 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water channels and zipper structures in Schiff base-like Cu(II) and Ni(II) mononuclear complexes†
Charles
Lochenie
ab,
Stephan
Schlamp
a,
Antoine P.
Railliet
b,
Koen
Robeyns
b,
Birgit
Weber
a and
Yann
Garcia
*b
aInorganic Chemistry II, Universität Bayreuth, Universitätsstraße 30, NW 1, 95440 Bayreuth, Germany
bInstitute of Condensed Matter and Nanosciences, Molecules, Solids and Reactivity (IMCN/MOST), Université Catholique de Louvain, Place L. Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: yann.garcia@uclouvain.be; Fax: +32 10472330; Tel: +32 10472826
First published on 5th June 2014
Abstract
The crystal structures of four Cu(II) complexes and one Ni(II) complex bearing a square planar N2O2 coordination sphere are discussed. In all cases a distorted square planar coordination sphere is observed that is independent of the metal centre and the ligand and is only influenced by the packing of the molecules in the crystal. For the less distorted copper complexes metal–aromatic interactions are observed, while no significant intermolecular interactions are found for the nickel complexes. This can be explained by the different electronic character of Cu(II) and Ni(II) and is also reflected in the differences in the solvatochromism of these complexes.
Introduction
Numerous complexes of Schiff base ligands are investigated due to their high potential as catalysts,1,4,5 biological agents,2,6 model compounds for active centres of metallo-enzymes3 (e.g. galactose oxidase7 for Cu(II)), magnetic materials,3etc. Many of the crystal structures of Cu(II) complexes show either a (distorted) square planar or a square pyramidal coordination sphere.5,6,8–10 In some cases interesting crystal packings such as “zipper” structures are observed.10 Another interesting facet is the solvatochromism of such copper complexes that can be observed in both solution and the solid state.9,11 Here we report X-ray crystal structures of a N2O22−-coordinating Schiff base-like ligand and its Cu(II) and Ni(II) complexes, the synthesis of which was first described in 1966.12 The influence of the crystallisation solvent on the crystal packing of the copper complexes explains why solvatochromism was observed in solution but not for the analogous nickel complexes.12Results and discussion
Synthesis of the complexes
The synthesis pathway as well as the structures of both ligands and complexes are shown in Scheme 1. Complexes can be obtained by mixing H2Lx with a metal acetate in methanol. After stirring at room temperature, complexes precipitate to 1 and 2.12 Refluxing the reaction mixture was proven unnecessary by comparison of the IR spectra of the refluxed and stirred compounds. The [CuL1] (1) and [CuL2] (3) complexes were both obtained as a brown powder and the [NiL1] (2) complex as an orange powder. The solutions of the nickel complex have the same orange colour, whereas for the copper complex different shades of brown were observed, from very dark brown (in CDCl3) to brown (in methanol, ethanol, water or THF). The pyridine solutions are all green.12 | ||
| Scheme 1 Synthesis pathway for the synthesis of 1, 2 and 3. | ||
Crystal structures
 | ||
| Fig. 1 An ORTEP14 drawing of the asymmetric unit with thermal ellipsoids shown at the 50% probability level. | ||
 | ||
| Scheme 2 Equilibrium between the keto–enamine tautomer (left) and the imino–enol tautomer (right). | ||
An equilibrium between these two forms can lead to thermo- and photochromism.15 The reported C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths (between 1.21 and 1.25 Å) indicate that the keto–enamine form is favoured (a CSD search on similar substructures yielded an average value of 1.219 Å for CO and 1.423 Å for C–OH). This is in agreement with the X-ray structures of a similar ligand,16 and in contrast to other Schiff base ligands like salen (N,N′-ethylenebis(salicylimine)), where the imino–enol form is preferred. The ligand is not planar and the angles between the planes, calculated through the ‘arms’ of the ligand, is 39.89°. Two intra-molecular hydrogen bonds are located between the enamine and ketone functions. Hydrogen bond distances and angles for all reported crystal structures are reported in Table 1. The crystal packing is shown in the ESI, Fig. S1.†
O bond lengths (between 1.21 and 1.25 Å) indicate that the keto–enamine form is favoured (a CSD search on similar substructures yielded an average value of 1.219 Å for CO and 1.423 Å for C–OH). This is in agreement with the X-ray structures of a similar ligand,16 and in contrast to other Schiff base ligands like salen (N,N′-ethylenebis(salicylimine)), where the imino–enol form is preferred. The ligand is not planar and the angles between the planes, calculated through the ‘arms’ of the ligand, is 39.89°. Two intra-molecular hydrogen bonds are located between the enamine and ketone functions. Hydrogen bond distances and angles for all reported crystal structures are reported in Table 1. The crystal packing is shown in the ESI, Fig. S1.†
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A | Type |
|---|---|---|---|---|---|
| a 1 + x, y, z. b 1/2 + x, 1/2 − y, 1 − z. c 1/2 − x, 1/2 + y, z. d − 1 + x, y, z. e 1 − x, −y, 1 − z. | |||||
| H2L1·0.5dioxane | |||||
| N6–H6⋯O2 | 0.88 | 1.87 | 2.561(4) | 134 | Intra |
| N6–H6⋯O2 | 0.88 | 2.35 | 2.740(4) | 107 | Intra |
| N13–H13⋯O17 | 0.88 | 1.90 | 2.558(4) | 130 | Intra |
| (1·2H2O) | |||||
| O31–H31A⋯O2a | 0.82 | 2.53 | 3.224(3) | 144 | |
| O31–H31A⋯O17a | 0.82 | 2.41 | 3.146(3) | 150 | |
| O31–H31B⋯O32b | 0.82 | 1.97 | 2.789(3) | 178 | |
| O32–H32A⋯O23c | 0.82 | 2.11 | 2.932(3) | 176 | |
| O32–H32B⋯O31d | 0.82 | 1.98 | 2.792(3) | 176 | |
| (2·MeOH) | |||||
| O101–H101⋯O23e | 0.84 | 1.99 | 2.826(6) | 179 | |
Single crystals of 3, although prepared in methanol, did not include any solvent molecules unlike the nickel complex 2·MeOH. The asymmetric unit of the five structures is shown in Fig. 1. Selected bond lengths and angles are reported in Table 2. Both the copper and nickel centres sit in a square planar N2O2 coordination sphere. The average coordination bond lengths are 1.91 Å (Cu–O and Cu–N), whereas for 2·MeOH, the bond lengths are shorter with 1.84 Å (Ni–N) and 1.85 Å (Ni–O), which can be solely attributed to the smaller covalent radius of Ni (124 pm) with respect to Cu (132 pm). In the case of 1·CHCl3 the square planar coordination sphere is slightly distorted with both coordinating oxygen atoms slightly above and below the N–Cu–N plane. The distortion can be appraised through the sum of the angles in the coordination sphere. In a perfect square planar geometry the sum of the angles is ∑ = 720° whereas for a tetrahedral coordination geometry ∑ = 656.82°. In the case of 1·CHCl3, ∑ = 705.6° indicating some distortion, however, it is close to the theoretical value for a square planar geometry. For the other complexes, an increase is noticed with ∑ = 712.6° and 712.9° for 1·xsolv and 3, respectively; ∑ = 714.1° for 2·MeOH and ∑ = 715.6° for 1·2H2O, indicating a decrease in the distortion of the square planar geometry. Another way to evaluate the distortion is to measure the angle between the N–M–N and the O–M–O planes. For 1·CHCl3 this angle is 11.0° and it decreases in the order 1·xsolv (5.5°), 2·MeOH and 3 (both 4.4°) to 1·2H2O (1.4°) displaying the same sequence as that obtained for the sum of the angles.
Cu, Ni)
| Bonds | 1·CHCl3 | 1·2H2O | 1·xsolv |
|---|---|---|---|
| M–O2 | 1.905(3) | 1.9081(18) | 1.909(2) |
| M–O17 | 1.897(3) | 1.9153(18) | 1.911(2) |
| M–N6 | 1.906(3) | 1.915(2) | 1.914(2) |
| M–N13 | 1.910(4) | 1.911(2) | 1.907(2) |
| 2·MeOH | 3 | ||
| M–O2 | 1.848(3) | 1.9191(15) | |
| M–O17 | 1.852(3) | 1.9143(16) | |
| M–N6 | 1.836(3) | 1.9051(18) | |
| M–N13 | 1.843(3) | 1.9114(18) | |
| Angles | 1·CHCl3 | 1·2H2O | 1·xsolv |
| O2–M–O17 | 89.39(13) | 89.44(7) | 89.10(9) |
| O2–M–N6 | 92.99(14) | 92.47(8) | 92.55(9) |
| O17–M–N13 | 92.52(14) | 92.64(8) | 92.49(10) |
| N6–M–N13 | 86.05(15) | 85.45(8) | 85.98(9) |
| O2–M–N13 | 173.74(16) | 177.92(8) | 174.52(10) |
| O17–M–N6 | 170.93(18) | 177.64(8) | 177.93(10) |
| 2·MeOH | 3 | ||
| O2–M–O17 | 85.98(13) | 89.32(7) | |
| O2–M–N6 | 93.68(14) | 93.16(7) | |
| O17–M–N13 | 93.45(14) | 92.27(7) | |
| N6–M–N13 | 87.04(15) | 85.40(8) | |
| O2–M–N13 | 177.67(14) | 176.40(7) | |
| O17–M–N6 | 176.31(14) | 176.32(8) |
 | ||
| Fig. 2 Superposition of adjacent complexes from within a stack, superposed by pair fitting of the central metal atoms and the primary coordination sphere of both molecules (10 pairs). The top left view shows a similar orientation; the right side view shows the intermolecular distance. Carbon atoms are drawn in magenta for 1·2H2O, in cyan for 1·xsolv, in green for 1·CHCl3 and in yellow for 2·MeOH. | ||
Despite the variety in the space groups and unit cell parameters observed for 1 and 2, all show a similar packing behaviour. The stacks of parallel oriented molecules are inclined with respect to the stacking direction and looking at the stacks one observes a similar orientation of the molecules. When water or methanol was used to crystallize the complexes, the solvent molecules are confined to one-directional channels alongside the columns of 1. In the case of CHCl3 these channels are too small to accommodate the solvent atoms and the columns of 1 are pushed away leaving a layer of CHCl3, resulting in the elongation of the unit cell parameters (~27 Å for 1·CHCl3 as opposed to ~23 Å for the remaining complexes of 1 and 2). This might suggest that π–π or metal–π interactions are responsible for the order within the stacks and that the choice of the solvent influences the position of the columns relative to one another, where the water or methanol channels stabilize the packing through hydrogen bonds (see Table 1). The intermolecular distance in the 3rd direction is controlled by the inclination of the complexes with respect to the stacks (Table 3). Water channels within the packing of 1 propagate as a 41 screw chain along the [100] axis. One of the hydrogen atom bonds to the stacked complexes while the other one bonds to the next water molecule. Fig. 3 shows the unit cell packing of 1·CHCl3 and 1·2H2O; the packing diagrams of the other complexes can be found in the ESI (S1–S10†).
| Inclination (°) | Stacking distance (A) | Twist angles (°) | |
|---|---|---|---|
| a All planes were calculated through all non-hydrogen atoms of the complexes. | |||
| 1·CHCl3 | 75.71(3) | 3.229 and 3.513 | −92.0 |
| 1·H2O | 57.27(2) | 3.312 and 3.440 | −97.7 |
| 1·xsolv | 71.66(2) | 3.299 and 3.334 | −89.4 |
| 2·MeOH | 70.52(3) | 3.317 and 3.405 | −91.7 |
| 3 | 79.40 | 3.275 and 3.353 | 180 |
 | ||
| Fig. 3 Left: unit cell packing of 1·CHCl3; right: 1·2H2O as seen along the stacking direction. The larger CHCl3 molecules prevent a denser packing resulting in the expansion of the unit cell parameters (vertical). The intermolecular distance across the figure is controlled by the inclination of the complexes with respect to the stacking direction. | ||
While the copper complex 3 also stacks in (loose) piles, the other complexes are rotated 180° with respect to one another.
The molecule is slightly distorted given that the angle between the benzyl ring plane and the N2O2 plane of 3.9°. This distortion could be due to the intermolecular metal–aromatic interactions present in the packing. No further π–π stacking or hydrogen bonds are observed.
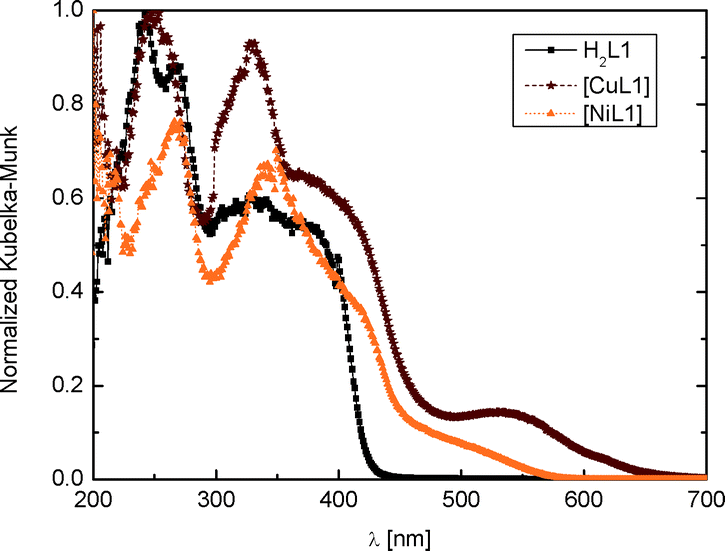 | ||
| Fig. 4 Electronic spectra of H2L1, 1 and 2. | ||
 | ||
| Fig. 5 Temperature dependence of the magnetic susceptibility measurements of the powder samples 1 and 3 between 300 K and 2 K. The 1/χMvs. T plots are given. | ||
Conclusions
The present structural report complements the previous article by Wolf and Jäger which described the synthesis of Schiff base complexes with Cu(II) and Ni(II).12 Indeed, these authors proposed that a penta-coordinated Cu(II) complex can be obtained by a coordination bond between the metal centre and a ketone function from a neighbouring complex. No such bond was found in the four crystal structures of the copper complexes which we have determined. Their hypothesis was built on the fact that the dx2−y2 orbital of copper in a square planar geometry is too high in energy and populating this orbital with one electron is not worth the stabilisation that the square planar geometry would bring to the complex. Indeed, in the crystal structure of 1·CHCl3, we observed a distorted square planar geometry. This distortion will lower the symmetry and thus decrease the energy of the dx2−y2 orbital. In the crystal structures of 1·2H2O, 1·xH2O·yMeOH and 3, the distortion of the square planar coordination sphere is significantly weaker. Here metal–aromatic interactions between the copper centre and the benzyl ring of a neighbouring complex were observed, which can also lower the symmetry of the complex and therefore reduce the energy of the dx2−y2 orbital. This is in line with the structure of the Ni(II) complex, 2·MeOH, where the coordination sphere is close to an ideal square planar one without significant interactions between the complex molecules. As the dx2−y2 orbital for this complex with one less d-electron is empty, its strong destabilisation has no effect on the total energy. The different electronic character of the copper and the nickel complexes also explain their different tendencies to show solvatochromism.12 In solution, the copper centre can interact with the solvent molecules. This leads to a distortion of the square planar geometry and will lower the energy of the dx2−y2 orbital. The strength of this interaction depends on the donating ability of the solvent and influences the splitting of the 3d orbitals of the copper centre. For the nickel complex no such interactions are necessary and no solvatochromism is observed.Experimental
Synthesis
All reagents were of reagent grade and used without further purification. All solvents were of analytical grade and used without further purification.Syntheses of ligands H2L1 and H2L2 were performed as described.12 X-ray quality single crystals of H2L1·0.5dioxane were obtained as yellow needles from vapour–vapour diffusion between a solution of the ligand in dioxane and water. UV-vis H2L1 (PTFE): λmax = 241, 268 and 328 nm (Fig. 4).
[CuL1] (1):12 copper acetate monohydrate (0.39 g) and H2L1 (0.66 g) were vigorously stirred in methanol (30 mL) overnight at room temperature,9 resulting in a brown powder which was filtered, washed twice with ethanol (15 mL) and dried under vacuum. Yield: 0.6 g (75%). IR (KBr): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 1652 (CO, COCH3), 1569 (CO, COCH3) cm−1. CHN (%): exp. (theo.) for C18H18CuN2O4: C, 55.21 (55.45); H, 4.38 (4.65); N, 7.10 (7.18). UV-vis (PTFE): λmax = 251, 328 and 536 nm.
= 1652 (CO, COCH3), 1569 (CO, COCH3) cm−1. CHN (%): exp. (theo.) for C18H18CuN2O4: C, 55.21 (55.45); H, 4.38 (4.65); N, 7.10 (7.18). UV-vis (PTFE): λmax = 251, 328 and 536 nm.
Three solvates were obtained as single crystals suitable for X-ray crystallography: 1·CHCl3 crystals were obtained from trichloromethane as brown needles, 1·2H2O crystals were obtained from THF as brown prisms and 1·xH2O·yMeOH from a MeOH–EtOH mixture.
[NiL1] (2):12 nickel acetate tetrahydrate (0.50 g) and H2L1 (0.66 g) were vigorously stirred in methanol (30 mL) overnight at room temperature.9 The resulting orange powder was filtered, washed twice with ethanol (15 mL) and dried under vacuum. Yield: 0.6 g (75%). IR (KBr): = 1653 (CO, COCH3), 1573 (CO, COCH3) cm−1. CHN (%): exp. (theo.) for C18H18NiN2O4: C, 56.15 (55.98); H, 4.71 (4.59); N, 7.27(7.30). UV-vis (PTFE): λmax = 266 and 340 nm.
Single crystals suitable for X-ray crystallography of 2·MeOH were obtained from solvothermal synthesis with a saturated solution of 2 in methanol. The solution was heated to 80 °C for one day in a solvothermal bomb then left to cool overnight in a turned-off oven. Orange needles were collected.
[CuL2] (3):12 copper acetate monohydrate (4.65 g) and H2L2 (10 g) were dissolved in methanol (175 mL).9 The solution was then refluxed for 2 min. After cooling, the brown precipitate was filtered and dried in vacuum. Single crystals suitable for X-ray crystallography were obtained from the mother liquor in the fridge as violet crystals. Yield: 9.28 g (82%). CHN (%): exp. (theo.) for C20H22CuN2O6: C, 51.81 (51.63); H, 5.14 (5.11); N, 5.49 (5.54). MS (DEI+) [m/z(%)]: 509 (100) [M]+, 464 (15), 345 (50), 273 (28).
Physical measurements
IR spectra were recorded using a Shimadzu FTIR-84005 spectrometer using KBr discs at room temperature. Electronic spectra in the solid state were recorded with a CARY 5E spectrophotometer using polytetrafluoroethylene (PTFE) as a reference. CHN analyses were performed at MEDAC Ltd (UK). Mass spectra were recorded with a Jeol MS-700 device. The ionisation method used was DEI+. Magnetic measurements were performed with a Quantum Design MPMSXL-5 SQUID magnetometer. The measurements were carried out at 0.7 T for 1 and 0.05 T for 2 in sweep mode (10 K min−1) between 300 K and 10 K and in settle mode (2 K min−1) between 10 K and 2 K. The data were corrected for the magnetisation of the sample holder and the diamagnetism of the ligand using tabulated Pascal's constants.Single crystal X-ray structure determination
Crystals were mounted on a nylon loop with paratone or grease and flash-cooled in a N2 gas stream. The data were collected using MoKα (λ = 0.71073 Å) radiation using a MAR345 image plate except for 3 which was measured using a KappaCCD detector. The structures were solved by direct methods using SHELXS13 and refined by full least-squares on |F2| using SHELXL97.13 All of the non-hydrogen atoms were refined with anisotropic temperature factors. The hydrogen atoms were placed in calculated positions in a riding model with the isotropic temperature factors fixed at 1.2Ueq of the parent atoms (1.5 for methyl groups). The crystallographic and refinement data are summarised in Table 4.| H2L1·0.5dioxane | 1·CHCl3 | 1·2H2O | |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b ωR2 = [∑[w(Fo2 − Fc2)2]/∑w(Fo2)2]1/2, w = 1/[σ2(Fo2) + (aP)2 + bP], where P = [Fo2 + 2(Fc2)]/3. | |||
| Formula | C18H20N2O4, 0.5(C4H8O2) | C18H18N2O4Cu, CHCl3 | C18H18N2O4Cu, 2(H2O) |
| FW (g mol−1) | 372.41 | 509.25 | 425.93 |
| System | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | C2/c | Aba2 | Pbca |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 |
| a (Å) | 7.4692(8) | 27.317(3) | 8.025(1) |
| b (Å) | 22.575(2) | 22.2583(14) | 19.28(1) |
| c (Å) | 22.4145(14) | 6.9575(5) | 23.39(1) |
| β (°) = 91.242(6) | |||
| Volume (Å3) | 3778.6(6) | 4230.4(6) | 3620.0(2) |
| Z | 8 | 8 | 8 |
| T (K) | 150 | 150 | 150 |
| F(000) | 1584 | 2072 | 1768 |
| 2θ (°) | 47.06 | 50.76 | 50.48 |
| Refl. collected | 9812 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 226 226 |
|
| Unique/>2sigma(I) | 2692/2057 | 3875/3207 | 3421/3129 |
| Parameters | 248 | 266 | 260 |
| R 1 (all)a | 0.0691 (0.0899) | 0.0596 (0.0766) | 0.0346 (0.0383) |
| ωR 2 (all)b | 0.1648 (0.1808) | 0.1213 (0.1310) | 0.0950 (0.0987) |
| GooF | 1.075 | 1.026 | 1.028 |
| 1·xsolv | 2·MeOH | 3 | |
| Formula | C18H18N2O4Cu | C18H28N2O4Ni, (C H4 O) | C22H26N2O8Cu |
| FW (g mol−1) | 389.88 | 417.08 | 510.00 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/c |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 |
| a (Å) | 11.2596(2) | 11.1128(5) | 6.74840(10) |
| b (Å) | 23.9363(4) | 23.9480(10) | 21.9827(3) |
| c (Å) | 6.9871(2) | 7.1300(3) | 16.0526(2) |
| β (°) | 103.591(2) | 103.599(5) | 105.1720(10) |
| Volume (Å3) | 1830.38(7) | 1844.31(14) | 2298.37(6) |
| Z | 4 | 4 | 4 |
| T (K) | 150 | 150 | 200 |
| F(000) | 804 | 872 | 1060 |
| 2θ (°) | 50.48 | 50.48 | 54.946 |
| Refl. collected | 16627 |
11832 |
19693 |
| Unique/>2sigma(I) | 3403/3144 | 3349/2289 | 5269 |
| Parameters | 231 | 250 | 302 |
| R 1 (all)a | 0.0408 (0.0438) | 0.0537 (0.0941) | 0.0401 (0.0656) |
| ωR 2 (all)b | 16627 (0.1134) |
0.1136 (0.1353) | 0.0960 (0.1067) |
| GooF | 1.060 | 1.037 | 1.038 |
Acknowledgements
This work was funded by the Fonds National de la Recherche Scientifique (FNRS) and the University of Bayreuth.Notes and references
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS PubMed.
- (a) T. Fasina, O. Ogundele, F. Ejiah and C. Dueke-Eze, Int. J. Biol. Chem., 2012, 6, 24–30 CrossRef CAS; (b) D.-D. Qin, Z.-Y. Yang and B.-D. Wang, Spectrochim. Acta, Part A, 2007, 68, 912–917 CrossRef PubMed.
- (a) B. Weber and E.-G. Jäger, Eur. J. Inorg. Chem., 2009, 4, 465–477 CrossRef; (b) T. M. Pfaffeneder, S. Thallmair, W. Bauer and B. Weber, New J. Chem., 2011, 35, 691–700 RSC; (c) S. Schlamp, B. Weber, A. D. Naik and Y. Garcia, Chem. Commun., 2011, 47, 7152–7154 RSC.
- O. Bienemann, A.-K. Froin, I. dos Santos Vieira, R. Wortmann, A. Hoffmann and S. Herres-Pawlis, Z. Anorg. Allg. Chem., 2012, 638, 1683–1690 CrossRef CAS.
- R. N. Patel, V. L. N. Gundla and D. K. Patel, Polyhedron, 2008, 27, 1054–1060 CrossRef CAS PubMed.
- X. Dong, Y. Li, Z. Li, Y. Cui and H. Zhu, J. Inorg. Biochem., 2012, 108, 22–29 CrossRef CAS PubMed.
- M. Orio, O. Jarjayes, H. Kanso, C. Philouze, F. Neese and F. Thomas, Angew. Chem., Int. Ed., 2010, 49, 4989–4992 CrossRef CAS PubMed.
- (a) T. N. Mandal, S. Roy, A. K. Barik, S. Gupta, R. J. Butcher and S. K. Kar, Polyhedron, 2008, 27, 3267–3274 CrossRef CAS PubMed; (b) A. Biswas, M. G. B. Drew and A. Ghosh, Polyhedron, 2010, 29, 1029–1034 CrossRef CAS PubMed; (c) K. Sundaravel, E. Suresh and M. Palaniandavar, Inorg. Chim. Acta, 2009, 362, 199–207 CrossRef CAS PubMed.
- W.-K. Dong, X.-N. He, H.-B. Yan, Z.-W. Lv, X. Chen, C.-Y. Zhao and X.-L. Tang, Polyhedron, 2009, 28, 1419–1428 CrossRef CAS PubMed.
- P. K. Bhaumik, S. Jana and S. Chattopadhyay, Inorg. Chim. Acta, 2012, 390, 167–177 CrossRef CAS PubMed.
- M. M. Bhadbhade and D. Srinivas, Inorg. Chem., 1993, 32, 5458–5466 CrossRef CAS.
- L. Wolf and E.-G. Jäger, Z. Anorg. Allg. Chem., 1966, 346, 76–91 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) C. K. Johnson and M. N. Burnett, ORTEP-III, Oak-Ridge National Laboratory, Oak-Ridge, TN, 1996 Search PubMed; (b) L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- (a) F. Robert, P. L. Jacquemin, B. Tinant and Y. Garcia, CrystEngComm, 2012, 14, 4396–4406 RSC; (b) D. A. Safin, K. Robeyns and Y. Garcia, RSC Adv., 2012, 2, 11379–11388 RSC; (c) D. A. Safin and Y. Garcia, RSC Adv., 2013, 3, 6466–6471 RSC.
- W. Bauer, T. Ossiander and B. Weber, Z. Naturforsch., B: J. Chem. Sci., 2010, 65b, 323–328 Search PubMed.
- M. Friedrich, J. C. Gálvez-Ruiz, T. M. Klapötke, P. Mayer, B. Weber and J. J. Weigand, Inorg. Chem., 2005, 44, 8044–8052 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. CCDC 995462–995467 for H2L1·0.5dioxane, 1·CHCl3, 1·2H2O, 1·xsolv, 2·MeOH and 3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00710g |
| This journal is © The Royal Society of Chemistry 2014 |