 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt(II), iron(II), zinc(II) and palladium(II) complexes of di-topic 4′-{4-[bis(2-pyridyl)aminomethyl]phenyl}-2,2′:6′,2′′-terpyridine. Synthetic and X-ray structural studies†
David J.
Bray
a,
Jack. K.
Clegg
ab,
Katrina A.
Jolliffe
*a and
Leonard F.
Lindoy
*a
aSchool of Chemistry F11, The University of Sydney, NSW 2006, Australia. E-mail: kate.jolliffe@sydney.edu.au; len.lindoy@sydney.edu.au
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, St Lucia, QLD 4072, Australia. E-mail: j.clegg@uq.edu.au
First published on 9th May 2014
Abstract
The di-topic terpy/dpa hybrid ligand, 4′-{4-[bis(2-pyridyl)aminomethyl]phenyl}-2,2′:6′,2′′-terpyridine (L), incorporating both 2,2′:6′,2′′-terpyridine (terpy) and 2,2′-dipyridylamino domains has been synthesised. The metal complexes [CoLCl2], [ZnLCl2], [ZnL(CH3CO2)2], [Fe(L)2(PF6)2] and [Fe(L)(HL)](PF6)0.5(ClO4)2.5 have been isolated. X-ray structures of all five structures show that the respective metal ions preferentially bind to the terpy site of L, with the dpa site in the first four complexes remaining empty. In the fifth complex, which was isolated following the attempted reaction of silver perchlorate with [Fe(L)2(PF6)2], one dpa site is protonated. The crystal packing of all these species is dominated by extensive π-interactions. The 5-coordinate zinc(II) and cobalt(II) complexes are isostructural and exhibit a 3D network of interactions involving π-stacking as well as CH⋯Cl and CH⋯N hydrogen bonding. Similar interactions occur in the packing of [ZnL(CH3CO2)2] and [Fe(L)(HL)](PF6)0.5(ClO4)2.5 giving rise to 3D arrays. Reaction of palladium(II) with L yields a product of stoichiometry Pd2LCl4 for which the evidence suggests that palladium binds at both the terpy and dpa domains. Based on this result, addition of [Pd(CH3CN)2Cl2] to [Fe(L)2](PF6) in acetonitrile yielded a heterometallic complex in which the “free” dpa sites of [Fe(L)2](PF6) are now occupied by palladium ions to form a heterometallic product of type [FePd2(L)2Cl4](PF6)2·1.5CH3CN.
Introduction
Di-, tri- and poly-pyridyl based ligands systems have been widely used as components in metallo-supramolecular chemistry, reflecting the coordination versatility of this heterocyclic amine ligand class.1,2 For example, the metal ion chemistry of 2,2′:6′,2′′-terpyridine (terpy) and its derivatives has received a great deal of attention over many decades3–6 as has 2,2′-dipyridylamine (dpa) and its extended (secondary amine-substituted) derivatives.7,8 Thus, in the latter case a range of bridging groups have been employed to connect from two to four dpa units via their respective secondary amine sites for use in metal coordination/metallo-supramolecular studies and a variety of such systems have now been investigated in some detail.7,8 As an example, in previous studies we have investigated the interaction of copper(II),9 palladium(II)9 and silver(I)10,11 with such linked dpa ligands, with our focus being on the supramolecular aspects of the resulting solid state structures. As an extension of these studies we now report the synthesis of the new linked “hybrid” ligand 4′-{4-[di-(2-pyridyl)aminomethyl]phenyl}-2,2′:6′,2′′-terpyridine (L) incorporating both dpa and 2,2′:6′,2′′-terpyridine domains together with an investigation of its interaction with selected metal ions.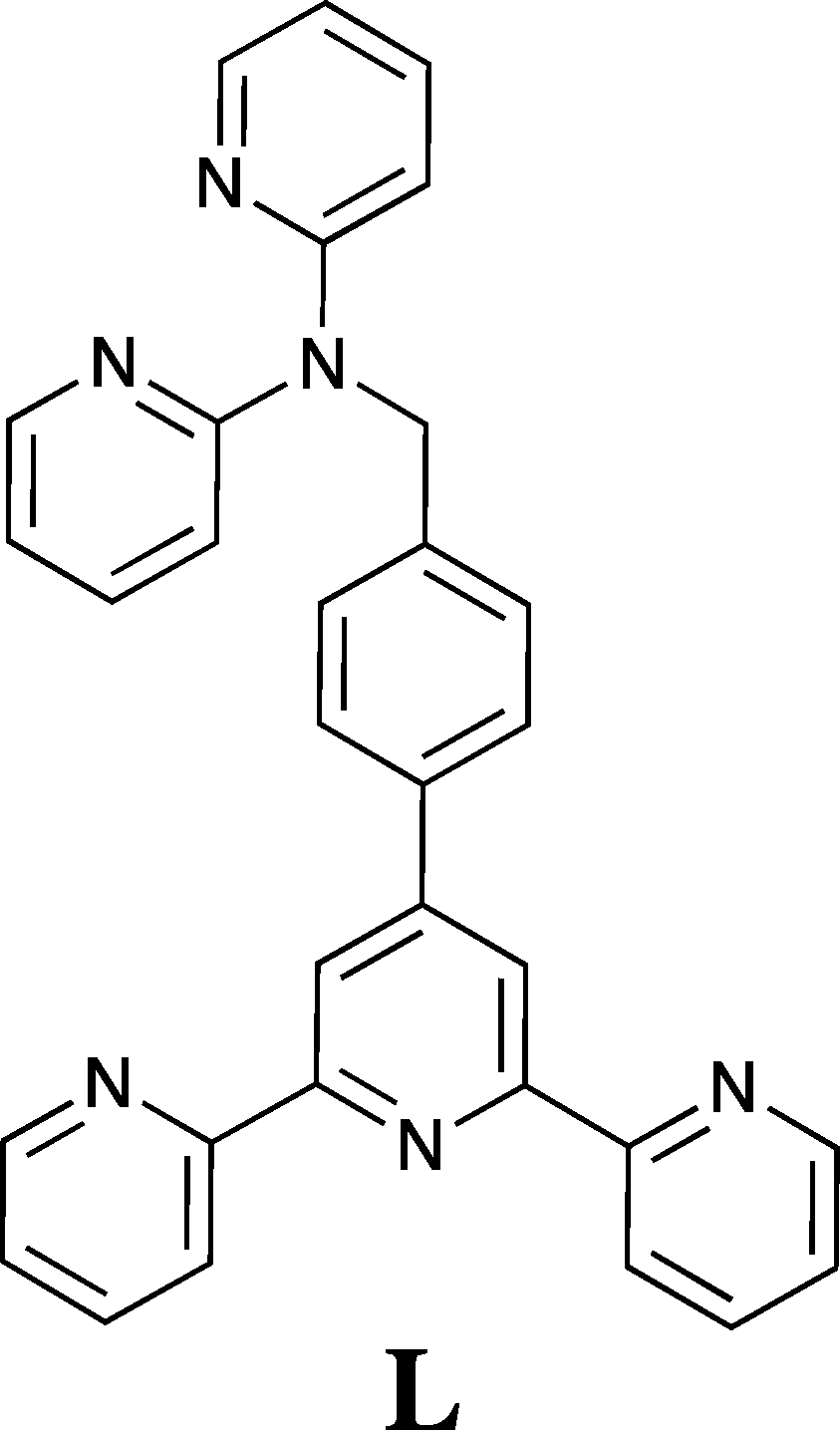
Experimental
NMR spectra were recorded on a Bruker Avance DPX200 or DPX300 spectrometer. Electrospray mass spectra (ESI-MS) were obtained on a Finnigan LCQ-8 spectrometer. FTIR spectra were determined on a Bio-Rad FTS-40 spectrometer. All commercially available reagents were used as received.Crystal structure determinations
Data were collected on either a Bruker-Nonius APEX2-X8-FR591 diffractometer employing graphite-monochromated Mo-Kα generated from a rotating anode (0.71073 Å) with ω and ψ scans to approximately 56° 2θ or a Bruker SMART 1000 diffractometer employing graphite-monochromated Mo-Kα generated from a sealed tube (0.71073 Å) with ω scans to approximately 56° 2θ. Data integration and reduction were undertaken with SAINT and XPREP12 and subsequent computations were carried out using the WinGX-32 graphical user interface.13 Multi-scan empirical absorption corrections were applied to the data using the program SADABS.14 Gaussian absorption corrections were applied using XPREP.12 Structures were solved by direct methods using SIR9715 then refined and extended with SHELXL-97.16 Unless otherwise stated, ordered non-hydrogen atoms were refined anisotropically while partial occupancy non-hydrogen atoms were refined isotropically. Hydrogen atoms attached to carbon atoms were included in idealised positions and a riding model was used for their refinement. Hydrogen atoms bound to solvent water molecules that could not be located in the difference Fourier map were not included in the model. CCDC 988029–988033.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2), a 8.2650(10), b 11.1740(10), c 16.397(2) Å, α 91.485(5), β 91.295(5), γ 94.105(5)°, V 1509.5(3) Å3, Dc 1.436 g cm−3, Z 2, crystal size 0.300 by 0.250 by 0.150 mm, colour green, habit block, temperature 150(2) Kelvin, λ(MoKα) 0.71073 Å, μ(MoKα) 0.825 mm−1, T(SADABS)min,max 0.753, 0.884, 2θmax 72.80, hkl range −13 13, −18 18, −27 27, N 70
(#2), a 8.2650(10), b 11.1740(10), c 16.397(2) Å, α 91.485(5), β 91.295(5), γ 94.105(5)°, V 1509.5(3) Å3, Dc 1.436 g cm−3, Z 2, crystal size 0.300 by 0.250 by 0.150 mm, colour green, habit block, temperature 150(2) Kelvin, λ(MoKα) 0.71073 Å, μ(MoKα) 0.825 mm−1, T(SADABS)min,max 0.753, 0.884, 2θmax 72.80, hkl range −13 13, −18 18, −27 27, N 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 587, Nind 14630 (Rmerge 0.0342), Nobs 11328 (I > 2σ(I)), Nvar 391, residuals‡R1(F) 0.0377, wR2(F2) 0.1063, GoF (all) 1.055, Δρmin,max −0.484, 0.969 e− Å−3.
(#2), a 8.4229(5), b 11.1372(6), c 17.7581(11) Å, α 79.110(3), β 79.382(4), γ 84.189(3)°, V 1604.02(16) Å3, Dc 1.378 g cm−3, Z 1, crystal size 0.477 by 0.061 by 0.043 mm, colour colourless, habit needle, temperature 150(2) Kelvin, λ(MoKα) 0.71073 Å, μ(MoKα) 0.970 mm−1, T(SADABS)min,max 0.539, 0.950, 2θmax 57.08, hkl range −11 11, −14 14, −23 23, N 39311, Nind 7979 (Rmerge 0.0630), Nobs 4912 (I > 2σ(I)), Nvar 378, residuals§R1(F) 0.0583, wR2(F2) 0.1841, GoF (all) 1.020, Δρmin,max −0.687, 1.154 e− Å−3.
(#2), a 13.7825(9), b 14.8655(10), c 18.2258(11) Å, α 70.210(2), β 79.959(3), γ 64.388(3)°, V 3166.7(4) Å3, Dc 1.418 g cm−3, Z 4, crystal size 0.300 by 0.250 by 0.075 mm, colour colourless, habit plate, temperature 150(2) Kelvin, λ(MoKα) 0.71073 Å, μ(MoKα) 0.826 mm−1, T(SADABS)min,max 0.846, 0.940, 2θmax 60.14, hkl range −19 19, −20 20, −19 25, N 84652, Nind 18409 (Rmerge 0.0495), Nobs 13160 (I > 2σ(I)), Nvar 851, residuals¶R1(F) 0.0437, wR2(F2) 0.1179, GoF (all) 1.030, Δρmin,max −0.377, 0.731 e− Å−3.
587, Nind 14630 (Rmerge 0.0342), Nobs 11328 (I > 2σ(I)), Nvar 391, residuals‡R1(F) 0.0377, wR2(F2) 0.1063, GoF (all) 1.055, Δρmin,max −0.484, 0.969 e− Å−3.
(#2), a 8.4229(5), b 11.1372(6), c 17.7581(11) Å, α 79.110(3), β 79.382(4), γ 84.189(3)°, V 1604.02(16) Å3, Dc 1.378 g cm−3, Z 1, crystal size 0.477 by 0.061 by 0.043 mm, colour colourless, habit needle, temperature 150(2) Kelvin, λ(MoKα) 0.71073 Å, μ(MoKα) 0.970 mm−1, T(SADABS)min,max 0.539, 0.950, 2θmax 57.08, hkl range −11 11, −14 14, −23 23, N 39311, Nind 7979 (Rmerge 0.0630), Nobs 4912 (I > 2σ(I)), Nvar 378, residuals§R1(F) 0.0583, wR2(F2) 0.1841, GoF (all) 1.020, Δρmin,max −0.687, 1.154 e− Å−3.
(#2), a 13.7825(9), b 14.8655(10), c 18.2258(11) Å, α 70.210(2), β 79.959(3), γ 64.388(3)°, V 3166.7(4) Å3, Dc 1.418 g cm−3, Z 4, crystal size 0.300 by 0.250 by 0.075 mm, colour colourless, habit plate, temperature 150(2) Kelvin, λ(MoKα) 0.71073 Å, μ(MoKα) 0.826 mm−1, T(SADABS)min,max 0.846, 0.940, 2θmax 60.14, hkl range −19 19, −20 20, −19 25, N 84652, Nind 18409 (Rmerge 0.0495), Nobs 13160 (I > 2σ(I)), Nvar 851, residuals¶R1(F) 0.0437, wR2(F2) 0.1179, GoF (all) 1.030, Δρmin,max −0.377, 0.731 e− Å−3.
Formula C64H53F12FeN12O2.50P2, M 1375.97, monoclinic, space group P21/n (#14), a 13.964(2), b 14.768(2), c 31.254(4) Å, β 102.320(9)°, V 6296.8(15) Å3, Dc 1.451 g cm−3, Z 4, crystal size 0.300 by 0.150 by 0.100 mm, colour purple, habit blade, temperature 150(2) Kelvin, λ(MoKα) 0.71073 Å, μ(MoKα) 0.382 mm−1, T(SADABS)min,max 0.705, 0.963, 2θmax 41.60, hkl range −13 13, −14 14, −31 31, N 27263, Nind 6373 (Rmerge 0.0874), Nobs 4043 (I > 2σ(I)), Nvar 856, residuals||R1(F) 0.0571, wR2(F2) 0.1599, GoF (all) 1.035, Δρmin,max −0.254, 0.571 e− Å−3.
618, Nind 14651 (Rmerge 0.0547), Nobs 9266 (I > 2σ(I)), Nvar 859, residuals**R1(F) 0.0823, wR2(F2) 0.2357, GoF (all) 1.004, Δρmin,max −0.914, 1.005 e− Å−3.
Ligand synthesis
Metal complex synthesis
:1) (15 mL) was added to L (0.5 g, 1 mmol) in dichloromethane/methanol (1:1; 20 mL). A dark purple solution formed immediately and this was heated and stirred until the volume was reduced to 20 mL. Water (10 mL) was added to this solution followed by saturated aqueous NH4PF6. This resulted in precipitation of a purple product, which was filtered off, washed with ether and dried. Yield, 0.6 g (90%). MS (ESI) m/z = 1185 [M − PF6]+. 1H NMR (acetonitrile-d3, 300 MHz, 300 K), δ: 5.70 (s, 4H, CH2), 6.99–7.09 (m, 8H), 7.17 (d, 4H), 7.31 (d, 4H), 7.70 (t, 4H), 7.81 (d, 4H), 7.90 (t, 4H) 8.22 (d, 4H), 8.37 (d, 4H), 8.60 (d, 4H), 9.15 (s, 4H). 13C NMR (acetonitrile-d3, 75 MHz, 300 K), δ: 50.4, 114.3, 121.2, 123.4, 126.9, 127.5, 128.2, 134.7, 137.3, 138.4, 142.9, 147.8, 150.0, 152.7, 156.7, 157.7, 159.9. IR (KBr disc, cm−1) 1581 m, 1468 m, 1432 m, 1376 w, 851 s, 839 s, 783, m, 557 m. X-ray quality crystals of [Fe(L)2(PF6)2]·2.5H2O were obtained by the slow diffusion of diethyl ether into an acetonitrile solution of the title complex.
:1; 10 mL). The resulting solution was heated and stirred for 30 min resulting in formation of a pale yellow precipitate. This product was filtered off and dried under vacuum. Yield, 30 mg (86%). MS (ESI) m/z = 811 (M − Cl)+. Found: C, 39.39; H, 3.53; N, 8.99. C32H24Cl4N6Pd2·7H2O requires: C, 39.59; H, 3.94; N, 8.66%.
Results and discussion
The difunctional ligand L containing terpy and dpa moieties was obtained in 70% yield by the direct reaction of dpa with previously reported 4′-(4-bromomethylphenyl)-2,2′:6′,2′′-terpyridine17 in dimethylformamide in the presence of KOH.The reaction of L with cobalt(II) chloride in acetonitrile/dichloromethane resulted in dark green crystals of [CoLCl2]·0.25DCM·0.5H2O whose X-ray structure (Fig. 1) showed that the cobalt cation coordinates at the terpy site (in a second experiment a similar product showing the absence of coordination at the dpa site was also obtained even when a several-fold excess of CoCl2 was employed for the reaction). The cobalt(II) centre is five-coordinate, adopting a distorted trigonal bipyramidal geometry (Addison τ parameter18 = 1.19) with the Ni(1) and N(3) pyridine nitrogens occupying the axial positions and the remaining pyridyl group and two chloride ions in the equatorial positions. The coordination bond lengths and angles of the cobalt(II) centre are similar to other cobalt(II) chloride terpyridyl complexes of type [Co(terpy)Cl2].19
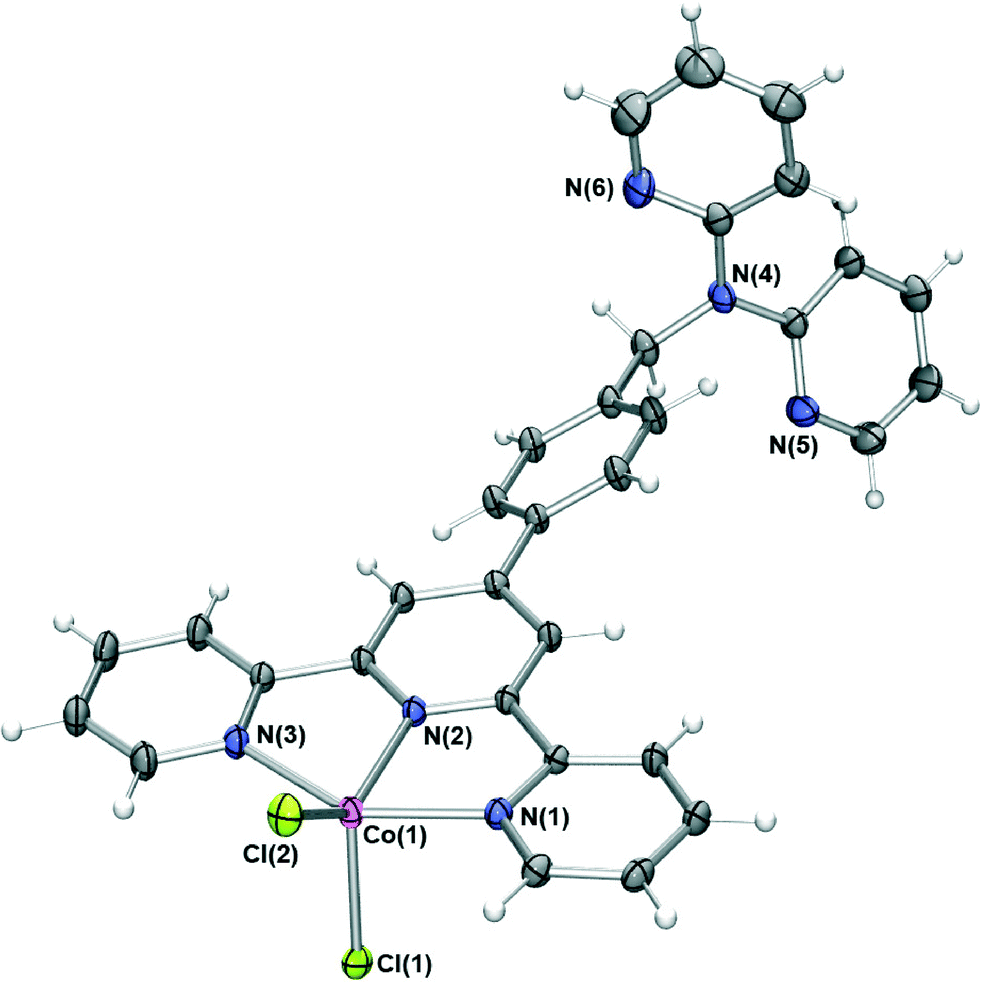 | ||
| Fig. 1 X-ray structure of [CoLCl2]·0.25DCM·0.5H2O. Solvent molecules have been removed for clarity. Thermal ellipsoids are shown at the 50 percent probability level. | ||
Adjacent complexes are involved in a complex web of intermolecular interactions forming an infinite three dimensional network; the close to planar “tolylterpy” sections of the molecule undergo a series of offset face-to-face arene–arene interactions indicated by ring centroid–ring centroid separations of 3.5–3.7 Å forming a one-dimensional stack that extends parallel to the crystallographic āb vector (Fig. 2). This arrangement is further stabilised by a series of hydrogen bonds between aromatic protons and coordinated chloride ions (CH⋯Cl distances of between 3.0 and 3.2 Å). More of these hydrogen bonds (CH⋯Cl distances of between 2.81 and 2.86 Å) and further arene–arene interactions bridge these one-dimensional chains resulting in two-dimensional sheets. Each of these sheets is further bridged by interactions involving the dpa part of the ligand. The N(5)-containing dpa pyridine acts as a hydrogen bond acceptor from an adjacent terpy pyridyl hydrogen donor (C(14)H⋯N(5) = 2.6 Å) and is also involved in edge-to-face π stacking, forming a three-dimensional network of interactions.
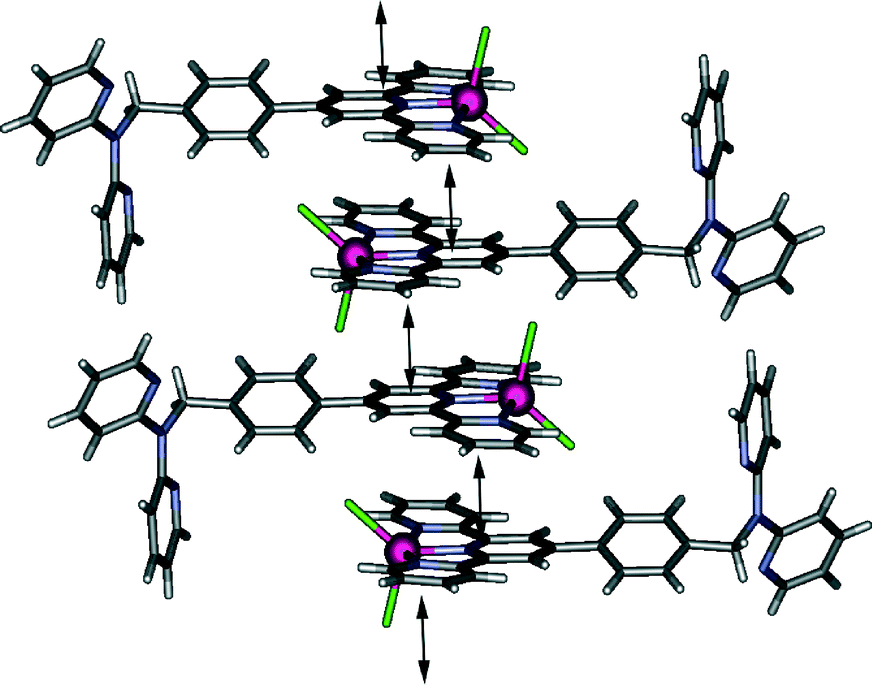 | ||
| Fig. 2 Representation of the one-dimensional stack that propagates parallel to the crystallographic āb vector in [CoLCl2]·0.25DCM·0.5H2O. Double headed arrows indicate the presence of arene–arene interactions. | ||
Interaction of zinc(II) chloride with L under similar conditions to those employed for the cobalt(II) complex (with methanol substituted for acetonitrile) resulted in formation of a pale yellow species of type [ZnLCl2]. This 5-coordinate zinc(II) complex is isostructural (Fig. 3) with the cobalt(II) complex described above and forms the same three-dimensional network of interactions consisting of arene-stacking and both CH⋯Cl and CH⋯N hydrogen bonding. Indeed, interactions of this type appear to dominate the crystal packing of each of [M(L′)Cl2] (where L′ is any ligand incorporating the “tolylterpy” motif); similar crystal packing arrangements to those found in the present study for both the zinc(II) and cobalt(II) complexes of L are present in each of the eight other [M(L′)Cl2] structures in the literature.20
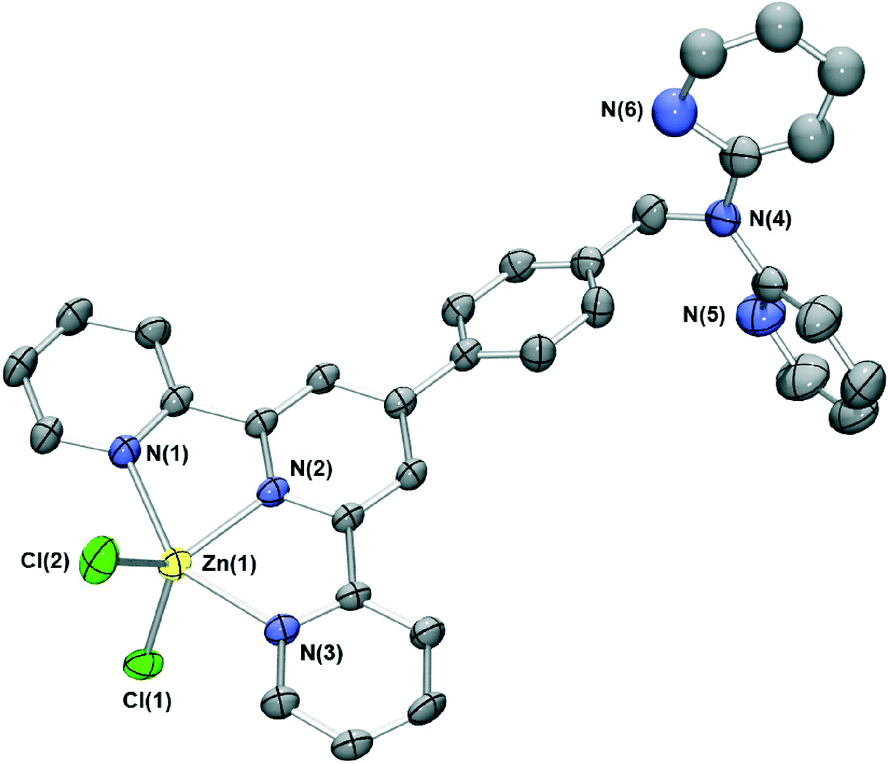 | ||
| Fig. 3 X-ray structure of [ZnLCl2]·0.7MeOH·0.3H2O. Solvent molecules and protons have been removed for clarity. Thermal ellipsoids are shown at the 50 percent probability level. The pyridyl group comprising N(4) exhibits minor positional disorder and was modelled over two sites. For clarity only one of these is shown. | ||
Reaction of zinc acetate in acetonitrile with L in dichloromethane resulted in yellow crystals whose X-ray structure is shown in Fig. 4. The zinc centre again has a distorted trigonal bipyramidal geometry with the metal bound to the terpy domain of L; the remaining two coordination sites are occupied by oxygens from monodentate acetate ions. Once again two pyridyl group occupy the axial positions, with the third and the anions in the equatorial plane, an arrangement that has been observed in two other [Zn(terpyR)(OAc)2] structures reported previously.21 Unlike the two chloride-bound complexes above, [ZnL(CH3CO2)2] crystallises with two chemically identical but crystallographically distinct molecules in the asymmetric unit.
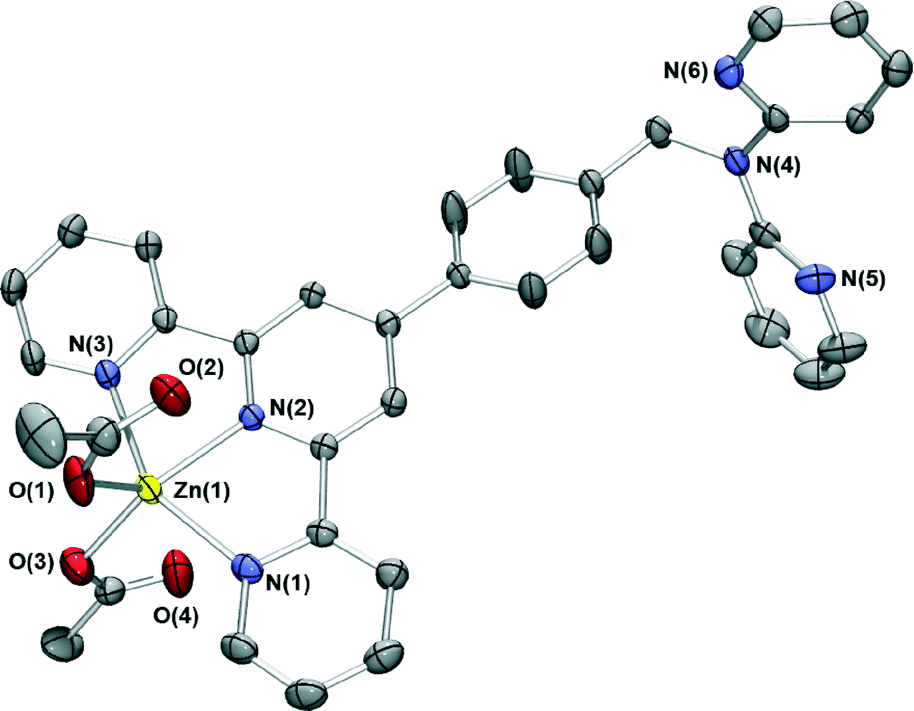 | ||
| Fig. 4 X-ray structure of [ZnL(CH3CO2)2]. Only one of the two chemically identical but crystallographically unique complexes in the asymmetric unit are shown for clarity. Thermal ellipsoids are at the 50 percent probability level. | ||
The dpa site is again not coordinated to a metal ion. In a similar fashion to the two chloride complexes, the relatively planar “tolylterpy” sections of the ligands lie closely above and below each other forming infinite one-dimensional chains which are further stabilised by CH⋯O hydrogen bonds. These chains interact with adjacent stacks through further dpapyridylH⋯O hydrogen bonds.
Reaction of ferrous ammonium sulfate with L followed by addition of ammonium hexafluorophosphate led to isolation of a purple product of type [Fe(L)2(PF6)2]. The nature of this compound was investigated by mass spectrometry, NMR and IR spectroscopy as well as by an X-ray structure determination (Fig. 5). The ESI mass spectrum of this product showed a parent peak corresponding to [Fe(L)2(PF6)]+ and the X-ray structure determination showed that the iron(II) is six coordinate with the two terpy units of two molecules of L coordinated in the commonly observed bis-terpy octahedral motif. The dpa units are, once again, not coordinated. The crystal packing of the complex is dominated by arene–arene interactions and aromatic CH⋯F hydrogen bonds resulting in a three-dimensional array, with the uncoordinated dpa groups participating in a series of edge-to-face and offset face-to-face arene–arene stacking. The “tolylterpy” parts of the molecule interact with each other in a ‘terpy embrace’ motif similar to that observed previously in the iron(II) and ruthenium(II) complexes of 4′-phenylterpy22 and 4′-pyridylterpy.23
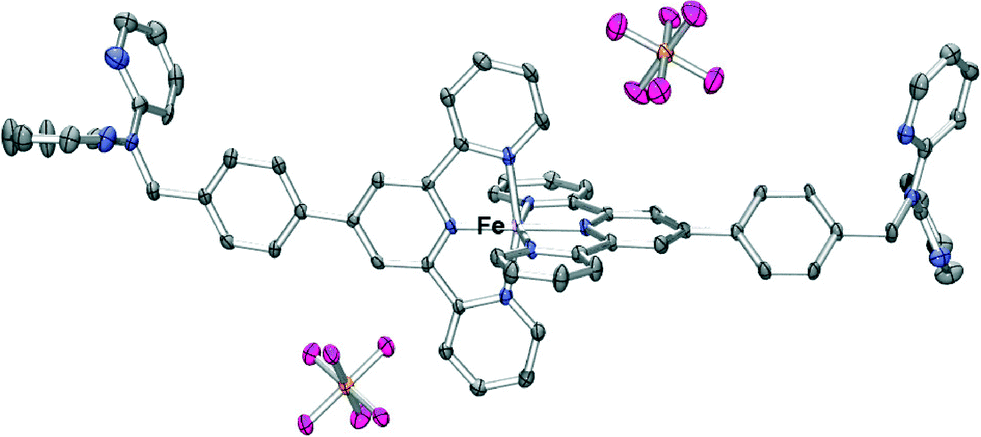 | ||
| Fig. 5 X-ray structure of [Fe(L)2(PF6)2]·2.5H2O. Solvent water molecules and protons have been removed for clarity. Thermal ellipsoids are shown at the 30 percent probability level. | ||
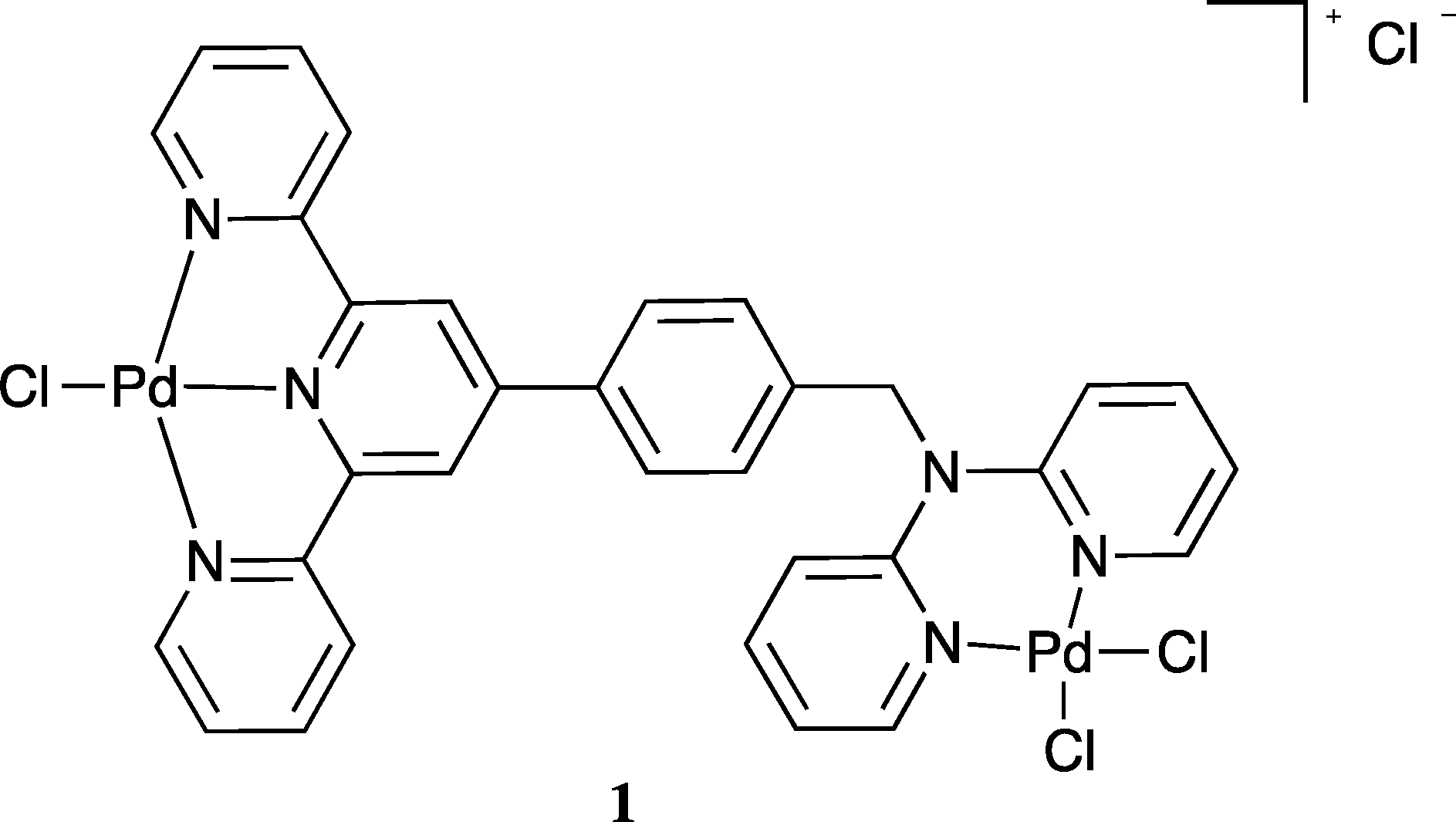
The strong coordination of palladium(II) to both dpa9,24,25 and terpy26 has been well documented and this provided a motivation for investigating the interaction of this metal ion with L. Reaction of L in dichloromethane/methanol (1:1) with palladium(II) chloride in 2 M hydrochloric acid resulted in a pale yellow precipitate. Elemental analysis of the isolated solid gave a Pd:L ratio of 2:1. The mass spectrum also revealed a parent peak corresponding to two palladium and one L in solution, in accord with formation of a homo-dinuclear species of type 1. Unfortunately crystals of 1 suitable for an X-ray diffraction study were not obtained.
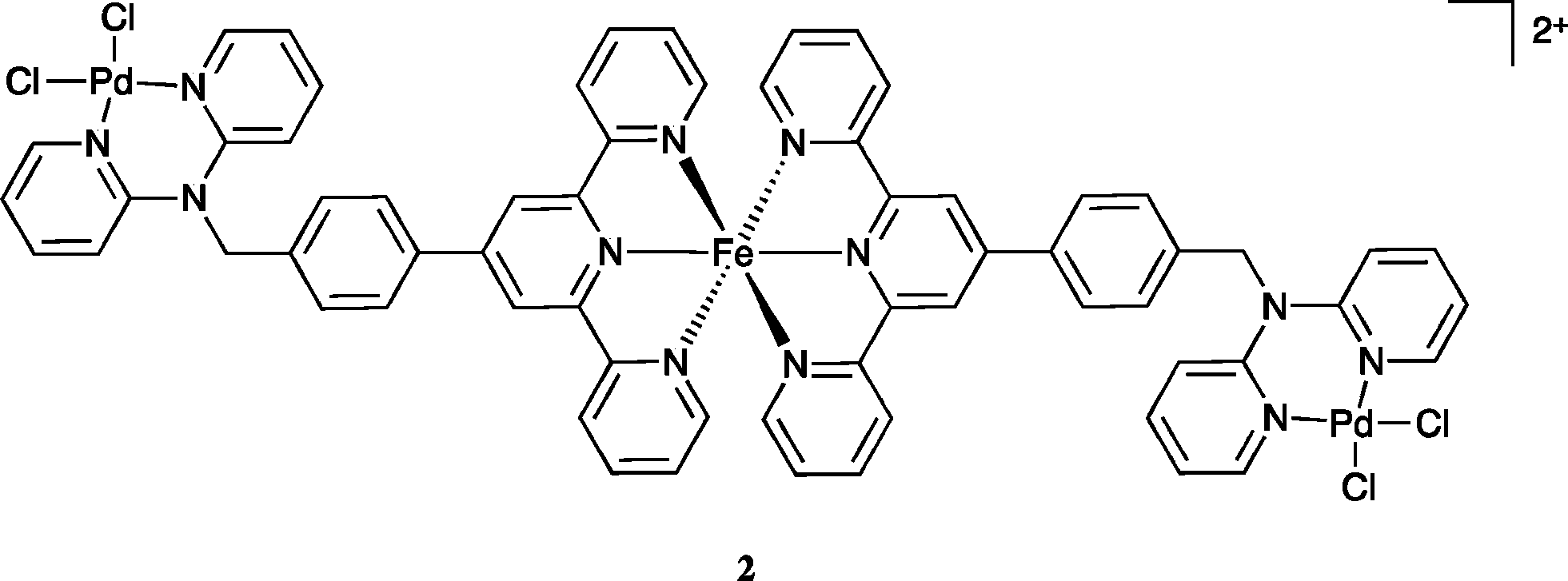
In view of the above result bis(acetonitrile)dichloropalladium(II) in acetonitrile was added to [Fe(L)2](PF6)2 in acetonitrile and the warm solution stirred for 30 minutes. The solution remained dark purple. Ether vapour was then diffused into the reaction mixture to yield a dark purple precipitate. The 1H NMR spectrum of this product exhibited a noticeable shift of the (single) methylene proton signal relative to the starting complex (in keeping with the occurrence of palladium coordination at both dpa sites). The mass spectrum revealed a parent peak corresponding to [FePd2L2Cl4PF6]+; such a stoichiometry corresponds to a structure of type 2. Further, the isotopic distribution of this ion (Fig. 6) is in agreement with a trinuclear structure and the elemental analysis of the isolated product also corresponded to the proposed stoichiometry. Unfortunately, crystals suitable for an X-ray structure determination were again not obtained; however, on the basis of the above evidence the product was assigned the trinuclear structure 2.
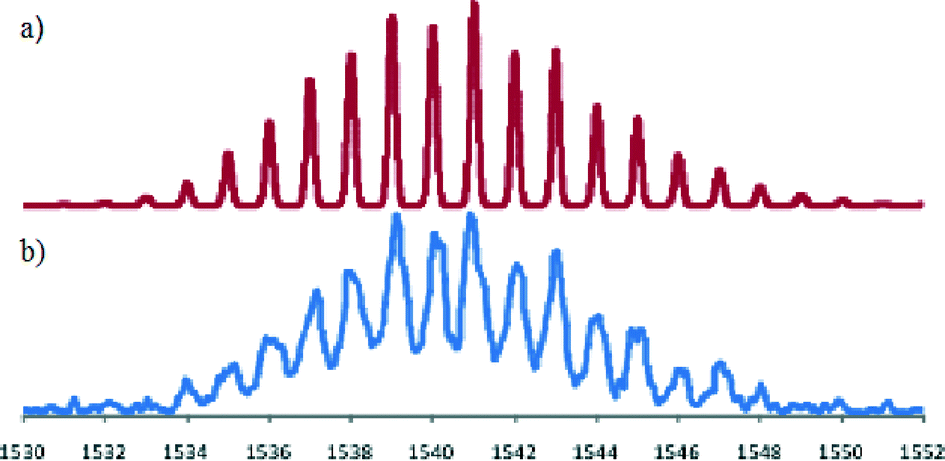 | ||
| Fig. 6 (a) Predicted mass spectral distribution for [FePd2L2Cl4PF6]+. (b) Observed isotopic distribution for this ion in the ESI mass spectrum of [(L)2FePd2(L)2Cl4](PF6)2. | ||
Based on the success of this study in forming a heterometallic complex we investigated the possibility of using [Fe(L)2](PF6)2 as a metallo-ligand for the formation a heterometallic coordination polymer in conjunction with silver(I). Our choice of silver was based on our previous success in coordinating silver(I) with dpa ligands11 and the well established propensity for silver(I) to form coordination complexes with nitrogen donor ligands when other metal ions have not been successful.27 Accordingly, an acetonitrile solution of [Fe(L)2](PF6)2 was mixed with one equivalent of AgClO4·H2O in methanol to yield a small number of purple crystals suitable for diffraction studies, following diffusion of ether vapour into the resulting purple solution. X-ray analysis revealed a mixed-anion complex of composition [Fe(L)(HL)](PF6)0.5(ClO4)2.5·H2O·1.5MeOH (Fig. 7).
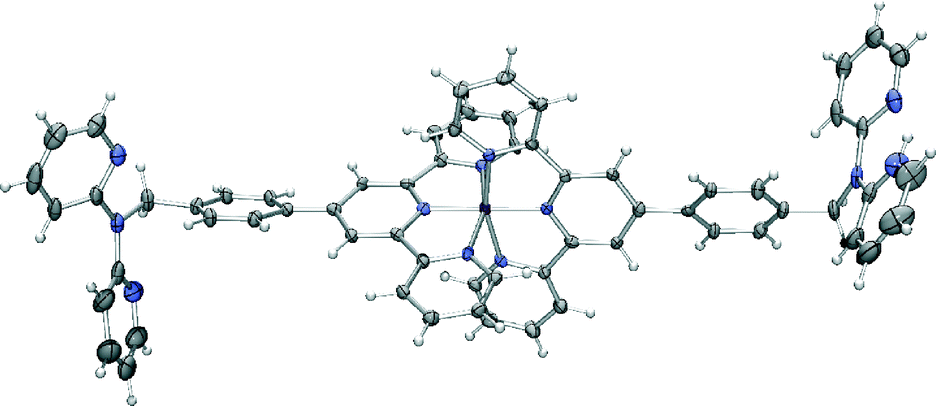 | ||
| Fig. 7 X-ray structure of [Fe(L)(HL)](PF6)0.5(ClO4)2.5·H2O·1.5MeOH. Solvent molecules and anions have been removed for clarity. Thermal ellipsoids are shown at the 50 percent probability level. | ||
In this complex, the iron(II) centre adopts the expected octahedral geometry, bound to one terpyridine domains from each ligand, one dpa moiety is protonated and both of these remain uncoordinated. The crystal packing is again dominated by π-interactions. In this case the bis-terpyridine groups form a classical one-dimensional terpyridine embrace motif28 with the network again extended to three dimensions by further π-stacking involving the dpa groups.
Conclusions
A new di-functional hybrid terpy/dpa ligand L along with five crystal structures of its metal derivatives are reported. In each complex the metal ion preferentially binds to the terpyridine moiety, with the dpa site remaining uncoordinated and the crystal packing dominated by extensive π-interactions. Contrasting with the above, reaction of palladium(II) with L gives a complex for which the evidence indicates that this metal ion binds at both the terpy and dpa domains. Based on this result, addition of [Pd(CH3CN)2Cl2] to [Fe(L)2](PF6) in acetonitrile yielded a heterometallic Fe/Pd (1:2) complex in which the evidence indicates that both non-metal bound dpa sites of [Fe(L)2](PF6) are now occupied by palladium ions to form a species of type [FePd2(L)2Cl4](PF6)2·1.5CH3CN. An initial attempt to form a related Fe(II)/Ag(I) species led instead to a silver-free [FeL2H]3+ species in which protonation of one of the two unoccupied dpa sites of dpa has occurred. Further studies involving L directed towards the synthesis of other heteronuclear species are planned for the future.
Acknowledgements
We thank the Australian Research Council for support.Notes and references
- C. R. K. Glasson, L. F. Lindoy and G. V. Meehan, Coord. Chem. Rev., 2008, 252, 940 CrossRef CAS PubMed.
- C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553 CrossRef CAS PubMed.
- E. C. Constable, Chem. Soc. Rev., 2007, 36, 246 RSC.
- (a) S. C. Yuan, H. B. Chen and H. C. Wang, Prog. Chem., 2009, 21, 2132 CAS; (b) C. Bazzicalupi, A. Bencini, A. Bianchi, A. Danesi, E. Faggi, C. Giorgi, S. Santarelli and B. Valtancoli, Coord. Chem. Rev., 2008, 252, 1052 CrossRef CAS PubMed; (c) H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373 RSC.
- (a) U. S. Schubert, H. Hofmeier and G. R. Newcome, Modern Terpyridine Chemistry, Wiley-VCH, Weimheim, 2006 Search PubMed; (b) I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834 CrossRef CAS PubMed.
- (a) A. R. Stefankiewicz, M. Walesa-Chorab, J. Harrowfield, M. Kubicki, Z. Hnatejko, M. Korabik and V. Patroniak, Dalton Trans., 2013, 42, 1743 RSC; (b) M. L. Saha, K. Mahata, D. Samanta, V. Kalsani, J. Fan, J. W. Bats and M. Schmittel, Dalton Trans., 2013, 42, 12840 RSC.
- See for example: (a) S. Youngme, J. Phatchimkun and N. Chaichit, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m602 CAS; (b) S. Youngme, J. Phatchimkun, N. Chaichit, C. Pakawatchai, I. Mutikainen and U. Turpeinen, J. Coord. Chem., 2006, 59, 1813 CrossRef CAS; (c) H. Chowdhury, S. H. Rahaman, R. Ghosh, S. K. Sarkar, M. Corbella and B. K. Ghosh, Inorg. Chem. Commun., 2006, 9, 1276 CrossRef CAS PubMed; (d) S. Youngme, J. Phatchimkun, U. Suksangpanya, C. Pakawatchai, G. A. van Albada, M. Quesada and J. Reedijk, Inorg. Chem. Commun., 2006, 9, 242 CrossRef CAS PubMed; (e) R. Carballo, B. Covelo, N. Fernández-Hermida, A. B. Lago and E. M. Vázquez-López, Z. Anorg. Allg. Chem., 2007, 633, 1791 CrossRef CAS; (f) R. Carballo, A. Castiñeiras, B. Covelo, A. B. Lago and E. M. Vázquez López, Z. Anorg. Allg. Chem., 2007, 633, 687 CrossRef CAS; (g) S. Youngme, T. Chotkhun and N. Chaichit, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, m59 CAS; (h) S. Youngme, P. Phuengphai, N. Chaichit, I. Mutikainen, U. Turpeinen and B. M. Murphy, J. Coord. Chem., 2007, 60, 131 CrossRef CAS; (i) S. Youngme, J. Phatchimkun, C. Pakawatchai, S. Prabpaic and P. Kongsaereec, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, m45 CAS; (j) S. Youngme, J. Phatchimkun, U. Suksangpanya, C. Pakawatchai, N. Chaichit, P. Kongsaeree, J. Krzystek and B. Murphy, Polyhedron, 2007, 26, 871 CrossRef CAS PubMed.
- C. J. Sumby, Coord. Chem. Rev., 2011, 255, 1937 CrossRef CAS PubMed.
- (a) Q. Z. Zha, C. X. Ding, X. Rui and Y. S. Xie, Cryst. Growth Des., 2013, 13, 4583 CrossRef CAS; (b) R. Y. Tan, Z. B. Wang, Y. Li, D. J. Kozera, Z. H. Lu and D. T. Song, Inorg. Chem., 2012, 51, 7039 CrossRef CAS PubMed; (c) T. M. Ross, B. Moubaraki, S. R. Batten and K. S. Murray, Dalton Trans., 2012, 41, 2571 RSC; (d) J. Ni, K. J. Wei, Y. Z. Min, Y. W. Chen, S. Z. Zhan, D. Li and Y. Z. Liu, Dalton Trans., 2012, 41, 5280 RSC; (e) C. X. Ding, X. Li, Y. B. Ding, S. W. Ng and Y. S. Xie, Cryst. Growth Des., 2012, 12, 3465 CrossRef CAS; (f) T. M. Ross, B. Moubaraki, D. R. Turner, G. A. Halder, G. Chastanet, S. M. Neville, J. D. Cashion, J. F. Letard, S. R. Batten and K. S. Murray, Eur. J. Inorg. Chem., 2011, 1395 CrossRef CAS; (g) G. Gupta, S. Park, S. S. Lee and J. Kim, Z. Anorg. Allg. Chem., 2011, 637, 1516 CrossRef CAS; (h) S. K. Mittal, P. Kumar, S. K. A. Kumar and L. F. Lindoy, Int. J. Electrochem. Sci., 2010, 5, 1984 CAS.
- B. Antonioli, D. J. Bray, J. K. Clegg, K. Gloe, A. Jager, K. A. Jolliffe, O. Kataeva, L. F. Lindoy, P. J. Steel, C. J. Sumby and M. Wenzel, Polyhedron, 2008, 27, 2889 CrossRef CAS PubMed.
- (a) B. Antonioli, J. K. Clegg, D. J. Bray, K. Gloe, K. Gloe, H. Hesske and L. F. Lindoy, CrystEngComm, 2006, 8, 748 RSC; (b) B. Antonioli, D. J. Bray, J. K. Clegg, K. Gloe, O. Kataeva, L. F. Lindoy, J. C. McMurtrie, P. J. Steel, C. J. Sumby and M. Wenzel, Dalton Trans., 2006, 4783 RSC.
- Bruker. SMART, SAINT and XPREP: Area detector control and data integration and reduction software, Bruker Analytical X-Ray Instruments Inc. Madison, Wisconsin, USA, 1995 Search PubMed.
- L. J. Farrugia, WinGX-32: System of Programs for Solving, refining and analysing single crystal X-ray diffraction data for small molecules, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- G. M. Sheldrick, SADABS: Empirical absorption and correction software, University of Göttingen, Institűt fűr Anorganische Chemie der Universität, Tammanstrasse 4D-3400 Göttingen, Germany, 1999 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giocavazzo, A. Guagliardi, A. G. C. Moliterni, G. Polidori and S. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, SHELX-97: Programs for crystal structure analysis, University of Göttingen, Institűt fűr Anorganische Chemie der Universität, Tammanstrasse 4, D-3400 Göttingen, Germany, 1998 Search PubMed.
- O. Johansson, M. Borgström, R. Lomoth, M. Palmblad, J. Bergquist, L. Hammatström, L. Sun and B. Akermark, Inorg. Chem., 2003, 42, 2908 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- (a) Y. Sato, Y. Nakayama and H. Yasuda, J. Organomet. Chem., 2005, 689, 744 CrossRef PubMed; (b) E. Goldschmied and N. C. Stephenson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 1867 CrossRef CAS.
- (a) R. Zibaseresht and R. M. Hartshorn, Dalton Trans., 2005, 3898 RSC; (b) Y.-L. Chen, B.-Z. Li, P. Yang and J.-Z. Wu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m238 CAS; (c) L. M. Hinkle, V. G. Young Jnr and K. R. Mann, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, m62 CAS; (d) C. Duboc, V. Astier-Perret, H. Chen, J. Pecaut, R. H. Crabtree, G. W. Brudvig and M.-N. Collomb, Inorg. Chim. Acta, 2006, 359, 1541 CrossRef CAS PubMed; (e) H. Zhou, F. Zhou, P. Wu, Z. Zheng, Z. Yu, Y. Chen, Y. Tu, L. Kong, J. Wu and Y. Tian, Dyes Pigm., 2011, 91, 237 CrossRef CAS PubMed; (f) C. Carruthers, T. K. Ronson, C. J. Sumby, A. Westcott, L. P. Harding, T. J. Prior, P. Rizkallah and M. J. Hardie, Chem. – Eur. J., 2008, 14, 10286 CrossRef CAS PubMed; (g) H. Chen, R. Tagore, S. Das, C. Incarvito, J. W. Faller, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2005, 44, 7661 CrossRef CAS PubMed; (h) D. Xia, L. Liu, X. Ma, J. Ma and J. Yao, Z. Kristallogr., 2012, 133, 227 Search PubMed.
- (a) Z. Ma, Y. Cao, Q. Li, M. F. C. G. da Silva, J. J. R. F. da Silva and A. J. L. Pombeiro, J. Inorg. Biochem., 2010, 104, 704 CrossRef CAS PubMed; (b) W. Li and Z. G. Lu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1672 CAS.
- (a) J. C. McMurtrie and I. Dance, CrystEngComm, 2009, 11, 1141 RSC; (b) J. C. McMurtrie and I. Dance, CrystEngComm, 2010, 12, 3207 RSC.
- J. E. Beves, D. J. Bray, J. K. Clegg, E. C. Constable, C. E. Housecroft, K. A. Jolliffe, C. J. Kepert, L. F. Lindoy, M. Neuberger, D. J. Price, S. Schaffner and F. Schaper, Inorg. Chim. Acta, 2008, 361, 2582 CrossRef CAS PubMed.
- S. Fakih, W. C. Tung, D. Eierhoff, C. Mock and B. Krebs, Z. Anorg. Allg. Chem., 2005, 631, 1397 CrossRef CAS.
- Y. Wanga, Y. Mizubayashi, M. Odoko and N. Okabe, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, M67 Search PubMed.
- P. Castan, F. Dahan, S. Wimmer and F. L. Wimmer, J. Chem. Soc., Dalton Trans., 1990, 2679 RSC.
- J. E. Beves, E. C. Constable, C. E. Housecroft, C. J. Kepert and D. J. Price, CrystEngComm, 2007, 9, 456 RSC.
- (a) J. C. McMurtrie and I. Dance, CrystEngComm, 2005, 7, 216 RSC; (b) J. C. McMurtrie and I. Dance, CrystEngComm, 2005, 7, 230 RSC.
Footnotes |
| † CCDC 988029–988033. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00414k |
| ‡ R1 = ∑‖Fo| − |Fc‖/∑|Fo| for Fo > 2σ(Fo); wR2 = (∑w(Fo2 − Fc2)2/∑(wFc2)2)1/2 all reflections. w = 1/[σ2(Fo2) + (0.0501P)2 + 0.5408P] where P = (Fo2 + 2Fc2)/3. |
| § R1 = ∑‖Fo| − |Fc‖/∑|Fo| for Fo > 2σ(Fo); wR2 = (∑w(Fo2 − Fc2)2/∑(wFc2)2)1/2 all reflections. w = 1/[σ2(Fo2) + (0.1018P)2 + 0.9483P] where P = (Fo2 + 2Fc2)/3. |
| ¶ R1 = ∑‖Fo| − |Fc‖/∑|Fo| for Fo > 2σ(Fo); wR2 = (∑w(Fo2 − Fc2)2/∑(wFc2)2)1/2 all reflections. w = 1/[σ2(Fo2) + (0.0523P)2 + 1.5804P] where P = (Fo2+2Fc2)/3. |
| || R1 = ∑‖Fo| − |Fc‖/∑|Fo| for Fo > 2σ(Fo); wR2 = (∑w(Fo2 − Fc2)2/∑(wFc2)2)1/2 all reflections. w = 1/[σ2(Fo2) + (0.08283P)2 + 4.8947P] where P = (Fo2 + 2Fc2)/3. |
| ** R1 = ∑‖Fo| − |Fc‖/∑|Fo| for Fo > 2σ(Fo); wR2 = (∑w(Fo2 − Fc2)2/∑(wFc2)2)1/2 all reflections. w = 1/[σ2(Fo2) + (0.1437P)2] where P = (Fo2 + 2Fc2)/3. |
| This journal is © The Royal Society of Chemistry 2014 |