
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereospecific generation of homochiral helices in coordination polymers built from enantiopure binaphthyl-based ligands†
M.
Crespo Alonso
a,
M.
Arca
a,
F.
Isaia
a,
R.
Lai
a,
V.
Lippolis
a,
S. K.
Callear
b,
M.
Caricato
c,
D.
Pasini
 *c,
S. J.
Coles
d and
M. C.
Aragoni
*a
*c,
S. J.
Coles
d and
M. C.
Aragoni
*a
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Cittadella Universitaria, S.S. 554 bivio Sestu, 09042 Monserrato Cagliari, Italy. E-mail: aragoni@unica.it
bISIS Facility, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxfordshire OX11 0PF, UK
cDepartment of Chemistry and INSTM Research Unit, University of Pavia, Viale Taramelli 10, 27100 Pavia, Italy
dUK National Crystallography Service, Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Southampton, SO17 1BJ, UK
First published on 4th July 2014
Abstract
The novel enantiopure dipyridyl spacer 2,2′-dimethoxy-1,1′-binaphthyl-3,3′-bis(4-pyridyl-amido) (R)-L has been designed as a robust source of axial chirality to obtain helical coordination polymers. The reaction of (R)-L and the differently substituted dithiophosphato complexes [Ni((RO)2PS2)2] [R = Me (1), Et (2)] efficiently yielded coordination polymers (1·L)∞ and (2·L)∞, respectively, consisting of helical chains in which the nickel(II) ions of the [Ni((RO)2PS2)2] units are bridged by the enantiopure L ligands. The obtained polymers differ in terms of the configuration at the metal centres, which is trans and cis for (1·L)∞ and (2·L)∞, respectively. The cis configuration in (2·L)∞ generates a further element of chirality around the metal center, which occurs stereospecifically, as only one enantiomeric form is present, with homochiral helices packed with opposite screw sense in the crystal. The electronic and structural features of L, (1·L)∞, and (2·L)∞ have been investigated by means of DFT theoretical calculations, and the theoretical results have been compared with the experimental ones coming from single-crystal X-ray diffraction. The cis/trans isomerism displayed by the metal centers in (1·L)∞ and (2·L)∞ has been tentatively explained on the basis of the results of theoretical calculations performed on hypothetical pentacoordinated intermediates.
Introduction
The preparation of coordination polymers by the self-assembly of neutral metal complexes and donor molecules via coordination bonds or secondary bonding interactions is an emerging topic in the field of crystal engineering.1 In this context, we have started a synthetic program based on the ability of neutral dithiophosphonato2 and dithiophosphato3 NiII complexes to act as building blocks for the predictable assembly of inorganic coordination polymers. Due to their coordinative unsaturation, the NiII ions in these square planar complexes tend to complete their coordination sphere through the binding of monodentate donor molecules, such as pyridine, to yield octahedral complexes.4 Therefore, by using suitable N-L-N bidentate bipyridyl-based spacers, we prepared 1D coordination polymers of the [Ni(ROpdt)2(N-L-N)]∞ type.5 The primary structural motif of the polymers has been proved to depend mainly on the features of the pyridyl-based spacers such as length, rigidity, number and orientation of the donor atoms,5,6 whereas the –OR substituents on the phosphorus atoms influence the final 3D architecture through hydrogen bonds and face-to-face or edge-to-face π–π interactions.5 This means that the use of suitable chiral rigid di-topic ligands featuring twisted bridging sites might induce the formation of chiral helical coordination polymers when linked to the dithiophosphonato/dithiophosphato NiII complexes.The construction of infinite metal-containing helices7,8 and the incorporation of chirality into metal–organic frameworks9 are areas of growing importance; on one side, there is interest in building novel supramolecular architectures able to recognize (enantio)selectively suitable inclusion guests.10 On the other, the introduction of chirality can be exploited as a tool for creating organization and function at the nanoscale.11 Although either achiral or racemic ligands can be used to form chiral helical chains, the overall chirality of the product is less predictable due to the formation of racemates containing a mixture of both the enantiomeric forms.12 On the other hand, the use of enantiopure organic ligands does not necessarily lead to helices with the same handedness.13
We have previously reported the incorporation of binaphthyl-based building blocks both for the rapid construction of macrocycles as chiroptical sensors and for nanoscale structuring.14 Binaphthyl-based synthons are popular in the recent literature; given their robustness, they are frequently used to impart or transfer chiral information, not only in the field of asymmetric synthesis and catalysis15 but also in materials science.16 The basic binaphthyl moiety can be conveniently functionalized in various positions, among which 4,4′ and 6,6′ positions are the most frequent ones, although access to the 3,3′ positions is also well documented (Scheme 1).17 The presence of at least two suitable metal coordination sites (for example, pyridine) is mandatory for the formation of coordination polymers. Moreover, the presence of amide functionalities can be a powerful hydrogen-bonding tool for the stabilization of the resulting assembled nanostructure, as testified by the important roles played by amide groups in the field of foldamers,18 or in the design of assembled architectures as artificial ion channel mimics.19 Here, we report on the design, synthesis and characterization of the novel helicoidal coordination polymers obtained by the reaction of the enantiopure spacer 2,2′-dimethoxy-1,1′-binaphthyl-3,3′-bis(4-pyridyl-amido) (R)-L (Scheme 1) with the differently substituted dithiophosphato complexes [Ni((RO)2PS2)2] [R = Me (1), Et (2)].
 | ||
| Scheme 1 Top: (R)-L ligand showing the intramolecular hydrogen bonding interactions and numbering of binaphthyl moiety (left) and the dithiophosphato complexes 1 and 2 (right). Bottom: synthesis of (R)-L ligand. | ||
Results and discussion
The binaphthyl-based building block (R)-L was designed with the aim of introducing an axially chiral spacer capable of bridging the metal connecting sites and imparting a helicoidal shape to the resultant coordination polymer. The functionalities are positioned in such a way (Scheme 1, top left) that the NH amide group is locked in an S(6)-type hydrogen-bonded system20 with the neighboring phenol ether in the 2,2′ positions. This is a further element of rigidity in order to efficiently transfer the chiral information and twisting derived from the binaphthyl chiral axis to the overall coordination polymers. The (R)-L ligand was synthesized in two steps from optically pure (R)-2,2′-dimethoxy-1,1′-binaphthyl-3,3′-dicarboxylic acid under nonracemizing conditions. The precursor could be obtained in the enantiopure form after a published four-step procedure21 (including an enantioresolution step), starting from commercially available 2-naphthol. The dicarboxylic acid was subsequently activated as the acid chloride, and then amidation in the presence of 4-dimethylamino pyridine (excess), with triethylamine as the non-nucleophilic acid scavenger, resulted in the title compound (R)-L in moderate yields after purification by column chromatography (Scheme 1, bottom).1H NMR spectroscopy in CDCl3 revealed the presence of a sharp signal for the NH proton resonances of the amide functionalities, indicating effective hydrogen bonding in noncompeting solvents, as previously reported for structurally related molecular skeletons.22
The reactions of the dithiophosphato complexes [Ni((RO)2PS2)2] [R = Me (1); Et (2)] and the difunctional L ligand with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio were performed in a 1:1 mixture of CH2Cl2 and the corresponding ROH alcohols obtaining the coordination polymers [(1·L)·0.5H2O]∞ (from now on reported as (1·L)∞) and (2·L)∞, respectively, in good yields (see Experimental), all having microanalytical data corresponding to 1:1 adducts between the nickel complex and the L donor. Single crystals of (1·L)∞ and (2·L)∞ were obtained by layering an alcoholic solution of L on a CH2Cl2 solution of the relevant dithiophosphato complex.
:1 molar ratio were performed in a 1:1 mixture of CH2Cl2 and the corresponding ROH alcohols obtaining the coordination polymers [(1·L)·0.5H2O]∞ (from now on reported as (1·L)∞) and (2·L)∞, respectively, in good yields (see Experimental), all having microanalytical data corresponding to 1:1 adducts between the nickel complex and the L donor. Single crystals of (1·L)∞ and (2·L)∞ were obtained by layering an alcoholic solution of L on a CH2Cl2 solution of the relevant dithiophosphato complex.
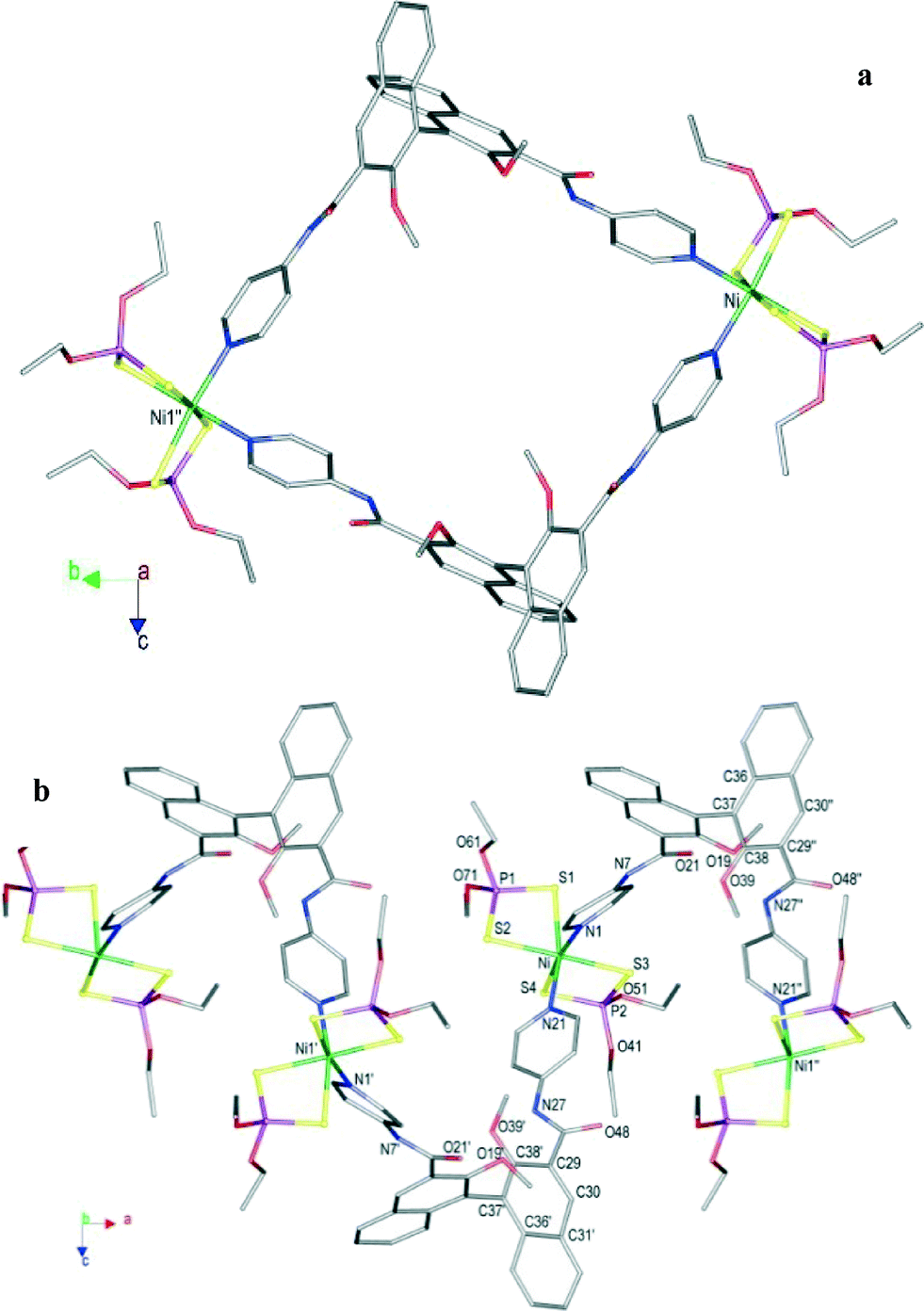
Single-crystal X-ray diffraction has been performed for both compounds; crystallographic data and selected bond lengths and angles for (1·L)∞ and (2·L)∞ are reported in Tables 1 and 2, respectively. Compounds (1·L)∞ and (2·L)∞ are helically shaped polymers formed by coordination of the binaphthyl-based spacer L to the NiII ions of the square planar complexes 1 and 2. The [Ni((RO)2PS2)2] units bridged by L and the resulting helices –L–[Ni(ROdtp)2]–L–[Ni(ROdtp)2]–L– are shown in Fig. 1 and 3 for (1·L)∞ and (2·L)∞, respectively.
| [(1·L)·(0.5H2O)]∞ | (2·L)∞ | |
|---|---|---|
| a w = 1/[\s2(Fo2) + (0.0550P)2]; P = (Fo2 + 2Fc2)/3. b Due to relatively poor data quality for (2·L)∞ the Flack parameter cannot be accurately refined and therefore the correct absolute structure cannot be reliably determined. | ||
| Empirical formula | C38H38N4NiO8P2S4 H2O | C42H46N4NiO8P2S4 |
| M | 936.64 | 983.74 |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P21 | P212121 |
| a (Å) | 9.6133(2) | 15.332(2) |
| b (Å) | 45.6542(12) | 8.6055(10) |
| c (Å) | 10.5801(3) | 34.795(4) |
| α (°) | 90 | 90 |
| β (°) | 113.202(1) | 90 |
| γ (°) | 90 | 90 |
| Volume (Å3) | 4267.92(19) | 4590.8(10) |
| Z | 2 | 4 |
| D calc (Mg m−3) | 1.458 | 1.423 |
| μ (mm−1) | 0.781 | 0.729 |
| θ min–max (°) | 2.9–27.1 | 3.0–25.0 |
| T (K) | 120 | 120 |
| Refl. collected/unique | 44414/18504 |
26414/7285 |
| Refl. obs. (I > 2σI) | 13551 |
4710 |
| R/RINT | 0.0587/0.066 | 0.1495/0.158 |
| wR2 | 0.1325 | 0.3147 |
| Min/max res. D. (e A−3) | −0.36, 0.75 | −0.73, 1.16 |
| GoF | 1.019 | 1.162 |
| Flack par | 0.048(12) | 0.27(6)b |
| (1·L)∞ | |||
|---|---|---|---|
| Ni1–S1 | 2.5385(15) | Ni2–S5 | 2.5196(16) |
| Ni1–S2 | 2.5001(15) | Ni2–S6 | 2.4738(17) |
| Ni1–S3 | 2.5490(15) | Ni2–S7 | 2.4647(16) |
| Ni1–S4 | 2.4900(15) | Ni2–S8 | 2.5232(16) |
| Ni1–N1 | 2.093(4) | Ni2–N2 | 2.074(5) |
| Ni1–N3 | 2.085(4) | Ni2–N4 | 2.084(4) |
| S1–P1 | 1.983(2) | S5–P3 | 1.985(2) |
| S2–P1 | 1.9675(19) | S6–P3 | 1.974(2) |
| S3–P2 | 1.983(2) | S7–P4 | 1.986(2) |
| S4–P2 | 1.9731(19) | S8–P4 | 1.987(2) |
| N1–Ni1–N3 | 179.05(16) | N2–Ni2–N4 | 179.63(17) |
| S1–Ni1–S2 | 81.70(5) | S5–Ni2–S6 | 82.03(5) |
| S1–Ni1–S3 | 178.74(5) | S5–Ni2–S7 | 96.23(53) |
| S1–Ni1–S4 | 97.48(5) | S5–Ni2–S8 | 178.42(6) |
| S2–Ni1–S3 | 99.42(5) | S6–Ni2–S7 | 178.25(6) |
| S2–Ni1–S4 | 179.00(5) | S6–Ni2–S8 | 99.43(5) |
| S3–Ni1–S4 | 81.40(5) | S7–Ni2–S8 | 82.32(5) |
| S1–Ni1–N1 | 89.70(12) | S5–Ni2–N2 | 91.19(13) |
| S1–Ni1–N3 | 89.48(12) | S5–Ni2–N4 | 88.71(12) |
| S2–Ni1–N1 | 90.78(12) | S6–Ni2–N2 | 90.38(13) |
| S2–Ni1–N3 | 88.65(12) | S6–Ni2–N4 | 89.26(12) |
| S3–Ni1–N1 | 90.88(12) | S7–Ni2–N2 | 89.50(13) |
| S3–Ni1–N3 | 89.96(12) | S7–Ni2–N4 | 90.86(12) |
| S4–Ni1–N1 | 89.79(12) | S8–Ni2–N2 | 89.40(13) |
| S4–Ni1–N3 | 90.77(12) | S8–Ni2–N4 | 90.71(12) |
| S1–P1–S2 | 113.06(9) | S5–P3–S6 | 111.75(10) |
| S3–P2–S4 | 112.32(9) | S7–P4–S8 | 111.49(9) |
| O1–P1–O2 | 96.8(2) | O5–P3–O6 | 94.5(3) |
| O3–P2–O4 | 97.4(2) | O7–P4–O8 | 95.5(2) |
| (2·L)∞ | |||
|---|---|---|---|
| Ni1–S1 | 2.513(6) | Ni1–N21 | 2.094(14) |
| Ni1–S2 | 2.499(5) | S1–P1 | 2.007(9) |
| Ni1–S3 | 2.514(5) | S2–P1 | 1.949(7) |
| Ni1–S4 | 2.538(6) | S3–P2 | 2.010(7) |
| Ni1–N1 | 2.092(15) | S4–P2 | 1.989(9) |
| N1–Ni–N21 | 91.6(5) | S3–Ni–S4 | 80.86(19) |
| S1–Ni–S2 | 79.66(19) | S3–Ni–N1 | 90.6(4) |
| S1–Ni–S3 | 98.4(2) | S3–Ni–N21 | 94.5(4) |
| S1–Ni–S4 | 93.28(16) | S4–Ni–N1 | 171.4(5) |
| S1–Ni–N1 | 87.4(4) | S4–Ni–N21 | 89.7(4) |
| S1–Ni–N21 | 167.0(4) | S1–P1–S2 | 108.5(3) |
| S2–Ni–S3 | 174.43(17) | S3–P2–S4 | 110.0(3) |
| S2–Ni–S4 | 93.99(19) | O61–P1–O71 | 92.8(8) |
| S2–Ni–N1 | 94.6(4) | O41–P2–O51 | 99.9(7) |
| S2–Ni–N21 | 87.6(4) | ||
 | ||
| Fig. 1 (a) Helical chain structure of (1·L)∞ with a selected atom numbering scheme; (b) view of one spiral along the 010 direction. H atoms have been omitted for clarity; symmetry codes: ′, −1 − x, 0.5 + y, −z; ′′, −1 − x, −0.5 + y, −z. | ||
The structure of compound (1·L)∞ consists of a homochiral right-handed helical chain in which the nickel(II) ions of the [Ni((MeO)2PS2)2] units are bridged by the enantiopure L ligands (Fig. 1). The polymer crystallizes in the monoclinic P21 chiral space group, with two [Ni((MeO)2PS2)2] units, two L ligands, and one water molecule in the asymmetric unit. The coordination environment around both the two symmetry-independent nickel ions results in a distorted octahedron with four sulphur atoms from two bidentate (MeO)2PS2 units on the equatorial plane and two nitrogen atoms from the bridging ligands in a trans fashion occupying the axial positions, with N–Ni–N angles of 179.05(16)° and 179.63(17)° (Table 2).
It is interesting to note that the coordination environments around Ni1 and Ni2 differ in terms of the orientation of the methoxy substituents at the P atoms, all pointing at the pyridine rings bonded to the same coordination core, with the exception of MeO(1) and MeO(4) which are engaged in strong H bonds with the amide groups of the adjacent helices (see the following). The binaphthyl moieties feature torsion angles of 71.1(7)° (C78–C77–C97–C98) and 114.5(6)° (C38–C37–C57–C58) along the pivotal 1,1′-bond joining the two naphthyl units, similar to those found in analogous 2,2′-dimethoxy-1,1′-binaphthyl molecules, normally ranging from 70° to 113°.‡ However, the dihedral angle between the ring planes is quite consistent at 71.84° and 71.17° for the C37–C57 and C77–C97 binaphthyl moieties, respectively. The two spacers differ in terms of the orientation of the 2,2′-methoxy substituents, which are convergent in the case of MeO(O39)/MeO(O59) and divergent in the case of MeO(O79)/MeO(O99) (Fig. 1b).
The spirals run parallel along the b direction involving a crystallographic 2-fold screw axis (Fig. 1), with a helical pitch of 45.65 Å, coincident with the b-axis length, and intertwine with each other in both the a and the c directions in a densely interlocked architecture stabilized by an intricate net of H bonds, mainly involving the amido groups, the water molecules and both the binaphthyl- and P-methoxy substituents [MeO (O79), MeO(O1), and MeO(O4), respectively; see Table 3]. The packing of adjacent spirals leads to a compact tridimensional network similar to the arrangement of partially embedded parallel springs running along b and shifted in the ac plane and intertwined in order to reciprocally occupy the empty space of one spiral with the spires of the adjacent ones (Fig. 2).
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| (1·L)∞ | ||||
| O1W–H1W⋯O6a | 0.86(4) | 2.21(4) | 2.960(6) | 146(4) |
| N27–H27⋯O1b | 0.90(5) | 2.15(4) | 3.003(6) | 156(4) |
| N87–H87⋯O4c | 0.87(5) | 2.16(5) | 3.022(6) | 172(4) |
| C4–H4B⋯O79b | 0.98 | 2.41 | 3.373(7) | 167 |
| C23–H23⋯O1b | 0.95 | 2.54 | 3.316(7) | 140 |
| C26–H26⋯O88d | 0.95 | 2.32 | 3.149(6) | 146 |
| C35–H35⋯O8e | 0.95 | 2.39 | 3.260(7) | 152 |
| C46–H46⋯O68f | 0.95 | 2.29 | 3.048(7) | 136 |
| C55–H55⋯O2b | 0.95 | 2.43 | 3.255(6) | 146 |
| C66–H66⋯O48d | 0.95 | 2.38 | 3.123(7) | 134 |
| C86–H86⋯O28g | 0.95 | 2.38 | 3.218(7) | 147 |
| (2·L)∞ | ||||
|---|---|---|---|---|
| Symmetry codes:a −1 − x, 0.5 + y, −1 − z.b 1 + x, y, z.c −1 + x, y, z.d 1 + x, y, 1 + z.e x, y, −1 + z.f −2 − x, −0.5 + y, −1 + z.g −1 + x, y, −1 + z.h x, 1 + y, z.i −0.5 + x, 0.5 – y, 2 − z.j −0.5 + x, 1.5 − y, 2 − z.k x, −1 + y, z.l 2 − x, 1.5 + y, 1.5 − z. | ||||
| N7–H7⋯S1h | 0.88 | 2.53 | 3.396(15) | 169 |
| N27–H62⋯O21i | 0.88 | 2.16 | 2.976(19) | 154 |
| N27–H62⋯O39j | 0.88 | 2.49 | 2.913(19) | 111 |
| C3–H3⋯O39k | 0.95 | 2.39 | 3.18(2) | 141 |
| C5–H5⋯S1h | 0.95 | 2.85 | 3.645(18) | 141 |
| C33–H33⋯O71l | 0.95 | 2.54 | 3.45(3) | 162 |
 | ||
| Fig. 2 Packing views of intertwining helices along the 010 (a) and 100 (b) directions. H atoms have been omitted and the spirals have been pointed out by using different colors for clarity. | ||
 | ||
| Fig. 3 View of one spiral of (2·L)∞ along the 100 (a) and 010 directions (b) with a selected atom numbering scheme. H atoms have been omitted for clarity; symmetry codes: ′, −0.5 + x, 1.5 − y, 2 − z; ′′, 0.5 + x, 1.5 − y, 2 − z. | ||
The structure of compound (2·L)∞ consists of one-dimensional left-handed helical chains in which the nickel(II) atoms of the [Ni(EtOpdt)2] units are bridged by the enantiopure L ligands (Fig. 3). The polymer crystallizes in the orthorhombic P212121 chiral space group, with one [Ni((EtO)2PS2)2] unit and one L ligand in the asymmetric unit. The coordination environment around the nickel ion results in a distorted octahedron with four sulphur atoms from two bidentate (EtO)2PS2 units and two nitrogen atoms from the pyridine rings of two bridging L ligands disposed in a cis configuration with a N–Ni–N angle of 91.6(5)° (Table 2). The binaphthyl moiety features a torsion angle of 97(2)° (C16–C17–C37–C38) and a dihedral angle of 76.55° between the two ring systems. The spirals run parallel along the a direction involving a crystallographic 2-fold screw axis (Fig. 3), with a helical pitch of 15.33 Å, coincident with the a-axis length. Homochiral helices pack with opposite screw sense in the crystal (top and bottom helices in Fig. 4a).
 | ||
| Fig. 4 (a) View of the helices (2·L)∞ running along the −100 (top) and 100 (bottom) directions; (b) packing view along the a axis showing alternating planes formed by intertwining spirals running along the −100 (yellow) and 100 (blue) directions. H atoms have been omitted for clarity. | ||
Spirals with the same orientation intertwine with each other and pack in a quite compact arrangement formed by stacking planes formed by helices running either along 100 or −100 (blue and yellow in Fig. 4b, respectively). It is interesting to point out that, different from what was previously observed for (1·L)∞, the crystal packing of (2·L)∞ does not involve the P atom substituents given that spirals intertwine through H bonds involving the amido groups, the MeO substituents and the pyridine rings of the binaphthyl ligands and the S1 atom coordinated to the metal ion. The planes formed by differently oriented helices pack on each other leaving small empty spaces of about 120 Å3, comprising 2.6% of the cell volume.
DFT calculations
An insight into the electronic features of (1·L)∞ and (2·L)∞ can be provided by theoretical calculations carried out at the DFT level. As model compounds, the monomer units 1·2L and 2·2L, featuring the central nickel(II) ion in trans and cis coordination geometries, respectively, have been considered. The isolated complex 1 shows metric parameters very similar to those found in the few examples deposited in the Cambridge Structural Database.23 In particular, the Ni–S and P–S bond distances (2.266 and 2.007 Å, respectively) are only slightly longer than those that have been experimentally structurally characterised.24A natural bond analysis (NBA) carried out on 1 at the optimised geometry has shown the Ni centre to be positively charged (QNi = +0.212). Wiberg bond indices25 reflect as expected the average P–S bond orders larger than unity (WBIPS = 1.173) and remarkably strong Ni–S bonds (WBINiS = 0.642). Notably, the electronic structure of 1 in its ground state shows a low-lying calculated virtual molecular orbital (MO), namely, Kohn–Sham LUMO+3, almost exclusively composed of the 3dz2 atomic orbital (AO) of the metal ion. This KS-MO is therefore available to receive electron density from Lewis σ-donors, such as pyridine, to coordinatively saturate the nickel(II) centre and yield an octahedral trans-disposed complex. Accordingly, the L ligand is calculated to show two filled MOs (HOMO-2 and HOMO-3), constituting the in-phase and out-of-phase combinations of the pyridine σ-type lone pairs localised on the N donor atoms (NBO charge, −0.503 e).
In order to investigate the isomerism of the complexes derived from the reaction of bis(dithiophosphate) nickel(II) complexes with pyridine (Py) donors, the structures of both cis and trans isomers of 1·2Py were optimised. The two isomers were almost isoenergetic, with their total electronic energies differing by only 1 kcal mol−1 in the gas phase (Fig. 5). The optimised metric parameters§ clearly show a remarkable trans effect exerted by the pyridine ligand. The hypothetical pentacoordinated species 1·Py was optimised with both square pyramidal and trigonal bipyramidal coordination arrangements at the metal centre, the former structure being more stable by about 11 kcal mol−1. It is conceivable that a pseudorotation26 or turnstile rotation mechanism in the intermediate pentacoordinate complex27 might be involved in the formation of the final cis or trans isomers. This mechanism could explain the isomerism experimentally found in the case of (1·L)∞ and (2·L)∞. The metric parameters optimised for 1·2L and 2·2L are very close to those calculated for 1·2Py (cis and trans isomers, respectively) at the same level of theory and can be finely compared with those determined in the crystal structure of (1·L)∞ and (2·L)∞ (Table 4).
 | ||
| Fig. 5 Sums of the total electronic energies calculated for the systems 1 + 2Py, 1·Py (trigonal bipyramid coordination) + Py, 1·Py (square-pyramid coordination) + Py, and 1·2Py (cis and trans isomers) relative to the most stable complex. Ball and stick drawings of the metal complexes at their optimised geometries are also depicted. | ||
| 1·2L | 2·2L | |||
|---|---|---|---|---|
| d | WBI | d | WBI | |
| Ni–S | 2.569 | 0.333 | 2.540 | 0.339 |
| 2.528 | 0.354 | 2.534 | 0.348 | |
| 2.530 | 0.333 | 2.540 | 0.339 | |
| 2.518 | 0.362 | 2.534 | 0.348 | |
| Ni–N | 2.116 | 0.244 | 2.111 | 0.256 |
| 2.116 | 0.247 | 2.111 | 0.256 | |
In particular, the Ni–N bonds show Wiberg indices typical of polarised covalent bonds (average values, 0.245 and 0.256 for 1·2L and 2·2L, respectively). Notably, NBA shows a remarkably more positive charge on nickel (+0.712 and +0.709 e for 1·2L and 2·2L, respectively) as compared to that calculated for the same atom in 1. This suggests a more polarised nature of the bonds between the dithiophosphato ligands and the metal centre in the octahedral complexes with respect to 1. This is reflected in a remarkable lengthening in Ni–S bond lengths, verified experimentally by X-ray diffraction measurements. The weakening in the Ni–S bonds is also pointed out by the relevant bond indices (average value, 0.346).
Conclusions
The new binaphthyl-based 2,2′-dimethoxy-1,1′-binaphthyl-3,3′-bis(4-pyridyl-amido) (R)-L building block has been deliberately designed with the aim of creating an axially chiral spacer capable of bridging the metal connecting sites and imparting a helical shape to the resultant coordination polymer. DFT calculations performed on the free ligand have shown the donor ability of the pyridine σ-type lone pairs localised on the N donor atoms towards the metal centres. The subsequent reaction of the neutral dithiophosphato complexes [Ni((RO)2PS2)2] [R = Me (1), Et (2)] as coordinatively unsaturated metal building blocks and the enantiopure (R)-L as a spacer has allowed the assembly of the two novel helicoidal coordination polymers (1·L)∞ and (2·L)∞.Although the starting Ni(II) complexes 1 and 2 feature similar square planar geometries, the obtained polymers differ in terms of the configuration of the pyridine ligands bound to the metal, which is trans and cis for (1·L)∞ and (2·L)∞, respectively, thus generating in (2·L)∞ a further element of chirality at the octahedral coordinated metal ion. The generation occurs stereospecifically, as only one enantiomeric form is present in the corresponding helices. This difference is also reflected in the resulting polymers that feature different helical pitches (45.65 and 15.33 Å for (1·L)∞ and (2·L)∞, respectively) and different screw sense. Notwithstanding the inner differences between the polymers, their helical nature confirms that the primary structure of the polymers is directly controlled by the choice of spacers. Therefore, chiral rigid di-topic ligands featuring twisted bridging sites such as (R)-L can be used to prepare chiral helical coordination polymers when linked to the dithiophosphato NiII complexes.
DFT calculations performed on the cis and trans isomers of 1·2Py and on the hypothetical pentacoordinated species 1·Py (in both square planar and trigonal bipyramidal geometries) taken as model compounds showed that the two isomers are almost isoenergetic. Different intermolecular interactions ensuing from the nature of the P-substituents in the dithiophosphato complexes can be evoked to explain the structural diversities in the topology and in the final 3D-architecture of the polymer.
Further work is in progress in our laboratory with the aim of studying the effects of the building blocks on the resulting structures in order to reach an extensive control of the ensuing nanoscale products.
Experimental
Materials and methods
All commercially available compounds were used as received. Bis[O-alkyl-dithiophosphato]Ni complexes {[Ni((RO)2PS2)2], R = Me (1), Et (2)}28 and (R)-2,2′-dimethoxy-1,1′-binaphthyl-3,3′-dicarboxylic acid21 were synthesised according to previously reported procedures. THF and CH2Cl2 were dried on CaH2 and distilled before use. 1H and 13C NMR spectra were recorded at 25 °C in CDCl3 on 200 or 300 MHz NMR spectrometers using the residual solvent signal as the internal standard. Mass spectra were recorded using an electrospray ionization spectrometer. Analytical thin-layer chromatography was performed on silica gel, chromophore-loaded, commercially available plates. Flash chromatography was carried out using silica gel (pore size, 60 Å, 230–400 mesh). Optical rotations were measured on a polarimeter with a sodium lamp (λ = 589 nm) and are reported as follows: [α]D (c = g (100 mL)−1, solvent). Elemental analyses were performed with an EA1108 CHNS-O Fisons instrument. FT-infrared spectra were recorded on a Thermo Nicolet 5700 spectrometer at room temperature using a flow of dry air. Middle IR spectra (resolution, 4 cm−1) were recorded using KBr pellets with a KBr beam splitter and KBr windows.X-ray diffraction
X-ray structure determinations and crystallographic data for compounds (1·L)∞ and (2·L)∞ were collected at 120(2) K by means of combined phi and omega scans on a Bruker-Nonius Kappa CCD area detector situated at the window of a rotating anode (graphite Mo-Kα radiation for (1·L)∞ and 10 cm confocal mirrors for (2·L)∞, λ = 0.71073 Å). The structures were solved by direct methods, SHELXS-97, and refined on F2 using SHELXL-97.29 Anisotropic displacement parameters were assigned to all non-hydrogen atoms. Hydrogen atoms were included in the refinement, but thermal parameters and geometry were constrained to ride on the atom to which they are bonded. The data were corrected for absorption effects using SADABS V2.10.30 A possible disordered water molecule was found in (1·L)∞: on modeling the Q-peak as an oxygen atom, the site is found to be approximately 1/4 occupied. It is not possible to be certain whether this is indeed a water molecule or just an artifact. For compound (2·L)∞ the R factor is slightly higher than usual due to the crystal only diffracting weakly as indicated by the data collection resolution despite using a rotating anode X-ray generator. However, the gross connectivity of the structure can be unambiguously determined and it is reliable enough to permit geometric comparisons. An ISOR restraint was used for the non-positive definite C25 thermal modeling, and DELU and SIMU restraints were also employed globally. The carbon and hydrogen atom Ueq(max)/Ueq(min) ratio is larger than expected due to the slight rotation of the methyl group C63 creating a larger ellipsoid, which could not be modeled as disordered. Both structures have been deposited with the Cambridge Crystallographic Data Centre (deposition numbers CCDC 1002587 and CCDC 1002588 for (1·L)∞ and (2·L)∞, respectively).DFT calculations
Quantum mechanical DFT calculations were performed on 1, L, pyridine (Py), 1·2Py (cis and trans isomers), the two possible conformers of the hypothetical intermediate 1·Py, 1·2L and 2·2L. Calculations were carried out by adopting the mPW1PW31 hybrid functional. Schäfer, Horn and Ahlrichs pVDZ basis sets were used for all atomic species.32 In all cases, geometry optimisations were followed by frequency calculations aimed to verify the nature of the energy minima. For all the optimised molecules, a full NBO33 analysis was carried out. The program Molden 5.0 (ref. 34) was used to investigate the charge distributions and molecular orbital shapes.Syntheses
Acknowledgements
Financial support from the University of Pavia and MIUR (PRIN 2004 to MCA and DP, and PRIN2009 A5Y3N9 to DP) is gratefully acknowledged. The authors would like to thank EPSRC for funding the UK National Crystallography Service. Dr. Riccardo Montis (Università degli Studi di Cagliari, Italy) is kindly acknowledged for the useful discussion.Notes and references
- M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257(7–8), 1282–1305 CrossRef CAS PubMed; O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43(8), 1166–1175 CrossRef PubMed; C. J. Sumby, Aust. J. Chem., 2013, 66(4), 397–400 CrossRef; J. Reedijk, Chem. Soc. Rev., 2013, 42(4), 1776–1783 RSC; L. Brammer, Chem. Soc. Rev., 2004, 33, 476–489 RSC; S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251(21–24), 2490–2509 CrossRef PubMed.
- M. Arca, A. Cornia, F. A. Devillanova, A. C. Fabretti, F. Isaia, V. Lippolis and G. Verani, Inorg. Chim. Acta, 1997, 262, 81–84 CrossRef CAS; W. E. van Zyl and J. D. Woollins, Coord. Chem. Rev., 2013, 257(3–4), 718–731 CrossRef PubMed.
- I. Haiduc, Handbook of Chalcogen Chemistry, ed. F. A. Devillanova, Royal Society of Chemistry, 2006, pp. 593–643 Search PubMed.
- M. C. Aragoni, M. Arca, F. Demartin, F. A. Devillanova, C. Graiff, F. Isaia, V. Lippolis, A. Tiripicchio and G. Verani, J. Chem. Soc., Dalton Trans., 2001, 2671–2677 RSC; L. Bolundut, I. Haiduc, E. Ilyes, G. Kociok-Köhn, K. C. Molloy and S. Gómez-Ruiz, Inorg. Chim. Acta, 2010, 363(15), 4319–4323 CrossRef CAS PubMed.
- M. C. Aragoni, M. Arca, N. R. Champness, A. V. Chernikov, F. A. Devillanova, F. Isaia, V. Lippolis, N. S. Oxtoby, G. Verani, S. Z. Vatsadze and C. Wilson, Eur. J. Inorg. Chem., 2004, 10, 2008–2012 CrossRef CAS; M. C. Aragoni, M. Arca, N. R. Champness, M. De Pasquale, F. A. Devillanova, F. Isaia, V. Lippolis, N. S. Oxtoby, G. Verani and C. Wilson, CrystEngComm, 2005, 7(60), 363–369 RSC; M. C. Aragoni, M. Arca, M. Crespo, F. A. Devillanova, M. B. Hursthouse, S. L. Huth, F. Isaia, V. Lippolis and G. Verani, CrystEngComm, 2007, 9(10), 873–878 RSC.
- L. Monnereau, M. Nieger, T. Muller and S. Bräse, Adv. Funct. Mater., 2014, 24, 1054–1058 CrossRef CAS.
- H.-F. Chow, K.-N. Lau, Z. Ke, Y. Liang and C.-M. Lo, Chem. Commun., 2010, 46(20), 3437–3453 RSC; E. C. Constable, N. Hostettler, C. E. Housecroft, N. S. Murray, J. Schönle, U. Soydaner, R. M. Walliser and J. A. Zampese, Dalton Trans., 2013, 42(14), 4970–4977 RSC; C. Piguet, M. Borkovec, J. Hamacek and K. Zeckert, Coord. Chem. Rev., 2005, 249(5–6), 705–726 CrossRef CAS PubMed.
- T.-T. Luo, H.-C. Wu, Y.-C. Jao, S.-M. Huang, T.-W. Tseng, Y. Wen, G.-H. Lee, S.-M. Peng and K.-L. Lu, Angew. Chem., Int. Ed., 2009, 48(50), 9461–9464 CrossRef CAS PubMed; Y. Cui, S.-J. Lee and W. Lin, J. Am. Chem. Soc., 2003, 125, 6014–6015 CrossRef PubMed; D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38(4), 273–282 CrossRef PubMed; B. Joarder, A. K. Chaudhari and S. K. Ghosh, Inorg. Chem., 2012, 51(8), 4644–4649 CrossRef PubMed.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 957–968 CrossRef CAS PubMed; L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef PubMed.
- W. Xuan, M. Zhang, Y. Liu, Z. Chen and Y. Cui, J. Am. Chem. Soc., 2012, 134(16), 6904–6907 CrossRef CAS PubMed; A. D. Cutland-Van Noord, J. W. Kampf and V. L. Pecoraro, Angew. Chem., Int. Ed., 2002, 41, 4667–4670 CrossRef PubMed.
- Chem. Soc. Rev., 2009, 38(3) CrossRef CAS PubMed, whole issue; H. Wang, Chirality, 2010, 22(9), 827–837 CrossRef CAS PubMed; J. K. Okuma, R. Itoyama, A. Sou, N. Nagahora and K. Shioj, Chem. Commun., 2012, 48(90), 11145–11147 RSC; M. Caricato, A. K. Sharma, C. Coluccini and D. Pasini, Nanoscale, 2014, 6, 7165–7174 RSC.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2(5), 353–361 CrossRef CAS PubMed; J. Zhang, S. Chen, T. Wu, P. Feng and X. Bu, J. Am. Chem. Soc., 2008, 130(39), 12882–12883 CrossRef PubMed; E. Yashima, K. Maeda, H. Lida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109(11), 6102–6211 CrossRef PubMed; K. K. Bisht and E. Suresh, Inorg. Chem., 2012, 51(18), 9577–9579 CrossRef PubMed.
- M. Albrecht, Chem. Rev., 2001, 101(11), 3457–3497 CrossRef CAS PubMed; Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22(37), 4112–4135 CrossRef PubMed; D. Pijper and B. L. Feringa, Soft Matter, 2008, 4(7), 1349–1372 RSC; H. Cao, X. Zhu and M. Liu, Angew. Chem., Int. Ed., 2013, 52(15), 4122–4126 CrossRef PubMed.
- A. Bencini, C. Coluccini, A. Garau, C. Giorgi, V. Lippolis, L. Messori, D. Pasini and S. Puccioni, Chem. Commun., 2012, 48, 10428–10430 RSC; A. Moletti, C. Coluccini, D. Pasini and A. Taglietti, Dalton Trans., 2007, 1588–1592 RSC; C. Coluccini, A. Mazzanti and D. Pasini, Org. Biomol. Chem., 2010, 8, 1807–1815 Search PubMed; C. Coluccini, D. Dondi, M. Caricato, A. Taglietti, M. Boiocchi and D. Pasini, Org. Biomol. Chem., 2010, 8, 1640–1649 Search PubMed; M. Caricato, C. Coluccini, D. Dondi, D. A. Vander Griend and D. Pasini, Org. Biomol. Chem., 2010, 8, 3272–3280 Search PubMed; M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112(2), 1196–1231 CrossRef CAS PubMed; T. J. Ward, Anal. Chem., 2006, 78(12), 3947–3956 CrossRef PubMed; P. S. Bhadury, Y. Yao and Y. He, Curr. Org. Chem., 2012, 16(15), 1730–1753 CrossRef; D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395–456 CrossRef; H. Pellissier, Adv. Synth. Catal., 2012, 354(2–3), 237–294 CrossRef.
- L. Pu, Acc. Chem. Res., 2012, 45(2), 150–163 CrossRef CAS PubMed; Q. Li, H. Guo, Y. Wu, X. Zhang, Y. Liu and J. Zhao, J. Fluoresc., 2011, 21(6), 2077–2084 CrossRef PubMed; J. Jiao, X. Liu, X. Mao, J. Li, Y. Cheng and C. Zhu, New J. Chem., 2013, 37(2), 317–322 RSC.
- C. Coluccini, A. Castelluccio and D. Pasini, J. Org. Chem., 2008, 73, 4237–4240 CrossRef CAS PubMed; J. M. Brunel, Chem. Rev., 2005, 105, 857–898 CrossRef PubMed.
- I. Huc, Eur. J. Org. Chem., 2004, 17–29 CrossRef CAS; Y. Baudry, G. Bollot, V. Gorteau, S. Litvinchuk, J. Mareda, M. Nishihara, D. Pasini, F. Perret, D. Ronan, N. Sakai, M. R. Shah, A. Som, N. Sorde, P. Talukdar, D.-H. Tran and S. Matile, Adv. Funct. Mater., 2006, 16, 169–179 CrossRef.
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- D. J. Cram, R. Helgeson, S. C. Peacock, L. J. Kaplan, L. A. Domeier, P. Moreau, K. Koga, J. M. Mayer, Y. Chao, M. G. Siegel, D. H. Hoffman and G. D. Y. Sogah, J. Org. Chem., 1978, 43, 1930–1946 CrossRef CAS; M. Asakawa, H. M. Janssen, E. W. Meijer, D. Pasini and J. F. Stoddart, Eur. J. Org. Chem., 1998, 983–986 CrossRef; C. Xin, S. Da, D. Dong, J. Liu and R. Wei, Tetrahedron: Asymmetry, 2002, 13, 1937–1940 CrossRef.
- S. Colombo, C. Coluccini, M. Caricato, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2010, 66, 4206–4211 CrossRef CAS PubMed; M. Caricato, N. J. Leza, C. Gargiulli, G. Gattuso, D. Dondi and D. Pasini, Beilstein J. Org. Chem., 2012, 8, 967–976 CrossRef PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- Ni–S1 2.218, Ni–S2 2.225, P–S1 1.984, P–S2 1.979, CDS refcode DMTPON, V. Kastalsky and J. F. McConnell, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem, 1969, 25, 909–915 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- R. S. Berry, J. Chem. Phys., 1960, 32, 933–938 CrossRef CAS PubMed.
- M. E. Cass, K. K. Hii and H. S. Rzepa, J. Chem. Educ., 2006, 83, 336 CrossRef CAS.
- M. C. Aragoni, M. Arca, M. Crespo, F. A. Devillanova, M. B. Hursthouse, S. L. Huth, F. Isaia, V. Lippolis and G. Verani, Dalton Trans., 2009, 2510–2520 RSC.
- G. M. Sheldrick, SHELX suite of programs for crystal structure solution and refinement, Univ. of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SADABS V2.10, 2003 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1002587 and 1002588. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce01101e |
| ‡ The range has been calculated from the dihedral torsion angles found in molecules containing analogous 2,2′-dimethoxy-1,1′-binaphthyl fragments (47 structures, 74 fragments, average value = 87.8°) from a search in the Cambridge Structural Database System version 5.35, 2014. |
| § Selected optimised distances for the cis isomer of 1·2Py: Ni–N, 2.131 Å; Ni–S (trans to N) 2.539; Ni–S (trans to S) 2.516 Å. Selected optimised distances for the trans isomer of 1·2Py: Ni–N, 2.129 Å; Ni–S, 2.526 Å. |
| This journal is © The Royal Society of Chemistry 2014 |