acetate oxygen atom O100 is 2.69(1) Å distant from atom O61iii of an {Zn2(OAc)4} unit (symmetry code iii = −1 − x, −1/2 + y, 3/2 − z) consistent with a hydrogen bond (H100⋯O61iii = 1.85 Å; O100–H100⋯O61iii = 171°). Secondly, the planarity of the 4,2′:6′,4′′-tpy is inconsistent with protonation of atom N2 (the non-coordinated nitrogen atom). To the best of our knowledge, the pKa values of the conjugate acids of 4,2′:6′,4′′-tpy have not been determined. For [Hpy]+ (py = pyridine), pKa = 5.1439 but this is highly dependent upon solvent.40 Tocher and coworkers have shown that crystallizations of pyridine with a number of simple organic acids lead to either salts or co-crystals, and if the difference between pKa values of acid and [Hpy]+ is small, predictions of salt formation are ambiguous.39 For the bidentate ligand 6, the only site available for protonation is the central pyridine nitrogen, and therefore 2,4,6-triphenylpyridine is a more appropriate model than pyridine. The pKa of [H(2,4,6-triphenylpyridine)]+ (determined in 70% EtOH solution) is 1.82,41 and the pKa of coordinated [H6]+ should be closer to this value than to that of [Hpy]+. This is consistent with the observation of acetic acid (rather than acetate and protonated 4,2′:6′,4′′-tpy) in the lattice of [{Zn2(OAc)4(6)·MeCO2H}n] (pKa of MeCO2H = 4.75).
 | ||
| Fig. 9 [{Zn2(OAc)4(6)·MeCO2H}n]: (a) accommodation of the n-hexoxy chain in the V-shaped pocket of an 4,2′:6′,4′′-tpy unit in an adjacent chain forces the chains apart (compare Fig. 6). (b) Packing of sheets (green, red, blue) and inclusion of MeCO2H molecules (yellow). | ||
The π-stacking motifs which define the interactions between the sheets in [{Zn2(OAc)4(6)·MeCO2H}n] do not replicate those in the polymers discussed above. Centrosymmetric pairs of ligands 6 stack with π-interactions between phenyl and pyridine (with N1) rings (Fig. 10a); the angle between the ring planes = 11.9° and centroidPh⋯planepy distance = 3.36 Å. These interactions are augmented by pyridine⋯pyridine (N1/N2iv rings, symmetry code iv = −x, 1 − y, 2 − z) interactions at an intercentroid separation of 3.57 Å, which result in an infinite stack of ligands (Fig. 10b).
 | ||
| Fig. 10 π-Stacking of centrosymmetric pairs of ligands in [{Zn2(OAc)4(6)·MeCO2H}] involving pyridine (with N1) and phenyl rings and (b) additional pyridine⋯pyridine (N1/N2) interactions. | ||
In [{4Zn2(OAc)4(7)·3H2O}n], the n-heptoxy chains in the two independent ligands adopt a folded and near-extended conformation, respectively. This is reminiscent of [{2Zn2(OAc)4(5)·2H2O}n] and, as in the latter, the zigzag chains in [{4Zn2(OAc)4(7)·3H2O}n] assemble into sheets with two characteristic inter-chain separations (d = 12.579(7) and 16.869(7) Å; d is defined in Fig. 6a). The shorter and longer separations are, respectively, associated with accommodation of the folded and extended chains (as shown in Fig. 8a for [{2Zn2(OAc)4(5)·2H2O}n]) in which values of d are 12.521(6) and 15.719(6) Å. The differences between the separation of the chains in the two structures has little effect on the overall packing. This involves discrete tetradecker π-stacks comprising three pairs of bpy/bpy interactions, each stack being terminated by two {Zn2(OAc)4} units (Fig. 11). A comparison of Fig. 8b and 11 shows the similarity of the packing in [{2Zn2(OAc)4(5)·2H2O}n] and [{4Zn2(OAc)4(7)·3H2O}n]. For the central bpy/bpy interaction (Fig. 11, yellow space-filling representations), the interplane separation is 3.35 Å, while for the outer bpy/bpy interaction (Fig. 11, green to yellow), the angle between the least squares planes through the bpy units is 7.5° and the centroid⋯plane distance is 3.26 Å.
 | ||
| Fig. 11 Discrete tetradecker π-stack in [{4Zn2(OAc)4(7)·3H2O}n]; pyridine rings (space-filling representation) coloured green or yellow are attached, respectively, to n-heptoxyphenyl units containing extended or folded chains. | ||
Discrete complexes formed between Zn(OAc)2·2H2O and ligands 5 and 8–10
The molecular structure of [Zn2(OAc)4(5)2] (Fig. 12) resembles that reported for [Zn2(OAc)4(L)2] where L = 4′-(4-dodecoxyphenyl)-4,2′:6′,4′′-tpy.23 [Zn2(OAc)4(5)2] crystallizes in the P space group with half of the molecule in the asymmetric unit; all bond metrics (Fig. 12 caption) are unexceptional. Each ligand 5 is monodentate and binds through atom N1 to an axial site of the paddle-wheel {Zn2(OAc)4} unit. The n-pentoxy group adopts an extended conformation (Fig. 12). The 4,2′:6′,4′′-tpy domain is virtually planar (the angles between the planes of the rings containing N1/N2 an N2/N3 are 1.3 and 6.5°). The phenyl ring is twisted 25.0° out of the plane of the central pyridine ring, consistent with minimizing H⋯H interactions. The molecules pack into sheets (Fig. 13a) with the pendant pyridine ring containing N3 directed into the V-shaped cavity defined by the pyridine rings containing N1ii and N2ii and the phenyl ring with C16ii (symmetry code ii = 1 + x, y, z) of an adjacent ligand 5; the shortest N⋯HC contact is 2.47 Å. Fig. 13a shows the interdigitation of the n-pentoxy chains, and also illustrates that the sheets contain voids between phenyl units of adjacent chains. These pockets accommodate the methyl groups of two of the four acetato-ligands from the next sheet (Fig. 13b).
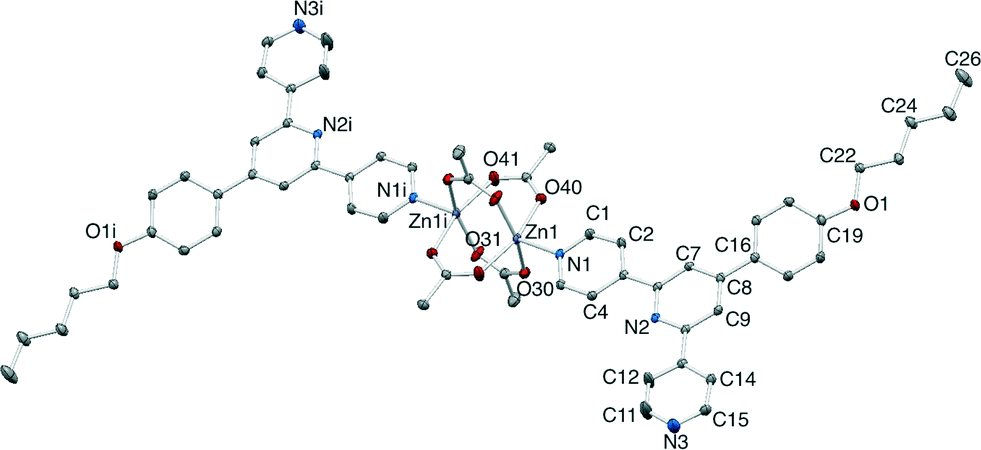 | ||
| Fig. 12 Molecular structure of [Zn2(OAc)4(5)2] with ellipsoids plotted at 40% probability level and H atoms omitted. Symmetry code i = 1 − x, 1 − y, 1 − z. Important bond parameters: Zn1–O40 = 2.0606(13), Zn1–O30 = 2.0555(14), Zn1i–O41 2.0232(14), Zn1i–O31 2.0414(14), Zn1–N1 = 2.0401(15), Zn1⋯Zn1i = 2.9114(5) Å; N1–Zn1–Zn1i = 177.72(5)°. | ||
 | ||
| Fig. 13 (a) Packing of molecules of [Zn2(OAc)4(5)2] into a sheet, and (b) filling of the cavities in each sheet with methyl residues (space-filling representation) of {Zn2(OAc)4} units from adjacent sheets. | ||
For ligands 8–10 with the longest alkoxy chains, single crystals of discrete complexes were obtained from reactions with zinc(II) acetate. The structures of [Zn2(OAc)4(8)2], [Zn2(OAc)4(9)2] and [Zn2(OAc)4(10)2] are analogous to that of [Zn2(OAc)4(5)2] and are shown in Fig. S9–S11.† Each complex crystallizes in the triclinic space group P. The asymmetric unit of [Zn2(OAc)4(8)2] contains a whole molecule and the independent ligands differ in the conformations of the n-octoxy chains (Fig. S9†). Molecules of [Zn2(OAc)4(9)2] and [Zn2(OAc)4(10)2] are centrosymmetric and both contain chains in fully extended conformations. The ligand in [Zn2(OAc)4(10)2] is disordered and the 4-decoxyphenyl units were modelled over two sites of occupancies 49 and 51%. Bond parameters for the three complexes are as expected (see captions to Fig. S9–S11†). In the three complexes, the deviation from planarity of the 4,2′:6′,4′′-tpy units vary with angles between the planes of adjacent pyridine rings lying in the range 0.5 to 20.4°; the phenyl substituents are twisted with respect to the central pyridine ring by between 24.3 and 26.4°. The molecular packing in each of [Zn2(OAc)4(8)2], [Zn2(OAc)4(9)2] and [Zn2(OAc)4(10)2] replicates that in [Zn2(OAc)4(5)2] (Fig. 13). Scheme 4 defines a parameter x which can be used to quantify the effect of increasing the length of the alkoxy chain. The Zn⋯Zn separation, x, varies from 34.802(2) Å in [Zn2(OAc)4(5)2], 39.089(2) Å in [Zn2(OAc)4(8)2], 39.005(3) Å in [Zn2(OAc)4(9)2] to 41.285(2) Å in [Zn2(OAc)4(10)2]; the latter value is not affected by the disorder in the structure. In each compound, packing of sheets involves the accommodation of methyl residues of {Zn2(OAc)4} units from adjacent sheets into the hollows in each sheet, as shown in Fig. 13b.
 | ||
| Scheme 4 Defined here for [Zn2(OAc)4(5)2], the Zn⋯Zn separation, x, quantifies the effect of increasing the alkoxy chain (see text) within a sheet. | ||
Conclusions
The synthesis and characterization of 4′-(4-ROC6H4)-4,2′:6′,4′′-tpy ligands 1–10 (Scheme 1) have been described. The results of crystallization experiments combining zinc(II) acetate with ligands (L) 1–10 reveal that coordination polymers [{Zn2(OAc)4(L)}n] or complexes [Zn2(OAc)4(L)2] are formed (Scheme 5). Polymers containing bidentate 4,2′:6′,4′′-tpy domains bridging {Zn2(OAc)4} paddle-wheel units are favoured for short alkoxy chains and discrete complexes [Zn2(OAc)4(L)2] containing monodentate 4,2′:6′,4′′-tpy domains are favoured for the longest chains. In the polymers, π-stacking interactions between aromatic (pyridine and/or phenyl) rings are dominant while packing of the discrete complexes features dominant van der Waals interactions between interdigitated chains.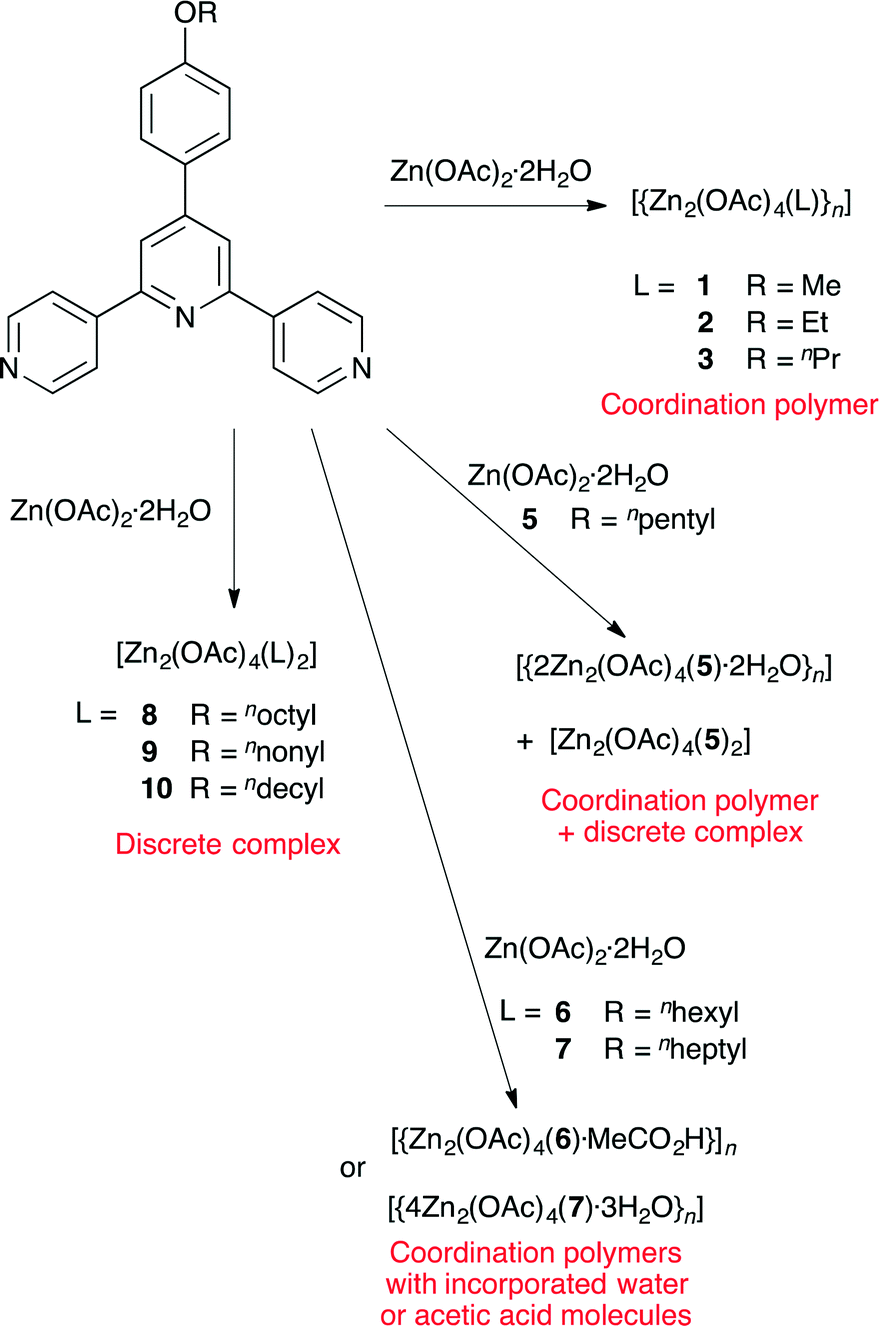 | ||
| Scheme 5 Overview of the formation of crystallographically proven coordination polymers and discrete complexes as a function of alkoxy chain length. | ||
Each polymer has a zigzag backbone and the chains nest together to generate planar sheets. The increasing length of the alkoxy substituent forces the polymer chains further apart within each sheet. In [{Zn2(OAc)4(3)}n] (n-propoxy group), the substituent adopts a folded conformation to compensate for its increased length and this permits the packing in [{Zn2(OAc)4(3)}n] to be essentially the same as in [{Zn2(OAc)4(1)}n] and [{Zn2(OAc)4(2)}n] (methoxy and ethoxy). Although the zigzag form of the polymer chain in [{2Zn2(OAc)4(5)·2H2O}n] replicates those in [{Zn2(OAc)4(1)}n] [{Zn2(OAc)4(2)}n] and [{Zn2(OAc)4(3)}n], lattice packing interactions are significantly different (isolated tetradecker π-stacks versus infinite π-stacked assemblies) and the wider separation of the polymer chains in a sheet results in the incorporation of water molecules. The assembly in [{2Zn2(OAc)4(5)·2H2O}n] (in which there are two independent ligands with different conformations) is also observed in [{4Zn2(OAc)4(7)·3H2O}n]. In contrast, the n-hexoxy-containing coordination polymer crystallizes with acetic acid in the lattice, and the zigzag polymer chains in [{Zn2(OAc)4(6)·MeCO2H}n] pack in a manner that is unique among the other polymers.
In conclusion, we have shown that the backbones of the zigzag polymer chains formed between 4′-(4-ROC6H4)-4,2′:6′,4′′-tpy ligands and {Zn2(OAc)4} units are structurally invariant, and that changes in the length of the alkoxy substituents can be accommodated up to a certain point by a combination of conformational changes of the alkyl groups, π-stacking interactions between arene rings and incorporation of H2O or MeCO2H molecules into the lattice. Once the alkoxy substituents reach a length of eight or more C atoms, the formation of discrete complexes appears to be preferred, presumably driven by favourable van der Waals interactions between interdigitated alkoxy groups. Although we do not have crystallographic support for the formation of [Zn2(OAc)4(6)2] and [Zn2(OAc)4(7)2], it is reasonable to conclude that the range between the n-pentoxy and n-heptoxy substituents represents an intermediate zone where the preference between the two structure types is marginal.
Acknowledgements
We thank the Swiss National Science Foundation, the European Research Council (Advanced Grant 267816 LiLo) and the University of Basel for financial support. Srboljub Vujovic is thanked for laboratory support.Notes and references
- C. E. Housecroft, Dalton Trans., 2014, 43, 6594 RSC.
- M. Barquín, J. Cancela, M. J. González Garmendia, J. Quintanilla and U. Amador, Polyhedron, 1998, 17, 2373 CrossRef.
- F. Yuan, Q.-E. Zhu, H.-M. Hu, J. Xie, B. Xu, C.-M. Yuan, M.-L. Yang, F.-X. Dong and G.-L. Xue, Inorg. Chim. Acta, 2013, 397, 117 CrossRef CAS PubMed.
- H.-N. Zhang, F. Yuan, H.-M. Hu, S.-S. Shen and G.-L. Xue, Inorg. Chem. Commun., 2013, 34, 51 CrossRef CAS PubMed.
- Y.-L. Gai, F.-L. Jiang, L. Chen, Y. Bu, M.-Y. Wu, K. Zhou, J. Pan and M.-C. Hong, Dalton Trans., 2013, 42, 9954 RSC.
- B. Xu, J. Xie, H.-M. Hu, X.-L. Yang, F.-X. Dong, M.-L. Yang and G.-L. Xue, Cryst. Growth Des., 2014, 14, 1629 CAS.
- M.-S. Wang, M.-X. Li, X. He, M. Shao and Z.-X. Wang, Inorg. Chem. Commun., 2014, 42, 38 CrossRef CAS PubMed.
- C. Liu, Y.-B. Ding, X.-H. Shi, D. Zhang, M.-H. Hu, Y.-G. Yin and D. Li, Cryst. Growth Des., 2009, 9, 1275 CAS.
- B.-C. Wang, Q.-R. Wu, H.-M. Hu, X.-L. Chen, Z.-H. Yang, Y.-Q. Shangguan, M.-L. Yang and G.-L. Xue, CrystEngComm, 2010, 12, 485 RSC.
- J. Song, B.-C. Wang, H.-M. Hu, L. Gou, Q.-R. Wu, X.-L. Yang, Y.-Q. Shangguan, F.-X. Dong and G.-L. Xue, Inorg. Chim. Acta, 2011, 366, 134 CrossRef CAS PubMed.
- Y.-Q. Chen, S.-J. Liu, Y.-W. Li, G.-R. Li, K.-H. He, Y.-K. Qu, T.-L. Hu and X.-H. Bu, Cryst. Growth Des., 2012, 12, 5426 CAS.
- Y.-Q. Chen, G.-R. Li, Z. Chang, Y.-K. Qu, Y.-H. Zhang and X.-H. Bu, Chem. Sci., 2013, 4, 3678 RSC.
- J. Heine, J. Schmedt auf der Günne and S. Dehnen, J. Am. Chem. Soc., 2011, 133, 10018 CrossRef CAS PubMed.
- E. C. Constable, G. Zhang, C. E. Housecroft and J. A. Zampese, CrystEngComm, 2011, 13, 6864 RSC.
- V. N. Dorofeeva, S. V. Kolotilov, M. A. Kiskin, R. A. Polunin, Z. V. Dobrokhotova, O. Cador, S. Golhan, L. Ouahab, I. L. Eremenko and V. M. Novotortsev, Chem. – Eur. J., 2012, 18, 5006 CrossRef CAS PubMed.
- D.-Y. Ma, D.-E. Sun and G.-Q. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m913 CAS.
- Y.-Q. Chen, G.-R. Li, Y.-K. Qu, Y.-H. Zhang, K.-H. He, Q. Gao and X.-H. Bu, Cryst. Growth Des., 2013, 13, 901 CAS.
- E. C. Constable, C. E. Housecroft, M. Neuburger, S. Vujovic, J. A. Zampese and G. Zhang, CrystEngComm, 2012, 14, 3554 RSC.
- J. Granifo, R. Gaviño, E. Freire and R. Baggio, J. Mol. Struct., 2014, 1063, 102 CrossRef CAS PubMed.
- E. C. Constable, C. E. Housecroft, S. Vujovic and J. A. Zampese, CrystEngComm, 2014, 16, 3494 RSC.
- J. Yoshida, S.-I. Nishikiori and H. Yuge, J. Coord. Chem., 2013, 66, 2191 CrossRef CAS.
- G. W. V. Cave and C. L. Raston, J. Supramol. Chem., 2002, 2, 317 CrossRef CAS.
- E. C. Constable, G. Zhang, C. E. Housecroft and J. A. Zampese, Inorg. Chem. Commun., 2012, 15, 113 CrossRef CAS PubMed.
- G. W. V. Cave and C. L. Raston, J. Chem. Soc., Perkin Trans. 1, 2001, 3258 CAS.
- Bruker Analytical X-ray Systems, Inc., APEX2, version 2 User Manual, M86-E01078, Madison, WI, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 389 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- J. Wang and G. S. Hanan, Synlett, 2005, 1251 CAS.
- H. S. Chow, E. C. Constable, C. E. Housecroft, M. Neuburger and S. Schaffner, Dalton Trans., 2006, 2881 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 380 CrossRef PubMed.
- G. W. V. Cave and C. L. Raston, J. Chem. Soc., Perkin Trans. 1, 2001, 3258 CAS.
- E. C. Constable, H.-J. Guntherodt, C. E. Housecroft, L. Merz, M. Neuburger, S. Schaffner and Y. Tao, New J. Chem., 2006, 30, 1470 RSC.
- G. W. V. Cave and C. L. Raston, Chem. Commun., 2000, 2199 RSC.
- E. C. Constable, G. Zhang, C. E. Housecroft, M. Neuburger and J. A. Zampese, CrystEngComm, 2010, 12, 2146 RSC.
- E. C. Constable, G. Zhang, E. Coronado, C. E. Housecroft and M. Neuburger, CrystEngComm, 2010, 12, 2139 RSC.
- E. C. Constable, C. E. Housecroft, M. Neuburger, J. Schönle, S. Vujovic and J. A. Zampese, Polyhedron, 2013, 62, 120 CrossRef PubMed.
- E. C. Constable, C. E. Housecroft, P. Kopecky, M. Neuburger, J. A. Zampese and G. Zhang, CrystEngComm, 2012, 14, 446 RSC.
- S. Mohamed, D. A. Tocher, M. Vickers, P. G. Karamertzanis and S. L. Price, Cryst. Growth Des., 2009, 9, 2881 CAS.
- L. Chmurzyński, J. Heterocycl. Chem., 2000, 37, 71 CrossRef.
- A. R. Katritzky and D. E. Leahy, J. Chem. Res., 1985, 28 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of precursors. Fig. S1–S11: 1H NMR and electronic spectra of ligands, structural figures and powder diffraction data. See DOI: 10.1039/c4ce01422g |
| This journal is © The Royal Society of Chemistry 2014 |