 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCan picolinamide be a promising cocrystal former?†
H. C. Stephen
Chan
*a,
Grahame R.
Woollam
b,
Trixie
Wagner
a,
Martin U.
Schmidt
c and
Richard A.
Lewis
a
aNovartis Institutes for BioMedical Research, 4002 Basel, Switzerland. E-mail: stephen.chan@novartis.com
bNovartis Pharma AG, 4002 Basel, Switzerland
cInstitut für Anorganische und Analytische Chemie der Universität Frankfurt, Max-von-Laue-Strasse 7, D-60438 Frankfurt, Germany
First published on 14th April 2014
Abstract
Eight novel cocrystals of picolinamide are reported in this study. Lattice energy calculations may help us to understand their relative stabilities versus those of the individual components. The stoichiometry of one cocrystal changed when heated. A polymorph of picolinamide was obtained alongside a cocrystal in a screening experiment.
Isonicotinamide (iso) and nicotinamide (nic) (Fig. 1) are popular cocrystal formers (CCFs). iso was used to demonstrate the importance of supramolecular interactions for cocrystal formation,1,2 whilst nic is vitamin B6 and is therefore of pharmaceutical relevance.3 Cocrystals of picolinamide (pic) (Fig. 1) are rarely seen in the literature,4 despite being a structural isomer of iso and nic. In a previous case study, a crystal structure prediction method showed that the predicted structures of 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pic:carbamazepine cocrystals are thermodynamically less stable when compared to the crystal forms of the pure components.5 The amide hydrogen and the heterocyclic nitrogen of pic are in close proximity. It was reported that their associated electrostatic potentials cancel each other out significantly, thus reducing the possibility of hydrogen bond formations.5 A survey of anhydrous and non-solvated cocrystals in the Cambridge Structural Database (CSD)6 resulted in 70 iso cocrystals and 49 nic cocrystals but only two pic cocrystals.‡ The intriguing statistics may imply that pic is less prone to cocrystallise with other molecules. To address this hypothesis, a number of carboxylic acids which have been reported to form cocrystals with iso and nic were selected for cocrystallisation attempts: salicylic acid (sal), S-mandelic acid (man), malonic acid (mal), succinic acid (suc), fumaric acid (fum), glutaric acid (glu) and adipic acid (adi) (Fig. 1).
:1 pic:carbamazepine cocrystals are thermodynamically less stable when compared to the crystal forms of the pure components.5 The amide hydrogen and the heterocyclic nitrogen of pic are in close proximity. It was reported that their associated electrostatic potentials cancel each other out significantly, thus reducing the possibility of hydrogen bond formations.5 A survey of anhydrous and non-solvated cocrystals in the Cambridge Structural Database (CSD)6 resulted in 70 iso cocrystals and 49 nic cocrystals but only two pic cocrystals.‡ The intriguing statistics may imply that pic is less prone to cocrystallise with other molecules. To address this hypothesis, a number of carboxylic acids which have been reported to form cocrystals with iso and nic were selected for cocrystallisation attempts: salicylic acid (sal), S-mandelic acid (man), malonic acid (mal), succinic acid (suc), fumaric acid (fum), glutaric acid (glu) and adipic acid (adi) (Fig. 1).
 | ||
| Fig. 1 Molecular diagrams of isonicotinamide (iso), nicotinamide (nic), picolinamide (pic), salicylic acid (sal), S-mandelic acid (man), malonic acid (mal), succinic acid (suc), fumaric acid (fum), glutaric acid (glu) and adipic acid (adi). | ||
Cocrystal samples were prepared by temperature cycling a slurry of pic and CCF in a defined molar ratio (TC) or by vapour diffusion (VD). Thermogravimetric analyses (TGA) were performed to rule out the possibility of solvate formation. The melting onsets and enthalpies of the samples were then measured by differential scanning calorimetry (DSC). Crystal forms obtained were characterised using Raman spectroscopy and X-Ray powder diffraction (XRPD) prior to structure determination by single crystal X-Ray diffraction (SXD). The TC method was found to yield 2:1 pic:fum, 2:1 pic:suc, 2:1 pic:glu, 2:1 pic:adi and 1:1 pic:adi cocrystals, whilst the VD method yielded 1:1 pic:sal, 1:1 pic:man and 1:1 pic:mal cocrystals. A polymorph of picolinamide‡ and the 1:1 pic:man cocrystal was discovered by SXD in the same cocrystal screening sample. Previous studies have described how polymorphs of individual components were occasionally discovered in attempted cocrystallisation experiments.7–9
In all cocrystals reported within this study, the primary amide of picolinamide forms a R22(8) hydrogen bonding motif with the carboxylic acid (Fig. 2a–c).10 The heterocyclic nitrogen, together with the amide, may adopt various hydrogen bonding patterns: a homomeric R22(10) motif in 1:1 pic:sal and 1:1 pic:mal (Fig. 2a), a heteromeric R22(10) motif in 1:1 pic:man (Fig. 2b) and a heteromeric R22(9) in 1:1 pic:adi (Fig. 2c). However, the prevailing pyridine:carboxylic acid heterosynthon in the CSD (Fig. 2d)10 was not observed, perhaps because the pyridine C–H is a weaker hydrogen bond donor than the amide N–H.
 | ||
| Fig. 2 The hydrogen bond patterns observed in (a) 1:1 pic:sal, (b) 1:1 pic:man, and (c) 1:1 pic:adi; (d) the pyridine:carboxylic acid heterosynthon commonly found in the CSD but not in the cocrystals obtained here. | ||
In 2:1 pic:glu, the glutaric acid molecule located on the 2-fold rotation axis has an unusual backbone torsion of 56.2° (Fig. 3). The succinic acid molecules in 2:1 pic:suc have two different configurations: the one located at the inversion center has an all-trans conformation, whilst the one located on the 2-fold rotation axis adopts the unusual backbone torsion angle of 69.0° as seen in 2:1 pic:glu (Fig. 3). Note that such an unusual conformation of the dicarboxylic acid molecules is not observed in the iso:glu, nic:glu, iso:suc and nic:suc cocrystals (CSD code: ULAWOT, NUKYEY, LUNNOD and DUZPAQ, respectively). The CSD search also illustrated that only 2.0% of the glutaric acid fragments have torsion angles between 54.0° and 58.5° whilst 7.3% of the succinic acid fragments have torsion angles between 67.5° and 72.0°.‡
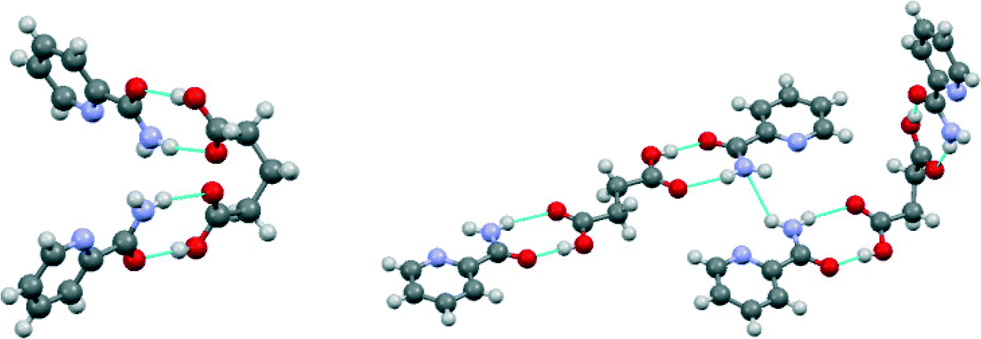 | ||
| Fig. 3 Conformations of the dicarboxylic acids and their hydrogen bonding in the 2:1 pic:glu (left) and the 2:1 pic:suc cocrystals (right). | ||
The melting point onset of 2:1 pic:adi was found to be 109 °C, whereas the DSC analysis of 1:1 pic:adi shows the first melt onset at 102 °C followed by an exothermic event, preceding the second endothermic melt onset at 107 °C (see the ESI†). Variable temperature X-Ray diffraction (VTXRD) showed that the characteristic diffraction peaks of 1:1 pic:adi at 15.5° and 19.0° 2θ are slowly diminished, whilst the peaks of 2:1 pic:adi at 11.5° and 14.5° 2θ appeared with increasing temperature. A significant loss of the crystallinity of the material at 110 °C was due to melting. However, the characteristic peaks of 2:1 pic:adi reappeared upon cooling. A subsequent sample was prepared by heating 1:1 pic:adi on a hot stage microscope to the point of full conversion. The post heating sample was then analysed by DSC and Raman spectroscopy. Only a single endothermic peak was recorded with an onset at 107 °C and the Raman spectrum was concordant with that of 2:1 pic:adi (see the ESI†). To investigate this phenomenon in detail, a separate VTXRD experiment was conducted at Goethe University in Frankfurt. A complete conversion from 1:1 to 2:1 pic:adi was confirmed at 95 °C (Fig. 4). However, a mixture of the individual components pic and adi was obtained after cooling the melt from 115 °C to room temperature (see the ESI†). The prolonged heating time together with higher attained temperature in the latter VTXRD experiment led to a completely molten material, and therefore, the converted 2:1 pic:adi was no longer available to seed the crystallisation from the melt.
 | ||
| Fig. 4 The diffractograms from the VTXRD experiment illustrate the temperature induced conversion from 1:1 pic:adi to 2:1 pic:adi. The characteristic peaks of the former at 15.5° and 19.0° were not observed at 95 °C. | ||
The relative stability of each cocrystal was calculated by comparing the minimised lattice energies per molar chemical unit with respect to their pure components (see the ESI,† Tables S1 & S2). The unit cell parameters and atomic coordinates of the experimental structures were optimised accordingly using the GRACE software11,12 which empirically corrects the dispersion energies for the Density Functional Theory (DFT) calculations implemented in VASP.13,14 The optimised crystal structures are concordant with the experimental structures, suggesting that the minimised lattice energies are reliable for comparison. The lowest energy polymorph of each coformer was taken as a reference. The pic polymorph discovered in a cocrystal screening sample is 0.27 kcal mol−1 less stable than the commercially available form according to the calculations. The eight pic cocrystals obtained experimentally were found to be more stable than the starting materials, implying that the formation of these cocrystals is thermodynamically favoured. This result is consistent with that of a previous study in which the same methodology was applied.15
The possibility to exchange the dicarboxylic acid molecules between the observed crystal packings of 1:1 pic:mal, 2:1 pic:suc, 2:1 pic:fum, 2:1 pic:glu, 2:1 pic:adi and 1:1 pic:adi was explored computationally. 24 proposed cocrystal structures were obtained by substituting an alternative acid molecule into one of the six crystal lattices and then optimised using the GRACE software. Their stabilities with respect to the pure components were calculated (Fig. 5). Substituting fum or adi resulted in unstable cocrystal structures. Substitutions of mal into 1:1 pic:adi, suc into 2:1 pic:fum, 2:1 pic:glu and 1:1 pic:adi, and glu into 2:1 pic:suc appear thermodynamically favourable. However, the five proposed structures are less stable when compared to their experimental counterparts obtained in this study, suggesting that the six experimental structures may have provided more optimal packings for the corresponding dicarboxylic acid molecules.
 | ||
| Fig. 5 The calculated relative stabilities of the six experimental structures (with asterisks) and 24 proposed structures of picolinamide:dicarboxylic acid cocrystals. The legend indicates the six observed cocrystal packings available for substitution with an alternative dicarboxylic acid molecule. | ||
Conclusions
It has been demonstrated that picolinamide has a strong potential to be a promising cocrystal former. The primary amide and the carboxylic acid moieties form a R22(8) ring which is in agreement with the CSD statistics. The heterocyclic nitrogen of picolinamide was previously reported to be less likely to form intermolecular hydrogen bonds. Our results show, however, a number of alternative hydrogen bonding motifs. Hence, the multicomponent hydrogen bond propensity tool from the CCDC Crystal Form Consortium (CFC) could aid the screening design.16 However, the lattice energy calculations show that, despite having very similar hydrogen bond connections to the experimental structures, none of the substituted picolinamide:dicarboxylic acid cocrystals are thermodynamically more stable than the experimental ones and that the less frequently observed conformations of the acid molecules are apparently stabilised within the crystal packings of 2:1 pic:glu and 2:1 pic:suc. Hence, no single type of predicted interaction, neither hydrogen bonding nor the molecular conformations independently, could offer the resulting crystal structures and stabilities. The result is a fine balance of all interactions which arise from the lattice environments. The serendipity of the outcome indicates that kinetics in the processes are still not fully understood and are a continuing challenge in computational chemistry – obtaining a mixture of a cocrystal and a metastable picolinamide polymorph in one of our experiments is a good example. Multi-component systems have more degrees of freedom and larger unit cells than single component systems, both of which make the calculations more challenging.
Acknowledgements
H. C. S. Chan would like to thank the Novartis Institutes for BioMedical Research for the Presidential Postdoctoral Fellowship, the Chemical and Pharmaceutical Profiling Group for providing the facilities for cocrystal preparation and characterisation, and Dr. Marcus Neumann (Avant-garde Materials Simulation, Freiburg) for his support in the QM lattice energy calculations. The authors would also like to thank Edith Alig (Goethe University, Frankfurt) for the VTXRD measurements. Dr. Peter Hunt and Dr. Rainer Wilcken (Novartis Institutes for BioMedical Research) are acknowledged for the discussions on research ideas.Notes and references
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240 CrossRef.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425 CrossRef PubMed.
- D. J. Berry, C. C. Seaton, W. Clegg, R. W. Harrington, S. J. Coles, P. N. Horton, M. B. Hursthouse, R. Storey, W. Jones, T. Friščić and N. Blagden, Cryst. Growth Des., 2008, 8, 1697 CAS.
- A. O. L. Évora, R. A. E. Castro, T. M. R. Maria, M. T. S. Rosado, M. R. Silva, J. Canothilho and M. E. S. Eusébio, CrystEngComm, 2012, 14, 8649 RSC.
- M. Habgood, M. A. Deij, J. Mazurek, S. L. Price and J. H. ter Horst, Cryst. Growth Des., 2010, 10, 903 CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- P. K. Thallapally, R. K. R. Jetti, A. K. Katz, H. L. Carrell, K. Singh, K. Lahiri, S. Kotha, R. Boese and G. R. Desiraju, Angew. Chem., Int. Ed., 2004, 43, 1149 CrossRef CAS PubMed.
- M. Rafilovich and J. Bernstein, J. Am. Chem. Soc., 2006, 128, 12185 CrossRef CAS PubMed.
- G. M. Day, A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2006, 54 RSC.
- P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499 CrossRef CAS PubMed.
- GRACE software from Avant-garde Materials Simulation, http://www.avmatsim.eu.
- M. A. Neumann and M.-A. Perrin, J. Phys. Chem. B, 2005, 109, 15531 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- H. C. S. Chan, J. Kendrick, M. A. Neumann and F. J. J. Leusen, CrystEngComm, 2013, 15, 3799 RSC.
- P. T. A. Galek, L. Fábián, W. D. S. Motherwell, F. H. Allen and N. Feeder, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 768 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental crystal data and the DFT-D optimised structures are available as cif files. The experimental details and results, together with the minimised lattice energies, are available as a pdf file. CCDC 977784–977792. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00265b |
| ‡ ConQuest version 1.15 was used for searching discrete anhydrous and non-solvated cocrystals of nicotinamide, isonicotinamide and picolinamide in the CSD version 5.34 with the May 2013 update. The polymorph of picolinamide found in a cocrystal sample corresponds to the metastable form I reported in ref. 4. The torsion angle distributions of glutaric acid and succinic acid fragments were surveyed using MOGUL 1.5. |
| This journal is © The Royal Society of Chemistry 2014 |