 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-crystal to single-crystal guest exchange and phase transformations in a porous metallocycle†
Marike
du Plessis
,
Vincent J.
Smith
and
Leonard J.
Barbour
*
Department of Chemistry and Polymer Science, University of Stellenbosch, Stellenbosch 7600, South Africa. E-mail: ljb@sun.ac.za; Fax: +27 21 808 3360; Tel: +27 21 808 3335
First published on 28th March 2014
Abstract
Single crystals of a previously reported porous metallocycle [Ag2L2](BF4)2·2CH3CN (1), where L is the ligand 1,4-bis(2-methylimidazol-1-ylmethyl)benzene, were grown from acetonitrile and immersed in different organic solvents. The crystals thus treated were subjected to single-crystal X-ray diffraction analysis, which revealed that the acetonitrile guest molecules had been replaced by the solvent that the compound was exposed to, yielding five different solvates: [Ag2L2](BF4)2·2(CH3)2CO (2), [Ag2L2](BF4)2·2CHCl3 (3), [Ag2L2](BF4)2·C6H6 (4), [Ag2L2](BF4)2·C6H4F2 (5), [Ag2L2](BF4)2·C7H8 (6). Thermogravimetric analysis supports these findings.
Introduction
The engineering of functional materials1a,b involves the rational design of targeted architectures to perform a specialised function when exposed to certain external stimuli (light, heat, pressure, solvent etc.). In particular, metal–organic frameworks and porous coordination networks have been receiving much attention over the past decade; with potential applications of such materials in sorption, separation, sensing and catalysis.2a–d When a functional single crystal is exposed to a stimulus that induces a structural or compositional change of the material it is often desirable that this change occurs as a single-crystal to single-crystal (SC–SC) transformation, not only for the sake of application, but also to facilitate investigation of the structure–property relationship using single-crystal X-ray diffraction (SCD) methods. Even though accounts of SC–SC transformations have escalated over the past decade they are still considered to be a rare and remarkable phenomenon.3a–g Various metal cation exchanges in coordination compounds have also been shown to occur in SC–SC fashion.4a–c In particular, most of these reports involve higher dimensional metal–organic framework compounds in contrast to the zero-dimensional complexes presented here.One of the target architectures for porous crystals investigated by our group is the “doughnut-shaped” metallocycle. The “doughnut” shape of the metallocycles prevents them from packing efficiently and results in the formation of crevices, cavities or channels in the packing arrangement. Naturally, owing to close-packing requirements the occurrence of empty space in the crystal structure is energetically unfavourable and therefore the available “space” is usually occupied by solvent molecules. Ideally, the porous phase can be obtained by removing the solvent molecules from the channels without disrupting the host framework. In this regard we have conducted a further investigation of the porous metallocycle previously reported by Barbour et al.5 The formation reaction of the metallocyclic complex [Ag2L2](BF4)2·2CH3CN (1) is shown in Scheme 1. We have reported that the guest molecules of 1 can be removed from the channels in a SC–SC fashion to yield a porous, gas sorbing material (1apohost)5 and elsewhere a series of eight solvates of the copper analogue of 1 was reported in an investigation of the effect of solvent templation on the formation of metallocyclic complexes.6 Most of the solvates in the latter case were prepared by removing the solvent from the channels of the as-synthesised crystals under reduced pressure, followed by dissolving the resulting apohost complex in different solvents. Slow evaporation of the solvent afforded the previously reported solvates.
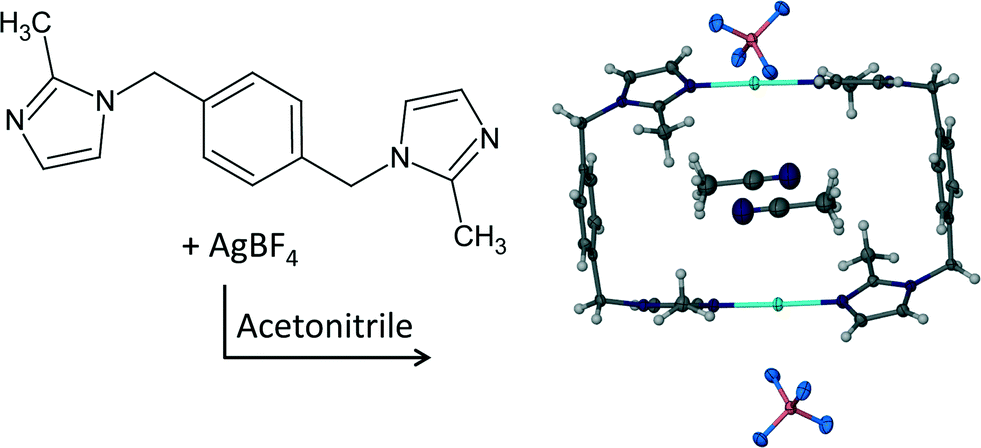 | ||
| Scheme 1 Formation of [Ag2L2](BF4)2·2CH3CN (1) where L = 1,4-bis(2-methylimidazol-1-ylmethyl)benzene. | ||
We now show that 1 can incorporate different solvent molecules into its channels in SC–SC transformations as opposed to these solvates being obtained by dissolving and recrystallising the apohost from different solvents. Furthermore, the transformation is accompanied by subtle changes, which result in a change in the space group and/or crystal system. It is quite unusual for metallocycles to undergo a SC–SC phase transformation5–8 as only a single case has been reported to date.9
Results and discussion
Crystal growth
The ligand L was prepared according to a procedure reported in the literature.10 All other chemicals and solvents were used as purchased without any further purification. Slow evaporation of a dilute acetonitrile solution containing the molecular building blocks in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio resulted in the formation of the metallocyclic complex [Ag2L2](BF4)2·2CH3CN (1). Diffraction quality single crystals thus obtained were used as a starting point towards the generation of the solvates 2–6, where the acetonitrile guest molecules of solvate 1 were exchanged with acetone, chloroform, benzene, 1,4-difluorobenzene and toluene, respectively, in a SC–SC fashion.
:1 ratio resulted in the formation of the metallocyclic complex [Ag2L2](BF4)2·2CH3CN (1). Diffraction quality single crystals thus obtained were used as a starting point towards the generation of the solvates 2–6, where the acetonitrile guest molecules of solvate 1 were exchanged with acetone, chloroform, benzene, 1,4-difluorobenzene and toluene, respectively, in a SC–SC fashion.
Crystal structures
The crystal structures 2–6 resulted from solvent exchange experiments with 1 as depicted in Scheme 2. SCD revealed that all of the solvates 2–6 have crystallographic inversion symmetry.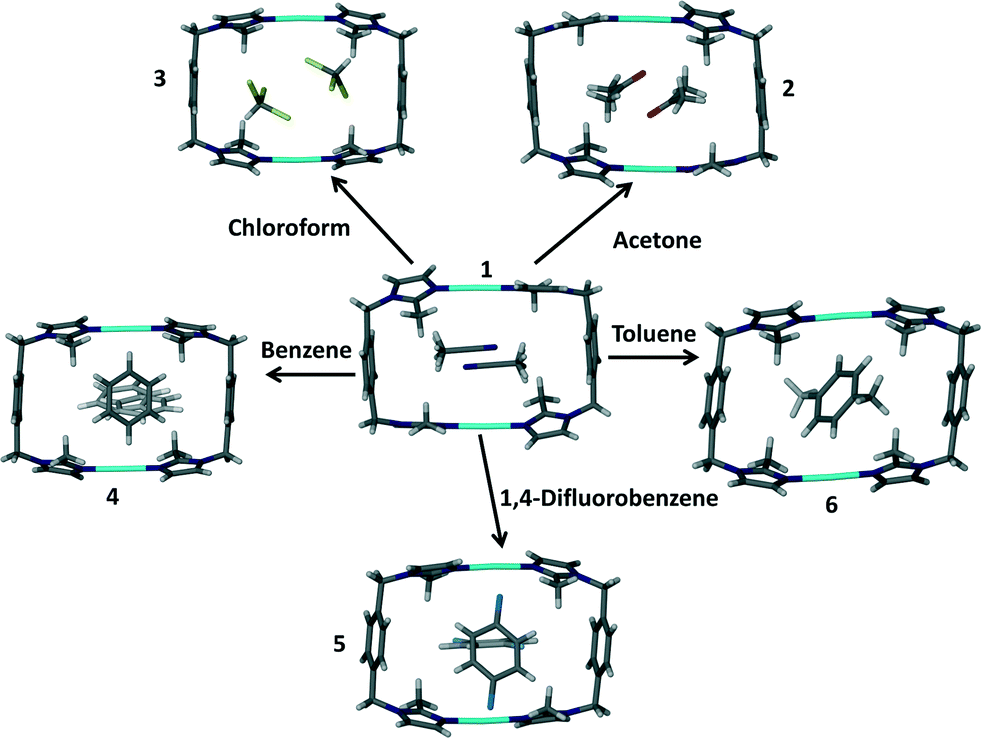 | ||
| Scheme 2 Formation of solvates 2–6 from the original solvate 1 by means of solvent exchange in SC–SC fashion. | ||
Using a glass pipette the prism shaped crystals of 1 were carefully removed from the mother liquor and allowed to dry on filter paper. The dry crystals were then immersed in the target solvent for periods ranging from two to eight weeks. In each case the crystals showed signs of cracking and deterioration such that it was no longer possible to subject the original crystal to SCD. This is in sharp contrast to other reported guest exchanges where the individual crystals generally remained intact.7 Although the bulk integrity of the crystals in the study reported here was not maintained, small single crystals could still be isolated by breaking the original single crystal into smaller fragments. Structure elucidation by means of SCD, together with thermogravimetric analysis (TGA) revealed unambiguously that the guest exchange process was successful. This process occurs with very little alteration of the host structure. As a result, structures 1 to 6 and 1apohost‡ are very similar with respect to the arrangement of the host molecules in the crystal structure and selected parameters are presented for comparison in Table 1. TGA results can be found in the ESI† and they support the findings elucidated by SCD methods.
| Structure | Guest | Host:guest |
Guest volumea/Å3 | Solvent accessible volumeb/Å3 | Intra Ag⋯Agc/Å | Inter Ag⋯Agd/Å | Metallocycle tilt angle (ε)e/° | Phenylene tilt angle (η)f/° | Imidazolyl tilt angle (θ)g/° |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated using a literature reported equation.11 b Solvent accessible void volume per unit cell as calculated by Platon, SQUEEZE.12 c Distance between Ag atoms belonging to the same metallocycle. d Distance between Ag atoms belonging to adjacent metallocycles. e Angle between the least squares plane through the metallocyclic complex and the stacking direction as calculated in Mercury.13 f Angle between the least squares plane through the benzene moiety of the metallocycle in 1 and that of the structure in question. g Angle between the least squares plane through the imidazole moiety of the metallocycle in 1 and that of the structure in question. | |||||||||
| 1apohost | — | — | — | 245 | 7.7 | 7.0 | 90 | 8.0 | 5.0 |
| 1 | Acetonitrile | 1:2 |
49 | 223 | 7.3 | 7.0 | 90 | 0 | 0 |
| 2 | Acetone | 1:2 |
67 | 235 | 7.5 | 7.1 | 87.5 | 8.5 | 5.0/4.5 |
| 3 | Chloroform | 1:2 |
72 | 237 | 7.7 | 7.1 | 86.7 | 4.8 | 4.0, 4.5/4.3 |
| 4 | Benzene | 1:1 |
81 | 238 | 7.4 | 7.1 | 88.6 | 2.8 | 1.7, 2.1/3.2 |
| 5 | 1,4-DFBz | 1:1 |
93 | 241 | 7.6 | 7.1 | 88.7 | 2.3/2.2 | 4.0/3.3 |
| 6 | Toluene | 1:1 |
99 | 208 | 7.5 | 7.0 | 84.1 | 7.6 | 2.1/6.3 |
[Ag2L2](BF4)2·2CH3CN (1)
It is appropriate to discuss the main features of the previously reported structure 1 to highlight similarities and to allow for comparison of this structure to the solvates 2–6. SCD revealed that the metallocyclic complex crystallises in the monoclinic space group C2/m. Two molecules of the ligand L in the C-shaped conformation share two Ag+ cations, thus forming a 2+ charged cyclic unit. The cyclic units stack in columns to form one-dimensional solvent-filled channels along the crystallographic c axis (Fig. 1). The charge is balanced by uncoordinated BF4− anions located in the interstitial spaces between metallocycles and outside the one-dimensional channel.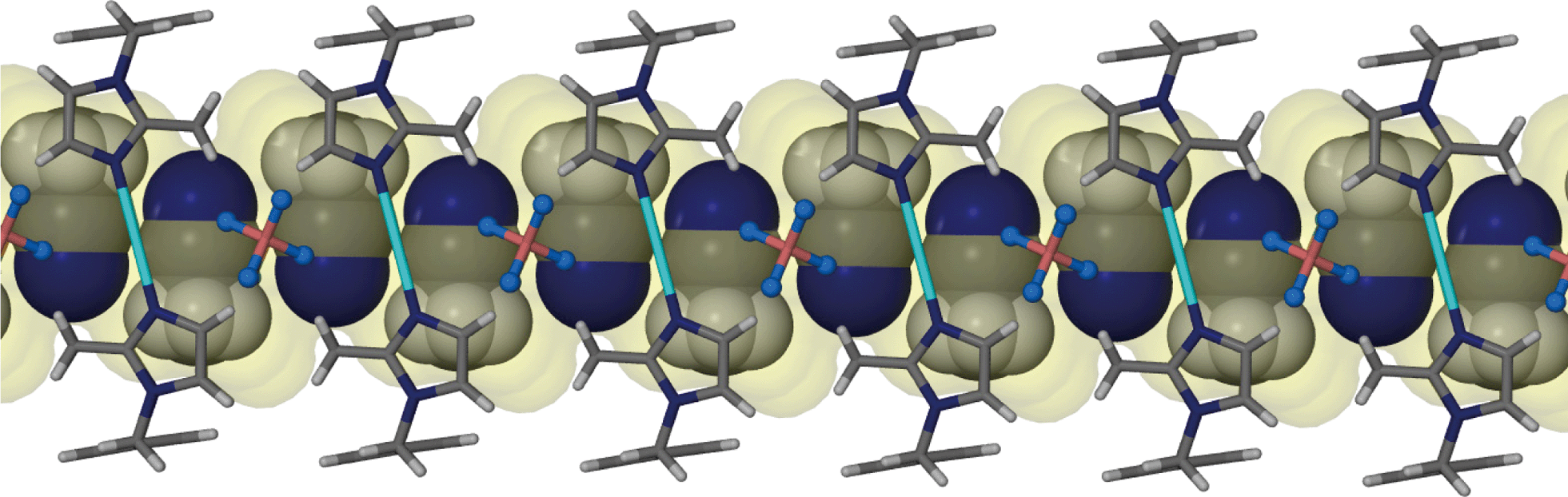 | ||
| Fig. 1 Perspective view perpendicular to a one-dimensional channel of solvate 1. Metallocyclic units that stack to form this channel are shown as capped stick models. The semi-transparent yellow Connolly surface was generated using a probe radius of 1.5 Å and it maps the solvent accessible volume inside the channel. Acetonitrile guest molecules are shown in space filling representation and BF4− anions are represented as ball and stick models. | ||
Acetonitrile molecules are situated in anti-parallel pairs along the infinite channel. Each pair of guest molecules is centred within a metallocycle resulting in a host-to-guest ratio of 1:2. In order to measure/show the subtle differences between the host metallocycles of 1 and 2–6, we define a metallocycle tilt angle (ε) between the least squares plane trough the metallocycle, and the stacking direction. In 1ε is perpendicular to the stacking direction. Furthermore we define a phenylene tilt angle (η) and an imidazolyl tilt angle (θ) as the angle between the least squares plane through the aromatic moiety and its corresponding moiety in the reference structure 1. These planes were calculated and the angles measured after overlaying each of the metallocycles 2–6 and 1apohost with the reference structure 1 using the molecule overlay function in Mercury.13
[Ag2L2](BF4)2·2(CH3)2CO (2)
It is interesting to note that replacement of the acetonitrile molecules in 1 by acetone molecules resulted in a change in crystallographic symmetry. Structure elucidation of 2 revealed that the space group transformed from monoclinic C2/m to triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Similarly to 1, the guest molecules in 2 are arranged in pairs with their carbonyl groups facing in opposite directions (Fig. 2). The distance between the silver cation and the carbonyl oxygen atom of the guest molecule (2.99 Å) is less than the sum of the van der Waals radii and is considered a close contact. However, it is questionable weather this interaction is significant enough to be structure directing.
. Similarly to 1, the guest molecules in 2 are arranged in pairs with their carbonyl groups facing in opposite directions (Fig. 2). The distance between the silver cation and the carbonyl oxygen atom of the guest molecule (2.99 Å) is less than the sum of the van der Waals radii and is considered a close contact. However, it is questionable weather this interaction is significant enough to be structure directing.
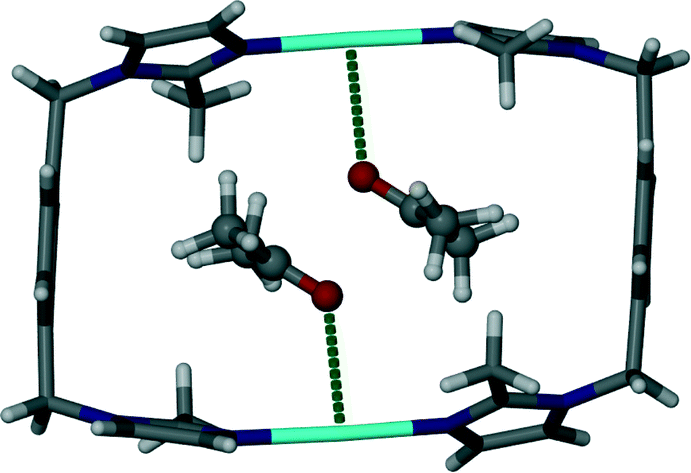 | ||
| Fig. 2 Capped stick representation of a metallocyclic complex of solvate 2. Acetone guest molecules are shown as ball and stick models and close contacts between the host and guest are illustrated as dotted green lines. | ||
The increase in guest volume from 49 Å3 for acetonitrile to 67 Å3 for acetone is accompanied by a corresponding decrease in ε. The tilt angles η and θ display a significant change in the shape of the host metallocycle, which is further adapted through increases in the inter- and intramolecular Ag⋯Ag distances. As expected, the increase in the volume of the guest gives rise to an increase in the guest accessible volume. Fig. 3 shows the one-dimensional solvent filled channel of 2.
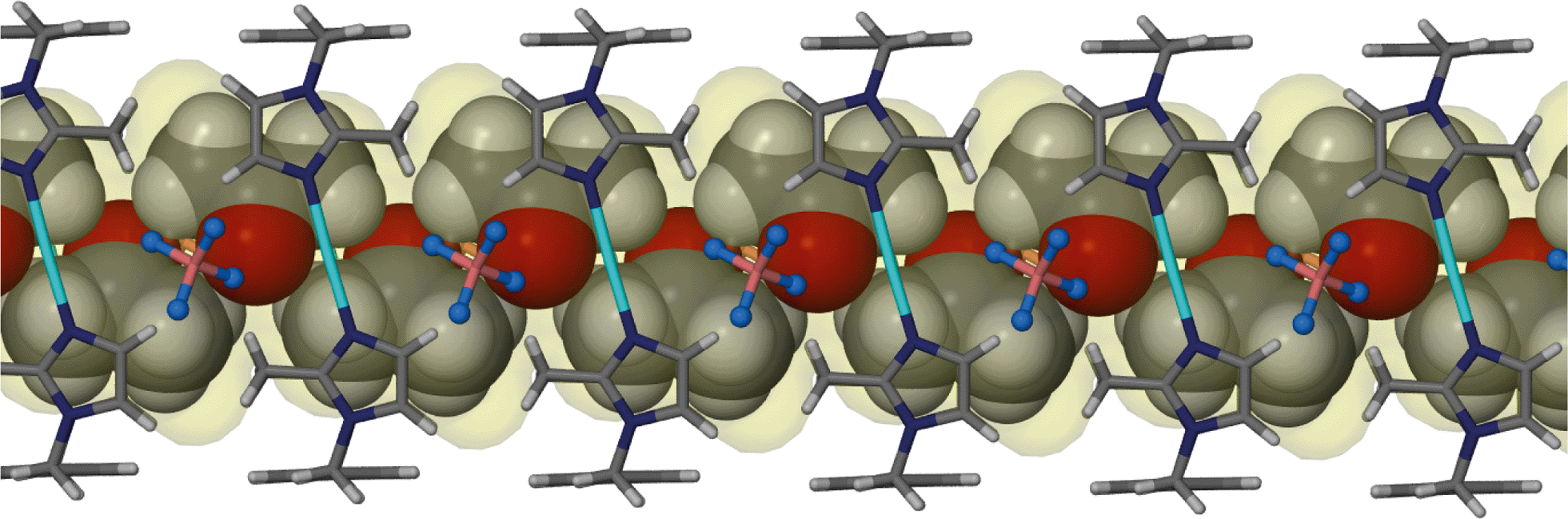 | ||
| Fig. 3 Perspective view perpendicular to a one dimensional channel of solvate 2. Acetone guest molecules are shown in space filling representation. | ||
[Ag2L2](BF4)2·2CHCl3 (3)
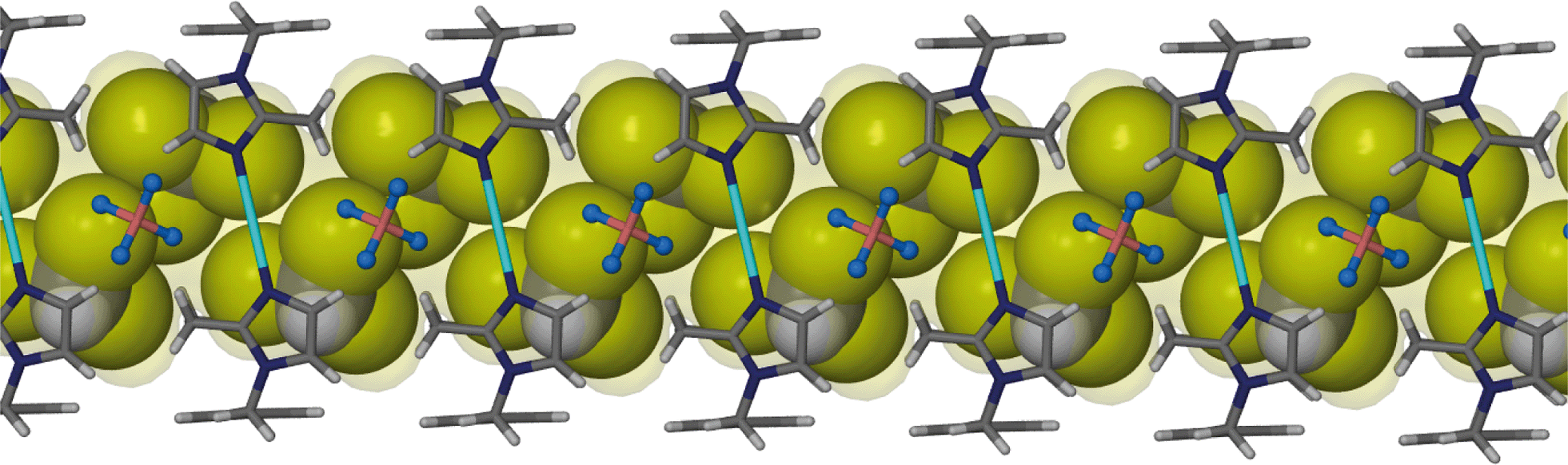
The chloroform solvate transforms from monoclinic C2/m to monoclinic P21/c. Similarly to solvates 1 and 2, two chloroform molecules are situated in the cavity of each metallocyclic unit and they face each other as shown in Fig. 4. In general the transformation from 1 to 3 follows similar changes to the transformation from 1 to 2.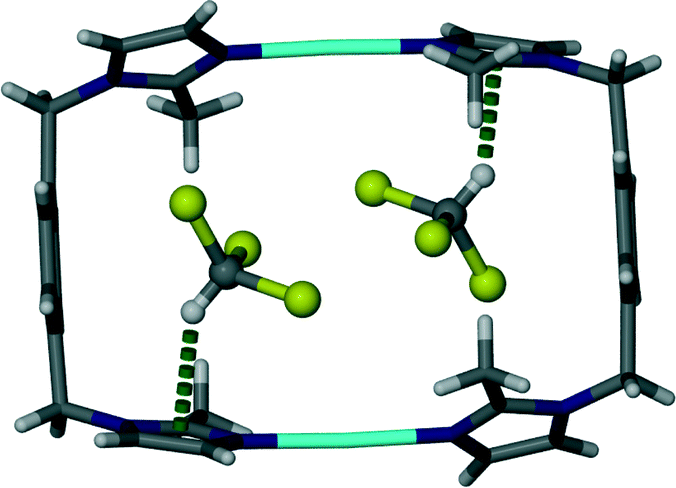 | ||
| Fig. 4 Capped stick representation of a metallocyclic complex of solvate 3. Chloroform guest molecules are shown as ball and stick models and weak host–guest interactions are illustrated as dotted green lines. | ||
Fig. 5 shows the hydrogen atoms of the guest protruding through the Connolly surface that maps the guest accessible volume of the infinite channel. This is an indication of a CH⋯π interaction between the chloroform hydrogen atom and the imidazole moiety of the ligand L. The distance between the carbon atom of the chloroform molecule and the centroid of the imidazole ring (2.59 Å) indicates the very close contact between these moieties.
 | ||
| Fig. 5 Perspective view perpendicular to a one dimensional channel of solvate 3. Chloroform guest molecules are shown in space filling representation. | ||
[Ag2L2](BF4)2·C6H6 (4)
Surprisingly, [Ag2L2](BF4)2 is also permeable to larger guest molecules such as benzene and other aromatic guests. The space group and crystal system for the benzene solvate is triclinic P. In contrast to solvates 1–3, 4 can only accommodate one guest molecule in the cavity of each metallocycle, resulting in a host:guest ratio of 1:1.
Although the guest volume increases from 72 Å3 for one chloroform molecule to 81 Å3 for a single benzene molecule, the solvent accessible volume calculated for solvates 3 and 4 are almost identical. While two molecules of chloroform occupy each cavity of the metallocycles in 3, only one benzene molecule occupies the corresponding space in 4. Therefore there is a relatively large amount of space available for the benzene guest molecules to orientate themselves within the one-dimensional channels and this may explain the disorder of the guest molecules. With reference to Fig. 6 and 7, benzene molecules are situated in the positions of the red molecules with a site occupancy of ca. 30% while the guest occupies the position of the green and orange molecules with occupancies of ca. 27 and ca. 13%, respectively. Note that the periodicity of the host in Fig. 7(C) and (D) is identical but there is a difference in the periodicity of the guest.
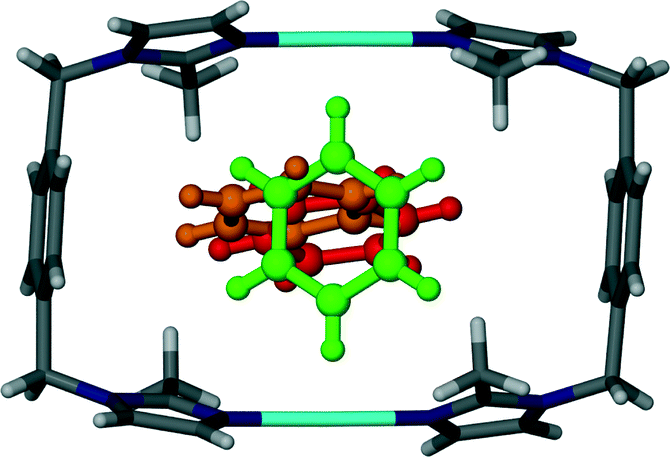 | ||
| Fig. 6 Capped stick representation of a metallocyclic complex of solvate 4. The benzene guest molecule is shown as ball and stick models in green, orange and red in three possible positions as a result of solvent disorder. | ||
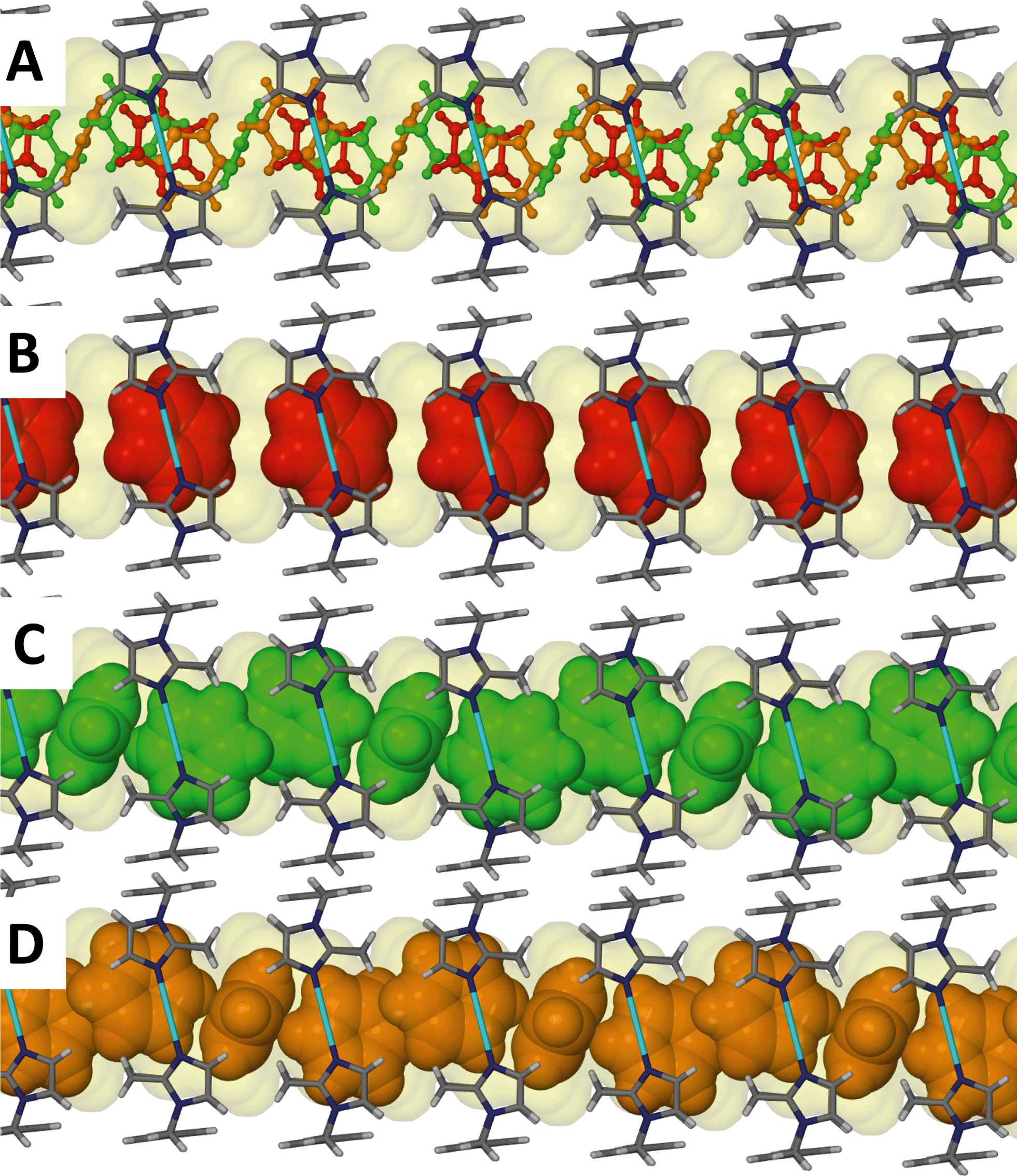 | ||
| Fig. 7 Perspective view perpendicular to a one dimensional channel of solvate 4. (A) Benzene guest molecules are shown as ball and stick models and, as a result of solvent disorder, the three possible positions of the guest molecules are represented by the red, green and orange molecules. (B) A space filling representation of the guest in the position of the red molecule. (C) A space filling representation of the guest in the position of the green molecule. (D) A space filling representation of the guest in the position of the orange molecule. Anions are omitted for clarity. | ||
[Ag2L2](BF4)2·C6H4F2 (5)
Analogous to 4, the difluorobenzene solvate 5 undergoes a transformation to the triclinic space group P and the guest molecules are disordered within the channel. The host:guest ratio for this metallocycle is also 1:1 with the guest molecule occupying two possible positions as shown in Fig. 8. The distribution of occupancies is 80% for the major position and 20% for the minor position. The molecule in the minor position is almost perpendicular to the molecule in the major position, as shown by the orange (major position) and green molecules (minor position) in Fig. 9. No significant interactions can be identified between the host metallocycles and the guest molecules.
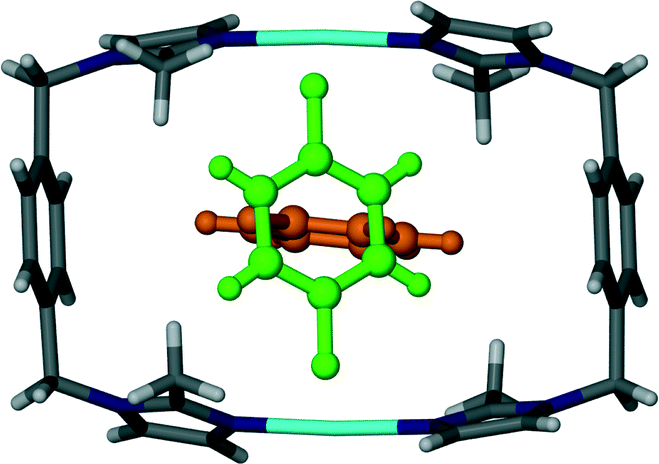 | ||
| Fig. 8 Capped stick representation of a metallocyclic complex of solvate 5. The 1,4-difluorobenzene guest molecule is shown as a ball and stick model in its position of primary occupancy (orange) and secondary occupancy (green). | ||
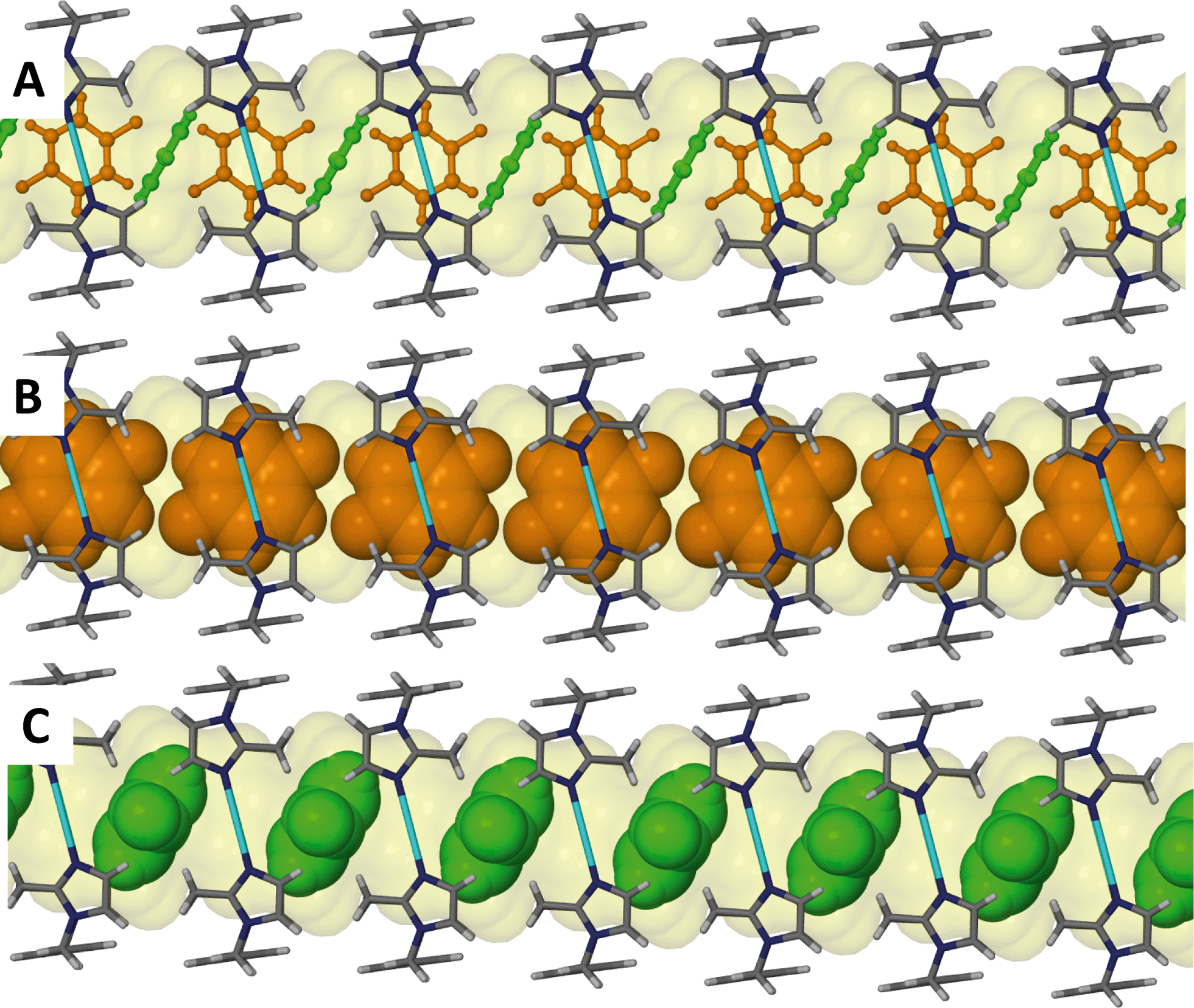 | ||
| Fig. 9 Perspective view perpendicular to a one dimensional channel of solvate 5. (A) 1,4-Difluorobenzene guest molecules are shown disordered over two positions as ball and stick models. (B) The primary position of the guest is shown by the orange coloured molecules in a space filling representation. (C) The secondary position of the guest is shown by the green coloured molecules in a space filling representation. Anions are omitted for clarity. | ||
[Ag2L2](BF4)2·C7H8 (6)
As in the case of 4 and 5, the toluene solvate (6) also undergoes a transformation to the triclinic space group P. Fig. 10 shows the guest molecule in a 1:1 ratio with the host metallocycle. The methyl group of the guest molecules is disordered over two positions of equal occupancy as shown by the orange and green coloured atoms in Fig. 11.
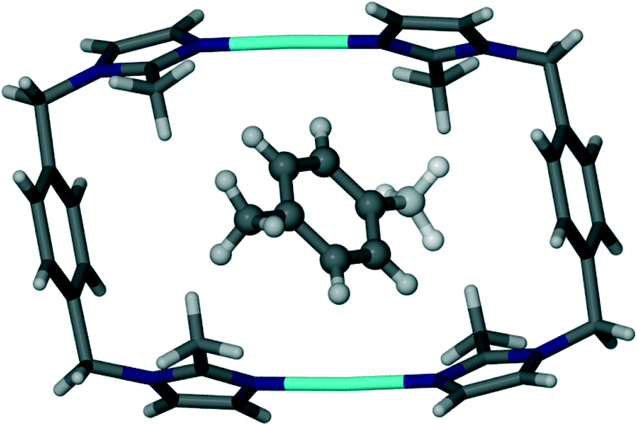 | ||
| Fig. 10 Capped stick representation of a metallocyclic complex of solvate 6. The toluene guest molecule is shown as a ball and stick model and the methyl group is disordered over two positions of equal occupancy. | ||
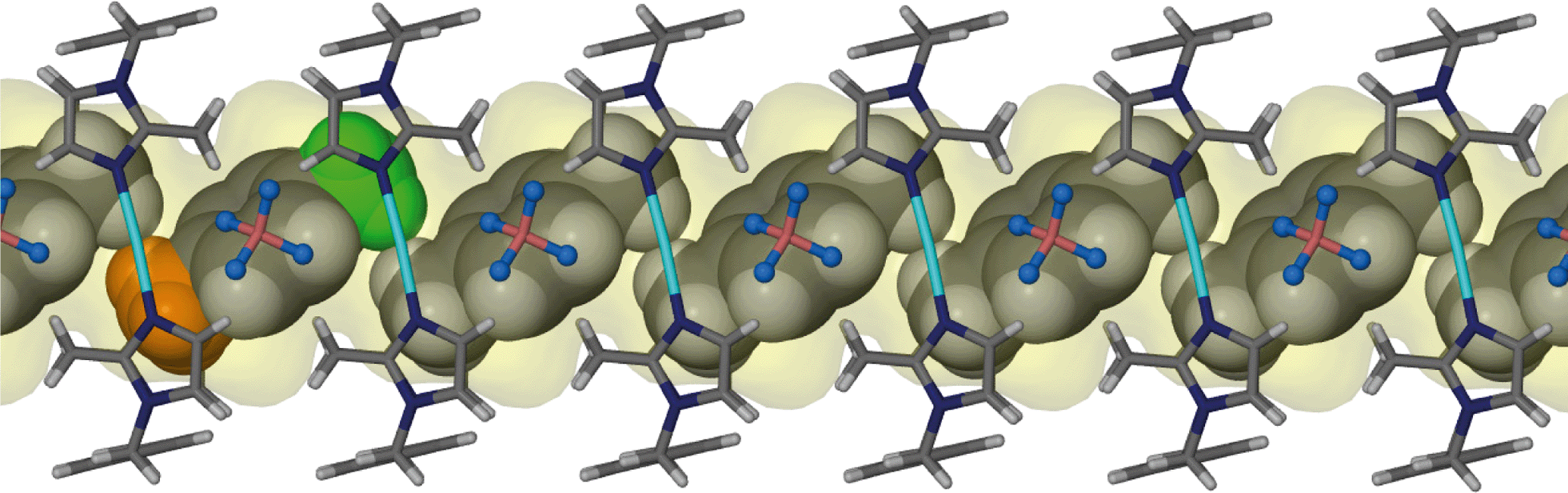 | ||
| Fig. 11 Perspective view perpendicular to a one dimensional channel of solvate 6. Disordered toluene guest molecules are shown in space filling representation. Green and orange coloured atoms represent the two equally occupied positions of the methyl group of the toluene guest. | ||
It is interesting to note that the toluene solvate deviates significantly from the overall trend of an increase in the solvent accessible volume with an increase in guest volume. Even though the van der Waals volume of toluene is the largest of the series of guest molecules, it has the smallest solvent accessible volume. Furthermore, ε in 6 is the largest of the deviations observed and the neighbouring metallocycles are stacked closest together, as is evident from the inter Ag⋯Ag distances. Despite the significant deviations in 6, there are no significant interactions between the host framework and the guest molecules.
Crystal packing and comparison of structures
A packing diagram of 1 viewed along the stacking direction of the metallocycles is compared to a similar view of the packing arrangement of 3 in Fig. 12. This view shows how π⋯π interactions stabilise the stacking of the cyclic units in a “brick wall” motif. As the new solvent molecules travel through the channels, displacing acetonitrile molecules in the transformation from 1 to 3, the metallocycle host framework needs to adapt its shape to accommodate the new guest molecules. This adjustment takes place, for example, in the form of tilting of the aromatic ring moieties and stretching or contracting of the metallocycle by adjusting the inter- and intramolecular Ag⋯Ag distance and the angles formed by the corners of the metallocycle (N–C–C). These changes are evident from a comparison of parameters given in Table 1 as well as in the ESI.†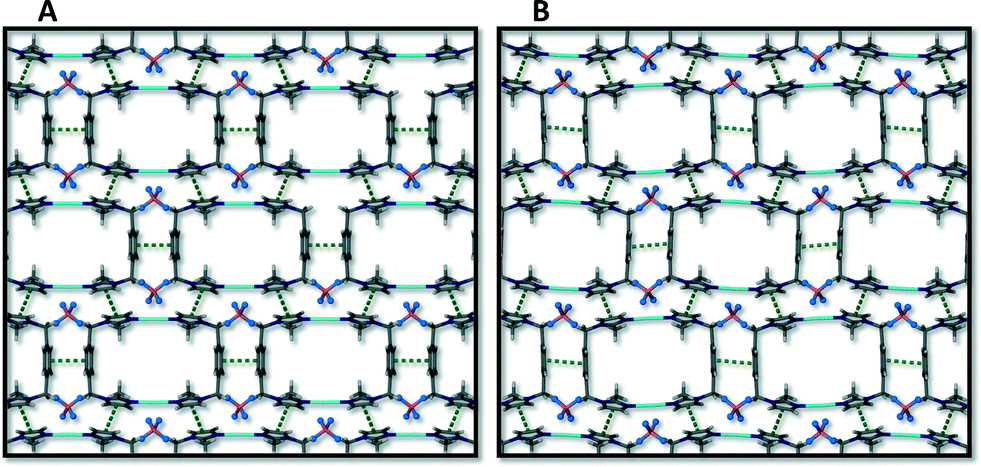 | ||
| Fig. 12 (A) Metallocyclic units of 1 stack in a brick wall packing arrangement that is stabilised by π⋯π interactions shown as dotted green lines. This view directly along the channels of 1 is compared to a similar view along the channels of 3 (B) to display the subtle difference in the shape and packing arrangement of the host framework after incorporation of a different guest into the channels of the host. | ||
When comparing the six solvates we find that the metallocyclic host frameworks are very similar. Upon closer inspection of the structural parameters it is evident that the host undergoes very subtle changes upon guest exchange. In the series of structures presented here, the volume of the guest molecule is gradually increased from 2–6 (67–99 Å3). A consequence of increasing the size of the guest molecule is that at some point the host:guest ratio has to decrease from 1:2 to 1:1 as observed by comparing the smaller, non-aromatic guests with the larger, aromatic guests. If we consider the aromatic- and non-aromatic guest molecules separately the intramolecular Ag⋯Ag distance displays a trend. There is an increase in this distance with an increase in the guest volume with the toluene solvate (6) being the exception. A similar observation is made when comparing the solvent accessible volume of the structures. It is interesting to note that the empty host structure has the largest void volume. It can be rationalised that the metallocycles of the apohost expand significantly since there is no guest to embrace. The other extreme is that the metallocycle collapses in on itself. Interestingly this may occur as a SC–SC transformation.7,9ε adjusts between 1 and 6 degrees while the intermolecular Ag⋯Ag distance changes only slightly (7.0–7.1 Å). η adjusts by as much as 8.5 degrees in 2. The largest values for η and θ are observed for 2 and 6, which can be rationalised since these are the solvates with the smallest and largest guests respectively in the series 2–6. The N–Ag–N angle ranges between 175.5° and 179.0° and deviates from linearity as the metallocycle adapts its shape to accommodate the guest. Further adjustments are observed in the corners of the metallocycles (the N–C–C angle ranges between 109.9° and 111.7°).
Conclusions
We have shown that the acetonitrile molecules in the channels of compound 1 can be exchanged with acetone, chloroform, benzene, 1,4-difluorobenzene and toluene molecules to yield solvates 2–6. The displacement of guest molecules results in minor adjustments in the packing arrangement and conformation of the host metallocycles in a SC–SC phase transformation.It could be postulated that limited weak interactions or rather, close contacts as observed in some of these solvates are responsible for the orientation of the guest molecules with respect to the host framework. However, none of the structures show host–guest or guest–guest interactions significant enough to direct and determine the position of the guest molecules in the host framework unequivocally. In fact, the contributions of these weak contacts are insignificant when compared to the many other factors that determine the crystal structure as Gavezzotti outlined in a recent article.14 We believe that incorporation of the various types of guest molecules into the channels of the host takes place via cooperative movement.15a,b During this process the host framework adapts to the shape of the guest molecules in a shape-fit manner.
Very little data exist in the literature of single-crystal replacement of guest molecules in zero-dimensional coordination compounds. It is envisioned that a large database of discrete coordination complexes incorporating different solvent molecules may provide sufficient information to assist in gaining more insight into the mechanism of guest exchange. Often the large solvent accessible space in MOFs presents difficulties with modeling guest molecules from SCD experimental data. In this regard, metallocyclic compounds are ideal candidates for SC–SC transformation studies with the aim of investigating structure–property relationships. We can, in effect, take “snapshots” of small organic molecules as they are captured by an appropriate crystalline host.
Acknowledgements
We thank the National Research Foundation of South Africa for financial support of this work.Notes and references
- (a) C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22 RSC; (b) G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS PubMed.
- (a) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275 RSC; (b) M. E. Medina, A. E. Platero-Prats, N. Snejko, A. Rojas, A. Monge, F. Gandara, E. Gutiérrez-Puebla and M. A. Camblor, Adv. Mater., 2011, 23, 5283 CrossRef CAS PubMed; (c) H.-L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351 RSC; (d) S. Ma, Pure Appl. Chem., 2009, 81, 2235 CrossRef CAS.
- (a) G.-C. Lv, P. Wang, Q. Liu, J. Fan, K. Chen and W.-Y. Sun, Chem. Commun., 2012, 48, 10249 RSC; (b) M. E. van der Boom, Angew. Chem., Int. Ed., 2011, 50, 11846 CrossRef CAS PubMed; (c) E. Y. Lee and M. P. Suh, Angew. Chem., Int. Ed., 2004, 43, 2798 CrossRef CAS PubMed; (d) H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844 CrossRef CAS PubMed; (e) Z.-M. Hao and X.-M. Zhang, Dalton Trans., 2011, 40, 2092 RSC; (f) S. Gosh, W. Kaneko, D. Kiriya, M. Ohba and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 8843 CrossRef PubMed; (g) G. Mehlana, G. Ramon and S. Bourne, CrystEngComm, 2013, 15, 9521 RSC.
- (a) M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172 CrossRef PubMed; (b) S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814 CrossRef CAS PubMed; (c) J. Tian, L. V. Saraf, B. Schwenzer, S. M. Taylor, E. K. Brechin, J. Liu, S. J. Dalgarno and P. K. Thallapally, J. Am. Chem. Soc., 2012, 134, 9581 CrossRef CAS PubMed.
- L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2005, 127, 13134 CrossRef PubMed.
- L. Dobrzańska, G. O. Lloyd and L. J. Barbour, New J. Chem., 2007, 31, 669 RSC.
- L. Dobrzańska, G. O. Lloyd, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2005, 45, 5856 CrossRef PubMed.
- (a) T. Jacobs and L. J. Barbour, CrystEngComm, 2013, 15, 1512 RSC; (b) T. Jacobs, G. O. Lloyd, J.-A. Gertenbach, K. K. Müller-Nedebock, C. Esterhuysen and L. J. Barbour, Angew. Chem., 2012, 124, 4997 CrossRef PubMed; (c) T. Jacobs and L. J. Barbour, Aust. J. Chem., 2010, 63, 573 CrossRef CAS.
- T. Jacobs and L. J. Barbour, New J. Chem., 2013, 37, 71 RSC.
- P. S. Mukherjee, S. Gosh and R. Chakrabarty, Dalton Trans., 2008, 1850 Search PubMed.
- Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Woo, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- A. Gavezzotti, CrystEngComm, 2013, 15, 4027 RSC.
- (a) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000 CrossRef CAS PubMed; (b) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: TGA results for 1apohost and 1–6, selected structure parameters, crystallographic data tables. CCDC 984255–984259. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00244j |
| ‡ Crystal data for 1apohost: C32H36Ag2B2F8N8, M = 922.05, colourless prism, 0.25 × 0.20 × 0.10 mm3, monoclinic, space group C2/m (no. 12), a = 14.8307(10), b = 20.5641(13), c = 7.0449(5) Å, β = 90.1180(10)°, V = 2148.5(3) Å3, Z = 2, Dc = 1.425 g cm−3, F000 = 920, Bruker APEX-II CCD, MoKα radiation, λ = 0.71073 Å, T = 100(2) K, 2θmax = 56.5°, 6760 reflections collected, 2555 unique (Rint = 0.0199). Final GooF = 1.071, R1 = 0.0267, wR2 = 0.0639, R indices based on 2430 reflections with I > 2sigma(I) (refinement on F2), 120 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.977 mm−1. Crystal data for 1: C36H42Ag2B2F8N10, M = 1004.16, colourless prism, 0.25 × 0.20 × 0.15 mm3, monoclinic, space group C2/m (no. 12), a = 14.9110(10), b = 20.1383(14), c = 7.0209(5) Å, β = 90.2930(10)°, V = 2108.2(3) Å3, Z = 2, Dc = 1.582 g cm−3, F000 = 1008, Bruker APEX-II CCD, MoKα radiation, λ = 0.71073 Å, T = 100(2) K, 2θmax = 56.4°, 6675 reflections collected, 2520 unique (Rint = 0.0203). Final GooF = 1.062, R1 = 0.0257, wR2 = 0.0625, R indices based on 2413 reflections with I > 2sigma(I) (refinement on F2), 139 parameters, 0 restraints. Lp and absorption corrections applied, μ = 1.004 mm−1. Crystal data for 2: C38H48Ag2B2F8N8O2, M = 1038.20, colourless prism, 0.17 × 0.11 × 0.10 mm3, triclinic, space group P Crystal data for 3: C34H38Ag2B2Cl6F8N8, M = 1160.78, colourless prism, 0.21 × 0.21 × 0.19 mm3, monoclinic, space group P21/c (no. 14), a = 7.0910(19), b = 20.479(6), c = 14.855(5) Å, β = 91.038(4)°, V = 2156.9(11) Å3, Z = 2, Dc = 1.787 g cm−3, F000 = 1152, Bruker APEX-II CCD, MoKα radiation, λ = 0.71073 Å, T = 100(2) K, 2θmax = 56.7°, 13 Crystal data for 4: C38H42Ag2B2F8N8, M = 1000.16, colourless prism, 0.20 × 0.19 × 0.18 mm3, triclinic, space group P Crystal data for 5: C38H40Ag2B2F10N8, M = 1036.14, colourless prism, 0.13 × 0.13 × 0.10 mm3, triclinic, space group P Crystal data for 6: C39H43Ag2B2F8N8, M = 1013.17, colourless prism, 0.17 × 0.13 × 0.11 mm3, triclinic, space group P |
| This journal is © The Royal Society of Chemistry 2014 |