 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lutidine adducts of urea: molecular mechanisms for twinning effects on cooling†
Christina
Taouss
and
Peter G.
Jones
*
Institut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Postfach 3329, D-38023 Braunschweig, Germany. E-mail: p.jones@tu-bs.de; Fax: +49 531 5387; Tel: +49 531 5382
First published on 6th May 2014
Abstract
The structures of three adducts between urea and 2,6- or 3,5-lutidine have been determined: urea![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2,6-lutidine (1:1) (1), urea:2,6-lutidine (2:1) (2) and urea:3,5-lutidine (2:1) (3). Adducts 1 and 3 each crystallize as different polymorphs depending on the temperature. In each of the five structures, the lutidine nitrogen acts as a hydrogen bond acceptor for both NH groups of a urea molecule, and the lutidine molecules form infinite stacks. The known structure of 1 in space group C2/c (1m), previously known only from photographic data in projection, was redetermined at 200 K. Both residues display crystallographic twofold symmetry. The urea substructure is a ribbon of [R22(8)] rings. Further cooling results in a reduction in symmetry to P
:2,6-lutidine (1:1) (1), urea:2,6-lutidine (2:1) (2) and urea:3,5-lutidine (2:1) (3). Adducts 1 and 3 each crystallize as different polymorphs depending on the temperature. In each of the five structures, the lutidine nitrogen acts as a hydrogen bond acceptor for both NH groups of a urea molecule, and the lutidine molecules form infinite stacks. The known structure of 1 in space group C2/c (1m), previously known only from photographic data in projection, was redetermined at 200 K. Both residues display crystallographic twofold symmetry. The urea substructure is a ribbon of [R22(8)] rings. Further cooling results in a reduction in symmetry to P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (structure 1t); crystals are non-merohedrally twinned by 180° rotation about (b*–c*), which is the same direction as the b axis of 1m. The qualitative structure remains almost unchanged, but the lutidine rings tilt slightly away from the twofold axis (which is lost on twinning). The three structures involving 2:1 adducts all display a urea substructure in the form of a corrugated layer, from which urea molecules project and form hydrogen bonds to the lutidines. The urea layers are formed from ribbons of [R22(8)] rings that are linked by lateral hydrogen bonds. However, the linkages are in all three cases subtly different. Adduct 2 crystallizes in Pnma with the lutidine and both ureas displaying mirror symmetry. Adduct 3 crystallizes in space group Abm2 (3o) at 173 K, with two lutidines and four ureas all displaying crystallographic mirror symmetry; on cooling to 100 K, the space group changes to Cc (3m) and the crystals form reticular pseudo-merohedral twins by 180° rotation about a*, probably as a consequence of the mutual displacement of the urea ribbons, parallel to their direction of propagation, to accommodate a change in hydrogen bonding.
(structure 1t); crystals are non-merohedrally twinned by 180° rotation about (b*–c*), which is the same direction as the b axis of 1m. The qualitative structure remains almost unchanged, but the lutidine rings tilt slightly away from the twofold axis (which is lost on twinning). The three structures involving 2:1 adducts all display a urea substructure in the form of a corrugated layer, from which urea molecules project and form hydrogen bonds to the lutidines. The urea layers are formed from ribbons of [R22(8)] rings that are linked by lateral hydrogen bonds. However, the linkages are in all three cases subtly different. Adduct 2 crystallizes in Pnma with the lutidine and both ureas displaying mirror symmetry. Adduct 3 crystallizes in space group Abm2 (3o) at 173 K, with two lutidines and four ureas all displaying crystallographic mirror symmetry; on cooling to 100 K, the space group changes to Cc (3m) and the crystals form reticular pseudo-merohedral twins by 180° rotation about a*, probably as a consequence of the mutual displacement of the urea ribbons, parallel to their direction of propagation, to accommodate a change in hydrogen bonding.
Introduction
We are interested in simple solvates of urea and thiourea, which we define as their adducts with compounds that are liquids at room temperature, and have recently reported the structures of urea:morpholine (1:1), urea:1,4-dioxane (1:1), thiourea:morpholine (4:3) and thiourea:1,4-dioxane (4:1);1 this article may also be consulted for a brief summary of the relevant literature. The 1:1 adduct of urea with 2,6-lutidine, henceforth adduct 1, was determined in space group C2/c from photographic data in projection,2 and it seemed worthwhile to repeat this structure using more modern methods, and to investigate the 2:1 adduct 2 whose existence, but not structure, was reported in the same paper. A related 1:1 adduct is that of urea with 2-picoline.3 We also wished to extend the series of urea adducts with liquid pyridine derivatives, and the obvious first choice was 3,5-lutidine. Here we report the structures of urea:2,6-lutidine (2:1) 2, two polymorphs of urea:2,6-lutidine (1:1) 1, and two polymorphs of urea:3,5-lutidine (2:1) 3. In each case the polymorph that is formed on cooling is twinned.
Twinning is a crystallographic topic that is often regarded as “difficult” and thus the preserve of a handful of experts,4 and some of the more mathematical treatments are indeed impenetrable to the everyday chemist. However, diffractometer software and structure refinement programs have now been improved to the extent that twinning has become a more accessible phenomenon, and the practising crystallographer can often acquire all the knowledge necessary for the successful analysis of most twinned structures. Less well understood is the question as to how twins are formed; for molecular structures, we are aware of few examinations5 of the molecular mechanism of twinning (although it is difficult to perform an exhaustive literature search and we may have missed other examples). Here, the relationships between the polymorph pairs of 1 and 3 are analysed in detail, which provides a basis for explaining the molecular mechanisms of the twinning phenomena.
Experimental
Crystal growth
Adduct 1 was obtained as single crystals by overlayering a solution of urea in methanol with 2,6-lutidine. Crystals of the 2:1 adduct 2 formed when a solution of urea in methanol and 2,6-lutidine was overlayered with diethyl ether. For 3,5-lutidine, we were only able to crystallize the 2:1 adduct 3, by allowing a solution of urea in 3,5-lutidine and methanol to stand for several days. All the adducts lose lutidine under ambient conditions.
X-ray crystallography
Unexpectedly, initial crystallographic investigations of the 1:1 adduct 1 of urea with 2,6-lutidine did not reveal the same cell as that found previously;2 at 100 K the diffraction patterns invariably showed split reflections, and could be indexed successfully as (non-merohedrally) twinned triclinic (1t) rather than monoclinic (1m). Despite the twinning, the structure was successfully determined. The structure of the 2:1 adduct 2 was registered in a straightforward manner. Crystals of the 2:1 adduct with 3,5-lutidine 3 were orthorhombic (3o) and showed a marked tendency to shatter at low temperatures (sometimes immediately, sometimes after a few hours); the structure determination of 3o was finally accomplished at 173 K. This led us to suppose that adduct 1 might behave in a similar fashion, and thus that the structure 1m might be successfully redetermined at somewhat higher temperatures; this was indeed done at 200 K. Finally, the structure of a monoclinic form of 3 (3m) was determined by cooling the orthorhombic form slowly to 100 K; many crystals shattered but one, although twinned, was successfully measured.
Details of intensity measurements and refinements are given in Table 1. Crystals were mounted in inert oil on glass fibres. Data were measured with an Oxford Diffraction Xcalibur E diffractometer using monochromated Mo Kα radiation or with an Oxford Diffraction Nova A diffractometer using mirror-focussed Cu Kα radiation; multi-scan absorption corrections were performed in some cases.6 The structures were solved with direct methods using SHELXS-97, and structure refinement was performed with full-matrix least-squares on F2 using SHELXL-977a or SHELXL-2012.7b NH hydrogens were refined freely and aromatic hydrogens using a riding model. Well-defined methyl groups were included as idealised rigid groups allowed to rotate but not tip, but the hydrogen sites were in some cases indistinct and were approximated where necessary as a hexagon of half-occupied hydrogen sites corresponding to rotational disorder; in other cases the methyl hydrogens were disordered over special positions (details are given in the deposited CIF files). Molecular graphics were prepared with XP.8
| Solvate | Urea:2,6-lutidine (1:1), 1m |
Urea:2,6-lutidine (1:1), 1t |
Urea:2,6-lutidine (2:1), 2 |
Urea:3,5-lutidine (2:1), 3o |
Urea:3,5-lutidine (2:1), 3m |
|---|---|---|---|---|---|
| Chemical formula | C8H13N3O | C8H13N3O | C9H17N5O2 | C9H17N5O2 | C9H17N5O2 |
| M r | 167.21 | 167.21 | 227.28 | 227.28 | 227.28 |
| Crystal system, space group | Monoclinic, C2/c | Triclinic, P |
Orthorhombic, Pnma | Orthorhombic, Abm2 | Monoclinic, Cc |
| Temperature (K) | 200 | 100 | 100 | 173 | 100 |
| a (Å) | 11.426(2) | 7.4126(6) | 8.0772(3) | 21.7368(15) | 8.5829(4) |
| b (Å) | 11.1168(9) | 7.6720(6) | 7.2986(3) | 7.2102(6) | 21.4843(8) |
| c (Å) | 7.4318(8) | 8.1731(6) | 20.4169(7) | 15.5904(15) | 7.2050(4) |
| α (°) | 90 | 88.391(6) | 90 | 90 | 90 |
| β (°) | 101.235(15) | 83.564(6) | 90 | 90 | 114.405(6) |
| γ (°) | 90 | 80.059(6) | 90 | 90 | 90 |
| V (Å3) | 925.9 | 454.92 | 1203.64 | 2443.4 | 1209.87 |
| d calc (g cm3) | 1.200 | 1.221 | 1.254 | 1.236 | 1.248 |
| Z | 4 | 2 | 4 | 8 | 4 |
| Radiation, wavelength | Mo Kα, λ = 0.71073 Å | Cu Kα, λ = 1.54184 Å | Mo Kα, λ = 0.71073 Å | Mo Kα, λ = 0.71073 Å | Cu Kα, λ = 1.54184 Å |
| F(000) | 360 | 180 | 488 | 976 | 488 |
| μ (mm−1) | 0.08 | 0.7 | 0.09 | 0.09 | 0.8 |
| Crystal size (mm) | 0.25 × 0.25 × 0.08 | 0.20 × 0.15 × 0.03 | 0.35 × 0.15 × 0.15 | 0.3 × 0.13 × 0.02 | 0.2 × 0.16 × 0.08 |
| Transmissions | No abs. corr. | 0.80–1.00 | No abs. corr. | 0.93–1.00 | 0.87–1.00 |
| 2θmax (°) | 50 | 152 | 58.7 | 52.7 | 153 |
| No. of measured and independent reflections | 5818, 819 | 3650 | 38287, 1710 |
21459, 1409 |
27700, 2403 |
| Completeness | 100% to 2θmax | 99.9% to 2θ 150° | 99.8% to 2θ 57° | 99.9% to 2θmax | 99.9% to 2θ 150° |
| R int | 0.033 | — | 0.029 | 0.060 | 0.052 |
| wR(F2) all refl., R1 [F > 4σ(F)], S(F2) | 0.134, 0.053, 1.04 | 0.107, 0.039, 1.05 | 0.110, 0.041, 1.06 | 0.089, 0.040, 1.04 | 0.132, 0.047, 1.08 |
| No. of parameters/restraints | 66/0 | 128/0 | 108/0 | 217/29 | 173/36 |
| Δρmax,min (e Å−3) | 0.27, −0.24 | 0.18, −0.20 | 0.45, −0.33 | 0.13, −0.18 | 0.36, −0.25 |
The atom numbering employed here does not always conform to the standard urea numbering, with N1–C2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N3, because of the frequent occurrence of crystallographic symmetry within the urea molecule. The urea hydrogens are numbered as H0x, whereby x is odd if the hydrogen is trans to OC across the N–C bond and even if it is cis.
O)–N3, because of the frequent occurrence of crystallographic symmetry within the urea molecule. The urea hydrogens are numbered as H0x, whereby x is odd if the hydrogen is trans to OC across the N–C bond and even if it is cis.
7b (but not in earlier versions of the program) by simply giving the twin matrix; the relative volume of the smaller component refined to 0.220(7). Because of the poorer data/parameter ratio of 3m (Cu radiation, non-centrosymmetric structure), its N–H and amino H⋯H distances were restrained to be equal, and the NH hydrogens were refined using a common displacement factor. For the non-centrosymmetric structure 3o, amino N⋯H distances were restrained to be equal. The anomalous dispersion of 3o with Mo Kα radiation was negligible; the Friedel pairs were merged and the Flack parameter is thus meaningless. For 3m, the anomalous scattering was just significant, and the absolute structure parameter was determined as 0.06(14).11 Attempts to allow for inversion twinning gave two additional BASF parameters (the relative volumes of the additional twinning components) not significantly different from zero and were therefore abandoned.
Results and discussion
Crystal structure of urea![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :2,6-lutidine (1:1), higher temperature form (1m)
:2,6-lutidine (1:1), higher temperature form (1m)
The redetermination of the structure of solvate 1m, measured at −73 °C, confirms the qualitative features reported previously.2 It crystallizes in C2/c with Z = 4. The formula unit is shown in Fig. 1, The atoms C1, O1, N11 and C14 lie on the twofold axis at x = 1/2, z = 3/4, so that both residues display crystallographic twofold symmetry. There is clearly considerable libration of the lutidine ring, and its dimensions should be interpreted with caution. Attempts to refine alternative models (in Cc; in P as a pseudomerohedral twin; or disordered in C2/c) led to no significant improvement.
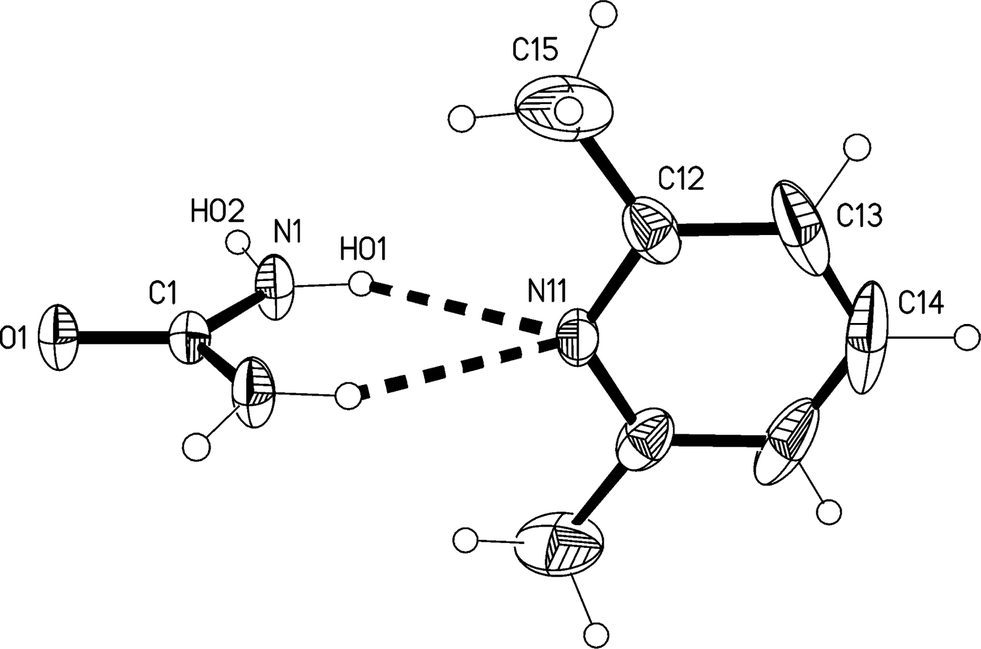 | ||
| Fig. 1 The formula unit of adduct 1m in the crystal. Ellipsoids correspond to 30% probability levels. Only the asymmetric unit is numbered. The dashed lines correspond to the classical hydrogen bonds within the formula unit. | ||
The molecular packing involves two classical hydrogen bonds, one of which is bifurcated, and one “weak” interaction (Table 2). The two-centre interaction N1–H02⋯O1 links the urea molecules to form a ribbon, consisting of linked [R22(8)] rings, parallel to the c axis (Fig. 2); such ribbons are a well-established feature of urea adducts.2,3,12 The bifurcated interaction (N1–H01)2⋯N11, part of an [R12(6)] ring, connects the lutidine residues peripherally to these ribbons, whereby the angle C1⋯N11⋯C14 is exactly 180° by symmetry. The weak interaction C14–H14⋯O1 links the ribbons to form layers parallel to the bc plane; the acceptor geometry at O1 is unusual, but this is sometimes observed for urea derivatives, especially where the oxygen is an acceptor of more than two hydrogen bonds,1 and even in urea itself.13 A view along the c axis (Fig. 3) shows ring stacking, with a vertical distance between rings of 3.71 Å and an offset of 0.9 Å.
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
|---|---|---|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: (i) −x + 1, −y, −z + 1; (ii) x, y + 1, z. | |||||
| 1 | N1–H01⋯N11 | 0.86(3) | 2.37(2) | 3.178(3) | 155(2) |
| 2 | N1–H02⋯O1i | 0.91(2) | 2.03(3) | 2.940(2) | 177(2) |
| 3 | C14–H14⋯O1ii | 0.95 | 2.51 | 3.460(4) | 180 |
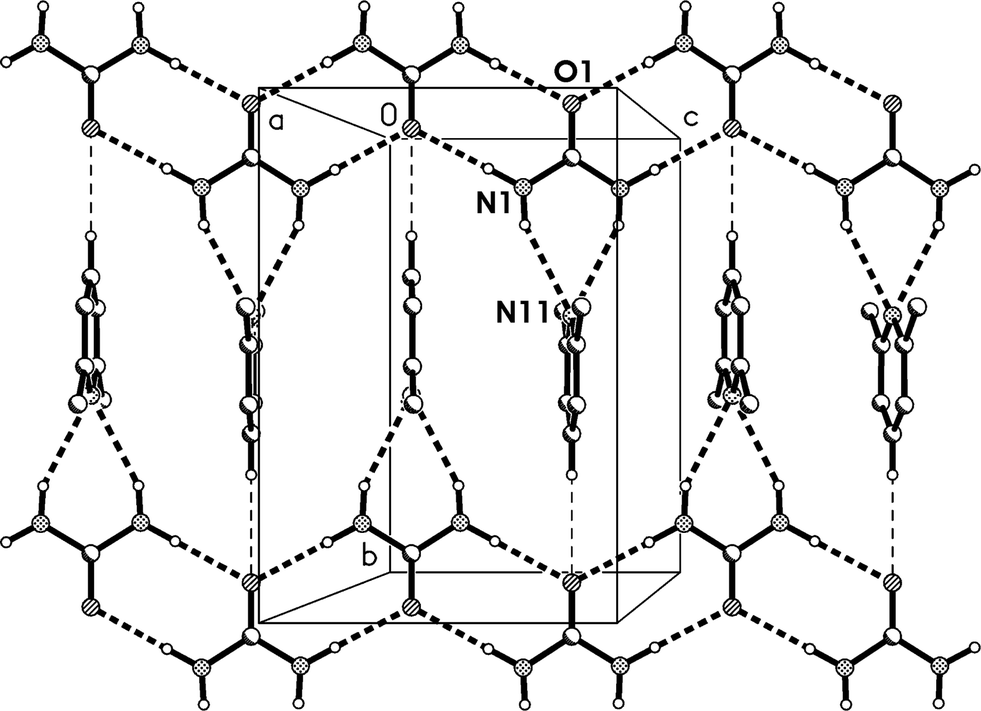 | ||
| Fig. 2 Packing diagram of adduct 1m with view direction perpendicular to the bc plane in the region x ≈ 1/2. Classical hydrogen bonds are drawn as thick and “weak” hydrogen bonds as thin dashed lines. | ||
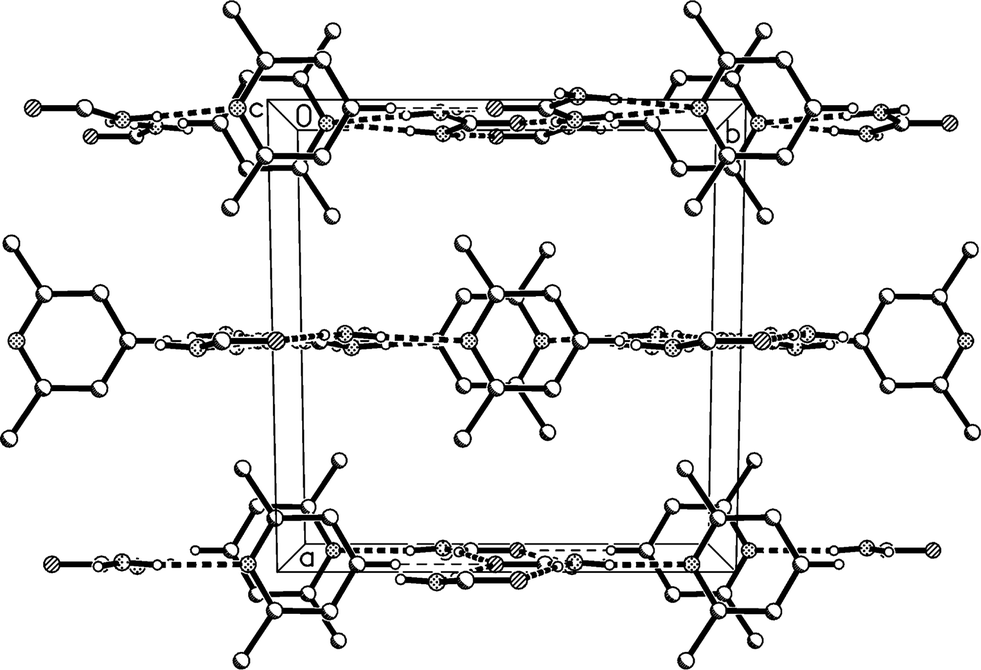 | ||
| Fig. 3 Packing diagram of adduct 1m. The view direction is parallel to the c axis. Classical hydrogen bonds are drawn as thick and “weak” hydrogen bonds as thin dashed lines. | ||
At this stage it is worth emphasising the two features that, with some modifications, are common to all five structures presented here. The first is the presence of ribbons of linked [R22(8)] rings. These are of an extremely simple form in 1m, being built up from just half a urea molecule in the asymmetric unit and being effectively planar (Fig. 2, 3). However, because the oxygens can accept a variable number of classical hydrogen bonds, the ribbons have in principle the ability to interlink; furthermore, they have some degree of torsional freedom and can twist both within a single ring, or along a ribbon (or both), or undergo twisting and/or translation with respect to a neighbouring and/or linked ribbon. The second is the lutidine stacking. A third such feature, relevant only for the 2:1 adducts, is discussed below.
Crystal structure of urea:2,6-lutidine (1:1), lower temperature form (1t)
The new polymorph 1t, measured at −173 °C, crystallizes in P with no imposed crystallographic symmetry and Z = 2; the structure is qualitatively similar to 1m, but the extreme libration is no longer observed. The formula unit is shown in Fig. 4. Corresponding to the loss of crystallographic symmetry, the two residues are now significantly tilted with respect to each other, with C2⋯N11⋯C14 171.17(3)°. A similar effect was noted for lithium acetylacetonate, which changes on cooling from orthorhombic to twinned monoclinic and thereby from a linear to a slightly curved chain structure.5a
 | ||
| Fig. 4 The formula unit of adduct 1t in the crystal. Ellipsoids correspond to 50% probability levels. The dashed lines correspond to the classical hydrogen bonds within the formula unit. | ||
The molecular packing is also closely similar to that of 1m, but with the following differences: the ribbon direction is parallel to the a axis, the layers are parallel to (011), and the tilting is incorporated as a feature of the packing (Table 3, Fig. 5). The lutidine rings stack with a spacing of 3.70 and an offset of 1.0 Å (Fig. 6).
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
|---|---|---|---|---|---|
| a Symmetry transformation used to generate equivalent atoms: (i) −x, −y + 2, −z; (ii) −x + 1, −y + 2, −z; (iii) x, y − 1, z + 1. | |||||
| 1 | N1–H01⋯N11 | 0.858(17) | 2.351(17) | 3.1482(14) | 154.9(14) |
| 2 | N3–H03⋯N11 | 0.896(17) | 2.345(17) | 3.1743(14) | 153.9(13) |
| 3 | N1–H02⋯O1i | 0.905(18) | 2.018(18) | 2.9225(13) | 177.3(14) |
| 4 | N3–H04⋯O1ii | 0.901(17) | 2.049(17) | 2.9493(13) | 177.0(13) |
| 5 | C14–H14⋯O1iii | 0.95 | 2.55 | 3.4396(10) | 156 |
 | ||
| Fig. 5 Packing diagram of adduct 1t with view direction perpendicular to (011) in the region z ≈ 1/4. Classical hydrogen bonds are drawn as thick and “weak” hydrogen bonds as thin dashed lines. | ||
 | ||
| Fig. 6 Packing diagram of adduct 1t with the view direction parallel to the a axis. Classical hydrogen bonds are drawn as thick and “weak” hydrogen bonds as thin dashed lines. | ||
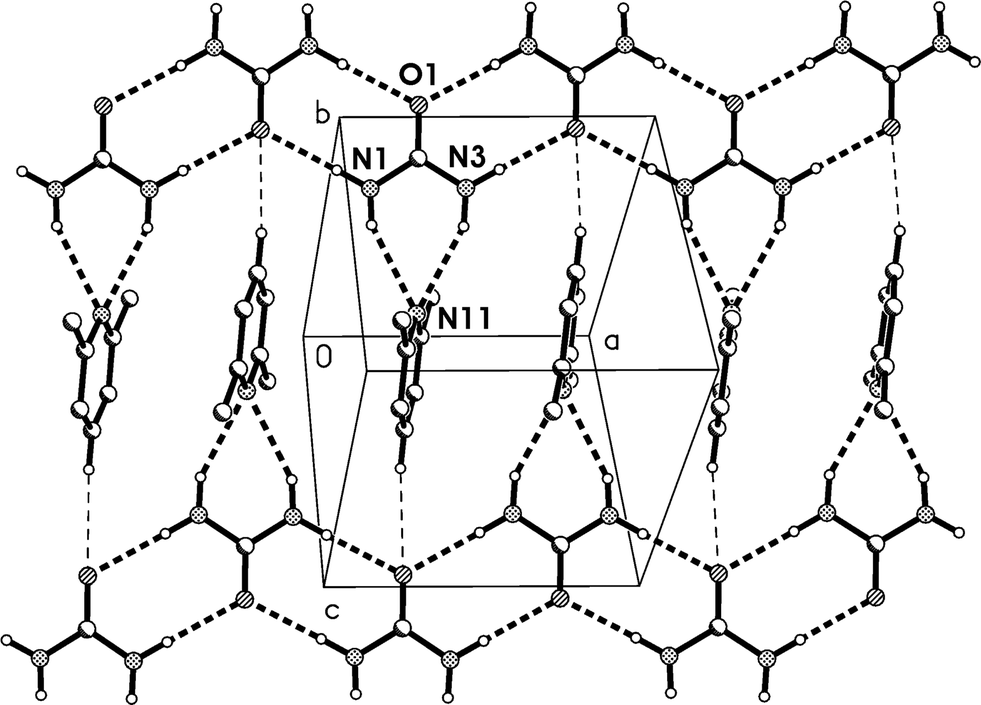
Relationship between the cells of 1t and 1m
The C-centred monoclinic cell of 1m can be converted into a triclinic cell approximately resembling that of 1t by the appropriate matrix:and the inverse matrix can be used to convert the 1t cell back, approximately, to 1m:
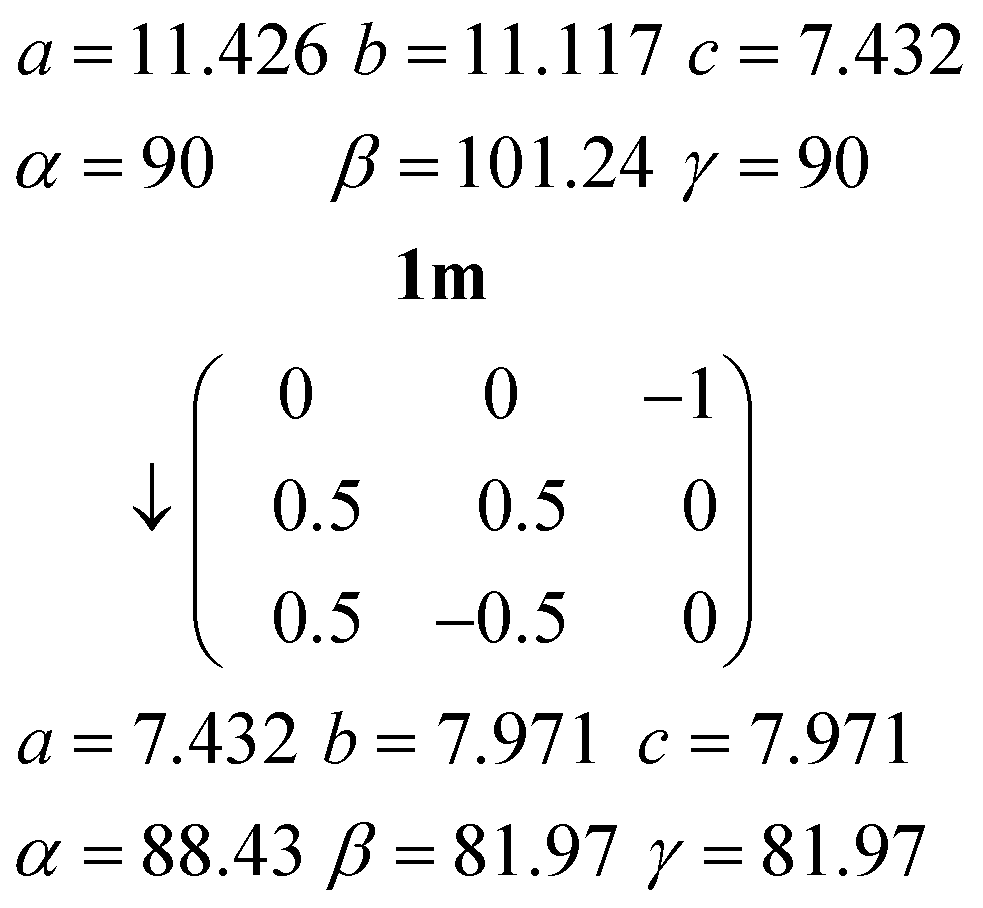
where cell constants are as usual in Å and degrees.
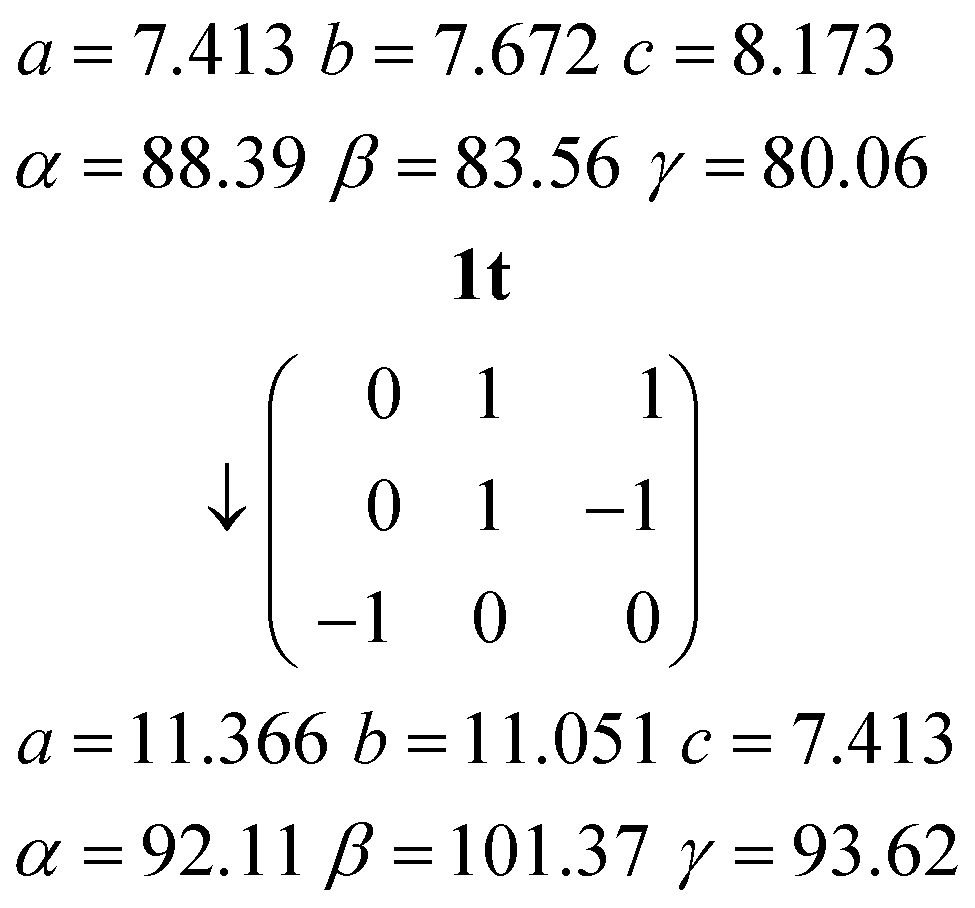
The relevant directions of both structures are then related in the same way; the urea ribbon direction of 1m (its c axis) is physically the same as that of 1t (its a axis), etc. The twinning may be interpreted as the reduction of libration in 1m by a tilting of the structure to either side of the monoclinic b axis, which then ceases to be a symmetry axis as the symmetry is lowered; the twinning components of 1t are related around the direction (b*–c*), which by the usual matrix methods is shown to be the same as the b axis of 1m. This axis changes from being a crystallographic axis to being a twin axis (the axis about which the twin law acts), which is a common feature of induced twinning. As a further check of the interrelationship, a fragment of the same crystal used to determine the structure of 1m was cooled to 100 K, whereupon only the twinned cell 1t could be found. The crystal did however not survive intact on re-warming, so the reversibility of the twinning was not tested.
The density of 1t (1.221 g cm−3) is greater than that of 1m (1.200 g cm−3), which might indicate better packing efficiency and higher lattice energy, but the values may not be strictly comparable because of the temperature difference.
Crystal structure of urea:2,6-lutidine (2:1)
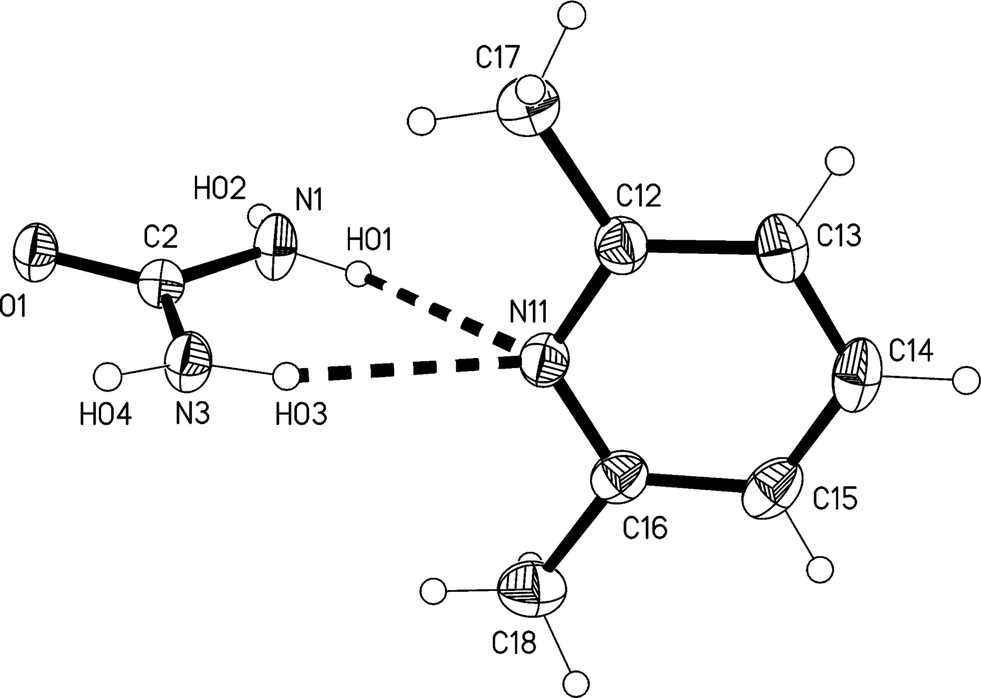
The 2:1 solvate 2, measured at −173 °C, crystallizes in Pnma with Z = 4. The asymmetric unit consists of a lutidine molecule lying in the mirror plane at z = 3/4 and two urea molecules #1 and #2 with their CO bonds (C1O1 and C2O2) lying in the mirror planes at z = 3/4 and 1/4 respectively (see Fig. 7). The bifurcated interaction of urea #1 to the lutidine is qualitatively the same as observed for the adducts 1m and 1t, with an inclination angle C1⋯N11⋯C14 of 170.44(7)°. The two ureas of the asymmetric unit are connected to form an [R22(8)] ring, but subtend an interplanar angle of 20.25(9)°.
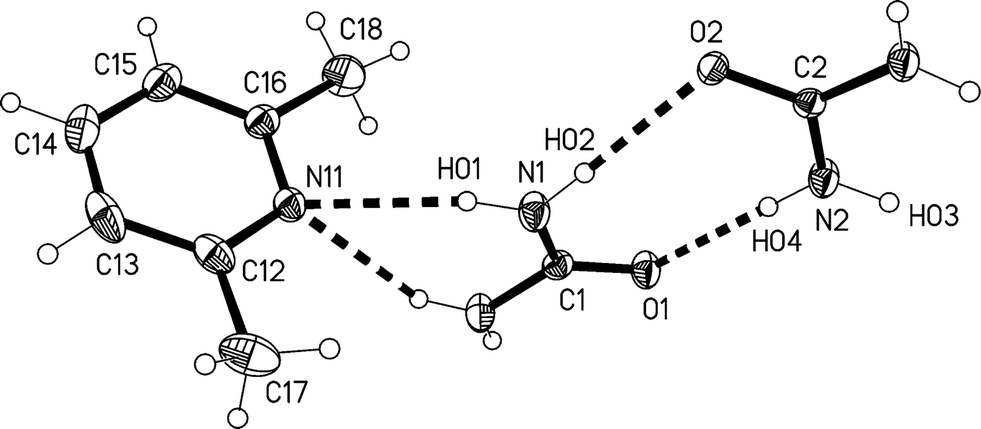 | ||
| Fig. 7 The formula unit of adduct 2 in the crystal. Ellipsoids correspond to 50% probability levels. Only the asymmetric unit is numbered. The dashed lines correspond to the classical hydrogen bonds within the formula unit. | ||
The residues pack to form corrugated layers of urea molecules parallel to the ab plane, perpendicular to which the lutidines are attached via urea molecules that project from the layers (Fig. 8a, Table 4). The urea layers contain two standard motifs: first, ribbons of [R22(8)] rings parallel to the b axis and secondly, chains of rings, graph set C(4)[R12(6)], that lie parallel to the a axis and link the ribbons. The chains involve only the second urea molecule, whereby O2 accepts four hydrogen bonds (two in the chain and two in the ribbon). Such chains of rings represent a robust supramolecular synthon in the structure of urea itself13 and of many N,N′-substituted ureas.14 As far as we are aware, such a two-dimensional urea substructure has not been observed before in any urea solvate.
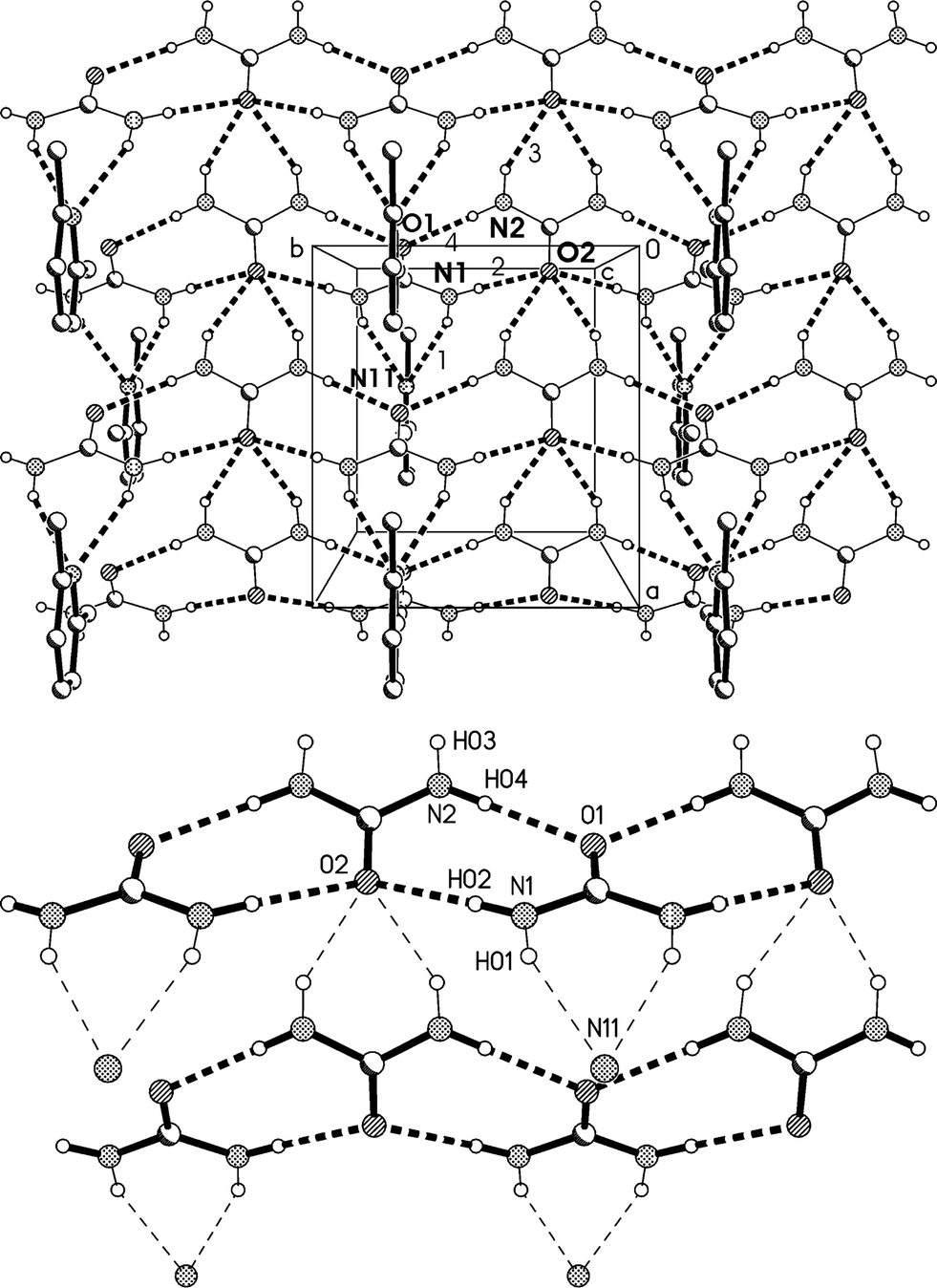 | ||
| Fig. 8 (a), Above: packing diagram of 2 with view direction parallel to the c axis in the region z ≈ 1/4; the mirror planes run vertically at y = 1/4 and 3/4. Hydrogen bonds are drawn as thick dashed lines and numbered according to Table 4. The urea ribbons run horizontally (e.g. at the top of the diagram) and the chains of rings vertically (e.g. immediately to the left of the cell). (b), Below: simplified version (see text). | ||
Fig. 9 presents a view of the packing parallel to the b axis, and shows the corrugated nature of the urea layers with the projecting urea molecules. The lutidine rings stack with a vertical spacing of 3.65 and an offset of 1.0 Å.
 | ||
| Fig. 9 Packing diagram of 2 with view direction parallel to the b axis. Hydrogen bonds are drawn as thin dashed lines. | ||
The urea layers represent a common feature of the 2:1 adducts 2 and 3 but, although broadly similar in form, nonetheless display some significant differences. In 2 the ribbons are of a very simple form, with mirror planes passing through each carbonyl group; the ribbons are linked by “extra” hydrogen bonds to the quadruple acceptor O2, whereby the linked ribbons show little or no mutual horizontal displacement (Fig. 8b). The simplicity of this structure means that it can be regarded as a standard for two-dimensional urea layers; the standard undergoes considerable modification in the structures of 3 (see below).
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
|---|---|---|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: (i) x − 1/2, y, −z + 1/2. | |||||
| 1 | N1–H01⋯N11 | 0.869(15) | 2.318(16) | 3.1248(15) | 154.4(13) |
| 2 | N1–H02⋯O2 | 0.871(16) | 2.102(16) | 2.9698(11) | 174.2(13) |
| 3 | N2–H03⋯O2i | 0.878(15) | 2.192(16) | 2.9963(13) | 152.1(14) |
| 4 | N2–H04⋯O1 | 0.870(15) | 2.018(16) | 2.8879(11) | 179.5(13) |
Crystal structure of urea:3,5-lutidine (2:1), higher temperature form (3o)
The higher temperature form 3o of the solvate 3, measured at −100 °C, crystallizes in the non-centrosymmetric orthorhombic space group Abm2 (ref. 15) with Z = 8. The asymmetric unit was chosen to form a complete unit with as many hydrogen bonds as possible; the two independent lutidine molecules then lie in the mirror plane at z = 3/4 and the four independent urea molecules lie with their CO bonds in the mirror planes at z = 1/4 (molecules #2 & #3) and 3/4 (molecules #1 & #4) (Fig. 10). Again, the bifurcated interactions of urea to lutidine are qualitatively the same as observed above, with inclination angles C1⋯N11⋯C14 170.9(2) and C4⋯N21⋯C24 162.7(2)°. The urea pairs that form the [R22(8)] rings subtend interplanar angles of 29.0(2) (ureas #1 & #2) and 35.0(2)° (ureas #3 & #4).
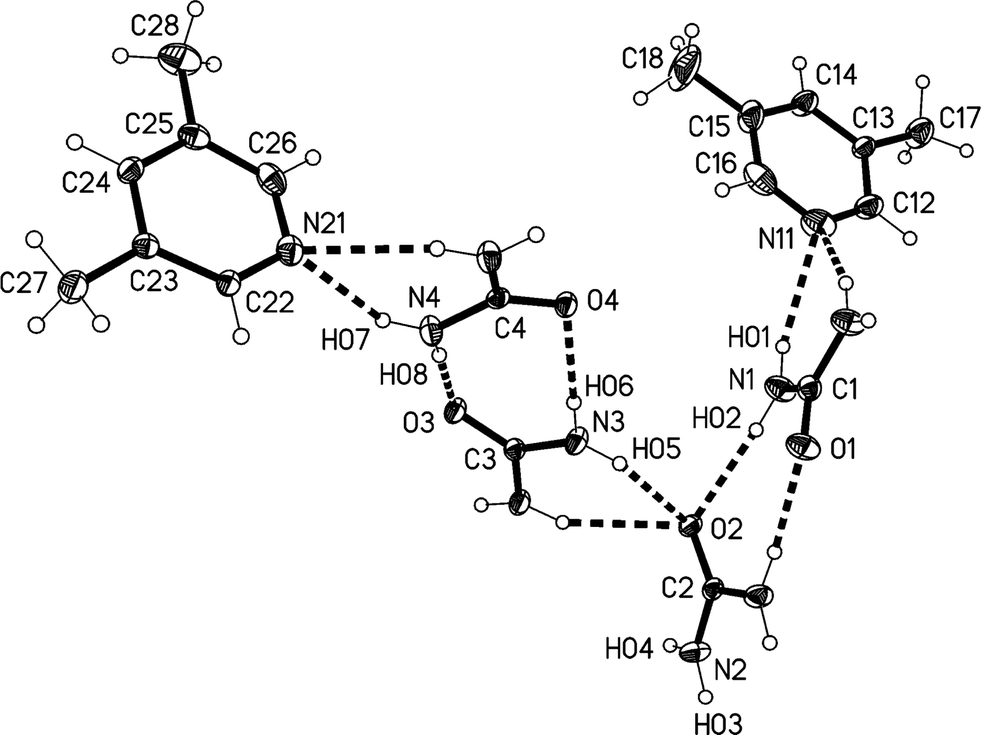 | ||
| Fig. 10 The double formula unit of adduct 3o in the crystal. Ellipsoids correspond to 30% probability levels. Only the asymmetric unit is numbered. The dashed lines correspond to classical hydrogen bonds. | ||
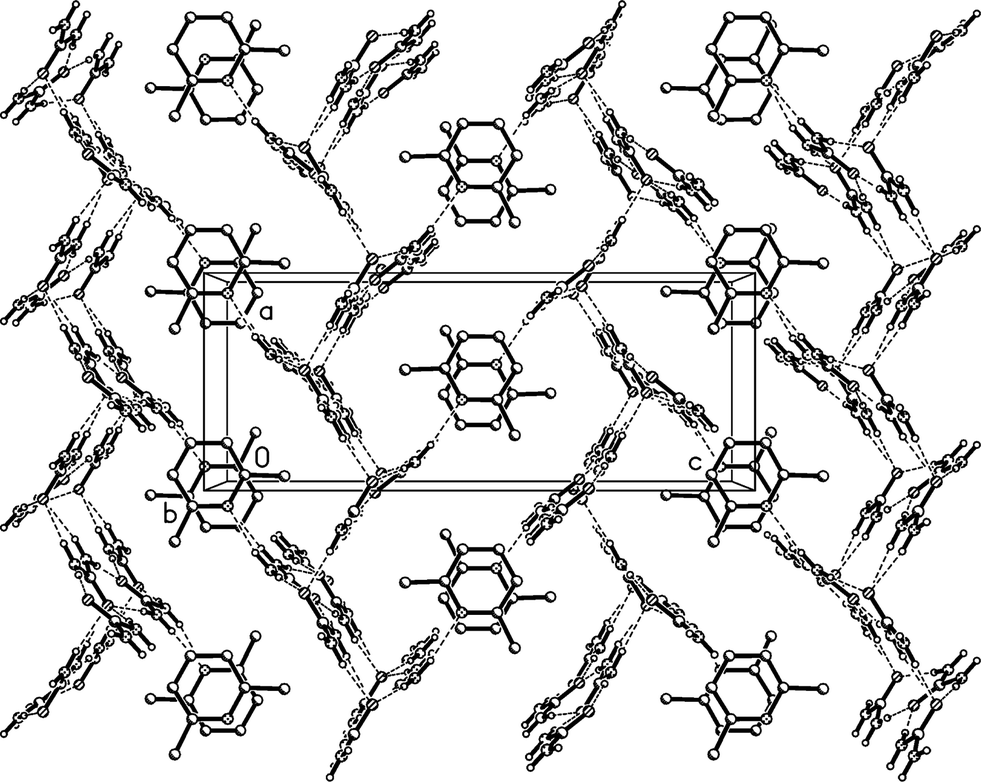
The residues pack in a way reminiscent of adduct 2 to form corrugated layers of urea molecules parallel to the bc plane (Fig. 11a, Table 5). There are two crystallographically distinct types of ribbons of [R22(8)] urea rings, one based on ureas #1 and #2, the other on ureas #3 and #4; they both run parallel to the b axis. Although a cursory inspection of the packing diagrams (especially Fig. 12) might suggest a packing closely analogous to that of 2, there are some important differences (Fig. 11b). First, the quadruple hydrogen bond acceptors O2 and O4 behave in a subtly different manner; although both accept two hydrogen bonds from the ribbon below them, O4 is formally on the “wrong” side of the [R22(8)] ring to do this (it is enabled to do so by the mutual tilting of the two ribbons involved). Secondly, there is a discontinuity after every second ribbon, whereby the following ribbons are shifted sideways by one ring; thus the urea [R12(6)] rings do not form a continuous chain as in adduct 2. Formally this is associated with the A-centring (compare the positions of the three pairs of urea [R12(6)] rings at bottom left, centre right, top left).
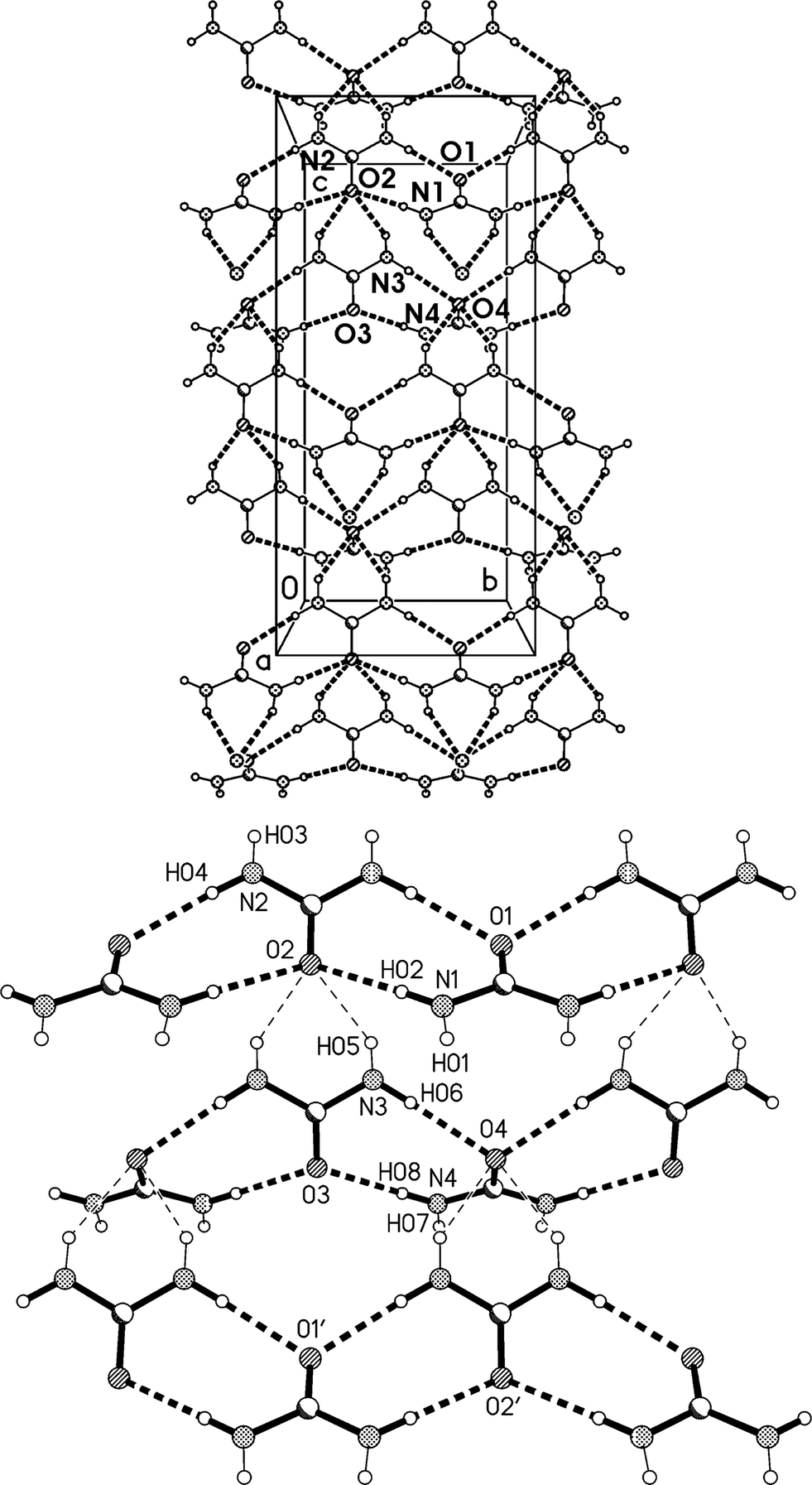 | ||
| Fig. 11 (a), Above: packing diagram of 3o with view direction parallel to the a axis in the region x ≈ 1/4; the mirror planes run vertically at y = 1/4 and 3/4. Hydrogen bonds are drawn as thick dashed lines. The urea ribbons run horizontally. Lutidines are represented only by the nitrogen atom N11 (the second lutidine lies behind this plane). (b), Below: simplified version without lutidines (see text). Primes indicate symmetry-equivalent atoms. | ||
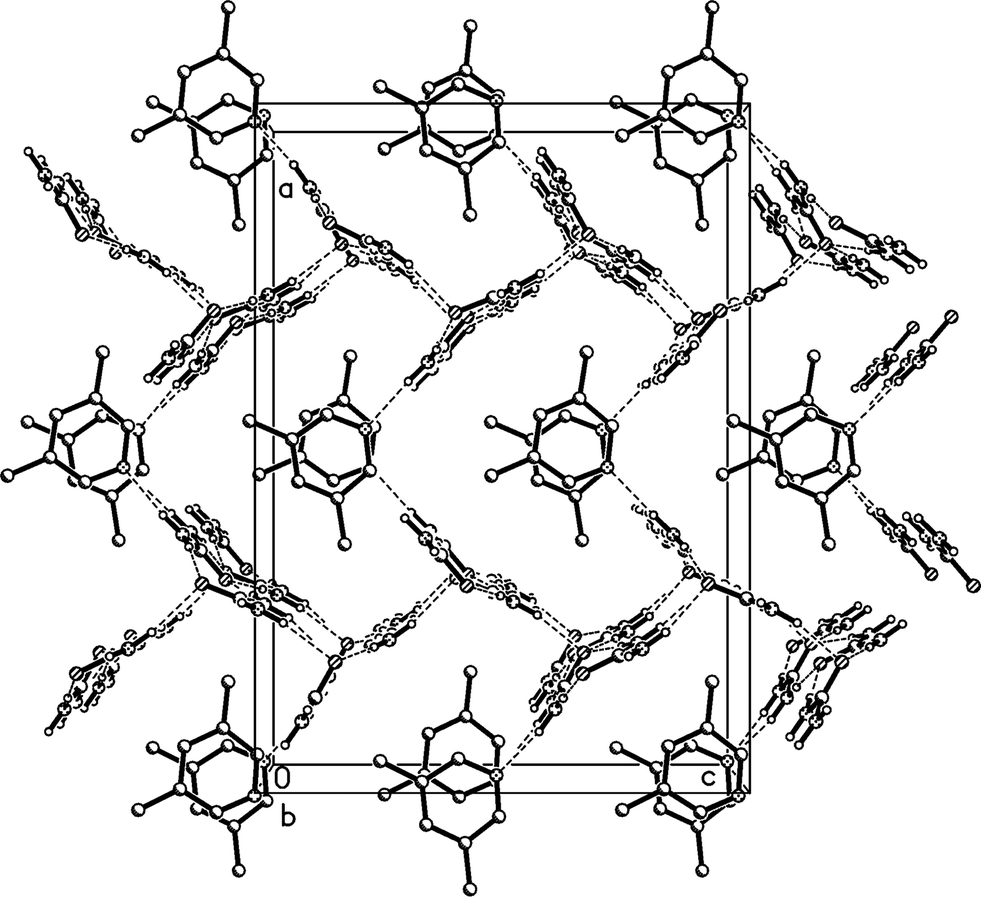 | ||
| Fig. 12 Packing diagram of 3o with view direction parallel to the b axis. Hydrogen bonds are drawn as thin dashed lines. Lutidine molecules #1 stack at x ≈ 1/2 and #2 at x ≈ 0, 1. | ||
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
|---|---|---|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: (i) x, y − 1/2, z + 1/2; (ii) x, y − 1, z. | |||||
| 1 | N1–H01⋯N11 | 0.878(16) | 2.33(2) | 3.161(4) | 157(3) |
| 2 | N1–H02⋯O2 | 0.873(16) | 2.119(16) | 2.988(3) | 174(2) |
| 3 | N2–H03⋯O4i | 0.876(17) | 2.29(2) | 3.072(3) | 148(3) |
| 4 | N2–H04⋯O1ii | 0.891(16) | 2.043(17) | 2.932(3) | 175(3) |
| 5 | N3–H05⋯O2 | 0.867(16) | 2.28(2) | 3.072(3) | 151(3) |
| 6 | N3–H06⋯O4 | 0.885(16) | 2.080(17) | 2.964(3) | 177(3) |
| 7 | N4–H07⋯N21 | 0.884(16) | 2.32(2) | 3.136(4) | 154(3) |
| 8 | N4–H08⋯O3 | 0.880(17) | 2.034(17) | 2.914(3) | 178(3) |
Fig. 12 presents a view of the packing parallel to the b axis, and again shows the corrugated nature of the urea layers with their projecting urea molecules. The lutidine rings stack with a vertical spacing of 3.605 and an offset of 0.5 Å (stacks of ring 1) or 0.9 Å (stacks of ring 2).
Crystal structure of urea:3,5-lutidine (2:1), lower temperature form (3m)
The lower temperature form 3m of the solvate 3, measured at −173 °C, crystallizes in the non-centrosymmetric monoclinic space group Cc, which has no special positions, with Z = 4. The asymmetric unit consists of one lutidine and two urea molecules (Fig. 13). The inclination angle C1⋯N11⋯C14 is 169.2(1)° and the angle between the urea planes is 34.0(2)°.
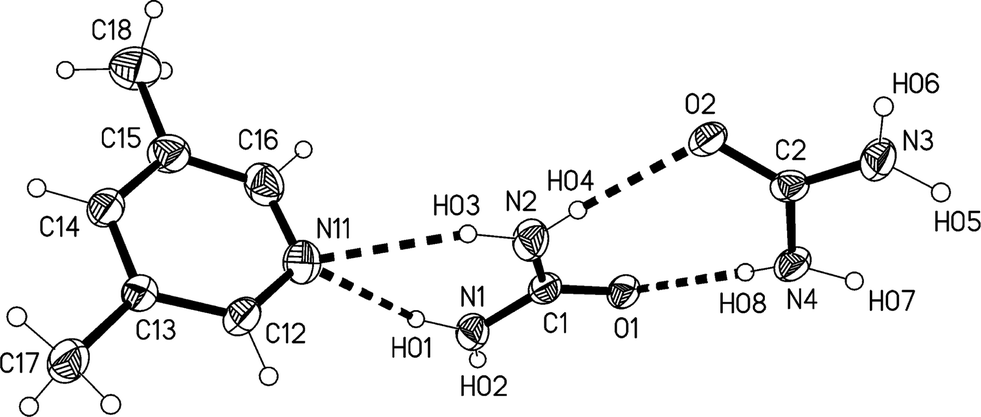 | ||
| Fig. 13 The formula unit of adduct 3m in the crystal. Ellipsoids correspond to 50% probability levels. The dashed lines correspond to classical hydrogen bonds. | ||
The residues again pack to form corrugated layers of urea molecules, now parallel to the ac plane, perpendicular to which the lutidines are attached (Fig. 14, Table 6). The ribbons of [R22(8)] urea rings run parallel to the c axis. In the previous two structures, the oxygen of the urea(s) coordinating to lutidine accepts two hydrogen bonds and the oxygen of the other urea(s) accepts four. In 3m all the urea oxygens accept three hydrogen bonds, and this entails another reorientation of the ribbons (Fig. 14b), which no longer lie approximately above and below each other (vertically in the two-dimensional representations given here) but are laterally staggered corresponding to the direction of the monoclinic c axis. The displacement should be equal to half the distance between the mirror planes or ¼b3o, some 1.8 Å.
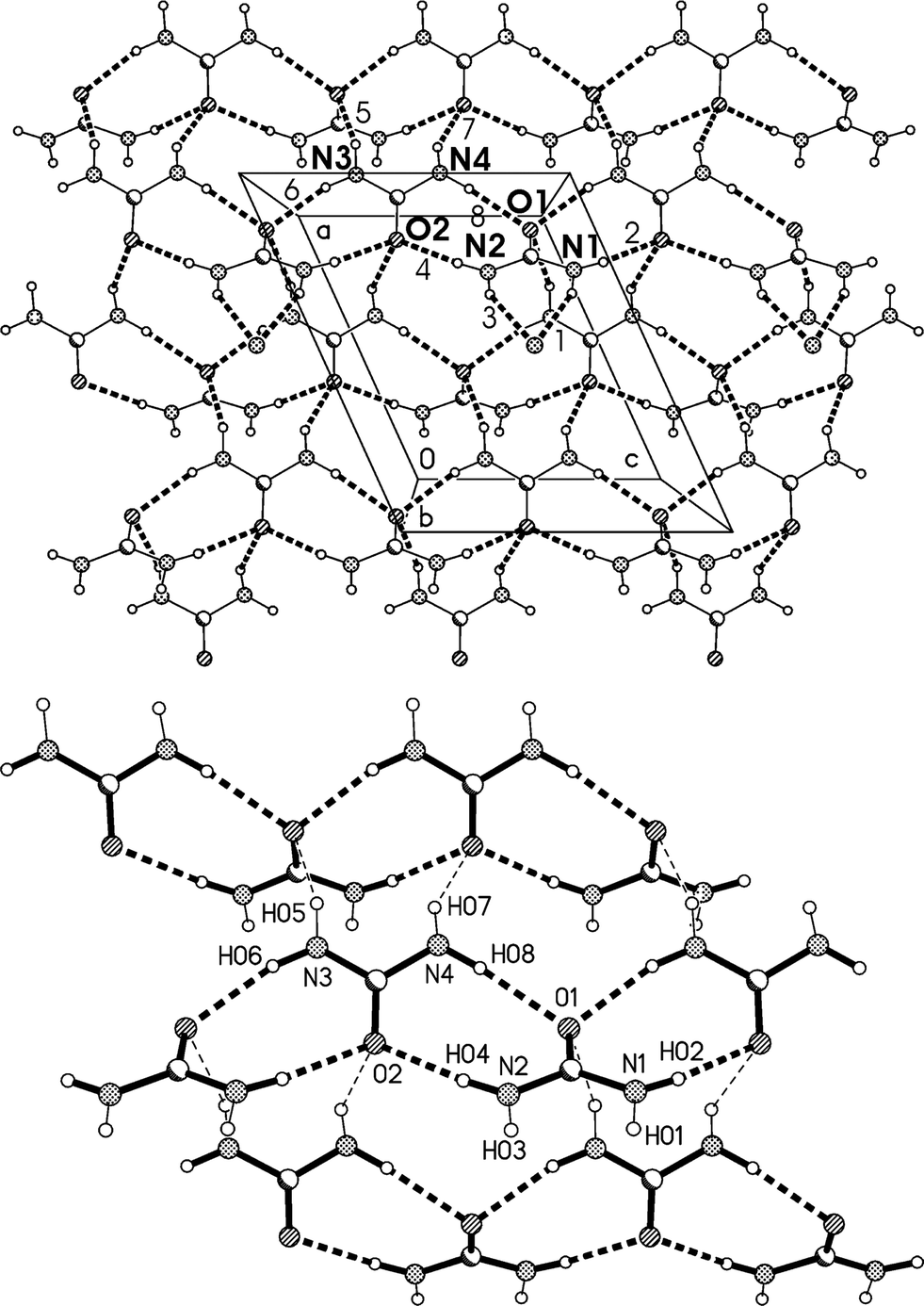 | ||
| Fig. 14 (a), Above: packing diagram of 3m with view direction parallel to the b axis in the region y ≈ 1/4. Hydrogen bonds are drawn as thick dashed lines and numbered according to Table 6. For clarity, only three lutidine nitrogens are shown (e.g. the acceptor of H bonds #1 and #3). The urea ribbons run horizontally. The a* axis (see text) runs vertically. (b), Below: simplified version without lutidines (see text). | ||
Fig. 15 presents a view of the packing parallel to the c axis; the lutidine rings stack with a vertical spacing of 3.61 and an offset of 0.7 Å, although successive rings are not exactly parallel (interplanar angle 2°).
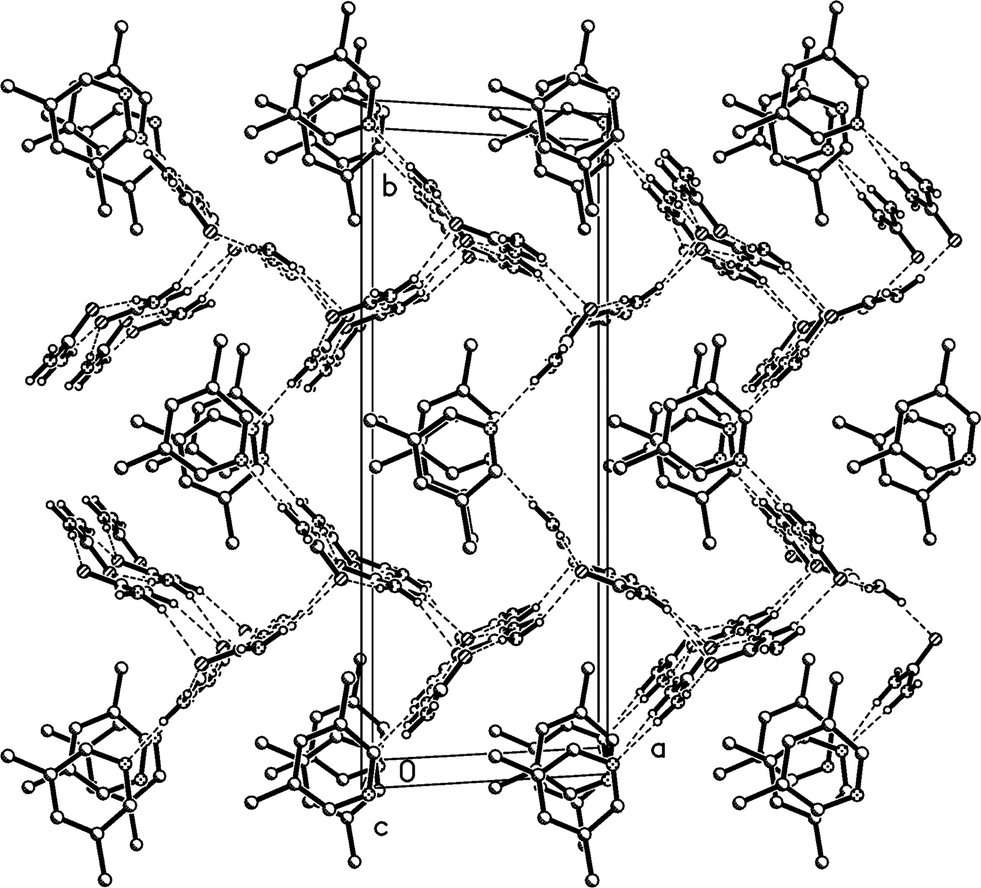 | ||
| Fig. 15 Packing diagram of 3m with view direction parallel to the c axis. Hydrogen bonds are drawn as thin dashed lines. All lutidine molecules are equivalent (cf.Fig. 12). | ||
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) | |
|---|---|---|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: (i) x, y, z + 1; (ii) x + 1/2, −y + 1/2, z − 1/2; (iii) x, y, z − 1; (iv) x + 1/2, −y + 1/2, z + 1/2. | |||||
| 1 | N1–H01⋯N11 | 0.90(2) | 2.26(3) | 3.113(4) | 158(4) |
| 2 | N1–H02⋯O2i | 0.89(2) | 2.06(2) | 2.939(4) | 168(4) |
| 3 | N2–H03⋯N11 | 0.91(2) | 2.32(3) | 3.165(4) | 153(4) |
| 4 | N2–H04⋯O2 | 0.90(2) | 2.07(3) | 2.948(4) | 168(4) |
| 5 | N3–H05⋯O1ii | 0.92(2) | 2.16(3) | 3.021(3) | 154(4) |
| 6 | N3–H06⋯O1iii | 0.92(2) | 2.03(3) | 2.926(4) | 167(4) |
| 7 | N4–H07⋯O2iv | 0.91(2) | 2.22(3) | 3.050(3) | 152(4) |
| 8 | N4–H08⋯O1 | 0.91(2) | 2.06(2) | 2.951(4) | 166(4) |
Relationship between the cells of 3m and 3o
The orthorhombic cell of 3o can be converted into a monoclinic cell resembling that of 3m by the appropriate matrix: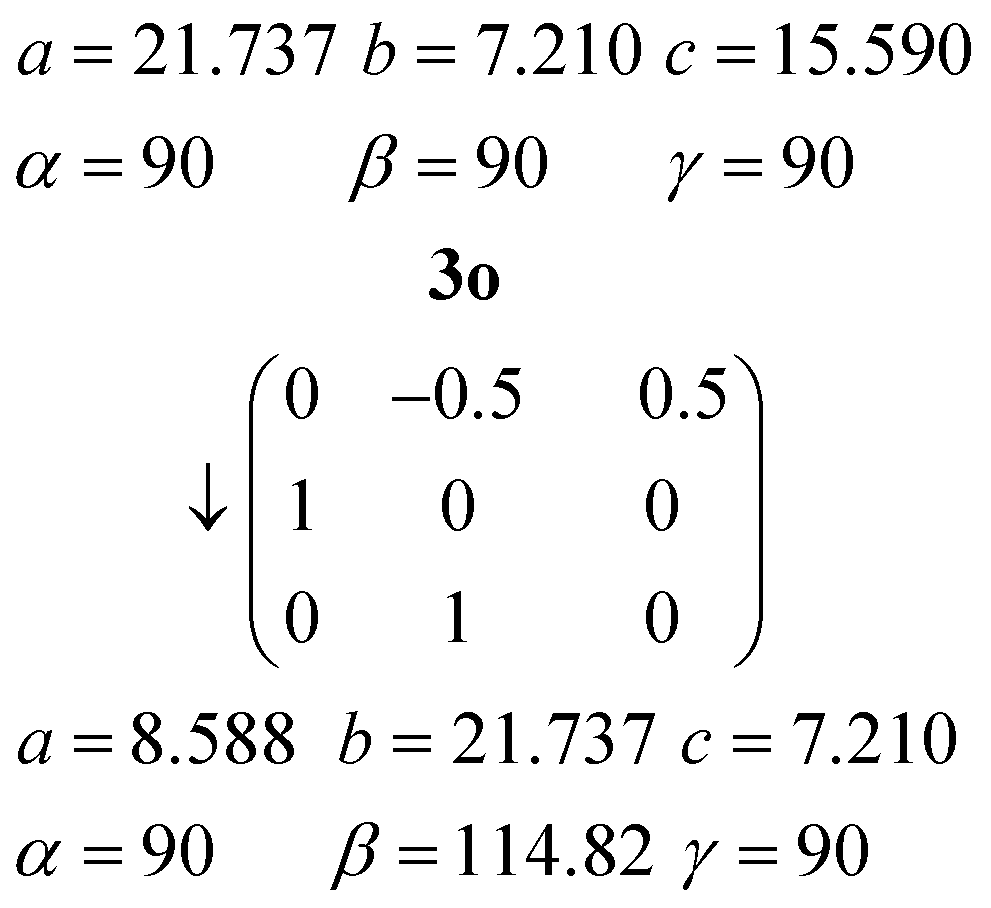
The new cell should however be primitive rather than centred (the expected appropriate subgroup of Abm2 would be Pc, not Cc).16a The “additional” C-centring in 3m renders e.g. all the lutidines and their stacks equivalent, in contrast to the lutidine stacks in 3o, which consist of two sets corresponding to the two independent molecules. Calculation suggests that the coordinates of the two independent lutidine molecules of 3o, transformed to 3m, are reasonably consistent with the C-centring in their x and y coordinates, but the z coordinates differ by approximately 0.23c3m. However, the urea ribbons and their accompanying lutidines are mutually displaced with respect to the 3o structure by a translation of ¼b3o, and this is the same axis. We thus conclude that the C-centring arises as a new symmetry operation between the previously independent formula units, induced by the “slippage” of the urea ribbons with their accompanying lutidines.16b
The inverse matrix can be used to convert the 3m cell back, approximately, to 3o:
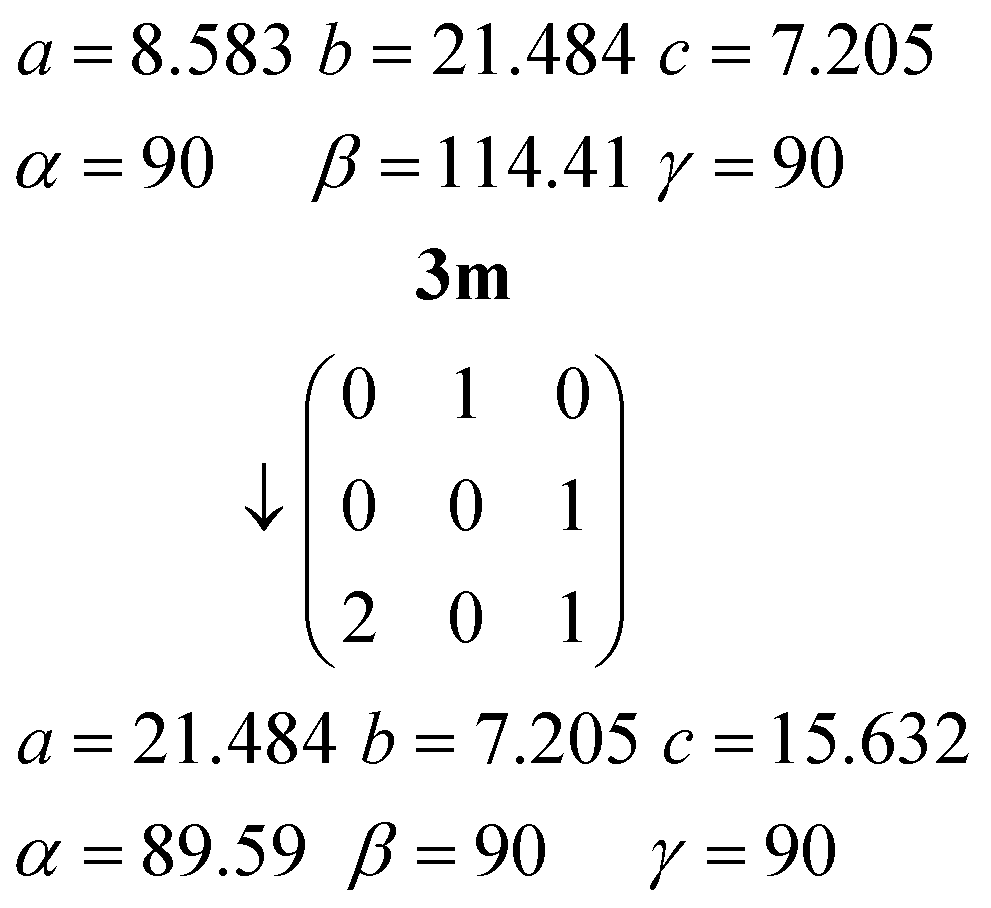
The twinning of 3m by 180° rotation about a* (previously the orthorhombic c axis direction, vertical in Fig. 14a) can be interpreted in terms of the lateral slippage between the urea ribbons in 3o taking place either to the left or to the right.
As for adducts 1, the density of 3m (1.248 g cm−3) is greater than that of 3o (1.236 g cm−3), but again this may be partially attributable to the temperature difference.
Conclusions
Reasonable explanations in terms of molecular displacements can be given for the twinning effects that are induced by cooling the adducts urea:2,6-lutidine (1:1) and urea:3,5-lutidine (2:1).
Acknowledgements
We are grateful to Dr. Regine Herbst-Irmer, University of Göttingen, for helpful discussions on twinning phenomena and in particular for advice on the use of new SHELXL versions in cases of reticular twinning.Notes and references
- C. Taouss, L. Thomas and P. G. Jones, CrystEngComm, 2013, 15, 6829–6836 RSC.
- J. D. Lee and S. C. Wallwork, Acta Crystallogr., 1965, 19, 311–313 CrossRef CAS.
- J. Ashurov, B. Ibragimov and S. Talipov, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o504 CAS.
- Twin categories and twin laws have been formalised by M. Nespolo and G. Ferraris, Acta Crystallogr., Sect. A: Found. Crystallogr., 2004, 60, 89–95 CrossRef PubMed , who also discuss previous classifications, and by H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1987, 43, 564–568 CrossRef . For general descriptions of the refinement of twinned structures see R. Herbst-Irmer and G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 443–449 CrossRef and R. Herbst-Irmer and G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 477–481 Search PubMed ; the latter gives notice of the improvements later to be implemented in SHELXL-2012.
- e.g. (a) C. A. Austin, B. D. Leskiw and M. Zeller, Phase Transitions, 2009, 82, 211–220 CrossRef CAS; (b) E. J. Chan, A. D. Rae and T. R. Welberry, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 509–515 CAS . Twinning may of course also be associated with order/disorder phenomena rather than simple molecular displacements, e.g. J. W. Bats, P. Schell and J. W. Engels, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o1028 Search PubMed . Temperature change can also lead to single-crystal to single-crystal transitions; for a recent example in a triazole complex of ZnBr2, see X. X. Wu, Y. Y. Wang, P. Yang, Y. Y. Xu, J. Z. Huo, B. Ding, Y. Wang and X. G. Wang, Cryst. Growth Des., 2014, 14, 477–490 Search PubMed Finally, cooling can lead to drastic mechanical stress and crystal destruction, as is often seen on shock-cooling of crystals for low temperature diffractometer measurements. Note added on revision: A referee has commented (paraphrased by us) “The observed twinning is a secondary effect that does not necessarily have to occur. Probably not all the crystals of the daughter phases are twinned”. We are happy to concur with the former comment. The latter may or may not be true in the cases described here; we looked at many (around 15) crystals of 1t in the hope of finding an untwinned crystal, but all were twinned. We only succeeded in cooling one crystal of 3o to 100 K, forming a twin of 3m.
- Oxford Diffraction, CrysAlis PRO, Oxford Diffraction Ltd., Yarnton, England, 2013 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, SHELXL-2012, a Program for refining Crystal Structures, University of Göttingen, Germany, 2012 Search PubMed.
- Siemens XP, version 5.03, Siemens Analytical X–Ray Instruments, Madison, Wisconsin, U.S.A, 1994 Search PubMed.
- For two-component twins, the diffractometer control program (here ref. 6) uses two orientation matrices for the data reduction and outputs the intensity data with reflections flagged as pure component 1 or 2 (the latter reflections may be omitted in some cases) or overlapped 1/2. The refinement program (here ref. 7) recognises this data format and refines a scale factor for the relative twin volumes. This is known in shorthand as the “HKLF5 method”, because “HKLF 5” is the command for reading the modified data format, but it has never been formally published, to the best of our knowledge.
- Or “reticular merohedry” if the overlap is exact; this is discussed for rhombohedral systems by R. Herbst-Irmer, in Crystal Structure Refinement, a Crystallographer's Guide to SHELXL, ed. P. Müller, IUCr/OUP, 2006, ch. 7 Search PubMed.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249–259 CAS.
- P. Fernandes, A. J. Florence, F. Fabbiani, W. I. F. David and K. Shankland, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o355 Search PubMed.
- For an interpretable packing diagram of urea, see S. Swaminathan, B. M. Craven and R. K. McMullan, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 300–306 CrossRef.
- R. Custelcean, Chem. Commun., 2008, 295–307 RSC (review article).
- The more modern name for this space group (no. 39) is Aem2: International Tables for X-Ray Crystallography, ed. T. Hahn, IUCr/Springer, 5th edn, 2005, vol. A Search PubMed.
- (a) U. Müller, Symmetriebeziehungen zwischen verwandten Kristallstrukturen, Vieweg + Teubner, 2012 Search PubMed; (b) We are grateful to Prof. Müller for helpful discussions and in particular for the comment that the residues at x3o ≈ 1/2 shift by ca. ± ¼b3o with respect to those at x3o ≈ 0 or 1.
Footnote |
| † CCDC 990312 (1m), 990308 (1t), 990309 (2), 990311 (3o), 990310 (3m) contain the supplementary crystallographic data for this paper. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00560k |
| This journal is © The Royal Society of Chemistry 2014 |