
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceO- vs. N-protonation of 1-dimethylaminonaphthalene-8-ketones: formation of a peri N–C bond or a hydrogen bond to the pi-electron density of a carbonyl group†
Nerea
Mercadal
a,
Stephen P.
Day
b,
Andrew
Jarmyn
a,
Mateusz B.
Pitak
c,
Simon J.
Coles
 c,
Claire
Wilson
d,
Gregory J.
Rees
b,
John V.
Hanna
*b and
John D.
Wallis
*a
c,
Claire
Wilson
d,
Gregory J.
Rees
b,
John V.
Hanna
*b and
John D.
Wallis
*a
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK. E-mail: john.wallis@ntu.ac.uk
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK. E-mail: j.v.hanna@warwick.ac.uk
cUK National Crystallography Service, Chemistry, University of Southampton, Highfield Campus, Southampton, SO17 1BJ, UK. E-mail: S.J.Coles@soton.ac.uk
dDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX1 0DE, UK. E-mail: claire.wilson@diamond.ac.uk
First published on 15th July 2014
Abstract
X-ray crystallography and solid-state NMR measurements show that protonation of a series of 1-dimethylaminonaphthalene-8-ketones leads either to O protonation with formation of a long N–C bond (1.637–1.669 Å) between peri groups, or to N protonation and formation of a hydrogen bond to the π surface of the carbonyl group, the latter occurring for the larger ketone groups (C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)R, R = t-butyl and phenyl). Solid state 15N MAS NMR studies clearly differentiate the two series, with the former yielding significantly more deshielded resonances. This is accurately corroborated by DFT calculation of the relevant chemical shift parameters. In the parent ketones X-ray crystallography shows that the nitrogen lone pair is directed towards the carbonyl group in all cases.
O)R, R = t-butyl and phenyl). Solid state 15N MAS NMR studies clearly differentiate the two series, with the former yielding significantly more deshielded resonances. This is accurately corroborated by DFT calculation of the relevant chemical shift parameters. In the parent ketones X-ray crystallography shows that the nitrogen lone pair is directed towards the carbonyl group in all cases.
Introduction
We are interested in mapping out the changes in electron distribution on the formation of a N–C bond via charge density measurements from X-ray diffraction studies on molecules containing N–C bonds of different lengths. Of particular interest are those bonds which are particularly long and thus represent bonds which are not fully formed. Bond formation is a fundamental process in chemistry, and its study underpins topics such as catalysis and enzyme action. The peri-naphthalene system has provided us with a series of zwitterionic materials such as 1–3 with N–C bonds in the range 1.612(2)–1.6536(14) Å,1–3 significantly longer than a typical unperturbed N–C bond of ca. 1.47 Å. We have also recently reported the structures of two salts of the aldehyde 4 in which protonation on oxygen has been accompanied by bond formation between the peri dimethylamino and aldehyde groups to form cation 5 which has N–C bond lengths in the range 1.624(4)–1.638(2) Å.3 A bond length as long as 1.71 Å has been reported in the tetrafluoroborate salt of the sterically hindered cation 6.4 We decided to investigate the structures of the salts formed from a series of naphthyl-8-ketones bearing a peri-dimethylamino group, 7–11, in which the electronic properties and size of the ketone group is varied: from methyl to isopropyl to t-butyl, and also phenyl and trifluoromethyl. It was envisaged that protonation would take place on O with formation of a N–C bond between peri groups to give structures such as 12, with the length of the new bond controlled by steric interactions between substituents at the terminal atoms of the bond. The resultant salts were studied by single X-ray crystallography, natural abundance solid state 1H, 13C and 15N MAS NMR spectrometry and DFT calculations of the relevant corresponding NMR chemical shifts. The crystal structures of the five ketones 7–11 were also determined to see how the electronic and steric properties of the acyl group affected the interaction with the peri-dimethylamino group, following on from the original crystal structure determination of the methyl ketone 75 by Dunitz et al., which demonstrated that the dimethylamino group was oriented so that its lone pair of electrons could interact with the carbonyl group.

Discussion
Structures of ketones
The ketones 7–11 were prepared by peri-lithiation of 1-dimethylaminonaphthalene followed by treatment with the appropriate anhydride. For most cases, crystals of ketones were grown by slow evaporation of solutions and crystal structures measured by X-ray diffraction at low temperature. A very fine needle shaped crystal of the low melting isopropyl ketone 8 was grown by sublimation and was measured using synchrotron X-radiation. Details of the ketones' molecular geometries are summarised in Table 1, their molecular conformations are shown in Fig. 1 and their crystal packings provided in the ESI.† The molecular conformations of the five ketones are very similar, with each dimethylamino group adopting pyramidal geometry and the lone pair directed towards the carbonyl carbon of the ketone (Fig. 1). As the ketone group increases in size on going from methyl to isopropyl to t-butyl ketone, the N⋯C separation increases from 2.5290(13) in 7 to 2.613(7) in 8 to 2.6859(13) and 2.6649(14) Å in 9. In the phenyl ketone 10 the N⋯C separation is similar to that in the methyl ketone, while the shortest N⋯C separation (2.424(2) Å) is in the most electron-deficient ketone which carries a trifluoromethyl group. The variation in this separation is achieved primarily by a change in the in-plane displacement of the ketone group. However, there is also an increasing displacement of the peri groups to either side of the naphthalene plane as the inter-group separation increases (Table 1).|
|
||||
|---|---|---|---|---|
| Compound, R | a/Å | b/Å | ϕ/° | τ 1/°b |
| 7, CH3 | 2.5290(13) | 1.2196(13) | 104.40(7) | 80.48(13) |
| 8, CHMe2 | 2.613(7) | 1.212(4) | 106.13(19) | 92.1(2) |
| 9, CMe3 | 2.6859(13) | 1.2193(12) | 101.58(6) | 100.75(12) |
| 2.6649(14) | 1.2165(12) | 100.97(8) | 84.18(12) | |
| 10, Ph | 2.5376(19) | 1.2200(17) | 107.10(11) | 83.70(19) |
| 11, CF3 | 2.424(2) | 1.213(2) | 107.26(12) | 80.2(2) |

| Compound, R | τ 2/°c | ΔC/Åd | Θ/°e | ΔNp/Å N, Cf |
|---|---|---|---|---|
|
a Ranges for angles: α: 122.39(4)–124.23(16)°; β: 115.35(16)–117.66(9)°; γ: 121.15(13)–123.56(9)°; δ: 121.51(15)–124.83(8)°; ε: 115.49(9)–118.13(17)°, further details in ESI.
b
τ
1: torsion: C2–C1–N1–C(H3), cis to CO.
c
τ
2: torsion: C2–C1–N1–C(H3), trans to CO.
d ΔC: deviation of C(O) atom from the plane of its three neighbouring atoms towards N(Me2).
e
Θ: angle between N⋯C vector and theoretical N lone pair axis.
f ΔNp: deviation of peri atoms, N(Me2) and C(O), from naphthalene ring's best plane.
|
||||
| 7, CH3 | −47.61(13) | 0.0942(10) | 16.4 | +0.108(1), −0.093(1) |
| 8, CHMe2 | −35.6(3) | 0.075(3) | 22.9 | +0.202(2), −0.159(3) |
| 9, CMe3 | −25.26(14) | 0.0909(9) | 28.1 | +0.247(1), −0.308(1) |
| −40.81(13) | 0.0942(11) | 27.0 | +0.003(1), −0.021(1) | |
| 10, Ph | −46.3(2) | 0.0722(14) | 17.1 | +0.151(1), −0.113(1) |
| 11, CF3 | −45.3(2) | 0.1252(16) | 20.4 | +0.049(1), +0.010(2) |
 | ||
| Fig. 1 Molecular structures of the ketones 7–11, showing the increase in displacements from the naphthalene plane as the Me2N⋯CO separation increases (top row), but the second molecule of 9 (bottom, left) shows minimal such displacements. The shortest Me2N⋯CO separation is in the trifluoromethyl ketone 11. | ||
Interestingly, for the t-butyl derivative, there are two independent molecules with similar Me2N⋯CO separations, but while one molecule shows the largest out of plane displacements for the N and C peri-substituent atoms in this series, the other molecule shows only minimal displacements. The similar N⋯C separation in the more planar form is realised by an increased outward splaying of the dimethylamino group and a widening of the external angle at the fusion between the two rings of the naphthalene framework. The overall optimisation of crystal packing in ketone 9 has led to the preference for adopting these two conformations, but illustrates the geometric consequences for the in-plane alignment of the two peri bonds. In all cases the carbonyl carbon atom is slightly pyramidalised with the carbon atom displaced towards the amino nitrogen atom by 0.072–0.125 Å (Table 1). While there is no clear trend, it is notable that the largest displacement is for the shortest N⋯C separation in the trifluoromethyl ketone. The dimethylamino groups are oriented such that the theoretical axes of their nitrogen atoms' lone pairs lie at 16–28° to their respective N⋯C vectors (Table 1), and for the two molecules of the t-butyl derivative the groups have maintained almost the same relative orientations. In contrast, when the substituents are oriented para rather than peri as in 13, an isomer of ketone 11, structural studies show strong interaction between substituents through the aromatic ring, and not through space. Further details are in the ESI.†
Preparation of salts of ketones 7–11
Treatment of ether solutions of ketones 7–11 with an ether solution of one of the acids, HCl, HBF4 or CF3SO3H, led to the precipitation of the corresponding salts. For each ketone, the most crystalline salts were studied by X-ray diffraction and natural abundance SS-NMR. The salts studied were: for 7 the hydrated chloride salt and the triflate salt, for 8 the BF4 salt, for 9 the triflate salt, for 10 the BF4 salt and for 11 a hydrated triflate salt. X-ray diffraction studies showed that the salts from ketones 7, 8 and 11 involved addition of the dimethylamino group to the protonated carbonyl group, whereas the salts from the t-butyl and phenyl ketones 9 and 10 are protonated on the dimethylamino group and there is a hydrogen bond between the N–H of the cation and the pi surface of the carbonyl group. The 15N SS-NMR studies identified a distinct difference of ca. 50 ppm in the chemical shift for the two series of compounds. The 13C SS-NMR spectra allowed identification of the carbonyl carbon resonance in the latter series of salts. The results are described in more detail below.Crystal structures of O-protonated salts of cations 7-H+, 8-H+ and 11-H+
The crystal structure of the four salts 7-H+·Cl−·H2O, 7-H+·CF3SO3−, 8-H+·BF4− and 11-H+·CF3SO3−·H2O involve formation of a bond between the peri groups and hydrogen bonding of the counterion or a molecule of water to the cation's hydroxyl group. The structures are shown in Fig. 2–4, and selected molecular geometry is given in Table 2. In the hydrated chloride salt of cation 7-H+ the chloride ion forms a hydrogen bond to the hydroxyl group of a cation (OH⋯Cl−: 2.17(3) Å), and chlorides are linked together with bridging water molecules (OH⋯Cl−: 2.41 & 2.48 Å) to form Cl−⋯H–O–H⋯Cl− chains along the c axis (Fig. 2). In contrast, the salts of 7-H+ with triflate and 8-H+ with tetrafluoroborate are not hydrated and there are just OH⋯[O–SO2CF3]− and OH⋯[F-BF3]− hydrogen bonds between cation and anion (1.80(8) and 1.82(3) Å respectively) (Fig. 2). In the triflate salt of 11-H+ a water molecule makes a hydrogen bond to the cation's hydroxyl group (OH⋯OH2: 1.68(2) Å), and then two such water molecules and two triflate ions form a cyclic hydrogen bonded motif (H–O–H⋯O–SO2CF3: 1.893(5) and 1.946(6) Å) (Fig. 3). Interestingly, the crystal packing segregates the trifluoromethyl groups from cations and anions away from the rest of the hydrocarbon based moieties (Fig. 4). The bond between the peri groups for the methyl and isopropyl ketone salts are remarkably long: 1.669(2), 1.6709(6) and 1.662(2) Å, and longer than for the aldehyde salt 5 (1.624(4)–1.638(2) Å). The salt from the more electron deficient trifluoromethyl ketone has the shortest N–C bond, 1.6375(18) Å.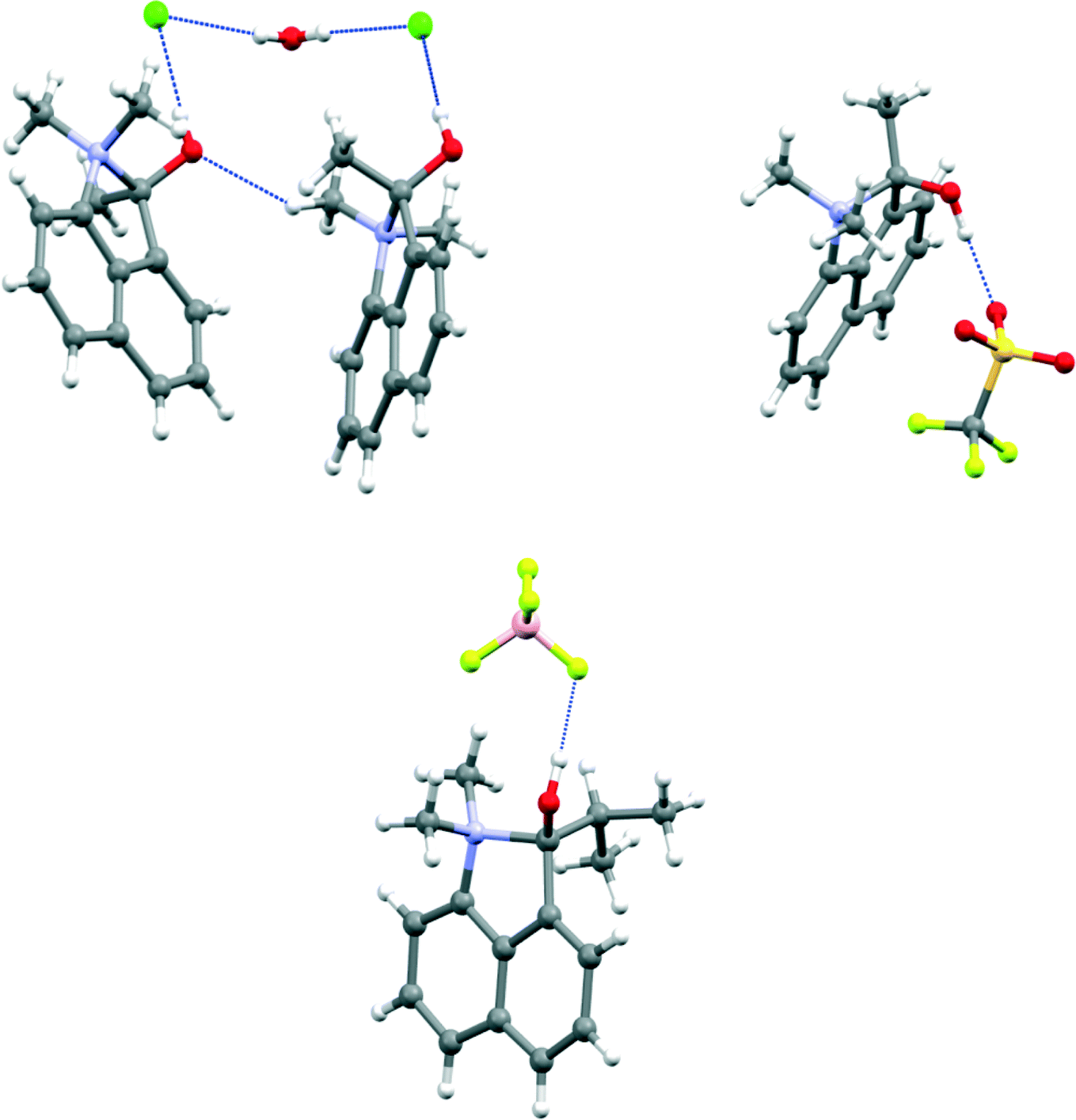 | ||
| Fig. 2 Molecular structures of (a) the hydrated chloride salt of the O-protonated methyl ketone 7-H+ showing the involvement of anions and water molecules in the hydrogen bonding pattern (left), (b) the triflate salt of 7-H+ (right) and (c) the tetrafluoroborate salt of the O-protonated isopropyl ketone 8-H+ (below). | ||
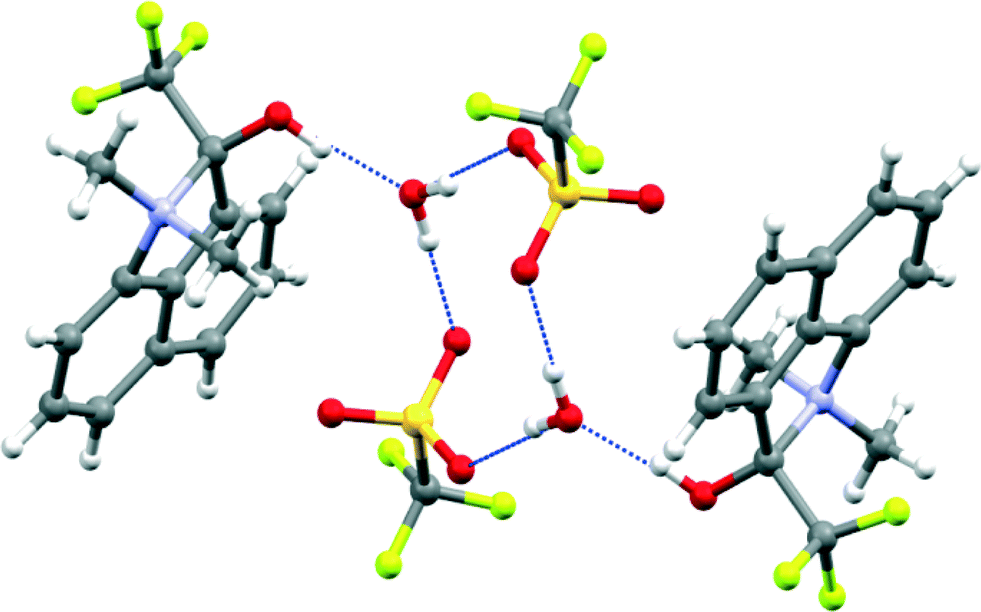 | ||
| Fig. 3 Hydrogen bonding linking together two cations with a cluster of two triflate anions and two water molecules in the crystal structure of 11-H+·triflate·H2O. | ||
 | ||
| Fig. 4 Space-filling view of the crystal packing of 11-H+·triflate·H2O viewed down the a axis, showing the segregation of the fluorous residues. | ||
|
|
||||
|---|---|---|---|---|
| Salt | a/Å | b/Å | N–Me/Å | c/Å |
| a Ranges for angles: α: 128.6(4)–129.49(17)°; β: 107.92(16)–109.3(3)°; γ: 113.6(4)–113.91(15)°; δ: 108.91(15)–109.95(14)°; ε: 130.39(16)–131.33(13)°, further details in ESI. | ||||
| 7-H+·Cl−·H2O | 1.669(2) | 1.364(2) | 1.497(2) & 1.503(2) | 1.513(2) |
| 7-H+·CF3SO3− | 1.670(6) | 1.415(6) | 1.490(5) & 1.511(6) | 1.482(6) |
| 8-H+·BF4− | 1.662(2) | 1.367(2) | 1.507(2) & 1.497(2) | 1.541(3) |
| 11-H+·CF3SO3−·H2O | 1.6375(18) | 1.3476(13) | 1.5037(18) & 1.5124(18) | 1.5452(19) |

The room temperature crystal structure of the trifluoroacetate salt of methyl ketone 7 has been reported,6 and shows the same type of O-protonated cation structure with a bond length of 1.679 Å between peri groups. Increased steric pressure between the N-methyl and C-isopropyl groups in 8-H+ compared to the corresponding interaction between two methyl groups in 7-H+ is relieved in part by an increase of the torsion about the peri-bond (from 23.6° to 30.8°) and by the greater length of the C–CHMe2 bond of 1.541(3) Å compared to 1.513(2) Å in 7-H+.
In 11-H+ the shorter bond between peri groups is accompanied by a long C–CF3 bond of 1.5452(19) Å so that the shortest intramolecular H⋯F contacts with an N-methyl group are 2.36 and 2.51 Å. For the ketones with t-butyl and phenyl groups, rather than form cyclised materials, protonation on the N atom is preferred. For the former, the N–C bond which would be formed in O-protonation mode would have to be longer and weaker than in 7-H+ and 8-H+, due to steric pressure from the t-butyl group, while for the latter, the carbonyl group is stabilised by conjugation with the phenyl group.
Crystal structures of N-protonated salts of cations 9-H+ and 10-H+
The crystal structures of salts 9-H+·CF3SO3− and 10-H+·BF4− show a cation formed by N-protonation with a hydrogen bond to the peri ketone group. There are two crystallographically unique cations and anions in the t-butyl ketone salt. The structures are shown in Fig. 5, with details of their geometries in Table 3. The relative orientations of the peri groups in all three cations are similar, and the hydrogen bonds between the protonated dimethylamino group and the carbonyl O atom lie in the range 1.77(2)–1.80(3) Å. The bond angle at the H atom lies in the narrow range 156(2)–159(2)°, and the angle at the O atom is in the range 98.6(8)–103.6(8)°. This is achieved by the carbonyl and N–H groups being directed to the same side of the naphthalene ring and then tilting towards each other. The two peri groups have to be splayed apart in the naphthalene plane, though this is limited by interactions with the ortho H atoms. The ketone is splayed out further than the dimethylammonium group; for the two unique molecules of 9-H+ the t-butyl group makes two (butyl)H⋯ortho H contacts as short as 2.08–2.11 Å, and one N-methyl group makes a H⋯H contact with its ortho H atom also in the same range. In 10-H+ the in-plane steric pressure from ortho-H atoms in less and only the N-methyl group makes a short H⋯H contact (2.16 Å). The naphthalene ring system is not strongly distorted from planarity and the peri substituent N and C atoms lie close to this plane; the largest deviations are for the peri C atoms in the two cations of 9-H+ (by 0.2–0.3 Å) which act to reduce steric pressure between the t-butyl group and an ortho H atom. | ||
| Fig. 5 Two views of the molecular structure and intramolecular hydrogen bond of (a) one of the two independent cations of 9-H+ (top), (b) the cation 10-H+ (bottom). | ||
|
|
||||
|---|---|---|---|---|
| Salt | CO/Å |
O⋯N/Å | NH⋯O/Å | N–H⋯O/° |
| 9-H+·CF3SO3− | 1.226(2) | 2.641(2) | 1.77(2) | 159(2) |
| 10-H+·BF4− | 1.233(2) | 2.658(3) | 1.78(3) | 157(2) |
| 1.229(3) | 2.660(3) | 1.80(3) | 156(2) | |
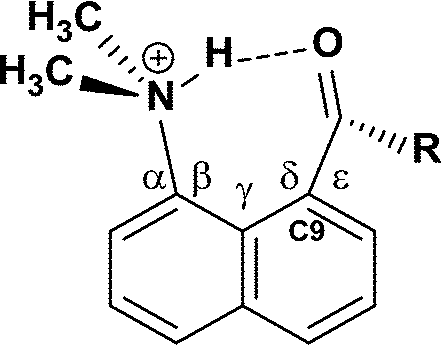
With regard to the hydrogen bonding in these two salts, the restrictions imposed by the peri groups lead to the H atom lying to one side of the carbonyl group, by 1.35–1.50 Å, and to the C(naphthalene)–CO⋯H torsion angle lying in the range 53.2(8)–61.5(8)°. This is very different from the preferred hydrogen bonding geometry to a carbonyl group in which the H atom lies in the carbonyl plane, out beyond the O atom, and with a CO⋯H angle in the range 110–180°.7 Thus, these structures provide a very nice model for hydrogen bonding to the underside of a carbonyl group, i.e. to the pi electron density rather than lone pair density. This is of particular interest given the proposal that hydrogen bonding to the pi surface of a carbonyl group promotes the kinetics of deprotonation alpha to the carbonyl group in various enzyme active sites.8 Hydrogen bonding from ammonium and substituted ammonium ions to the pi faces of alkenes, alkynes and benzene rings is well known.9,10 The energy of interaction of a N(+)–H bond with ethene has been estimated at ca. 10 kcal mol–1.10
In the solution NMR spectra for the salts of 9-H+ and 10-H+ the N–H group gives signals at δH 11.15 and 11.27 ppm respectively, and the carbonyl carbon atoms resonate at δC 221.3 and 205.0 ppm which are ca. 8 ppm downfield from the positions observed in the parent ketones (9: 213.0 and 10: 193.5 ppm) consistent with these salts retaining their N-protonated structures in solution. Solution NMR studies of the salts of 7-H+, 8-H+ and 11-H+ suggest that the closed ring structure is maintained in solution for the salts of the methyl and trifluoromethyl ketones, 7-H+·Cl (in CD3OD) and 7-H+·triflate and 11-H+·triflate (in (CD3)2CO), with the carbon atom attached to the hydroxyl and ammonium groups appearing at δC 124.8 and 122.5 ppm in the salts of the methyl ketone. In contrast, the salt of the more hindered isopropyl ketone exists in CD3CN as a 6.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of N-protonated to O-protonated forms, i.e. favouring the form not observed in the solid state, with the major species exhibiting a N–H resonance at δH 12.45 ppm and a carbonyl carbon resonance at δC 217.3 ppm. The balance between the two structural forms, in this case at least, is fine and dependent on the particular external environment, with the form observed in the solid state controlled by the relative stabilities of the crystal packing arrangements.
:1 mixture of N-protonated to O-protonated forms, i.e. favouring the form not observed in the solid state, with the major species exhibiting a N–H resonance at δH 12.45 ppm and a carbonyl carbon resonance at δC 217.3 ppm. The balance between the two structural forms, in this case at least, is fine and dependent on the particular external environment, with the form observed in the solid state controlled by the relative stabilities of the crystal packing arrangements.
Solid state NMR studies
The two sets of salts whose X-ray structures showed either ring formation with O-protonation or just N-protonation were studied by solid state 1H, 13C and 15N MAS NMR, to establish a method for determining between the two species, since as observed for 8-H+·BF4 the structure adopted in solution may be different. The measured 15N data are particularly diagnostic in discriminating between the two types of structures. The 15N CPMAS NMR data are shown in Fig. 6 and Table 4, and it can be observed that the 15N δiso values are highly correlated around two regions: δiso −334.6 and −334.5 ppm (for 9-H+·CF3SO3−, 10-H+·BF4−, respectively) and the more deshielded region of δiso −284.0 to −285.6 ppm (for 7-H+·Cl−·H2O, 7-H+·CF3SO3− and 11-H+·CF3SO3−·H2O). This observation unambiguously demonstrates the existence of two distinct N environments within this series. The chemical shifts of the latter group of salts is similar to that of the cation 5 formed by intramolecular attack of the dimethylamino group on a protonated aldehyde group (δiso −290 ppm)3 also determined by 15N MAS NMR methods. Previous studies have reported that solid trimethylammonium chloride, which exhibits moderately strong N–H⋯Cl hydrogen bonding, has a 15N shift of δiso −335.7 ppm for the (CH3)3NH+ cation.11 This has a comparable N environment to that of the N-protonated cations in 9-H+·CF3SO3−, 10-H+·BF4−, and this is reflected by the similar 15N δiso values (with small variations in δiso evident due to the difference in the electronegativity of the hydrogen bond acceptor). In contrast, the considerable downfield shift observed in the cations 7-H+·Cl−·H2O, 7-H+·CF3SO3− and 11-H+·CF3SO3−·H2O is due primarily to the electron withdrawing effect of the OH group. For comparison, the reported 15N isotropic chemical shift for solid tetramethylammonium iodide is δiso −342.2,12 but even for a neutral N atom in 2,2-dimethyl-1,3-oxazolidine, which contains an α-O atom, the 15N chemical shift occurs at δiso −305–−316 ppm.13 The additional positive charge of cations 7-H+ and 11-H+ contributes to their further downfield shifts. Interestingly, the 15N chemical shifts of cations 7-H+ and 11-H+ are closer to that in N,N-dimethylacetamide (δiso(solution) −282.2 ppm)14 in which the N lone pair is partially donated to a carbonyl group.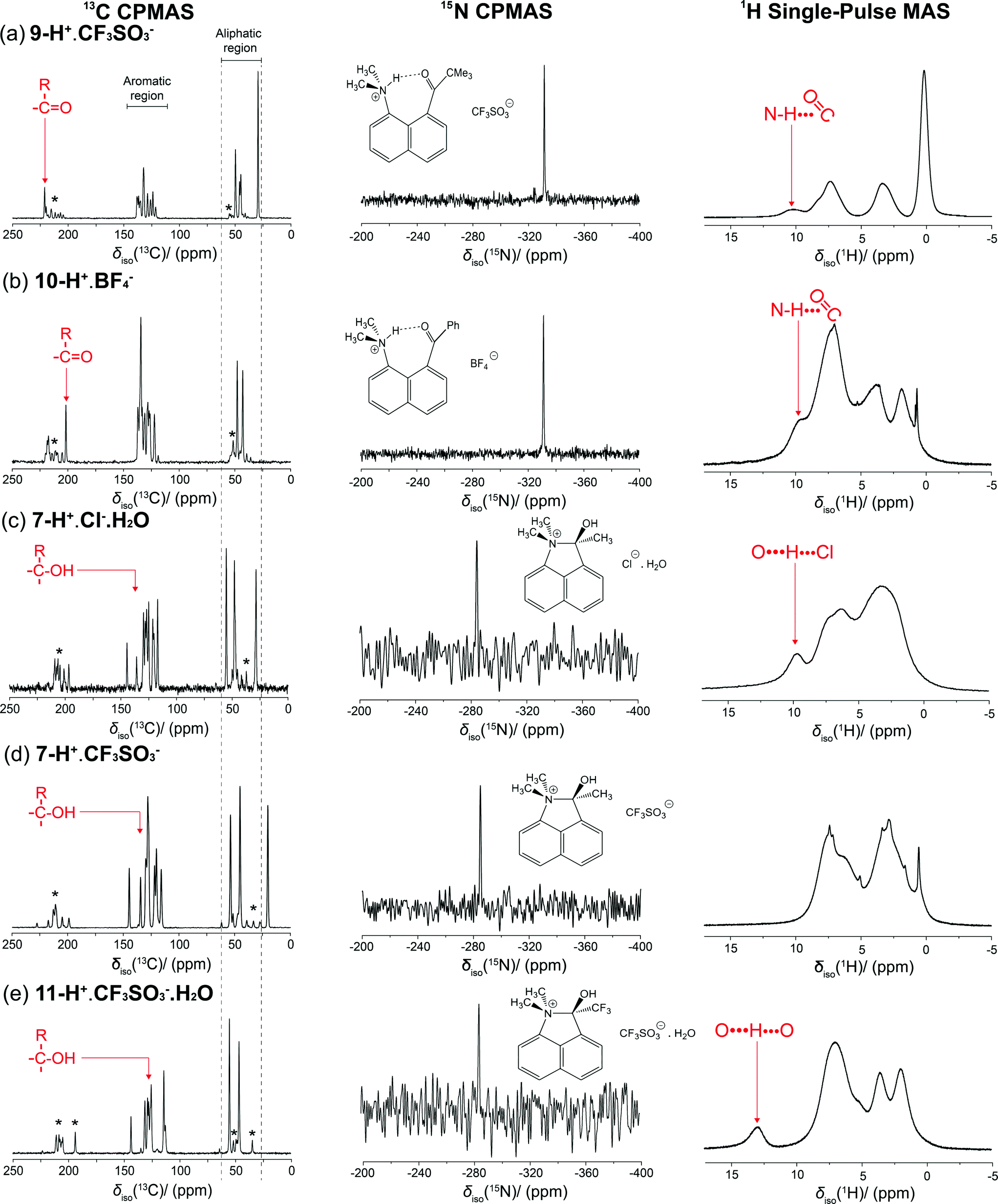 | ||
| Fig. 6 13C, 15N and 1H CPMAS NMR spectra for salts 9-H+·CF3SO3− and 10-H+·BF4− (upper) and the salts 7-H+·Cl−·H2O, 7-H+·CF3SO3− and 11-H+·CF3SO3−·H2O (lower). * represent the spinning sidebands. | ||
| 15N | Experiment δiso,mas (ppm)(±0.5) | CASTEP/PBE δiso (ppm) | CASTEP/SEDC δiso (ppm) |
|---|---|---|---|
| 9-H+·CF3SO3− | −334.7 | −344.9 | −344.3 |
| 10-H+·BF4− | −334.5 | −345.2 | −345.0 |
| 7-H+·Cl−·H2O | −284.0 | −290.3 | −288.9 |
| 7-H+·CF3SO3− | −284.8 | −289.9 | −288.0 |
| 11-H+·CF3SO3−·H2O | −285.6 | −288.3 | −286.0 |
| 13C | Experiment δiso,mas (ppm)(±0.5) | CASTEP/PBE δiso (ppm) | CASTEP/SEDC δiso (ppm) |
|---|---|---|---|
| a Tentative assignments based on the predicted shifts obtained from the GIPAW CASTEP calculation. | |||
| 9-H+·CF3SO3− | 201.3 | 205.3 | 206.5 |
| 10-H+·BF4− | 221.2 | 229.8 | 230.2 |
| 7-H+·Cl−·H2O | 136.0a | 136.6 | 138.3 |
| 7-H+·CF3SO3− | 134.8a | 136.3 | 139.8 |
| 11-H+·CF3SO3−·H2O | 127.7a | 127.2 | 130.0 |
| 1H | Experiment δiso,mas (ppm)(±0.5) | CASTEP/PBE δiso (ppm) | CASTEP/SEDC δiso (ppm) |
|---|---|---|---|
| 9-H+·CF3SO3− | 9.6 | 10.3 | 9.9 |
| 10-H+·BF4− | 10.2 | 10.7 | 10.6 |
| 7-H+·Cl−·H2O | 10.1 | 9.9 | 9.9 |
| 7-H+·CF3SO3− | 7.3 | 7.9 | 7.9 |
| 11-H+·CF3SO3−·H2O | 12.9 | 15.1 | 15.5 |
The 15N chemical shift parameters calculated via the GIPAW approach (see Table 4) closely mirror this observed demarcation in the δiso values according to the structural motif describing each system. The crystal structure data for 9-H+·CF3SO3− and 10-H+·BF4− suggest that there is protonation of the N site in these structures, and the implicit hydrogen bond remains after allowing the crystal structures to relax. This provides supporting evidence that the π-hydrogen bond arrangement in these particular systems is stable. As observed in Table 4, although the data trends are unambiguously reflected in the CASTEP data, the calculated 15N δiso values consistently represent a high field/lower frequency overestimation even after the dispersion correction is applied. The combined structure relaxation/dispersion correction (SEDC) approach only marginally improves the correlation between the measured and calculated 15N δiso data, with the shift differences for the hydrogen bonded systems being typically Δδiso ~10 ppm, whereas for the directly bonded systems it amounts to Δδiso ~0.5–5 ppm.
The 13C CPMAS NMR data are also displayed in Fig. 6 with the measured 13C δiso values summarized in Table 4. For the internally hydrogen bonded 9-H+·CF3SO3− and 10-H+·BF4− systems the corresponding ketone moiety is evidenced by the 13C shifts at δiso 221.2 and 201.3 ppm, respectively, which are very similar to those observed in their solution spectra. The large 13C shift difference of Δδiso ~20 ppm exhibited between the ketone C atoms reflects the differences in electron donation and bond strength between the pendant Ph and C(CH3)3 groups and the CO moiety. Of note is that these chemical shifts are 8.2 and 7.7 ppm downfield, respectively, from the carbonyl carbon signals observed in the solution spectra of the unprotonated ketones, suggesting an effect from the formation of the hydrogen bond to a positively charged group.
In contrast, for cations in 7-H+·Cl−·H2O, 7-H+·CF3SO3− and 11-H+·CF3SO3−·H2O the addition of the dimethylamino group to the protonated ketone group leads to a long N+–C(OH)R bond where the resultant quaternary C has 13C shifts in the range δiso 127–136 ppm; this phenomenon has been outlined by previous studies.3,15–17 In a similar fashion to the 15N study, the corresponding DFT calculated 13C δiso parameters corroborate the demarcation between the hydrogen bonded and directly bonded systems; however, in contrast to the 15N study, the CASTEP calculations supported by only the PBE functional give a superior correlation with the experimentally measured 13C CPMAS data in comparison to those undertaken with the SEDC scheme. From Table 4 it is evident that the SEDC scheme causes a divergence away from the PBE supported (and experimental) shifts by a factor of δiso ~1–3 ppm.
Previous computational and experimental studies of carbonyl and (in particular) carboxylic systems have shown that a shift to higher δiso values has largely been attributed to a strong dependence of the δ22 component of the second rank chemical shift anisotropy (CSA) tensor.16–19 In the principal axis frame of this tensor the δ22 component is aligned along the CO bond and is highly correlated to the overall strength of the hydrogen bond.18 It is worth noting that the GIPAW DFT calculations underpinning this work predict a similar alignment of the δ22 component of the CSA tensor along the CO bond in the ketone moiety for the 9-H+·CF3SO3− and 10-H+·BF4− salts suggesting that the large δiso values (δiso > 200 ppm) originate from the dominant contribution of the δ22 component of the CSA tensor; this phenomenon is particularly prevalent for the 10-H+·BF4− system. These DFT calculations also demonstrate that there is very little variation in the magnitude of δ33, as similarly observed in other studies.17–19
The 1H MAS NMR data acquired under single pulse conditions are also shown in Fig. 6. Generally, the 1H δiso value is a very direct entity for identifying and gauging the strength of hydrogen bonding as the extent to which the proton is deshielded correlates directly with the downfield shift in the 1H resonance. The 1H MAS spectra for 9-H+·CF3SO3− and 10-H+·BF4− show resonances located at δiso 9.5 and 10.3 ppm, respectively, that can be considered as representing weak hydrogen bonding. In solution the corresponding signals are δH 11.1–11.3 ppm. For the cyclised cations studied, only the hydrated 11-H+·CF3SO3−·H2O salt exhibits a characteristic downfield 1H signal at δiso ~13 ppm. This salt contains three hydrogen bonds, two between triflates and water (1.89 and 1.95 Å) and a shorter one (1.68 Å) between the cation's OH group and the water. For the hydrated chloride salt 7-H+·Cl−·H2O, with a OH–Cl− hydrogen bond and two hydrogen bonds between chloride and water, a resonance is observed at ~10 ppm, while for the corresponding triflate salt the signal is probably obscured by aromatic hydrogen signals. In all cases studied here, none correspond to particularly strong hydrogen bonding for which the 1H MAS NMR signal would be expected to be in the δiso 16–22 ppm range.17,19,20
From Table 4 a comparison of the experimental and DFT calculated 1H δiso values shows that CASTEP has again overestimated these shifts on the downfield side, even after the Semi-Empirical Dispersion Correction (SEDC) scheme is applied. As the extent of the downfield shift correlates with the strength of the hydrogen bond CASTEP appears to be consistently predicting slightly stronger hydrogen bonds than those observed experimentally. Conventionally, the greatest errors in the δiso calculations are found in those systems where hydrogen bonding has a more dominant role in the structural formation, which is consistent with CASTEP's inexact treatment of van der Waals' forces. However, given that the application of the SEDC scheme does not improve the results significantly this error is likely to be due to a systematic error originating from the condition of 0 K imposed on atomic positions for the calculations. Unaccounted for motional atomic effects due to thermal fluctuations (particularly of the hydrogen bonding proton) could be observed as an average nuclear environment with a weaker hydrogen bond in the NMR experiment.
Conclusion
X-ray crystallography and SS-NMR provide a powerful combination of techniques for studying the protonated salts of a series of 1-dimethylamino-naphthalenes with peri ketone groups, and have provided interesting structural models for incomplete bond formation between the two groups and for hydrogen bonding between a protonated dimethylamino group and the pi face of the carbonyl group. Three of the salts provide some of the longest N–C bonds known, and further insight into these incompletely formed N–C bonds will come from charge density measurements. The hydrogen bonding to the side of a carbonyl group, rather than to its terminal lone pairs, is an aspect which is not often considered in crystal engineering. It is of interest to see whether formation of attractive interactions or covalent bonds between functional groups could be used as a method for organising molecules in the solid-state.Experimental
Preparation of ketones
n-BuLi (18.7 ml, 2.5 M in hexane solution) was added to a stirred solution of 1-dimethylaminonaphthalene (2.00 g, 11.7 mmol) in dry ether (30 ml) under nitrogen at room temperature and left for 4 days during which time a yellow precipitate formed. The solution was carefully removed, and the yellow solid washed several times with dry ether under nitrogen. The solid was suspended in dry ether (30 ml), cooled to −78 °C and a solution of the appropriate acid anhydride (15 mmol) in dry ether (20 ml) added. The mixture was allowed to gradually warm to room temperature over 12 h, after which it was treated with methanol (30 ml) and the solvent evaporated. The residue was extracted with dichloromethane (2 × 50 ml), and the combined extracts washed with water (2 × 50 ml) and brine (1 × 50 ml) and then dried with anhydrous magnesium sulphate. Evaporation gave the crude product which was purified further by column chromatography with cyclohexane–ethyl acetate mixtures. Full characterisations of the products 7–11 are given in the ESI.† Products 8, 9 and 13 have been described before.21,22Preparation of salts
A stirred solution of the ketone (2 mmol) in dry ether (20 ml) under nitrogen was treated with a ca. 1 M solution of the acid (HCl, HBF4 or CF3SO3H) (2.5 mmol) in dry ether at room temperature to give a precipitate. After stirring for 30 min, the solid was filtered off, washed with ether and dried under vacuum to give the salt. Full characterisations of the salts 7-H+·Cl−·H2O, 7-H+·CF3SO3−, 8-H+·BF4−, 9-H+·CF3SO3−, 10-H+·BF4− and 11-H+·CF3SO3−·H2O are given in the ESI.†X-ray crystallography
Low temperature (100–150 K) X-ray diffraction data (Mo Kα) for compounds 7, 9, 9-H+·CF3SO3− and 11-H+·CF3SO3− were collected at the UK National Crystallography Centre,23 Southampton University on a Rigaku AFC12 diffractometer equipped with enhanced sensitivity (HG) Saturn724+ CCD detector mounted at the window of an FR-E+ SuperBright rotating anode generator (Mo Kα, λ = 0.71075 Å) with VHF Varimax optics (70 μm focus) using Crystal Clear software24 for data collection and reduction. For compounds 10, 11, 14, 7-H+·Cl−·H2O, 7-H+·CF3SO3−, 8-H+·BF4−, 10-H+·BF4− data were measured on an Agilent Xcalibur diffractometer equipped with a Sapphire detector at Nottingham Trent University using the CrysAlis-Pro software package.25 Diffraction data for 8 was performed with the use of synchrotron X-rays at Diamond Light Source UK,23 beamline I19 (λ = 0.6889 Å) on a Crystal Logic diffractometer and Rigaku Saturn 724+ detector equipped with an Oxford Cryosystems Cryostream and using Crystal Clear software.24 Structures were solved are refined using the SHELXS and SHELXL suite of programs26 using the XSEED interface.27 Molecular illustrations were made with Mercury.28 Crystal data are provided in Tables 5 and 6. Data is deposited at the Cambridge Crystallographic Data Centre with code numbers CCDC-984760–984765 and 984801–984806.| Parameters | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|
| a A room temperature structure has been reported.5 | |||||
| Formula | C14H15NO | C16H19NO | C17H21NO | C19H17NO | C14H12F3NO |
| M r | 213.27 | 241.32 | 255.36 | 275.34 | 267.25 |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/n | Pna21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | P21/n |
| a/Å | 8.3814(2) | 9.37(2) | 10.4848(6) | 9.0485(4) | 12.5626(8) |
| b/Å | 10.2345(2) | 9.34(2), | 12.0004(7) | 11.9670(5) | 7.4932(4) |
| c/Å | 13.8672(9) | 15.72(4) | 12.0396(8) | 14.4409(6) | 13.1887(9) |
| α/° | 90 | 90 | 73.685(5) | 90 | 90 |
| β/° | 105.897(8) | 90 | 85.342(6) | 107.512(5) | 98.319(6) |
| γ/° | 90 | 90 | 83.541(6) | 90 | 90 |
| V/Å3 | 1144.03(8) | 1376(5) | 1442.66(15) | 1491.24(11) | 1228.44(13) |
| Z | 4 | 4 | 4 | 4 | 4 |
| D calcd/g cm−3 | 1.24 | 1.16 | 1.18 | 1.23 | 1.44 |
| μ/mm−1 | 0.078 | 0.072 | 0.072 | 0.075 | 0.122 |
| T/K | 100 | 100 | 100 | 150 | 150 |
| Reflns, Rint | 2586, 0.024 | 3086, 0.056 | 6618, 0.029 | 3429, 0.026 | 2795, 0.034 |
| Reflns, with F2 > 2σ | 2297 | 2863 | 5418 | 2641 | 1992 |
| R(F, F2 > 2σ), wR(all) | 0.038, 0.100 | 0.048, 0.135 | 0.039, 0.106 | 0.056, 0.114 | 0.052, 0.102 |
| Crystals from | Ether | Sublimation in vacuo | Acetone | Ether | Acetone |
| Parameters | 7-H+·Cl−·H2Oa | 7-H+·CF3SO3–b | 8-H+·BF4− | 9-H+·CF3SO3–c | 10-H+·BF4− | 11-H+·CF3SO3−·H2O |
|---|---|---|---|---|---|---|
| a The positions of the water molecule's hydrogen atoms were constrained to be 0.85 Å from the O atom, and 1.34 Å apart. b There is some unresolved disorder in the triflate anion. c Crystal was a merohedral twin, twin law (1 0 1, 0 −1 0, 0 0 −1), refined BASF = 0.47. | ||||||
| Formula | C14H16NO·Cl·H2O | C14H16NO·CF3SO3 | C16H20NO·BF4 | C17H22NO·CF3SO3 | C19H18NO·BF4 | C14H13F3NO·CF3SO3·H2O |
| M r | 267.74 | 363.35 | 329.14 | 405.43 | 363.15 | 435.34 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/c | P21/c | P21/n | P21/c | Pbca | P21/c |
| a/Å | 11.3796(3) | 13.8475(10) | 6.2039(2) | 21.072(4) | 13.6452(5) | 10.3782(7) |
| b/Å | 10.5436(2) | 7.7466(4) | 10.6675(3) | 11.828(3) | 13.5547(4) | 17.4219(8) |
| c/Å | 11.0577(3) | 15.6785(9) | 23.5777(8) | 16.397(4) | 18.2545(8) | 9.9913(4) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 |
| β/° | 97.604(2) | 106.669(6) | 97.541(3) | 112.765(14) | 90 | 103.321(7) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 1315.06(6) | 1611.17(17) | 1546.88(8) | 3768.5(15) | 3376.3(2) | 1757.90(16) |
| Z | 4 | 4 | 4 | 8 | 8 | 4 |
| D calcd/g cm−3 | 1.35 | 1.49 | 1.41 | 1.43 | 1.43 | 1.65 |
| μ/mm−1 | 0.284 | 0.253 | 0.12 | 0.22 | 0.12 | 0.27 |
| T/K | 150 | 150 | 150 | 100 | 150 | 100 |
| Reflns, Rint | 3007, 0.023 | 3727, 0.042 | 3555, 0.024 | 8602, 0.024 | 3948, 0.031 | 4015, 0.027 |
| Reflns, with F2 > 2σ | 2500 | 2988 | 2809 | 7821 | 2983 | 3459 |
| R(F, F2 > 2σ), wR(all) | 0.042, 0.104 | 0.103, 0.225 | 0.058, 0.142 | 0.042, 0.088 | 0.077, 0.166 | 0.032, 0.078 |
| Crystals from | Acetonitrile | Acetonitrile | Acetonitrile | Acetone | Acetonitrile | Acetone |
1H, 13C and 15N MAS NMR measurements
Natural abundance solid state 13C and 15N MAS NMR measurements were performed at 14.1 and 11.7 T, respectively, using a Bruker Avance II+ −600 spectrometer (Larmor frequencies of ν0(1H) = 600.3 MHz and ν0(13C) = 150.9 MHz) and a Bruker Avance III-500 spectrometer (Larmor frequencies of ν0(1H) = 500.3 MHz and ν0(13C) = 50.7 MHz). All data were acquired at ambient temperatures. The 15N measurements were performed using a ramped 1H–15N cross-polarisation MAS (CPMAS) NMR experiment using a Bruker 3.2 mm triple channel (HXY) MAS probe operating at a spinning frequency of 12 kHz. The SPINAL-64 heteronuclear decoupling scheme was used during acquisition with a 1H decoupling field of ν1(1H) = 100.0 kHz being implemented. A minimum of 14000 transients were acquired for each spectrum, and an initial 1H π/2 pulse of 2.5 μs, a 1H–15N Hartmann–Hahn contact period of 1.0 ms and a recycle delay of 5.0 s were utilised in all 15N CPMAS NMR measurements. All 15N CPMAS NMR data were calibrated against the primary 15N reference of CH3NO2 in CHCl3via a secondary solid reference of 15N labelled histidine (three 15N shifts of δiso −333.1, −204.3 and −191.0 ppm), which was also used to establish the 1H–15N Hartmann–Hahn match condition. The corresponding 13C MAS NMR data were also acquired using a ramped 1H–13C CPMAS experiment and SPINAL-64 heteronuclear decoupling scheme during acquisition. A Bruker 4.0 mm dual channel (HX) MAS probe was used which implemented spinning frequencies of 8 and 12 kHz to clearly identify the sideband manifold defining the 13C chemical shift anisotropy. All 13C CPMAS data were calibrated against the primary 13C reference of TMS via a secondary solid reference of alanine (three 13C shifts of δiso 20.5, 51.0 and 177.8 ppm). A minimum of 8352 transients were acquired for each spectrum, and a 1H π/2 pulse width of 3 μs, a 1H–13C Hartmann–Hahn contact period of 1 ms and a recycle delay of 2 s were utilised for all 13C CPMAS NMR measurements. Additional 1H MAS NMR measurements were performed at 14.1 T and at ambient temperatures using a Bruker Avance II+ −600 spectrometer (Larmor frequencies of ν0(1H) = 600.3 MHz). A Bruker 2.5 mm dual channel (HX) MAS probe was used which delivered spinning frequencies of 30 kHz, and all 1H MAS NMR data were acquired using single pulse experiments. An excitation pulse of 2.5 μs and recycle delays of 10s were common to all measurements (although checks for slower relaxing H species were undertaken), and the reported 1H chemical shifts are directly referenced to the primary reference TMS.
Ab initio density functional theory calculations
Ab initio density functional calculations (DFT) calculations were performed using CASTEP 6.01,29 a DFT code that evokes the Kohn–Sham DFT formalism using a planewave description of the electronic wavefunction under a pseudopotential approximation. The pseudopotentials were generated on-the-fly using the Accelrys' Material Studio 5.5 software.30 Full details are supplied in the ESI.†Acknowledgements
We thank the EPSRC for grant (EP/E018203/1) from the Physical Organic Chemistry Initiative and for funding the UK National Crystallography Service including access to Synchrotron facilities at the Diamond Light Source, the EPSRC UK National Mass Spectrometry Facility for data, and the Chemical Database Service31 for access to the Cambridge Structural Database.32 We thank Nottingham Trent University for support for diffraction facilities, and the Erasmus program for supporting NM. JVH thanks EPSRC and the University of Warwick for partial funding of the solid state NMR infrastructure at Warwick, and acknowledges additional support for this infrastructure obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Projects 1 and 2), with support from Advantage West Midlands (AWM) and partial funding by the European Regional Development Fund (ERDF). JVH also acknowledges the facilities of HECToR, the UK's national high-performance computing service, which is provided by UoE HPCx Ltd. at the University of Edinburgh, Cray Inc. and NAG Ltd., and funded by the Office of Science and Technology through EPSRC's High End Computing Programme.References
- P. C. Bell and J. D. Wallis, Chem. Commun., 1999, 257–258 RSC.
- J. O'Leary, X. Formosa, W. Skranc and J. D. Wallis, Org. Biomol. Chem., 2005, 3, 3273–3283 Search PubMed.
- A. Lari, M. B. Pitak, S. J. Coles, G. J. Rees, S. P. Day, M. E. Smith, J. V. Hanna and J. D. Wallis, Org. Biomol. Chem., 2012, 10, 7763–7779 CAS.
- G. Dyker, M. Hagel, G. Henkel, M. Kockerling, C. Nather, S. Petersen and G. P. Schiemenz, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 1109–1116 CAS.
- W. B. Schweizer, G. Procter, M. Kaftory and J. D. Dunitz, Helv. Chim. Acta, 1978, 61, 2783–2808 CrossRef CAS.
- I. I. Schuster, A. J. Freyer and A. L. Rheingold, J. Org. Chem., 2000, 65, 5752–5759 CrossRef CAS PubMed.
- (a) R. Taylor, O. Kennard and W. Versichel, J. Am. Chem. Soc., 1983, 105, 5761–5766 CrossRef CAS; (b) P. Murray-Rust and J. P. Glusker, J. Am. Chem. Soc., 1984, 106, 1018–1025 CrossRef CAS.
- R. J. T. Houk, A. Monzingo and E. V. Anslyn, Acc. Chem. Res., 2008, 41, 401–410 CrossRef CAS PubMed.
- E. S. Stoyanov, I. V. Stoyanova and C. A. Read, Chem. – Eur. J., 2008, 14, 7880–7891 CrossRef CAS PubMed; T. Steiner and S. A. Mason, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 254–260 Search PubMed.
- M. Meot-Ner and C. A. Deakyne, J. Am. Chem. Soc., 1985, 107, 474–479 CrossRef CAS.
- G. H. Penner, R. Webber and L. A. O'Dell, Can. J. Chem., 2011, 89, 1036–1046 CrossRef CAS.
- T. Giavani, K. Johannsen, C. J. H. Jacobsen, N. Blom, H. Bildsoe, J. Skibsted and H. J. Jacobsen, Solid State Nucl. Magn. Reson., 2003, 24, 218–235 CrossRef CAS.
- B. C. Chen, W. von Philipsborn and K. Nagarajan, Helv. Chim. Acta, 1983, 66, 1537–1555 CrossRef CAS.
- G. J. Martin, J. P. Gouesnard, J. Dorie, C. Rabiller and M. L. Martin, J. Am. Chem. Soc., 1977, 99, 1381–1384 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- Z. Gu and A. McDermott, J. Am. Chem. Soc., 1993, 115, 4282–4285 CrossRef CAS.
- G. J. Rees, S. P. Day, A. Lari, A. P. Howes, D. Iuga, M. B. Pitak, S. J. Coles, T. L. Threlfall, M. E. Light, M. E. Smith, J. D. Wallis and J. V. Hanna, CrystEngComm, 2013, 15, 8823–8839 RSC.
- Z. Gu, R. Zambrano and A. McDermott, J. Am. Chem. Soc., 1994, 116, 6368–6372 CrossRef CAS.
- R. Gobetto, C. Nervi, M. R. Chierotti, D. Braga, L. Maini, F. Grepioni, R. K. Harris and P. Hodgkinson, Chem. – Eur. J., 2005, 11, 7461–7471 CrossRef CAS PubMed.
- P. A. Frey, Magn. Reson. Chem., 2001, 39, S190–S198 CrossRef CAS.
- C. Kiefl and A. Mannschreck, Synthesis, 1995, 1033–1037 CrossRef CAS PubMed.
- M. Hojo, R. Masuda and E. Okada, Tetrahedron Lett., 1987, 28, 6199–6200 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- Crystal Clear-SM Expert 2.0 r11, Rigaku, 2011 Search PubMed.
- CrysAlisPro, Agilent Technologies, Version 1.171.35.15 (release 03-08-2011 CrysAlis171 .NET) Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, X-Seed - A software tool for supramolecular crystallography, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- Accelrys Inc., 10188 Telesis Court, Suite 100 San Diego, CA 92121, USA.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, The United Kingdom Chemical Database Service, J. Chem. Inf. Comput. Sci., 1996, 36, 746–749 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 984760–984765, 984801–984806. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00981a |
| This journal is © The Royal Society of Chemistry 2014 |