
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal structure analyses facilitate understanding of synthesis protocols in the preparation of 6,6′-dibromo-substituted BINOL compounds†
Marco
Agnes
ab,
Alessandro
Sorrenti
a,
Dario
Pasini
 *b,
Klaus
Wurst
c and
David B.
Amabilino‡
*a
*b,
Klaus
Wurst
c and
David B.
Amabilino‡
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus Universitari de Bellaterra, 08193 Cerdanyola del Vallès, Catalonia, Spain. Fax: (+34) 93 5805729; E-mail: David.Amabilino@nottingham.ac.uk
bDepartment of Chemistry and INSTM Research Unit, University of Pavia, VialeTaramelli, 10-27100, Pavia, Italy. E-mail: dario.pasini@unipv.it
cInstitut für Anorganische Chemie, Innrain 80/82, A-6020 Innsbruck, Austria
First published on 11th September 2014
Abstract
A combination of crystallographic and spectroscopic techniques has been used in order to address thorough purification protocols for a series of atropisomeric 1,1′-binaphthalene-2,2′-diol (BINOL) derivatives to be used as building blocks for chiral nanoscale constructs. The compounds were obtained following an initial electrophilic aromatic bromination reaction at the 6,6′ position of BINOL, which was used in either its racemic or its enantiopure form. The presence of regioisomeric compounds, in which bromine atoms are located at different positions of the naphthalene skeletons, could be hinted at by classical spectroscopy (1H NMR and IR) and fully confirmed by single-crystal X-ray analysis of the solids formed during crystal growth of the crude products, where solid solutions are identified in part due to the presence of bromine that is a relatively heavy atom. If purification by recrystallization in suitable solvents is not performed, further chemical elaboration by means of alkylation on the 2,2′-phenolic positions produces compounds in which the regioisomeric impurities are still present. The set of five X-ray structures highlights interesting packing features extremely useful for the crystal engineering of nanostructures integrating these axially chiral building blocks.
Introduction
The translation of molecular chirality into supramolecular and nanoscale chirality has been the subject of intense research activity in recent years, and ordered chiral nanostructures have demonstrated potential for a number of bulk material applications.1 Substituted 1,1′-binaphthyl derivatives are a versatile class of compounds – chirality is due to atropisomerism because of hindered rotation around the bond linking the naphthyl groups – which have found applications in many different areas of chemistry.2 Their chirality is a result of the angle between the aromatic rings caused by steric interactions, and the restricted rotation around the naphthalene–naphthalene bond means that the enantiomers can be isolated in certain cases. The rigid structure and the C2 symmetry of the binaphthyl skeleton are key factors in the efficient transfer of chiral information. As a consequence of the favourable properties, these synthons have become attractive molecular modules in fields as diverse as asymmetric catalysis,3 chiral supramolecular recognition4 and crystal engineering.5 Recent applications as building blocks for polymers6 and for chiroptical sensing have also emerged.7 Perhaps the most common derivative amongst the class of substituted 1,1′-binaphthyl derivatives is 2,2′-dihydroxy-1,1′-binaphthyl (1, Fig. 1), often referred to as BINOL. Its basic skeleton can be conveniently functionalized in various positions; the most frequent are the 4,4′ and 6,6′ positions, but access to the 3,3′-positions is also well documented.2d | ||
| Fig. 1 Chemical structures of the compounds used in this study. Only (R) enantiomers are shown. | ||
The functionalization in the 6,6′ positions is most frequently achieved by electrophilic bromination on the naphthalene rings. A plethora of synthesis opportunities, such as arylation and vinylation reactions via Stille or Suzuki-type coupling, has then been utilized to substitute bromine atoms, thus synthesizing derivatives with specific functionalities.2d,6
During the course of a continuing project dealing with the use of BINOL-based building blocks for functional nanostructures,8 we turned our attention to 6,6′-disubstituted BINOL derivatives. In this paper, we report five novel crystal structures for a series of 6,6′-disubstituted BINOL derivatives, either in their racemic or in their enantiopure form. We describe how a combination of crystallographic and spectroscopic techniques has been used in a synergistic way in order to identify minor amounts of impurities and thus to set up thorough purification protocols to be used in the synthesis of these popular BINOL-based synthons. The results are obtained after bromination of the parent compound, and the identification of the impurities has been achieved due to the presence of bromine, a heavy atom easily identifiable by X-ray crystallography.
Results and discussion
Synthesis, NMR characterization and purification procedures
The compounds described in this paper are shown in Fig. 1. They were synthesized using protocols described in the literature or with minor modifications with respect to the reported procedures. The reactions were conducted on either racemic (RS)-1 or enantiopure (R)-1; both are commercially available compounds.Compound 2 was prepared by electrophilic bromination of compound 1.9 Derivatization at 2,2′ phenolic positions was conducted in two different ways under reaction conditions already established as non racemizing: a) protection with methoxyethoxymethyl (MEM) chloride to give compound 3 (ref. 10) and b) alkylation under mild basic conditions (Cs2CO3, DMF)11 to yield the diester compound 4.
To date, the bromination step to give 2 has been reported to be nearly quantitative and regiospecific for the 6,6′-positions as a consequence of the strong electron-donating character of the –OH groups in the 2,2′-positions.9§
However, the 1H NMR spectra of either enantiopure or racemic 2 after workup of the reaction showed some impurities in the aromatic region (Fig. 2a and S1†). The impurities could not be readily attributed to a specific organic compound; the few small peaks not covered by other resonance signals (a doublet at 8.52 ppm, a multiplet at 7.49 ppm and a doublet at 6.93 ppm) hinted at a regioisomer in which the bromine atoms are placed at different positions on the two naphthalene units.
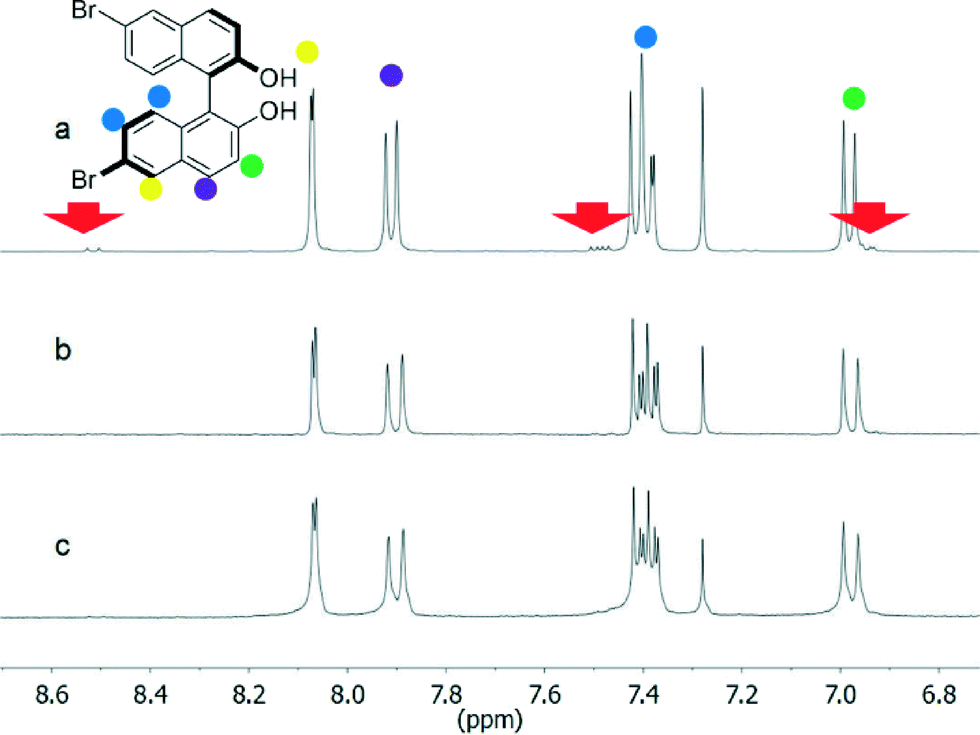 | ||
| Fig. 2 1H NMR spectra of (R)-2 before (a) and after crystallization from different mixtures of solvents: CH2Cl2/cyclohexane (b) and toluene/n-hexane (c). The red arrows indicate the impurity. | ||
Impurities could be estimated to be around 10–12% of the product and could not be removed by column chromatography. Complete purification of 2 (see NMR spectra, Fig. 2b and c and S1†) was achieved by following standard crystallization protocols, screening different mixtures of solvents and evaluating the purity of the products after crystallization by 1H NMR spectroscopy. All of the signals related to the impurities disappeared in the spectra taken after crystallization from the solvents specified in Table 1 (entries 1–3 for compounds 2). These crystallization solvent combinations can be therefore considered adequate for purification. This compound, and the other chiral compounds in this series, was characterised by circular dichroism spectroscopy that shows the expected exciton chirality for the enantiomer used in the synthesis (see the ESI†).
| Entry | Compound | Mixture of solventsa | Outcomeb |
|---|---|---|---|
a All solvents used were in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio.
b Purified or not purified from the presence of contaminating regioisomers. :1 volumetric ratio.
b Purified or not purified from the presence of contaminating regioisomers.
|
|||
| 1 | (RS)-2 | Toluene/cyclohexane | Purified |
| 2 | (R)-2 | CH2Cl2/cyclohexane | Purified |
| 3 | (R)-2 | Toluene/n-hexane | Purified |
| 4 | (RS)-3 | CHCl3/EtOH | Not purified |
| 5 | (RS)-4 | CH2Cl2/EtOH | Not purified |
IR spectroscopy was also informative and supported the presence of impurities in the initially isolated material (Fig. S2†). The peak at 734 cm−1 in the spectrum of (R)-2 immediately after the workup corresponds to stretching frequencies of C–Br bonds (usually in the region between 730 cm−1 and 750 cm−1) in different halogenated impurities. This result could also be consistent with a dibromosubstituted BINOL in which the two bromine atoms are at different positions of the naphthalene units. After purification by crystallization from the appropriate mixture of solvents (Table 1), the peak at 734 cm−1 is no longer present (Fig. S2,† bottom). The absence of this peak demonstrates that unsymmetrically halogenated products have been removed successfully in this crystallisation; for comparison, the C–Br bond in the monohalogenated (symmetrical) pure product 2 stretches at 670 cm−1.
When regioisomerically contaminated samples of 2 were characterised using the following synthesis steps without prior purification by crystallization, further purification of 3 or 4 by crystallization was not possible, and solid solutions of the different regioisomers were found by X-ray crystal structure analysis (vide infra). On the other hand, regioisomerically pure samples of compound 2 were able to generate, after chemical modifications, regioisomerically pure samples of 3 and 4.
Optical microscopy and X-ray structure analyses of single crystals
Crystals of the compounds (grown under the conditions shown in Table 1) were inspected using a polarising optical microscope, checking for the birefringence that should characterise single crystals. The morphologies of the crystals evidenced marked differences depending on the degree of regioisomeric purity of the compounds (Fig. 3 and ESI†). | ||
| Fig. 3 Morphology of crystals of 3. The micrograph on the left shows crystals obtained after chemical functionalization from samples of 2 contaminated with regioisomers, while the formation of much higher quality single crystals (on the right) was obtained after chemical functionalization starting from regioisomerically pure 2. | ||
It is interesting to note that although a large number of literature references can be found for the preparation of 2 (racemic or enantiopure), the X-ray structure of this compound has not been reported before. In previous papers, X-ray structures involving this compound were reported, but these compounds were crystallised by the addition of secondary molecules, forming crystals that are incorporated in the asymmetric unit.12
The crystal structure of (R)-2 shows the characteristic quasi-perpendicular disposition of the naphthyl rings (Fig. 4), where the torsion angle at the positions bearing the oxygen atoms is −105°. The compound crystallises in the P212121 space group with four identical molecules in the unit cell. The Flack parameter for this chiral compound is in accordance with the expected value. The molecules pack forming what can be described as interdigitated layers. Within the layers, O–H⋯O hydrogen bonds between hydroxyl groups in adjacent molecules (H⋯O distance 2.13 Å, O–H⋯O angle 164°) and C–H⋯π interactions between molecules (H⋯C distance 2.88 Å, C–H–C angle 123°) are present (see the table in the ESI† for details). Between the sheets, interactions between bromine atoms and protons on neighbouring molecules are apparent (C–Br⋯H–O, H⋯Br distance of 2.74 Å and C–Br⋯H–C, H⋯Br distance of 3.02 Å).
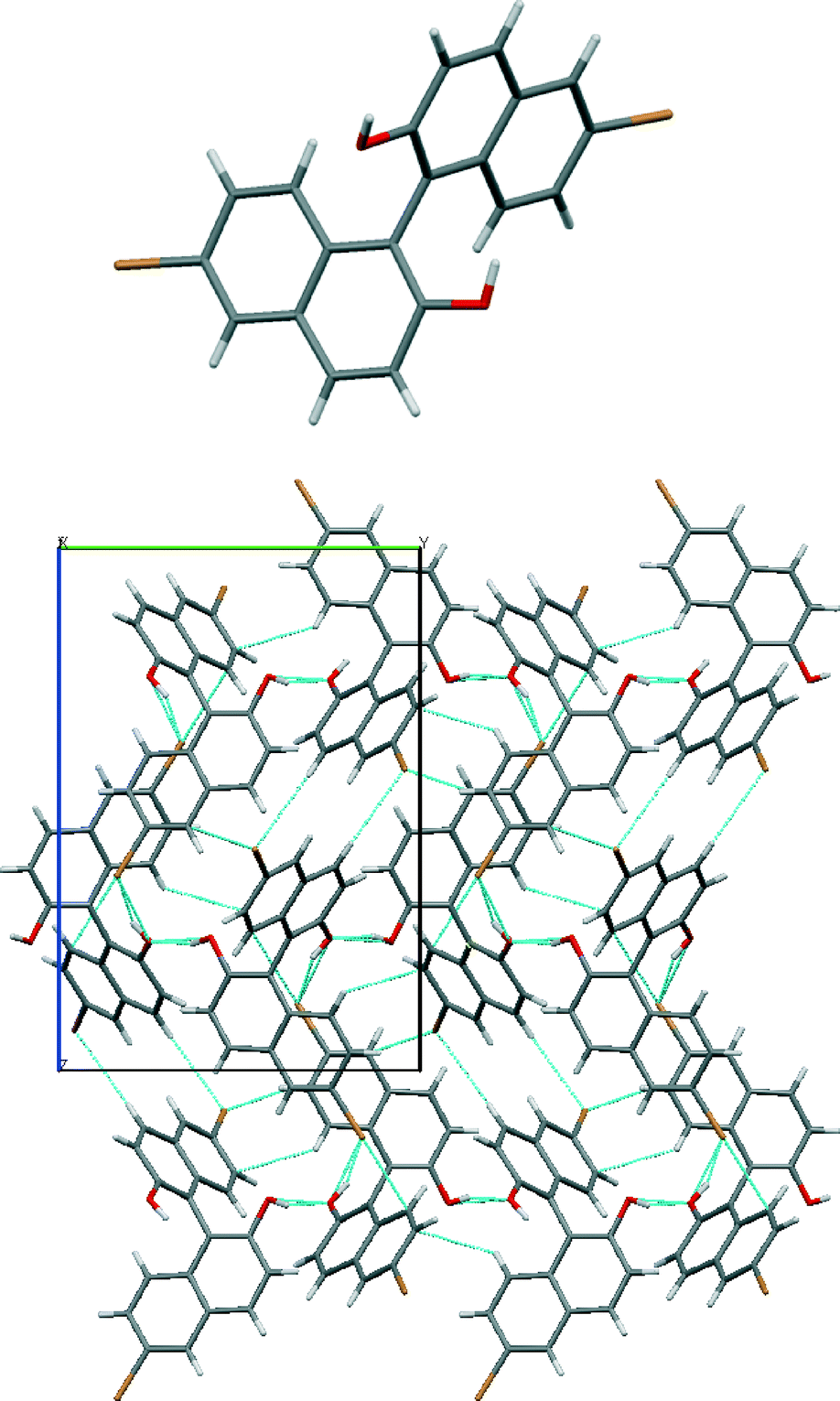 | ||
| Fig. 4 Views of the crystal structure of (R)-2. An image of the molecule (top) and its packing (below) seen along the a axis with short contacts indicated by light blue dotted lines. | ||
The crystal structure of (RS)-2 shows a similar quasi-perpendicular disposition of the naphthyl rings to the enantiopure compound, where the torsion angle at the positions bearing the oxygen atoms is somewhat smaller (±85°). The material is a true racemic compound in which the enantiomers are located at a well-defined position in the crystal lattice. The space group is P21/n. The structure is formed with homochiral tapes in which the molecules are joined through hydrogen bonds (Fig. 5; see the table in the ESI† for details), where consecutive molecules in the tape are joined through the O–H⋯O interaction between hydroxyl groups (H⋯O distance 2.15 Å, O–H⋯O angle 142°) and O–H⋯π interactions (H⋯C distances 2.70 and 2.82 Å). These homochiral tapes pack into racemic sheets where there is an alternation of chirality in each tape that makes up the layer (Fig. 6). The sheets are formed due to short contacts between bromine atoms and hydrogen atoms (C–Br⋯H–C, H⋯Br distance of 3.05 Å Br2–H5) and between a hydrogen atom and a carbon atom in the naphthyl system within the tape (consistent with C–H⋯π interaction, H⋯C distance 2.96 Å, C–H–C angle 158°, H18–C3A). These corrugated heterochiral sheets stack on top of one another in the crystal with short C–Br⋯H–C interaction (distance of 2.90 Å, Br2–H8), and π–π-interaction with a distance of 3.59 Å (C15–C17A).
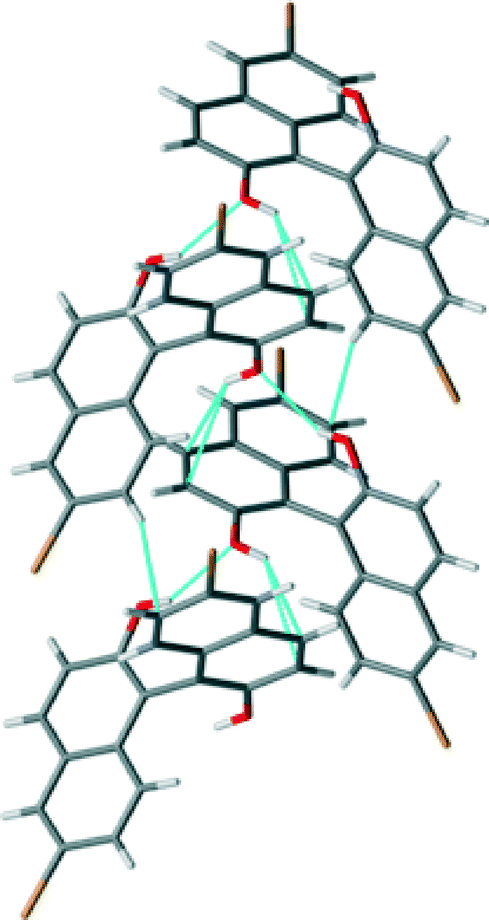 | ||
| Fig. 5 View of the homochiral tapes in the crystal structure of (RS)-2. Short contacts indicated by light blue dotted lines. | ||
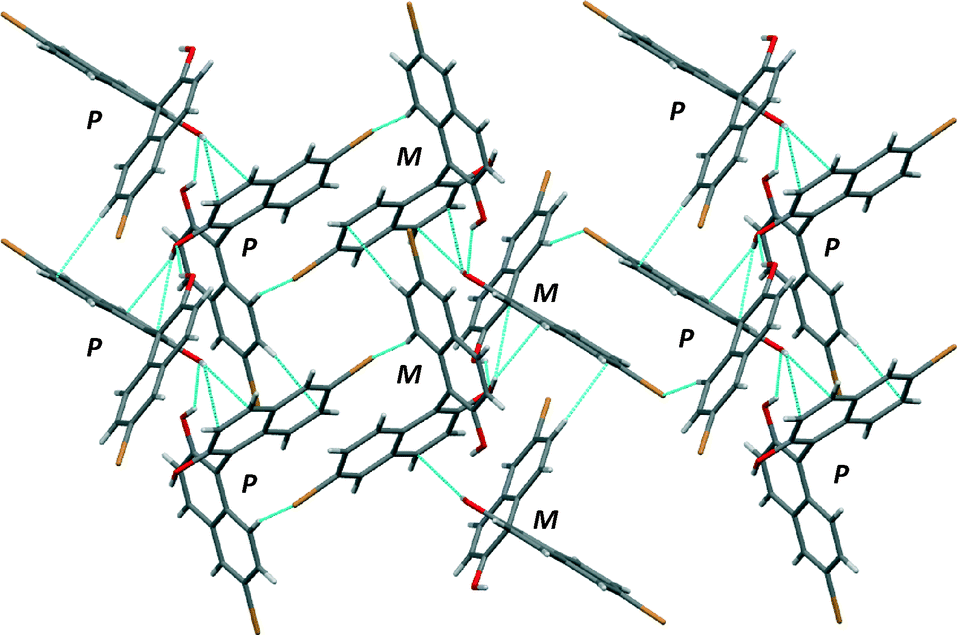 | ||
| Fig. 6 View of the heterochiral sheets in the crystal structure of (RS)-2. Short contacts indicated by light blue dotted lines. P and M correspond to the helical chirality of each molecule. | ||
Enantiopure (R)-3 – in which the oxygen atoms attached to the naphthyl moieties are now of ether type – stacks into crystals, forming dimers (Fig. 7). These two molecules have distinct conformations (the torsion angles between the naphthyl rings are −72° and −101°) and there are two non-equivalent dimers. In one dimer the distance between π systems is 3.34 Å at its smallest (there is slight splaying) and this interaction is accompanied by C–H⋯π interaction (H⋯C distance 2.80 Å, C–H–C angle 140°) from one of the methylene units in the chain attached to the binaphthyl moiety. In the other, the distance between π systems is 3.39 Å at its smallest (there is slight splaying) and this interaction is accompanied by C–H⋯π interaction (H⋯C distance 2.89 Å, C–H–C angle 161°) from one of the methylene units in the chain attached to the binaphthyl moiety. There are also C–H⋯O interactions present in both dimers (indicated in Fig. 7). These two dimers alternate in a chain that runs along the b axis of the crystal. Between these chains there are interactions between bromine and hydrogen atoms seen in the other compounds in this series.
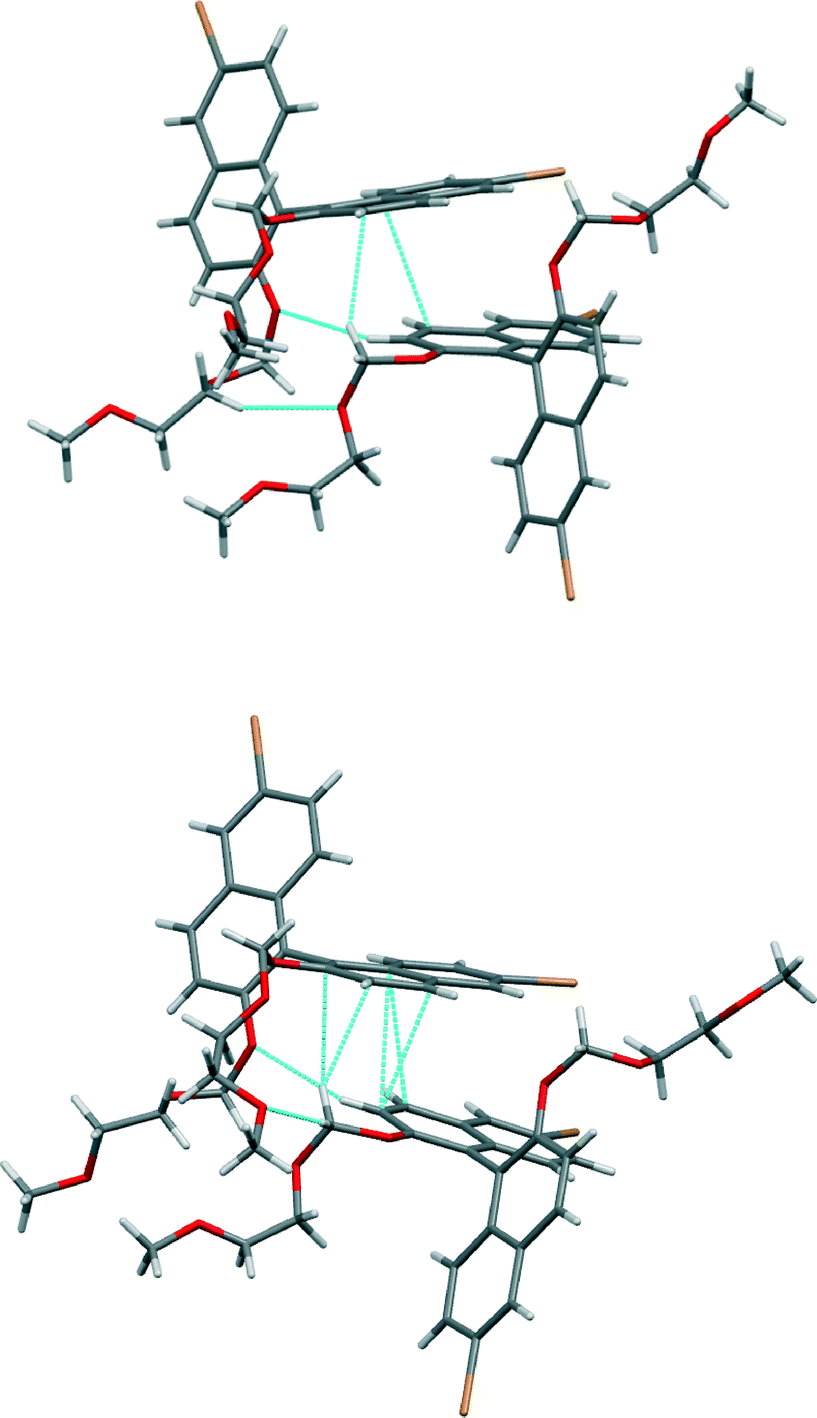 | ||
| Fig. 7 Views of the non-equivalent dimers in the crystal structure of (R)-3. Short contacts indicated by light blue dotted lines. | ||
In the racemic modification of 3, a true racemic compound is formed with a structure pertaining to the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and in which chains of alternating chirality zigzag down the a axis (Fig. 8). There are no significant interactions between π systems (in contrast to the enantiopure compound); instead the alternating R and S molecules are connected through a series of C–H⋯O and C–H⋯π interactions. Like chirality molecules are located “on top” of one another with the enantiomer making a non-covalent link between them through the bridging achiral side groups.
space group and in which chains of alternating chirality zigzag down the a axis (Fig. 8). There are no significant interactions between π systems (in contrast to the enantiopure compound); instead the alternating R and S molecules are connected through a series of C–H⋯O and C–H⋯π interactions. Like chirality molecules are located “on top” of one another with the enantiomer making a non-covalent link between them through the bridging achiral side groups.
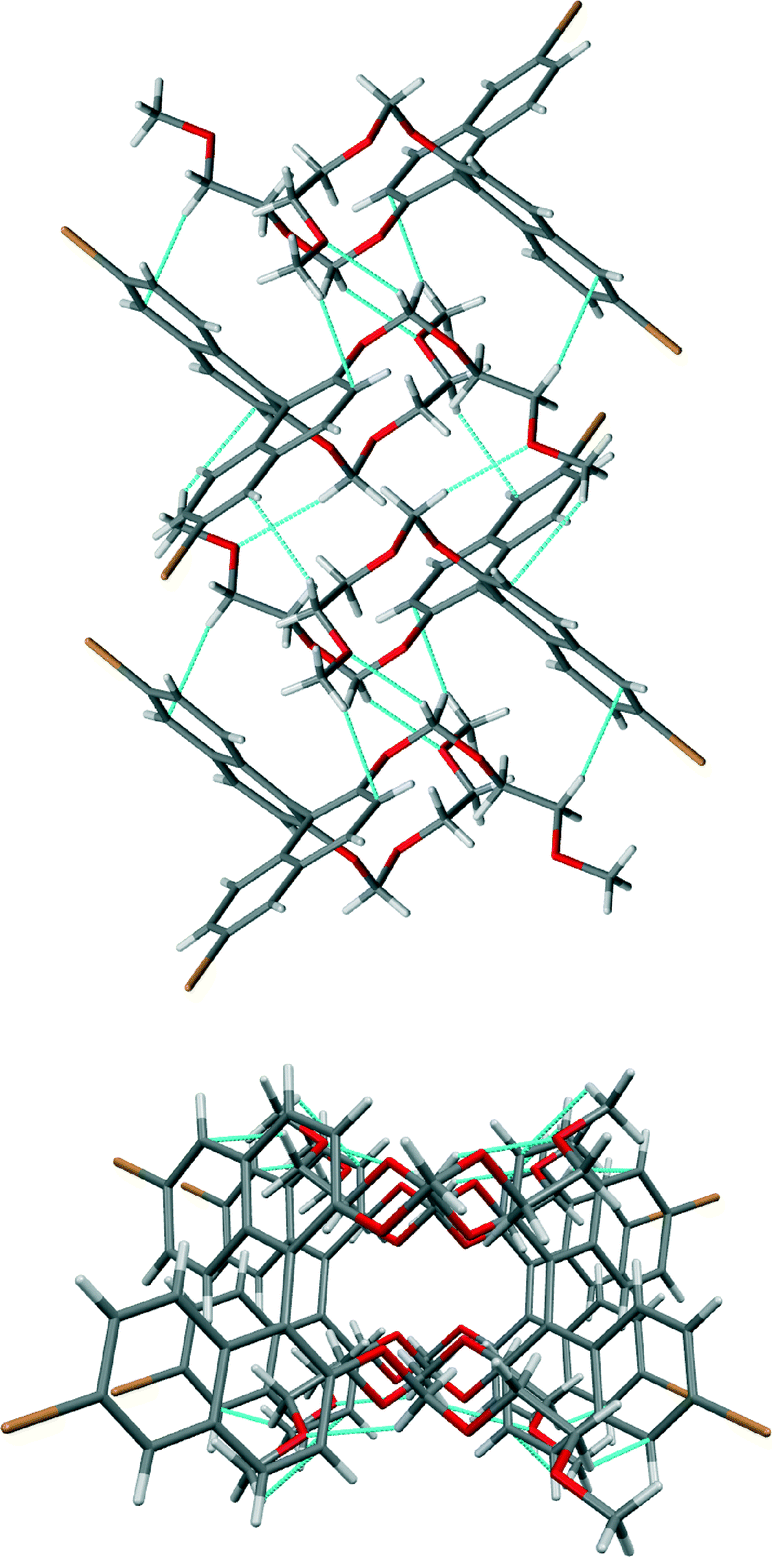 | ||
| Fig. 8 Views of the heterochiral chains in the crystal structure of (RS)-3. Top side-on and bottom images show a perspective view along the chains. Short contacts indicated by light blue dotted lines. | ||
The racemic form of 4, in common with 3, is a racemic compound where rows of one enantiomer line up next to a row of the other and where weak hydrogen bonds unite the molecules (Fig. 9). Again, the space group is P, but in this case the molecules form a sheet in the ab plane. As for 3, the achiral side groups link enantiomers non-covalently, and like enantiomers are atop one another presumably as propellers stack on top of one another. Although there is no direct contact between them, it is clear that one enantiomer in this position would not fit in a dense packing arrangement. It is therefore the interactions between these “stacks” that lead to the racemic compound. The shortest non-covalent bonds involve the carbonyl group that forms a trifurcated hydrogen bond with three C–H groups (H⋯O distances 2.43, 2.52 and 2.67 Å, C–H–C angles 174°, 157° and 134°) and the head-to-tail stacked carbonyl groups along the a-axis (C⋯O distance 3.05 Å, C–O–C angles 102°).
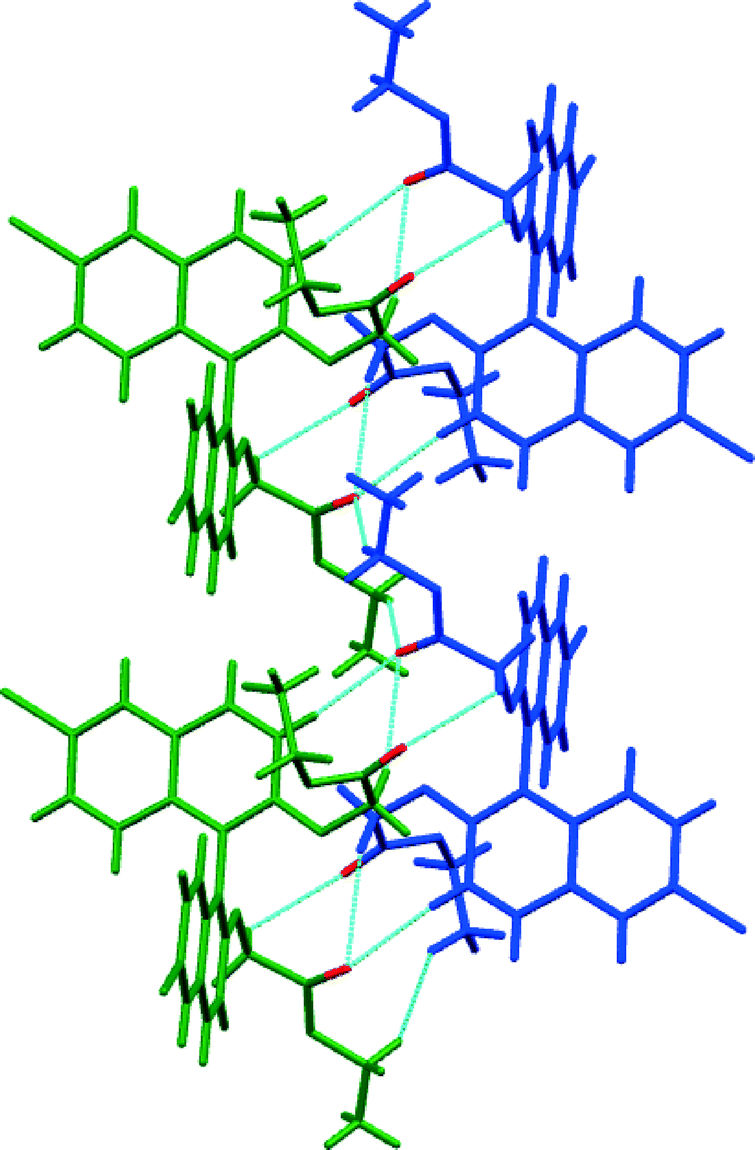 | ||
| Fig. 9 View of the heterochiral sheet in the crystal structure of (RS)-4. The chains shown run along the b axis; short contacts between carbonyl oxygen atoms (in red) and hydrogen atoms are indicated by light blue dotted lines. Enantiomers are shown in green and blue. | ||
Disorder of the bromine atoms in the structures
It was during the study of the crystals of (RS)-4 that the presence of regioisomers was discerned: the bromine atoms should formally be located at the 6 and 6′ positions assuming a totally selective reaction, but in the solid-state structure they were also evident (because of the electron density in the diffraction data) in low proportions at the 5 or 5′ positions (but not at both positions in the same molecule apparently). This occupational disorder also led to a positional disorder caused by the proximity of bromine atoms in the “wrong” positions in neighbouring molecules. This solid solution can be interpreted through the presence of four molecules, which are shown in Fig. 10. It is the main and desired 6,6′ isomer (84%, top in Fig. 10), a molecule resulting in disorder because of bromination at the Br2A position (6%, next to the top in Fig. 10), an isomer resulting in disorder because of bromination at Br1A (4%, next to bottom in Fig. 10) and a form of the desired isomer (6%, bottom in Fig. 10) with a more twisted conformation than the major component, a distortion that arises because of the interaction between Br2A of one molecule with Br1 of a neighbouring molecule in the cell.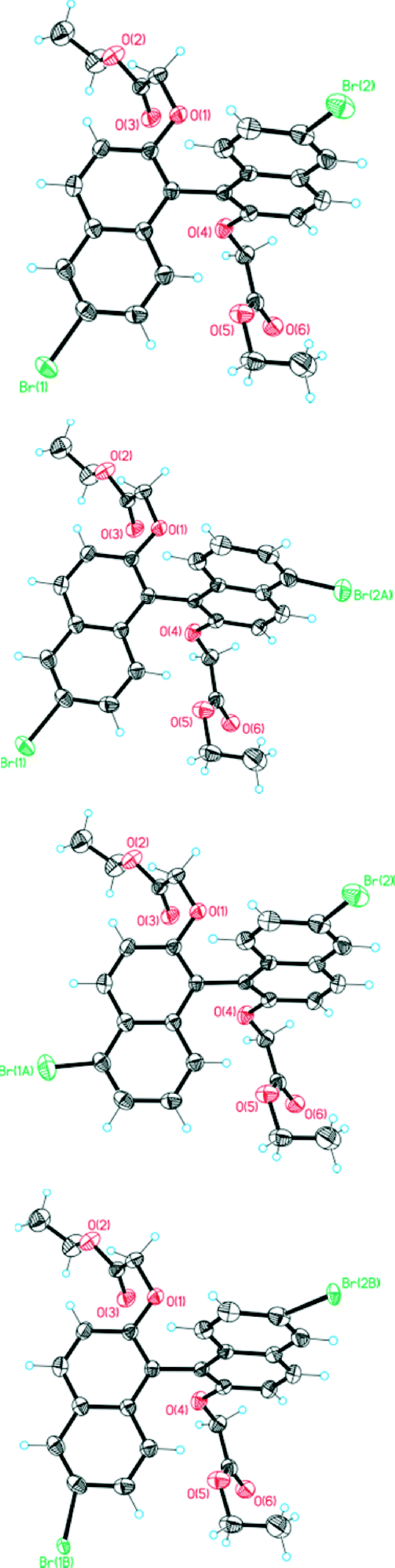 | ||
| Fig. 10 Four molecules present in the solid solution formed by (RS)-4 and impurities in it. The top image shows the main and desired compound. Below it different isomers of the compound with bromine atoms at 5 and 6 positions are shown, and the bottom image is the desired isomer with positional disorder caused by the proximity of bromine atoms from the impurities. | ||
In detail, the major bromine positions Br1 at C6 with an occupancy of 0.90 and Br2 at C17 with an occupancy of 0.88 come from the main compound. Br1A at C5 with an occupancy of 0.04 and Br2A at C16 with an occupancy of 0.06 come from the impurity of the isomeric compound. Within the cell, the Br2A position has a distance of 2.62 (and 2.67) Å to Br1 (and Br1A) of a neighbouring molecule. This distance is too close for comfort and the neighbouring molecule will be twisted. Therefore, positional disorder around Br1 and Br2 (twisting) occurs due to the additional Br1B and Br2B positions, each occupied with an occupancy of 0.06. The Br1A position also shows a short distance of 2.74 Å to the Br2B position, but because of the 4% and 6% distribution of these positions this plays no further role. Because only 6% of neighbouring molecules are twisted, this disorder could not be observed at the carbon atoms (low movement and anisotropic displacement parameters appear normal).
This disorder that results in a solid solution makes the purification of the compound at this stage of the synthesis of derivatives of these compounds unfeasible by crystallization.
Conclusions
The electrophilic aromatic bromination reaction of BINOL leads predominantly to reaction at 6,6′ positions and alternatively at 5,5′ positions. The presence of regioisomeric compounds was revealed definitively by single-crystal X-ray analysis of the solids formed during crystal growth of the crude products after subsequent reactions that showed the presence of solid solutions, a fact that was possible because bromine, being a heavy atom, is easily located by crystallography. Thus, the purification of 6,6′-dibromo-1,1′-binaphthalene-2,2′-diol is essential before further reactions are performed.In all of the racemates studied here, the racemic compound is the favoured form in the crystal. While homochiral associations can be picked out in the crystals – largely because of steric interactions between non-like enantiomers that lead to enantiophobicity13 at the stack level – the columns or layers come together in an alternate fashion. On the whole, the enantiopure compounds pack due to relatively weak interactions, and these are apparently overcome to produce the racemic compounds.
The structures provide important information on the design of new binaphthyl compounds that are synthetically easily accessed and that may lead to interesting properties as chiral materials.
Experimental section
Synthesis
The starting materials (RS)-1 (racemic) and (R)-1 ((+)-BINOL) with a purity of 99% were purchased from Sigma-Aldrich and used as received. All other commercially available compounds and reactants were used as purchased. THF and CH2Cl2 were dried and distilled before use. Compounds (R)- or (RS)-2 (ref. 9) (R)- or (RS)-3 (ref. 10) were prepared by following published procedures. Thin-layer chromatography was performed with silica gel on aluminium plates, and flash chromatography was performed with silica gel of 60 Å pore size. 1H NMR spectra were recorded using 300 MHz and 400 MHz spectrometers in CDCl3 solutions with the solvent residual protons or TMS as the internal standard. IR spectra were recorded using a Perkin Elmer spectrometer with an attenuated total reflection attachment. Mass spectra were recorded using a Bruker MALDI instrument in reflectron mode. Circular dichroism spectroscopy (Fig. S9†) was performed in 1 cm quartz cells by recording the spectra at 25 °C using a JASCO spectropolarimeter at a scanning speed of 50 nm min−1 and pure solvent spectra were subtracted.Crystallization procedure
All of the crystallization processes were performed by following the same procedure: first, the products (2, 3, 4) both in the enantiomeric (R) and racemic (RS) forms were isolated after the workup and dried under vacuum. Then, in small (3 mL) vials, a few milligrams (less than 10 mg) of the compounds were dissolved in 1 mL of a good dissolving solvent, such as halogenated solvents or toluene; 1 mL of a second solvent, in which the compounds were insoluble, was added to the vial very slowly, leaving the two solvents to equilibrate at the contact surface. The vials were left open and nice crystals were obtained after partial evaporation of the solvent mixture. The remaining solvent was removed with a Pasteur pipette and the crystals were washed with a few drops of the non-solvent. No more steps were necessary and all of the analyses were performed on the crystals as they precipitated.¶Optical imaging
Optical micrographs were obtained using an Olympus BX51 microscope in transmission mode using the variable angle polarising filters at 90° for the crossed polarised images, and were recorded using a DP20 digital camera.Acknowledgements
MA wishes to acknowledge an Erasmus Placement Fellowship from the University of Pavia supporting his stay in Barcelona. DP acknowledges support from MIUR (Programs of National Relevant Interest PRIN grant 2009-A5Y3N9), and, in part, from CARIPLO Foundation (2007–2009) and INSTM-Regione Lombardia (2010–2012 and 2013–2015). DBA thanks the MINECO, Spain, project CTQ2010-16339, and the DGR, Catalonia, project 2009 SGR 158.Notes and references
- (a) J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed; (b) L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342–356 RSC; (c) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed; (d) D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC; (e) M. Yanga and N. A. Kotov, J. Mater. Chem., 2011, 21, 6775–6792 RSC; (f) J. A. A. W. Elemans, I. De Cat, H. Xu and S. De Feyter, Chem. Soc. Rev., 2009, 38, 722–736 RSC; (g) J. Kumaki, S. Sakurai and E. Yashima, Chem. Soc. Rev., 2009, 38, 737–746 RSC; (h) J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC; (i) C. Noguez and I. L. Garzón, Chem. Soc. Rev., 2009, 38, 757–771 RSC; (j) F. Vera, J. L. Serrano and T. Sierra, Chem. Soc. Rev., 2009, 38, 781–796 RSC; (k) J. Clayden, Chem. Soc. Rev., 2009, 38, 817–829 RSC; (l) J. Crassous, Chem. Soc. Rev., 2009, 38, 830–884 RSC.
- (a) L. Pu, Chem. Rev., 1998, 98, 2405–2494 CrossRef CAS PubMed; (b) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3211 CrossRef CAS PubMed; (c) P. Kocovsky, S. Vyskocyl and M. Smrcina, Chem. Rev., 2003, 103, 3213–3245 CrossRef CAS PubMed; (d) J. M. Brunel, Chem. Rev., 2005, 105, 857–898 CrossRef CAS PubMed.
- (a) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef CAS PubMed; (b) B. Maciá Ruiz, K. Geurts, M. A. Fernández-Ibáñez, B. ter Horst, A. J. Minnaard and B. L. Feringa, Org. Lett., 2007, 9, 5123–5126 CrossRef PubMed; (c) E. F. Di Mauro and M. C. Kozlowski, Org. Lett., 2001, 3, 1641–1644 CrossRef CAS PubMed; (d) V. Rauniyar and D. G. Hall, Synthesis, 2007, 3421–3426 CAS; (e) A. Alexakis, F. Romanov-Michailidis and L. Guenee, Angew. Chem., Int. Ed., 2013, 52, 9266–9270 CrossRef PubMed; (f) C. Coluccini, A. Castelluccio and D. Pasini, J. Org. Chem., 2008, 73, 4237–4240 CrossRef CAS PubMed.
- (a) D. J. Cram, R. Helgeson, S. C. Peacock, L. J. Kaplan, L. A. Domeier, P. Moreau, K. Koga, J. M. Mayer, Y. Chao, M. G. Siegel, D. H. Hoffman and G. D. Y. Sogah, J. Org. Chem., 1978, 43, 1930–1946 CrossRef CAS; (b) A. S. Droz, U. Neidlein, S. Anderson, P. Seiler and F. Diederich, Helv. Chim. Acta, 2001, 84, 2243–2289 CrossRef CAS; (c) A. Bähr, A. S. Droz, M. Püntener, U. Neidlein, S. Anderson, P. Seiler and F. Diederich, Helv. Chim. Acta, 1998, 81, 1931–1963 CrossRef; (d) T. Kawase, T. Nakamura, K. Utsumi, K. Matsumoto, H. Kurata and M. Oda, Chem. – Asian J., 2008, 3, 573–577 CrossRef CAS PubMed; (e) Z.-B. Li, J. Lin, H.-C. Zhang, M. Sabat, M. Hyacinth and L. Pu, J. Org. Chem., 2004, 69, 6284–6293 CrossRef CAS PubMed; (f) Z.-B. Li, J. Lin and L. Pu, Angew. Chem., Int. Ed., 2005, 44, 1690–1693 CrossRef CAS PubMed; (g) J. Heo and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 941–944 CrossRef CAS PubMed; (h) A. Bencini, C. Coluccini, A. Garau, C. Giorgi, V. Lippolis, L. Messori, D. Pasini and S. Puccioni, Chem. Commun., 2012, 48, 10428–10430 RSC.
- (a) L. Izotova, B. Ibragimov, J. Ashurov, S. Talipov and E. Weber, Cryst. Growth Des., 2006, 6, 2523–2529 CrossRef CAS; (b) T. Tu, T. Maris and J. D. Wuest, Cryst. Growth Des., 2008, 8, 1541–1546 CrossRef CAS; (c) C. Coluccini, D. Dondi, M. Caricato, A. Taglietti, M. Boiocchi and D. Pasini, Org. Biomol. Chem., 2010, 8, 1640–1649 RSC; (d) M. Boiocchi, M. Bonizzoni, A. Moletti, D. Pasini and A. Taglietti, New J. Chem., 2007, 31, 352–356 RSC; (e) L. Q. Ma and W. B. Lin, Angew. Chem., Int. Ed., 2009, 48, 3637–3640 CrossRef CAS PubMed; (f) L. Ma, J. M. Falkowski, C. Abney and W. B. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed; (g) M. E. Belowich, Y. Y. Botros, T. Gasa, J. F. Stoddart, C. Valente, E. Choi, C. J. Doonan, Q. Li and O. M. Yaghi, Chem. Commun., 2010, 46, 4911–4913 RSC.
- A. Shockravi, A. Javadi and E. Abouzari-Lotf, RSC Adv., 2013, 3, 6717–6746 RSC.
- (a) M. Caricato, N. J. Leza, K. Roy, D. Dondi, G. Gattuso, L. S. Shimizu, D. A. Vander Griend and D. Pasini, Eur. J. Org. Chem., 2013, 6078–6083 CrossRef CAS; (b) M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef CAS PubMed; (c) M. Caricato, C. Coluccini, D. Dondi, D. A. Vander Griend and D. Pasini, Org. Biomol. Chem., 2010, 8, 3272–3280 RSC; (d) A. Moletti, C. Coluccini, D. Pasini and A. Taglietti, Dalton Trans., 2007, 1588–1592 RSC.
- (a) D. Pasini and M. Ricci, Curr. Org. Synth., 2007, 4, 59–80 CrossRef CAS; (b) M. Caricato, A. K. Sharma, C. Coluccini and D. Pasini, Nanoscale, 2014, 6, 7165–7174 RSC.
- Selected examples: (a) Y. Cui, O. R. Evans, W. B. Lin, H. L. Ngo and P. S. White, Angew. Chem., Int. Ed., 2002, 41, 1159–1162 CrossRef CAS; (b) K. H. Doetz and A. Minatti, Tetrahedron: Asymmetry, 2005, 16, 3256–3267 CrossRef PubMed; (c) Z. An, W. Huang, H. Shi, N. Shi, J. Yin, Z. An, R. Chen and H. Jiang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3868–3879 CrossRef CAS; (d) L. Chen, C.-Q. Li and M. Shi, J. Am. Chem. Soc., 2005, 127, 3790–3800 CrossRef PubMed; (e) G. Gao, J. Lan, X. Su, F. Yang, J. You and L. Yang, Org. Biomol. Chem., 2011, 9, 2618–2621 RSC; (f) Y. Harada, K. Onitsuka, S. Takahashi and F. Takei, Chem. Commun., 1998, 643–644 Search PubMed; (g) M. Rueping, E. Sugiono, A. Steck and T. Theissmann, Adv. Synth. Catal., 2010, 352, 281–287 CrossRef CAS; (h) K. Hattori and S. Miyano, J. Am. Chem. Soc., 1998, 120, 9086–9087 CrossRef; (i) G. C. Clarkson, C. L. North, M. Wills, Y. Xu, G. Docherty and G. Woodward, J. Org. Chem., 2005, 70, 8079–8087 CrossRef PubMed; (j) H. Ishitani, S. Kobayashi, H. Shimizu and Y. Yamashita, J. Am. Chem. Soc., 2002, 124, 3292–3302 CrossRef PubMed; (k) X. Liu, P. Wang, P. Wang, Q. Yang, Y. Yang and X. Liu, Chem. – Asian J., 2010, 5, 1232–1239 CrossRef CAS PubMed.
- A. Brethon, P. Hesemann, J. J. E. Moreau, L. Rejaud and M. W. C. Man, J. Organomet. Chem., 2001, 627, 239–248 CrossRef CAS.
- C. Coluccini, A. Mazzanti and D. Pasini, Org. Biomol. Chem., 2010, 8, 1807–1815 CAS.
- Selected examples: (a) Y. Imai, N. Tajima, T. Sato and R. Kuroda, Org. Lett., 2006, 8, 2941–2944 CrossRef CAS PubMed; (b) R. Kuroda, Y. Imai and T. Sato, Chirality, 2001, 13, 588–594 CrossRef CAS PubMed; (c) T. Kinuta, N. Tajima, T. Sato, R. Kuroda, Y. Matsubara and Y. Imai, J. Mol. Struct., 2010, 964, 27–30 CrossRef CAS PubMed.
- L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2007, 36, 941–967 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional NMR spectra, IR spectra of selected compounds, optical microscope images of crystals, CD spectra, and tables containing hydrogen bond data. CCDC 1007143, 1007144, 1007145, 1007146 and 1007147. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce01160k |
| ‡ Present address: School of Chemistry, The University of Nottingham, University Park, Nottingham NG7 2RD. |
| § A literature search revealed several published experimental procedures for the preparation of (R)-2 or (RS)-2 from 1; all of them report very similar reaction protocols (ref. 9): use of molecular bromine as the electrophile, dry CH2Cl2 as the solvent, low temperatures (−80 °C to −20 °C), and inert atmosphere. None of these papers mention regioisomeric impurities. |
| ¶ Crystal data for (R)-2, (RS)-2, (R)-3, (RS)-3 and (RS)-4: (R)-2: C20H12Br2O2, Mr = 444.12, orthorhombic, P212121, a = 8.2300(2) Å, b = 11.8142(3) Å, c = 17.1253(3) Å, V = 1665.11(7) Å3, Z = 4, T = 233 K, 10 (RS)-2: C20H12Br2O2, Mr = 444.12, monoclinic, P21/n, a =9.9622(3) Å, b = 8.3317(2) Å, c = 19.9628(5) Å, β = 98.208(1)°, V = 1639.98(8) Å3, Z = 4, T = 233 K, 10 (R)-3: C28H28Br2O6, Mr = 620.32, monoclinic, P21, a = 9.0274(2) Å, b = 20.4427(4) Å, c = 14.5383(3) Å, β = 99.274(1)°, V = 2647.89(10) Å3, Z = 4, T = 233 K, 17 (RS)-3: C28H28Br2O6, Mr = 620.32, triclinic, P (RS)-4: C28H24Br2O6, Mr = 616.29, triclinic, P |
| This journal is © The Royal Society of Chemistry 2014 |