 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of ferromagnetic cobalt nanoparticle tipped CdSe@CdS nanorods: critical role of Pt-activation†
Lawrence J.
Hill
a,
Nathaniel E.
Richey
a,
Younghun
Sung
b,
Philip T.
Dirlam
a,
Jared J.
Griebel
a,
In-Bo
Shim
c,
Nicola
Pinna
*d,
Marc-Georg
Willinger
e,
Walter
Vogel
f,
Kookheon
Char
*b and
Jeffrey
Pyun
*ab
aDepartment of Chemistry and Biochemistry, University of Arizona, 1306 E. University Blvd., Tucson, AZ 85721, USA. E-mail: jpyun@email.arizona.edu
bWorld Class University Program for Chemical Convergence for Energy and Environment, The National Creative Research Initiative Center for Intelligent Hybrids, School of Chemical and Biological Engineering, Seoul National University, Seoul 151-744, Korea. E-mail: khchar@snu.ac.kr
cDepartment of Nano and Electronic Physics, Kookmin University, Seoul, 136-702, Korea. Fax: +82 2 910 5170; Tel: +82 2 910 4757
dInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: nicola.pinna@hu-berlin.de
eDepartment of Inorganic Chemistry, Fritz Haber Institute of the Max Planck Society, Germany
fDepartment of Chemistry, National Central University, Taiwan
First published on 12th May 2014
Abstract
The synthesis of a ferromagnetic heterostructured material consisting of a CdSe@CdS nanorod attached to a single dipolar cobalt nanoparticle (CoNP) into a “matchstick” morphology is reported. CdSe@CdS nanorods were modified by an activation reaction with Pt(acac)2 which enabled selective one-sided deposition of a dipolar metallic CoNP-tip via the thermolysis of Co2(CO)8 in the presence of polystyrene ligands. Small (<2 nm) PtNP-tips on CdSe@CdS nanorods were found to be responsible for the selective deposition of CoNP-tips onto one terminus per nanorod. The influence of the Pt-activation step for cobalt tipping was investigated by examination of numerous conditions and characterization of intermediates and materials using transmission electron microscopy and synchrotron X-ray diffraction.
Introduction
Synthetic routes to multi-component inorganic nanocomposites have generated considerable interest as a route to complex materials with enhanced optical, electronic, magnetic, or catalytic properties.1–15 Heterostructured nanorods composed of a semiconducting nanorod coupled to a terminal metal nanoparticle “tip” are particularly interesting due to the ability to systematically tune the size of each domain, which allows for direct control of optoelectronic and magnetic material properties.1–3,16–24 The synthesis of cadmium selenide seeded cadmium sulfide nanorods with either one, or two terminal cobalt nanoparticles (i.e., CoNP-tipped CdSe@CdS) nanorods was recently reported using platinum nanoparticle tipped (PtNP-tipped) nanorods as activated intermediates amenable to cobalt deposition.25 Control over the number of CoNP-tips per nanorod was achieved by variation of the Pt-activation time and subsequent feed ratio of cobalt precursor (Co2(CO)8) used during cobalt deposition.Herein, we report on the synthesis of CdSe@CdS nanorods that incorporate a single dipolar CoNP tip per nanorod (Scheme 1). Using a multi-step synthesis, magnetic heterostructured nanorods were prepared with a single, dipolar, ferromagnetic CoNP (CoNP-tip) conjugated to a CdSe@CdS nanorod (hereafter referred to as a “matchstick”, versus a two-sided CoNP-tipped nanorod or “dumbbell”). The key step in this colloidal total synthesis was understanding and optimizing the nanorod activation step (Pt-activation) using Pt(acac)2, which enabled selective deposition of a single, dipolar, ferromagnetic CoNP-tip. While previous reports have demonstrated deposition of CoNP-tips onto semiconductor nanorods at one or both nanorod termini,25–29 the methodology in this report affords nanorod precursors for the formation of larger (diameter (D) > 20 nm) dipolar cobalt nanoparticles attached at primarily one terminus per nanorod. Dipolar cobalt domains on matchstick CoNP-tipped nanorods directed spontaneous formation of linear nanorod assemblies through magnetic interactions of the CoNP-tips,30–49 while nanorods bearing additional CoNP inclusions per nanorod were found to result in less ordered self-assembled architectures. This finding demonstrated that control over the morphology of dipolar CoNP-tipped products was a crucial factor in directing the self-assembly modes of these dipolar heterostructures. This report focuses on the synthetic methods required to obtain dipolar matchstick CoNP-tipped nanorods capable of linear assembly, while investigation of the self-assembly of these dipolar matchstick structures is reported elsewhere.50
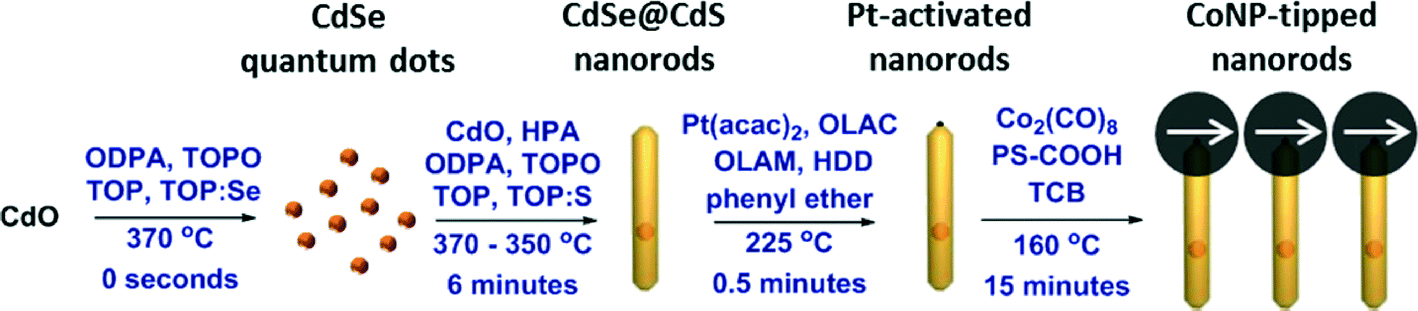 | ||
| Scheme 1 Synthesis of CoNP-tipped CdSe@CdS nanorods over four steps. CdSe quantum dots and CdSe@CdS nanorods were synthesized as reported elsewhere.25,53 CdSe@CdS nanorods were modified by a brief Pt-activation reaction which preferentially activated these nanorods towards cobalt deposition at the sulfur-rich nanorod terminus with minimal deposition of Pt onto the nanorod. Dipolar CoNP-tips were then deposited onto these activated CdSe@CdS nanorods at primarily one terminus per nanorod (matchstick morphology) and the CoNP-tips spontaneously assembled into chains due to magnetic interactions. Abbreviations: OLAC = oleic acid; OLAM = oleylamine; HDD = 1,2-hexadecanediol; PS-COOH = carboxylic acid terminated polystyrene38 (Mn = 8600 g mol−1; PDI = 1.07); TCB = 1,2,4-trichlorobenzene. Ligands are omitted for clarity. | ||
Experimental section
Chemicals
1,2,4-Trichlorobenzene (TCB) (>99%) was purchased from both Aldrich and Alfa Aesar and passed through a 0.2 micron syringe filter prior to use. 1,2-Dichlorobenzene (DCB) (>99%) was purchased from both Aldrich and Acros Organics and passed through a 0.2 micron syringe filter prior to use. 1,2-Hexadecanediol (HDD) (90%), toluene (99.5%), cadmium(II) oxide (CdO) (99.5%), selenium (99.999%), oleic acid (OLAC) (90%), oleylamine (OLAM) (70%), and 1-octadecene (ODE) (90%) were purchased from Aldrich. Octadecylphosphonic acid (ODPA) (97%), trioctylphosphine oxide (TOPO) (99%), trioctylphosphine (TOP) (90%), trioctylphosphine (TOP) (97%), hexylphosphonic acid (HPA) (97%) and dicobalt octacarbonyl stabilized with 5% hexanes (Co2(CO)8) were purchased from Strem. Platinum(II) acetylacetonate (Pt(acac)2) (98%) and diphenyl ether (DPE) (99%) were purchased from Acros Organics. Sulfur (>99.0%) was purchased from EMD, and absolute ethanol (EtOH) was purchased from Pharmco-Aaper. All chemicals were commercially available and used as received, unless otherwise noted.Electron microscopy
Low resolution bright field TEM images (those not labeled “high resolution”) were obtained on a Phillips CM12 transmission electron microscope (CM12) at 80 kV, or a Tecnai Spirit transmission electron microscope (FEI) at 100 kV using in house prepared carbon coated copper grids (Cu, hexagon, 200 mesh). Image analysis was performed using ImageJ software (Rasband, W.S., National Institutes of Health, http://rsb.info.nih.gov/ij/, 1997–2007) from bright field TEM images at a minimum of 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification by sizing a minimum of 100 particles (unless otherwise stated). High resolution TEM (HRTEM) and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images were acquired using a CM200FEG (Philips) microscope with a field emission gun operated at 200 kV or a Cs corrected FEI Titan operated at 300 kV.
000× magnification by sizing a minimum of 100 particles (unless otherwise stated). High resolution TEM (HRTEM) and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images were acquired using a CM200FEG (Philips) microscope with a field emission gun operated at 200 kV or a Cs corrected FEI Titan operated at 300 kV.
X-ray diffraction
XRD measurements were performed on the μSpot beam line at BESSY II synchrotron.51 A Si 111 Monochromator was used and an incident X-ray energy of 12.48 keV (0.9933 Å) was selected. The beam size was 100 μm. A polymer sample holder with 2 mm holes and a thickness of about 1 mm was used. Samples were filled into the holes and covered with Scotch tape. To protect the area detector (MAR-CCD) the primary beam was blocked by a 3.5 mm beam stop made of lead. Each sample was measured twice with an acquisition time of 150 s. The program Fit2D was used for integration of the images.Optical spectroscopy
UV-vis measurements were obtained using a Shimadzu Corporation UV-vis recording spectrophotometer (no. UV-2401PC, Kyoto Japan). CdSe quantum dot diameters were estimated from UV-vis data by using the correlation of quantum dot diameter with the low energy visible absorbance maximum reported by Peng et al.52 Fluorescence measurements were obtained using a Photon Technology International spectrometer (no. 3005, Weatherford TX).Synthesis of CdSe quantum dots and CdSe@CdS nanorods
CdSe quantum dots and CdSe@CdS nanorods were synthesized as previously reported,25 which was modified from the methods of Manna et al.53Synthesis of Pt-activated nanorods
Pt-activation of CdSe@CdS nanorods was conducted using a modification of the Pt-deposition conditions established by Mokari et al.54 and full synthetic details are provided in the ESI.† Briefly, CdSe@CdS nanorods (CdSe seed: D = 3.2 nm; nanorod: L = 66.0 nm, D = 5.3 nm, 43 mg, 20 wt-% organic content by TGA, approximately 8 nanomol nanorods) were weighed into a 20 mL vial and dispersed in 1,2-dichlorobenzene (1.2 mL) via vortex agitation, where sonication in certain cases was briefly applied (<2 min). A solution of platinum(II) acetylacetonate (1 mg in 0.1 mL DCB; 2.5 μmol) was then transferred to the vial containing the nanorod dispersion via syringe no more than 5 min prior to use (see below).Diphenyl ether (10 mL at 40 °C), oleic acid (0.2 mL; 0.61 mmol), oleylamine (0.2 mL; 0.63 mmol), and 1,2-hexadecanediol (43 mg; 0.17 mmol) were weighed into a 50 mL three-neck-round bottom flask equipped with a reflux condenser and 1" magnetic stir bar. The flask was fitted with rubber septa and a temperature probe was passed through a septum until it came into direct contact with the solution. The solution was then heated to 80 °C in vacuo for 30 min using a heating mantle and temperature controller with continuous stirring at 300 RPM and subsequently backfilled with Ar. The contents of the flask were then heated to 225 °C under argon, followed by injection via syringe of CdSe@CdS nanorods dispersed in a solution of Pt(acac)2/DCB (described above) to the reaction mixture. The heating mantle was removed from the reaction flask after 30 s from the injection of Pt(acac)2/nanorods/DCB. Toluene (10 mL) was injected via syringe into reaction mixture once the internal temperature of the medium reached 100 °C, and the orange dispersion was allowed to cool to room temperature prior to purification by centrifugation (ESI†). The synthesis yielded an orange powder (32 mg, 20.6 wt-% organic content from TGA).
Synthesis of CoNP-tipped CdSe@CdS nanorods
Full synthetic details are provided in the ESI.† Carboxylic acid-terminated polystyrene (PS-COOH) was synthesized by ATRP as previously reported and used for all cobalt deposition reactions (Mn = 8600 g mol−1 by GPC versus polystyrene; PDI = 1.07).38 A stock solution of Co2(CO)8 (300 mg, 0.9 mmol) dissolved in 1,2,4-trichlorobenzene (TCB, 3 mL) was prepared in a glass vial before setting up the cobalt deposition reaction. The vial was sealed with a screw top cap and left to stand under ambient conditions (20 min) before vigorous vortex agitation (no sonication) for several min to promote complete dissolution of all solids. The vial was then subjected to vortex agitation for an additional 2 min.Pt-activated CdSe@CdS nanorods (5 mg for 66.0 nm nanorods with 20.6% organic content by mass, approximately 0.95 nanomol) were loaded into a 20 mL vial containing PS-COOH (10 mg; 1.2 μmol), and TCB (1 mL) was added before the vial was fitted with a screw top polypropylene cap and subjected to 10 min sonication. A 10 mm magnetic stir bar was added to the vial after sonication, and the vial was fitted with a rubber septum. A temperature probe was passed through the septum until it came into direct contact with the reaction mixture before securing the septum with electrical tape. The reaction vessel was evacuated for 2 min and backfilled with argon before heating to 160 °C using a heating mantle and temperature controller with continuous stirring (1000 RPM). Upon reaching 160 °C, room temperature Co2(CO)8 dissolved in TCB (0.5 mL of the Co2(CO)8 stock solution described above; approximately 50 mg or 146 μmol of Co2(CO)8) was injected. The temperature was held at 160 °C for 15 min from the time of injection before the vial was removed from the heating mantle and allowed to cool to room temperature before purification by repeated magnetic collection steps (ESI†). The purified product was imaged via TEM and sized using ImageJ software (CoNP-tip diameter = 20 ± 4 nm; approximately 88% of dipolar CoNPs imaged carried a nanorod side chain). These CoNP-tipped matchsticks were sensitive to sonication which resulted in an increased number of shorter “broken” free nanorods being observed in sonicated samples. Dipolar CoNP-tipped nanorods were stored as dispersions in PS-COOH/TCB (2 mg mL−1) when intended for further use. The sample was dried strictly for yield determination to yield a black powder (43 mg, 66.6 weight-% organic content from TGA).
Results & discussion
Activation of CdSe@CdS nanorods toward deposition of dipolar CoNP-tips
Dipolar CoNP-tipped nanorods were synthesized over four steps using CdSe quantum dots and CdSe@CdS nanocrystal reagents, with the key step of the synthesis being nanorod activation with Pt(acac)2 (Scheme 1). CdSe quantum dots and CdSe@CdS nanorods were synthesized as previously reported,25,53 and these nanorods served as substrates for the deposition of metal NPs to prepare the target dipolar CoNP-tipped structure. Native CdSe@CdS nanorods were found to be ineffective precursors for the deposition of CoNP-tips by the thermolysis of Co2(CO)8 in DCB in the presence of polystyrene ligands, which resulted in the homogeneous nucleation of unattached CoNPs (Fig. 1a, d). This result indicated that unmodified nanorods were not reactive toward metallic cobalt deposition, and pointed to the need for additional activation of the terminal nanorod surface. The major challenge in the deposition of a single, dipolar, CoNP-tip per nanorod was then in finding conditions which selectively activated a single nanorod terminus with minimal activation of the opposing terminus, or lateral facets.25 | ||
| Fig. 1 TEM images of (a) CdSe@CdS nanorods used in this figure (Length = L = 64.1 + 5.4 nm, n = 100; D = 5.7 + 0.6 nm, n = 100; CdSe seed D = 2.5 nm by UV-vis52). (b) “Pt-activated” nanorods after 30 s Pt-activation. (c) PtNP-tipped nanorods after 8 min Pt-activation. (d) Product of cobalt deposition onto native CdSe@CdS nanorods shown in 1a. (e) Product of cobalt deposition onto “Pt-activated” nanorods shown in 1b. (f) Product of cobalt deposition onto PtNP-tipped nanorods shown in 1c. | ||
The terminal deposition of PtNPs, or very small Pt clusters was investigated as a route to activate nanorods towards the deposition of dipolar CoNP-tips, based on our previously reported methodology for the installation of smaller CoNP-tips.25 CdSe@CdS nanorods were therefore treated with Pt(acac)2, HDD, OLAC, and OLAM for 8 min at 225 °C in phenyl ether, where low concentrations of Pt(acac)2 (0.23 mM) resulted in nanorods having small PtNP-tips at a single terminus per nanorod, as observed by bright field TEM imaging (Fig. 1c). However, treatment of these Pt-activated CdSe@CdS nanorods with Co2(CO)8 in DCB at elevated temperature resulted in mixtures of matchstick and dumbbell tipped nanorods (i.e., CdSe@CdS nanorods with one, or two CoNP tips) (Fig. 1f, S-7, ESI†). Multiple attempts were made to alter this Pt-deposition reaction by variation of reaction time and concentration of Pt(acac)2 in an attempt to obtain PtNP-tipped nanorods amenable to the formation of dipolar CoNP-tipped matchsticks. These experiments produced one of two general results: either 1) PtNP-tipped nanorods were obtained which resulted in mixtures of CoNP-tipped products that did not efficiently form linear dipolar assemblies (Fig. 1c), or 2) CdSe@CdS nanorods appeared unmodified after Pt-deposition, as determined by bright field TEM imaging (Fig. 1b).
Determination of optimized Pt-activation conditions for nanorods to enable the deposition of a single dipolar CoNP tip required screening of Pt-activation times and concentrations of precursors followed by correlation of CoNP deposition products and morphology (i.e., matchstick vs. dumbbell). This effort was particularly challenging since the required Pt-activation steps were typically very short and did not afford PtNP tips that could be resolved by HRTEM. However, the effects of these short Pt-activation times were easily discerned by the morphology of CoNP tips deposited onto Pt-activated nanorods, where very short times tended to afford matchstick tipped nanorods, while longer activation times yielded dumbbell tipped nanorods. In this manner, optimized matchstick activation conditions (referred to as “Pt-activated nanorods”) were discovered which required a combination of low Pt precursor concentration (0.23 mM) and brief reaction time (30 s, see ESI†), as determined by characterization of the CoNP-tipped products by standard bright field TEM imaging (Fig. 1b, e). Note that this short Pt-activation time resulted in Pt-activated nanorods which were indistinguishable from the native nanorod precursors by standard low resolution bright field TEM (Fig. 1b). Additional control experiments confirmed the need for the low concentration of Pt(acac)2 used in these experiments (Fig. S-5, ESI†).
Dipolar matchstick CoNP-tipped nanorods
Despite optimization of Pt-activation conditions to afford dipolar matchstick CoNP-tipped nanorods, purification of the desired heterostructure from untipped nanorods was required (Fig. 1e). Samples were enriched in dipolar CoNP-tipped nanorods by use of a custom built magnetic probe which was inserted directly into the crude reaction product (ESI†) to enable collection of magnetic material present in these samples from a good solvent (TCB) in a reasonable amount of time (2 min). Collection of the magnetic material from a good solvent was desirable to prevent aggregation of CoNP-tipped nanorods, since these dipolar heterostructures could not be redispersed by sonication without resulting in mechanical fracture of nanorod side chains. Repeated magnetic collection steps resulted in samples highly enriched in dipolar CoNP-tipped nanorods, with minimal free nanorods (<10%) observed by TEM after purification (Fig. 3a). Approximately 88% of the dipolar CoNPs imaged in these samples carried a nanorod side chain.Upon magnetic purification, spatial and structural characterization of each colloidal component in the nanocomposite was conducted by HRTEM and power spectrum analysis (Fig. 2). Power spectrum analysis (defined as the square of the Fourier transform of the image data) of the high resolution TEM image (HRTEM, Fig. 2b) presented rings that were indexed to both the fcc phase of cobalt NPs and to the wurtzite modification of CdS. The inner spots of the power spectrum were assigned to the CdSe@CdS nanorods (the red ring overlaps with the 002 reflections of the wurtzite modification of CdS while just inside and just outside that ring the 100 and the 101 reflections, respectively, are also present) and the spots overlapping to the green ring were assigned to the fcc phase of the cobalt nanoparticles. To further prove this assignment, Fourier analysis was carried out by selecting the reflections close to the green and red ring (Fig. 2c and d, respectively). The reflections corresponding to Co-phase arose only from the NP backbone (Fig. 2c), which were constituted by ca. 20 nm Co particles made of smaller crystallites as clearly visible from the phase contrast image. Moreover, the position and the orientation of the CdSe@CdS nanorods arising from the backbone were clearly visible when selecting the most intense reflection of CdS only (Fig. 2d). The crystallite size was determined to be 4.2 nm from XRD line width analysis.50 XRD also clearly proved the unusual fcc structure of cobalt, as noted by the presence of fcc-type peaks 111 and 200, and the absence of the strongest hcp type 101 pattern.
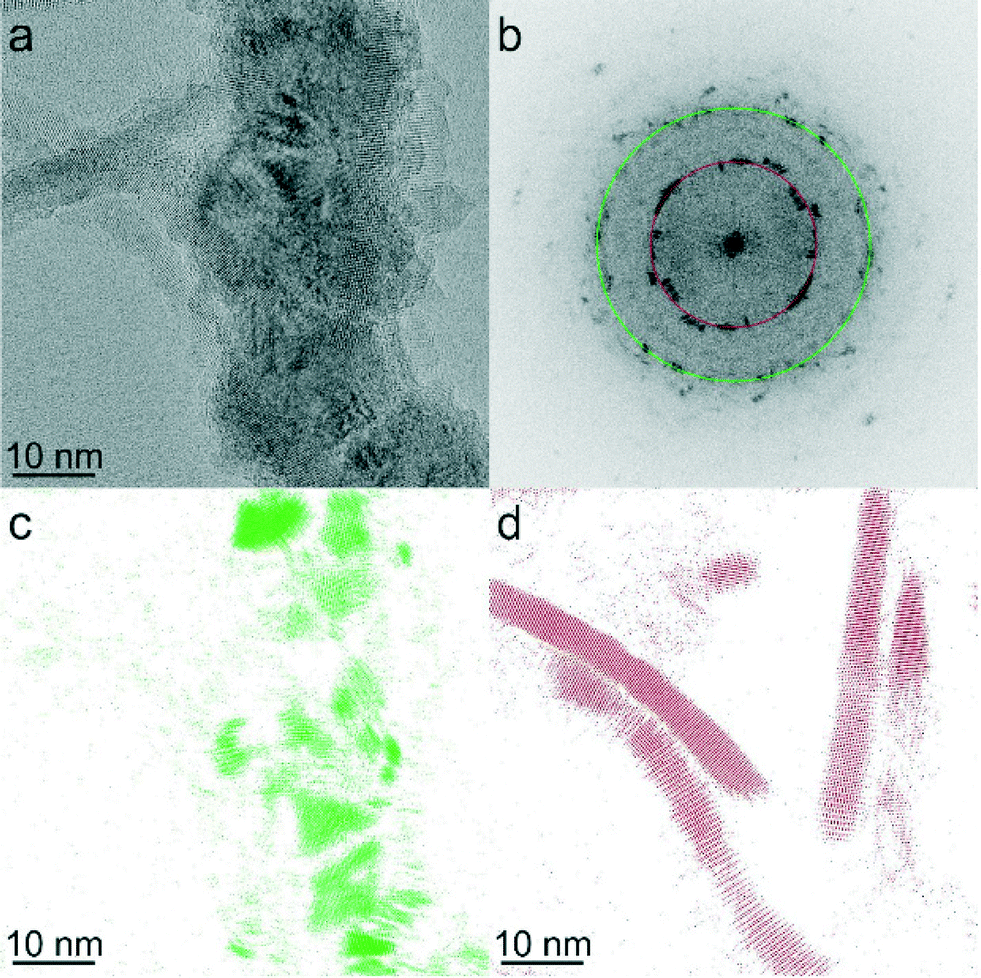 | ||
| Fig. 2 (a) HRTEM image of CoNP-tipped nanorods. (b) Power spectrum of the HRTEM image in Fig. 2a. (c) Fourier filtered image selecting the 111 reflections of the cubic modifications of cobalt (in green) from Fig. 2b, (d) Fourier filtered image selecting 100, 002, 101 reflections of the hexagonal modification of CdS (region closer to the red circle) from Fig. 2b. | ||
Effects of heterostructure morphology on the architecture of dipolar assemblies
As mentioned above, the ability to suppress the formation of nanorod side products with lateral CoNP inclusions and/or dumbbell CoNP-tipped nanorods enabled the synthesis of matchsticks that formed 1-D dipolar NP chains with nanorods carried into the assembly as densely grafted pendant functionalities (Fig. 3a). A striking feature of these dipolar matchstick nanorods and the resulting dipolar assemblies was the exclusive perpendicular orientation of CdSe@CdS nanorod side chains relative to the CoNP main chain and the magnetic easy axis of each dipolar NP unit. As a result of this efficient dipolar coupling, linear assemblies of dipolar matchstick CoNP-tipped nanorods frequently spanned several microns in length when cast onto carbon coated TEM grids for imaging (Fig. 3a). By comparison, mixtures containing dipolar matchstick nanorods with a fraction of either dumbbell nanorods, or laterally decorated nanorods were obtained from PtNP-tipped nanorods (which were activated for longer times, Fig. 1f). This mixture of CoNP-tipped nanorod morphologies tended to afford less ordered colloidal assemblies, particularly through branching from dumbbell nanorods, which pointed to the need for precisely controlled Pt-activation in achieving CoNP-tipped nanorods capable of 1-D assembly (Fig. 3b). These examples demonstrated how selective control of the CoNP-tipped structure of these “colloidal monomers” directly correlated with the architecture of the dipolar assemblies, as observed for other colloidal polymer systems.55–65 | ||
| Fig. 3 (a) Low resolution TEM image of dipolar CoNP-tipped matchstick nanorods prepared from optimized Pt-activated nanorods (from Fig. 1b), with higher resolution inset (DCoNP-tip = 22.4 ± 4.7 nm). (b) Low resolution TEM image of dipolar matchstick and dumbbell CoNP-tipped nanorod mixtures prepared from PtNP-tipped nanorods obtained from longer Pt-activation times (from Fig. 1c), with higher resolution inset (DCoNP-tip = 23.0 ± 4.8 nm). | ||
Characterization of Pt-activated CdSe@CdS nanorods
While the importance of Pt-activation was demonstrated by the previously described results, further investigation into this step was conducted to gain insight into the effects of this chemical modification to the CdSe@CdS nanorods. As alluded to earlier, a fundamental challenge in studying this Pt-activation step was the absence of discernible metallic PtNPs, or clusters deposited onto the CdSe@CdS nanorod, particularly since solution optical absorption and emission spectroscopies did not reveal any chemical changes to the parent nanorod (ESI† Fig. S-8 and S-9).Therefore, further investigation of the intermediate Pt-activated nanorod structure was conducted to determine if chemical, or structural changes of the nanorod could be observed via XRD or TEM. The incorporation of metallic platinum phases was not detected in XRD for Pt-activated nanorods subjected to optimized activation conditions (30 s Pt-activation), hence, CdSe@CdS nanorods were further subjected to longer Pt-activation times (5 min) to reveal at which point PtNP tips could be observed by conventional characterization methods, such as, TEM and XRD. Even after longer reaction times for Pt-activation (5 min), metallic platinum was still not detected in this sample by low resolution TEM or by XRD as a nanocrystal phase (Fig. 4). The patterns of both samples (activated for 30 s and 5 min) only showed the reflections of the wurtzite CdS nanorods with more intense and sharper 002 reflections due to the preferential growth along this crystallographic direction.
 | ||
| Fig. 4 XRD spectra of CdSe@CdS nanorods after 30 s activation (black line) and 5 min activation (red dashed line) scaled to similar CdSe@CdS intensity. Nanorod dimensions: L = 69.1 ± 5.5 nm, n = 109; D = 6.0 ± 0.5 nm, n = 105. | ||
To further investigate the morphological effects of the Pt-activation reaction on CdSe@CdS nanorods, HRTEM and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) were conducted on nanorods treated for brief (30 s, Fig. 5a) and extended (5 min, Fig. 5b) Pt-activation times. For optimized Pt-activated nanorods (30 s nanorod activation), the deposition of metallic PtNP-tips could not be imaged using HRTEM. However, HAADF-STEM of these Pt-activated nanorods suggested the deposition of very small PtNP-tips, as noted by faint particulate features on nanorod termini (Fig. 5a). Conversely, HAADF-STEM permitted imaging the incorporation of very small high Z-contrast features at the end of CdSe@CdS nanorods subjected to longer Pt-activation times (5 min; Fig. 5b). HRTEM imaging of the Pt-activated CdSe@CdS nanorods subjected to longer Pt-activation times (5 min) revealed the presence of low contrast very small NP features (D ≤ 1 nm) deposited onto the nanorod terminus (Fig. 5c). It is important to note that cobalt deposition experiments onto nanorods subjected to longer Pt-activation times (>30 s) yielded mixtures containing two-sided CoNP-tipped nanorods, which resulted in poorly defined self-assembled morphologies (Fig. 3b).
 | ||
| Fig. 5 (a) HAADF-STEM image of Pt-activated CdSe@CdS nanorods (30 s reaction time), with arrows pointing to few high atomic number atoms. (b) HAADF-STEM image of Pt-activated CdSe@CdS nanorods after 5 min reaction time, with arrows pointing to small (<1 nm) PtNP-tips. (c) HRTEM of Pt-activated CdSe@CdS nanorods after 5 min reaction time. The inset shows the power spectrum showing the 002 reflections of the nanorods in the center of the image. No additional reflections from Pt or other phases could be discerned. | ||
The collective findings of these imaging and variation in Pt-activation studies suggest that a trace amount of platinum that was below the resolution of conventional HRTEM was present on the optimized Pt-activated nanorods (30 s) which enabled subsequent one-sided deposition of dipolar CoNP-tips onto nanorods. Upon treatment of CdSe@CdS nanorods with longer Pt-activation times, the growth of a very small, but observable, PtNP-tip onto one side of the nanorod occurred, as confirmed by HAADF-STEM. However, concurrent chemical etching of the opposing nanorod terminus and lateral facets resulted in an increased tendency towards cobalt deposition to form “dumbbell” CoNP-tipped nanorods.25
Conclusion
The synthesis of dipolar heterostructured nanorods bearing a single CoNP-tip at one terminus per CdSe@CdS nanorod required subtle optimization of the reactivity of CdSe@CdS nanorods using a Pt-activation step. This variation in nanorod reactivity was achieved by leveraging the key Pt-activation step to activate nanorod termini toward the deposition of CoNPs to enable synthesis of the targeted nanomaterial. Complex heterostructured nanocomposites were thus synthesized using nanocrystal reagents as precursors with precise spatial control over heterostructure morphology. This precise control over heterostructure morphology on 1–2 nm length scales was shown to strongly impact the formation of hierarchical structures on mesoscopic length scales ranging several micrometers.Acknowledgements
This work was funded by the Division of Chemical Science, Geosciences and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (DE-FG03-02ER15753) (L.J.H., J.P.), the NSF (DMR-0645618 for N.E.R., J.P. and DMR-130792 for P.T.D., J.P.), the World Class University Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R31-10013) (Y.S.), and the Alfred P. Sloan Foundation (P.T.D., J.P.) are acknowledged for synthetic support of this work. K.C. acknowledges financial support from NRF through the National Creative Research Initiative Center for Intelligent Hybrids (2010-0018290). Dr. Ralf Bienert and Dr. Franziska Emmerling from Bundesanstalt für Materialforschung und -prüfung are acknowledged for XRD measurements.References
- U. Banin, Y. Ben-Shahar and K. Vinokurov, Chem. Mater., 2013, 26, 97–110 CrossRef.
- M. R. Buck and R. E. Schaak, Angew. Chem., Int. Ed., 2013, 52, 6154–6178 CrossRef CAS PubMed.
- R. Costi, A. E. Saunders and U. Banin, Angew. Chem., Int. Ed., 2010, 49, 4878–4897 CrossRef CAS PubMed.
- L. Carbone and P. D. Cozzoli, Nano Today, 2010, 5, 449–493 CrossRef CAS PubMed.
- C. de Mello Donega, Chem. Soc. Rev., 2011, 40, 1512–1546 RSC.
- C. Wang, C. Xu, H. Zeng and S. Sun, Adv. Mater., 2009, 21, 3045–3052 CrossRef CAS PubMed.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed.
- D. J. Milliron, R. Buonsanti, A. Llordes and B. A. Helms, Acc. Chem. Res., 2014, 47, 236–246 CrossRef CAS PubMed.
- H. Gu, R. Zheng, X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 5664–5665 CrossRef CAS PubMed.
- T. Teranishi, Y. Inoue, M. Nakaya, Y. Oumi and T. Sano, J. Am. Chem. Soc., 2004, 126, 9914–9915 CrossRef CAS PubMed.
- H. Gu, Z. Yang, J. Gao, C. K. Chang and B. Xu, J. Am. Chem. Soc., 2005, 127, 34–35 CrossRef CAS PubMed.
- L. Yanqiu, Z. Qiang, A. V. Nurmikko and S. Shouheng, Nano Lett., 2005, 5, 1689–1692 CrossRef PubMed.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS PubMed.
- S.-H. Choi, E.-G. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 2520–2521 CrossRef CAS PubMed.
- M. R. Buck, J. F. Bondi and R. E. Schaak, Nat. Chem., 2012, 4, 37–44 CrossRef CAS PubMed.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS PubMed.
- T. Mokari, C. G. Sztrum, A. Salant, E. Rabani and U. Banin, Nat. Mater., 2005, 4, 855–863 CrossRef CAS.
- P. D. Cozzoli and L. Manna, Nat. Mater., 2005, 4, 801–802 CrossRef CAS.
- P. D. Cozzoli, T. Pellegrino and L. Manna, Chem. Soc. Rev., 2006, 35, 1195–1208 RSC.
- G. Dukovic, M. G. Merkle, J. H. Nelson, S. M. Hughes and A. P. Alivisatos, Adv. Mater., 2008, 20, 4306–4311 CrossRef CAS.
- L. Amirav and A. P. Alivisatos, J. Phys. Chem. Lett., 2010, 1, 1051–1054 CrossRef CAS.
- S. Chakrabortty, J. A. Yang, Y. M. Tan, N. Mishra and Y. T. Chan, Angew. Chem., Int. Ed., 2010, 49, 2888–2892 CrossRef CAS PubMed.
- X. H. Li, J. Lian, M. Lin and Y. T. Chan, J. Am. Chem. Soc., 2011, 133, 672–675 CrossRef CAS PubMed.
- M. G. Alemseghed, T. P. A. Ruberu and J. Vela, Chem. Mater., 2011, 23, 3571–3579 CrossRef CAS.
- L. J. Hill, M. M. Bull, Y. Sung, A. G. Simmonds, P. T. Dirlam, N. E. Richey, S. E. DeRosa, I.-B. Shim, D. Guin, P. J. Costanzo, N. Pinna, M.-G. Willinger, W. Vogel, K. Char and J. Pyun, ACS Nano, 2012, 6, 8632–8645 CrossRef CAS PubMed.
- M. Casavola, V. Grillo, E. Carlino, C. Giannini, F. Gozzo, E. Fernandez Pinel, M. A. Garcia, L. Manna, R. Cingolani and P. D. Cozzoli, Nano Lett., 2007, 7, 1386–1395 CrossRef CAS PubMed.
- S. Deka, A. Falqui, G. Bertoni, C. Sangregorio, G. Poneti, G. Morello, M. De Giorgi, C. Giannini, R. Cingolani, L. Manna and P. D. Cozzoli, J. Am. Chem. Soc., 2009, 131, 12817–12828 CrossRef CAS PubMed.
- J. Maynadie, A. Salant, A. Falqui, M. Respaud, E. Shaviv, U. Banin, K. Soulantica and B. Chaudret, Angew. Chem., Int. Ed., 2009, 48, 1814–1817 CrossRef CAS PubMed.
- M. Casavola, A. Falqui, M. A. Garcia, M. Garcia-Hernandez, C. Giannini, R. Cingolani and P. D. Cozzoli, Nano Lett., 2009, 9, 366–376 CrossRef CAS PubMed.
- J. R. Thomas, J. Appl. Phys., 1966, 37, 2914–2915 CrossRef CAS PubMed.
- V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115–2117 CrossRef CAS PubMed.
- V. F. Puntes, D. Zanchet, C. K. Erdonmez and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 12874–12880 CrossRef CAS PubMed.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- D. L. Huber, Small, 2005, 1, 482–501 CrossRef CAS PubMed.
- J. Gao, B. Zhang, X. Zhang and B. Xu, Angew. Chem., Int. Ed., 2006, 45, 1220–1223 CrossRef CAS PubMed.
- B. D. Korth, P. Keng, I. Shim, S. E. Bowles, C. Tang, T. Kowalewski, K. W. Nebesny and J. Pyun, J. Am. Chem. Soc., 2006, 128, 6562–6563 CrossRef CAS PubMed.
- J. Pyun, Polym. Rev., 2007, 47, 231–263 CrossRef CAS.
- P. Y. Keng, I. Shim, B. D. Korth, J. F. Douglas and J. Pyun, ACS Nano, 2007, 1, 279–292 CrossRef CAS PubMed.
- J. J. Benkoski, S. E. Bowles, B. D. Korth, R. L. Jones, J. F. Douglas, A. Karim and J. Pyun, J. Am. Chem. Soc., 2007, 129, 6291–6297 CrossRef CAS PubMed.
- J. J. Benkoski, S. E. Bowles, R. L. Jones, J. F. Douglas, J. Pyun and A. Karim, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2267–2277 CrossRef CAS.
- P. Y. Keng, B. Y. Kim, I.-B. Shim, R. Sahoo, P. E. Veneman, N. R. Armstrong, H. Yoo, J. E. Pemberton, M. M. Bull, J. J. Griebel, E. L. Ratcliff, K. G. Nebesny and J. Pyun, ACS Nano, 2009, 3, 3143–3157 CrossRef CAS PubMed.
- M. M. Bull, W. J. Chung, S. R. Anderson, S.-J. Kim, I.-B. Shim, H.-J. Paik and J. Pyun, J. Mater. Chem., 2010, 20, 6023–6025 RSC.
- J. G. Railsback, A. C. Johnston-Peck, J. Wang and J. B. Tracy, ACS Nano, 2010, 4, 1913–1920 CrossRef CAS PubMed.
- B. Y. Kim, I.-B. Shim, Z. O. Araci, S. S. Saavedra, O. L. A. Monti, N. R. Armstrong, R. Sahoo, D. N. Srivastava and J. Pyun, J. Am. Chem. Soc., 2010, 132, 3234–3235 CrossRef CAS PubMed.
- B. Y. Kim, I.-B. Shim, O. L. A. Monti and J. Pyun, Chem. Commun., 2011, 47, 890–892 RSC.
- P. Y. Keng, M. M. Bull, I.-B. Shim, K. G. Nebesny, N. R. Armstrong, Y. Sung, K. Char and J. Pyun, Chem. Mater., 2011, 23, 1120–1129 CrossRef CAS.
- B. Y. Kim, S.-H. Yu, H. S. Kim, D.-C. Lee, I.-B. Shim, S. E. Derosa, Y.-E. Sung and J. Pyun, J. Mater. Chem., 2011, 21, 14163–14166 RSC.
- K. S. Khalil, A. Sagastegui, Y. Li, M. A. Tahir, J. E. S. Socolar, B. J. Wiley and B. B. Yellen, Nat. Commun., 2012, 3, 1798 Search PubMed.
- L. Hill and J. Pyun, ACS Appl. Mater. Interfaces, 2014 DOI:10.1021/am405786u.
- L. J. Hill, N. E. Richey, Y. Sung, P. T. Dirlam, E. Lavoie-Higgins, I.-B. Shim, N. Pinna, M.-G. Willinger, W. Vogel, J. J. Benkoski, K. Char and J. Pyun, ACS Nano, 2014, 8, 3272–3284 CrossRef CAS PubMed.
- O. Paris, C. Li, S. Siegel, G. Weseloh, F. Emmerling, H. Riesemeier, A. Erko and P. Fratzl, J. Appl. Crystallogr., 2007, 40, s466–s470 CrossRef CAS.
- W. W. Yu, L. H. Qu, W. Z. Guo and X. G. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- S. E. Habas, P. D. Yang and T. Mokari, J. Am. Chem. Soc., 2008, 130, 3294–3295 CrossRef CAS PubMed.
- Z. H. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS PubMed.
- K. Liu, Z. Nie, N. Zhao, W. Li, M. Rubinstein and E. Kumacheva, Science, 2010, 329, 197–200 CrossRef CAS PubMed.
- K. Liu, N. Zhao and E. Kumacheva, Chem. Soc. Rev., 2011, 40, 656–671 RSC.
- G. A. DeVries, M. Brunnbauer, Y. Hu, A. M. Jackson, B. Long, B. T. Neltner, O. Uzun, B. H. Wunsch and F. Stellacci, Science, 2007, 315, 358–361 CrossRef CAS PubMed.
- K. Nakata, Y. Hu, O. Uzun, O. Bakr and F. Stellacci, Adv. Mater., 2008, 20, 4294–4299 CrossRef CAS.
- A. Klinkova, H. Thérien-Aubin, R. M. Choueiri, M. Rubinstein and E. Kumacheva, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18775–18779 CrossRef CAS PubMed.
- K. Liu, A. Lukach, K. Sugikawa, S. Chung, J. Vickery, H. Therien-Aubin, B. Yang, M. Rubinstein and E. Kumacheva, Angew. Chem., Int. Ed., 2014, 10, 2648–2653 CrossRef PubMed.
- H. Wang, L. Chen, X. Shen, L. Zhu, J. He and H. Chen, Angew. Chem., Int. Ed., 2012, 51, 8021–8025 CrossRef CAS PubMed.
- M. X. Yang, G. Chen, Y. F. Zhao, G. Silber, Y. Wang, S. X. Xing, Y. Han and H. Y. Chen, Phys. Chem. Chem. Phys., 2010, 12, 11850–11860 RSC.
- L. H. Tan, S. X. Xing, T. Chen, G. Chen, X. Huang, H. Zhang and H. Y. Chen, ACS Nano, 2009, 3, 3469–3474 CrossRef CAS PubMed.
- X. Wang, G. Li, T. Chen, M. Yang, Z. Zhang, T. Wu and H. Chen, Nano Lett., 2008, 8, 2643–2647 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ce00680a |
| This journal is © The Royal Society of Chemistry 2014 |