DOI:
10.1039/C4CE00105B
(Paper)
CrystEngComm, 2014,
16, 4382-4389
Three metal–organic frameworks based on the semirigid V-shaped 5-(3-amino-tetrazole-5-phenoxy)-isophthalic acid ligand: syntheses, topological structures and properties†
Received
15th January 2014
, Accepted 1st February 2014
First published on 3rd February 2014
Abstract
The designed organic ligand 5-(3-amino-tetrazole-5-phenoxy)-isophthalic acid (H2atpia) has been successfully prepared and its coordination features have been explored. By the reaction of H2atpia with transition metals, three metal–organic frameworks (MOFs), namely [Cd(atpia)]2 (1), [Cu3O2(atpia)2] (2) and [Mn3(OH)2(atpia)3]·1.25H2O (3) have been synthesized under hydrothermal conditions. All the compounds were fully characterized by elemental analysis, IR spectroscopy, thermogravimetric analysis, power X-ray diffraction and single-crystal X-ray diffraction. These three compounds all display new topologies. Compound 1 exhibits a 3D (2,3,10)-connected framework with (4·62)2(412·618·811·104)(4)2 topology. Compound 2 possesses a 2D 3-nodal layer with (416·614·811·104)(43)2(4)2 topology. Compound 3 features a 2D (3,3,8,8)-connected structure with unprecedented (3·42)2(34·46·56·68·73·8) topology. The diverse structures of these three compounds demonstrate that the distinctive coordination modes of different metals have a significant impact on the construction of MOFs. Moreover, the photoluminescence properties of 1 and the magnetic properties of 2 and 3 have been studied and discussed.
Introduction
Over the past decade, crystal engineering concerning metal–organic frameworks (MOFs) has provoked great interest due to their intriguing network topologies1–4 and potential technological application such as magnetism, luminescence, adsorption and catalysis.5–8 The connectivity and symmetry of the constituting metal ion (or metal cluster) and organic ligand often govern the structural characteristics of metal–organic materials.9 Even a small change in the characteristics of the ligand can produce significant differences in the final structure. Therefore, the judicious choice and design of organic moieties is very important and there exists a great interest in the search for new bridging ligands.
Many construction features of ligands could affect the structural assembly process of these materials, such as the size and shape, the flexibility and the functional groups within the ligands. Large size ligands have been proven to be good choices to prepare MOFs with a high surface area and the development of synthetic strategies that allow the control of the pore size and pore geometry is particularly important.10 Moreover, the flexibility of the ligands plays an important role in building MOFs, showing not only versatile coordination abilities but also numerous coordination modes. For instance, in comparison with the rigid aromatic ligands, the semi-rigid V-shaped ligands possess more flexibility because of the free rotation of the benzene ring and therefore may build MOFs with more diverse structures. In addition, researchers spend the most time on the design and synthesis of multifunctional organic bridging ligands in order to obtain desired frameworks. Among them, MOFs with carboxylic acid groups which provide reliable coordination modes have been widely studied.11 Similar to the carboxylic acid group, tetrazole and their derivatives have attracted much attention because their nitrogen electron-donating atoms allow them to serve as either multidentate ligands or bridging building blocks in supramolecular assemblies.12,13 It can be foreseen that the building blocks containing both carboxyl and tetrazole moieties will generate rich and unpredictable structures.14 Furthermore, the introduction of substituent groups such as amine, methyl and nitro groups14d,15 attached to organic linkers may bring about changes of electron, space, solubility and configuration, which may result in significant conversion in the types of skeletons and functionalities. For all of the above reasons, it would be attractive to develop synthetic approaches towards new flexible MOF materials.
One strategy that has been employed for this purpose involves the use of large size semi-rigid polytopic ligands and such ligands may become a type of building block to form MOFs with novel topologies and properties.16 Here, we designed and synthesized the new ligand 5-(3-amino-tetrazole-5-phenoxy)-isophthalic acid (H2atpia, Scheme 1) as a multidentate bridging ligand, and successfully prepared three novel MOFs, [Cd(atpia)]2 (1), [Cu3O2(atpia)2] (2) and [Mn3(OH)2(atpia)3]·1.25H2O (3). The results show that these compounds possess interesting topological structures and functional properties.
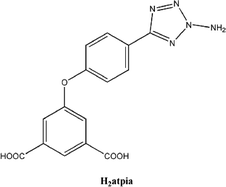 |
| | Scheme 1 A schematic representation of the H2atpia ligand. | |
Experiment section
Materials and measurements
All reagents were purchased commercially and used without further purification. The hydrothermal reaction was performed in a 15 mL Teflon-lined stainless steel bomb. The diffraction data collection was performed with Mo-Kα radiation (λ = 0.71073 Å) on a Rigaku RAXIS-RAPID area-detector diffractometer. The IR spectrum was measured in KBr pellets on a Bruker IFS-66V/S FT-IR spectrometer. The thermogravimetric measurement was performed on pre-weighed samples in a nitrogen stream using a Netzsch STA449C apparatus with a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) data were obtained using a Rigaku D/Max2550 automated diffractometer (Cu-Kα, 1.5418 Å). The excitation and luminescence spectra were performed on a FLUOROMAX-4 fluorescence spectrometer in the solid state at room temperature. The magnetic measurements of the compounds were carried out by the use of a Quantum Design SQUID MPMS-VSM magnetometer in the temperature range of 2–300 K for fields up to 5 T. 1H NMR was done on a Bruker Avance III spectrometer at 400 MHz.
The synthesis of dimethyl 5-(4-cyanophenoxy)-isophthalate (dcpi).
Using 10 g (0.05 mol) of 5-hydroxy-isophthalic acid dimethyl ester17 and following the procedure reported previously,18 14 g of dcpi was produced. Yield: 95%.
The synthesis of 5-(3-amino-tetrazole-5-phenoxy)-isophthalic acid (H2atpia).
Magnetically stirred mixtures of dcpi (2.0 g, 6.4 mmol), NH4Cl (0.45 g, 8.4 mmol) and NaN3 (0.55 g, 8.4 mmol) in anhyd DMF (15 mL) were heated to 140 °C for 48 h. The cooled mixtures were poured into H2O (100 mL) and acidified to pH = 2 with concentrated hydrochloric acid. After 12 h at 4 °C, the suspension was filtrated using a Büchner funnel, the residue was washed with H2O, H2O/EtOH (1/1) and H2O and dried. Recrystallisation from EtOH gave 1.8 g pure products. Yield: 80%. 1H NMR ([D6]DMSO, 400 MHz): δ = 7.28 (2H), 7.76 (2H), 7.96 (2H), 8.10 (2H), 8.27 (1H), 13.49 (2H) ppm. Elemental analysis: calcd for C15H11N5O5 (341.08): C, 52.79; H, 3.25, N, 20.52; found: C, 52.90; H, 3.29; N, 20.64. ESI-MS: m/z (100%) 341.0826 [M]+.
The synthesis of [Cd(atpia)]2 (1).
Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol) and H2atpia (0.0341 g, 0.1 mmol) were mixed in 5 mL distilled water. The mixture was placed in a Teflon-lined stainless steel vessel and heated at 180°C for 48 h, then cooled to room temperature over 24 h. Colorless crystals of 1 were obtained and collected by filtration, washed with water and then dried in air. Yield: 58% (based on H2atpia). Anal. calcd (%): C, 39.89; H, 2.01; N, 15.51. Found (%): C, 40.01; H, 2.06; N, 15.44. IR (KBr, cm−1): 3514, 3432, 3080, 1627, 1563, 1453, 1408, 1259, 1206, 1169, 1110, 976, 842, 790, 730, 521, 432.
The synthesis of Cu3O2(atpia)2 (2).
A similar reaction of Cu(NO3)2·3H2O (0.0362 g, 0.15 mmol) with H2atpia (0.0171 g, 0.05 mmol) in 5 mL distilled water gave 2 as green crystals (55% yield). Anal. calcd (%): C, 39.98; H, 2.01; N, 15.54. Found (%): C, 39.91; H, 2.07; N, 15.60. IR (KBr, cm−1): 3629, 3591, 3552, 3460, 3358, 3080, 1631, 1575, 1523, 1476, 1374, 1265, 1210, 1163, 1103, 999, 975, 936, 850, 772, 735, 514, 420.
The synthesis of [Mn3(OH)2(atpia)3]·1.25H2O (3).
A similar reaction of MnCl2·4H2O (0.0198 g, 0.1 mmol) with H2atpia (0.0341 g, 0.1 mmol) in 5 mL distilled water gave 3 as colorless crystals (60% yield). Anal. calcd (%): C, 43.90; H, 1.92; N, 17.07. Found (%): C, 43.76; H, 2.01; N, 16.98. IR (KBr, cm−1): 3656, 3492, 3460, 3086, 2531, 1710, 1645, 1592, 1468, 1421, 1303, 1242, 1200, 1093, 999, 960, 905, 850, 757, 728, 694, 498.
Single-crystal structure determination
The data collection and structural analysis of crystals 1–3 were performed on a Rigaku RAXIS-RAPID equipped with a narrow-focus, 5.4 kW sealed tube X-ray source (graphite-monochromated Mo-Kα radiation, λ = 0.71073 Å). The data processing was accomplished with the PROCESS-AUTO processing program. All of the data were collected at a temperature of 20 ± 2 °C. Direct methods were used to solve the structure using the SHELXL19 crystallographic software package. All non-hydrogen atoms were easily found from the difference Fourier map. All non-hydrogen atoms were refined anisotropically. The basic information pertaining to the crystal parameters and structure refinement is summarized in Table 1. Selected bond lengths and angles are listed in Table S1.†
Table 1 The crystal data and structure refinement for compounds 1–3
| Compound |
1
|
2
|
3
|
| Molecular formula |
C30H18Cd2N10O10 |
C60H38Cu6N20O25 |
C45H31.50Mn3N15O18.25 |
| Fw |
903.34 |
1820.34 |
1239.17 |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
|
a (Å) |
10.130(2) |
9.976(2) |
19.408(4) |
|
b (Å) |
10.646(2) |
9.999(2) |
38.069(8) |
|
c (Å) |
28.531(6) |
17.099(3) |
14.436(3) |
|
α (deg) |
90 |
85.38(3) |
90 |
|
β (deg) |
94.23(3) |
75.37(3) |
105.32(3) |
|
γ (deg) |
90 |
84.31(3) |
90 |
|
V (Å3) |
3068.5(11) |
1639.6(6) |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 287(4) 287(4) |
|
Z
|
4 |
1 |
8 |
|
D
c (g cm−3) |
1.955 |
1.844 |
1.600 |
|
μ (mm−1) |
1.465 |
2.013 |
0.815 |
|
F(000) |
1776 |
912 |
5020 |
| Reflections collected/unique |
29299/6976 |
16088/7333 |
47116/11191 |
|
R
int
|
0.0626 |
0.0305 |
0.0771 |
|
R indices [I > 2σ(I)] |
R
1 = 0.0394 |
R
1 = 0.0438 |
R
1 = 0.0652 |
| wR2 = 0.0804 |
wR2 = 0.1169 |
wR2 = 0.1651 |
|
R indices (all data) |
R
1 = 0.0518 |
R
1 = 0.0601 |
R
1 = 0.1106 |
| wR2 = 0.0859 |
wR2 = 0.1277 |
wR2 = 0.1917 |
Results and discussion
The crystal structures of [Cd(atpia)]2 (1)
The single-crystal X-ray diffraction analysis reveals that compound 1 is a 3D framework. The asymmetric unit of 1 has two cadmium(II) atoms and two crystallographically independent ligands. Cd1 is seven coordinated by six oxygen atoms (O1, O2, O3, O4, O8, O8A) from four ligands and one nitrogen atom from a different ligand, respectively, to furnish a capped octahedron geometry. Cd2 is hexacoordinate bonded by a nitrogen atom from the ligand and five oxygen atoms (O1, O6, O7, O9, O4A) from four diverse ligands (Fig. 1a). The bond lengths of Cd–O fall between 2.231(3) and 2.547(3) Å, which are similar to the documented Cd–O (carboxylate) distances [2.251–2.879 Å].20 The bond lengths of Cd–N are in the range of 2.503(4)–2.537(4) Å, which are in good agreement with previous studies.21 The ligands possess two modes in 1 (Fig. 1b). In mode 1, the ligand L2− links six Cd centers through the μ6:η1:η1:η1:η2:η1 coordination mode, in which two nitrogen atoms coordinate to two Cd2+ ions and two caboxylate groups connect four Cd2+ ions in μ3:η1:η2 and a chelate coordination fashion, respectively. In mode 2, the two diverse carboxyl groups both in the μ3:η1:η2 mode and Cd1 shares O1 with Cd2 to form a tetranuclear cadmium building unit (Fig. 1d).
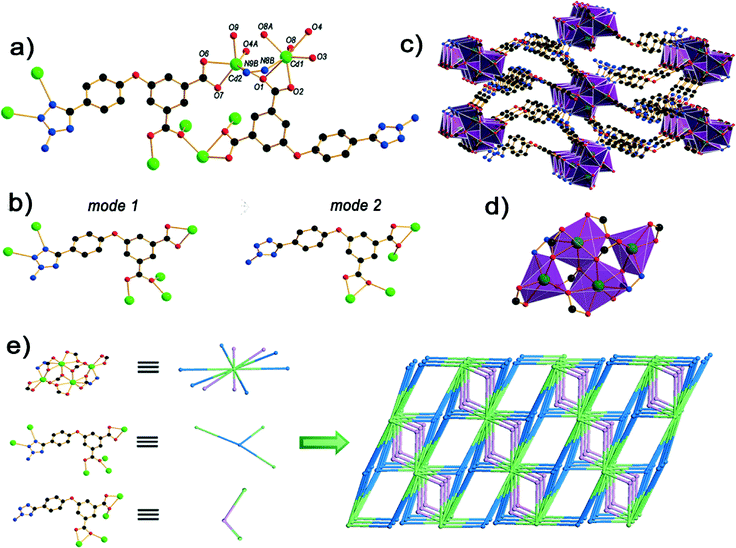 |
| | Fig. 1 (a) The coordination environment of Cd(II) and the bonding mode of H2atpia in the coordination polymer. All H atoms are omitted for clarity; (b) the two coordination modes in 1; (c) the 3D framework connected by neighboring Cd4 clusters in different directions; (d) the tetranuclear-like cadmium building unit; (e) the (2,3,10)-connected topology of 1. Black, C; red, O; blue, N; green, Cd. | |
To achieve better insight into the 3D framework structure of 1, topological analysis was carried out.22 Each simple framework in 1 can be topologically represented as a 3-nodal (2,3,10)-connected net by reducing the Cd4 clusters as a 10-connected node and the H2atpia ligands as 2-connected and 3-connected nodes. Notably, this topology with a vertex symbol of (4·62)2(412·618·811·104)(4)2 is unknown in previously reported metal–organic frameworks (Fig. 1e).
The crystal structures of Cu3O2(atpia)2 (2)
The single-crystal X-ray analysis reveals that compound 2 crystallizes in the triclinic space group P. The asymmetric unit of 2 has three copper(II) atoms, two bridging oxygen atoms and two crystallographically independent ligands. The water molecule within the framework is disordered. The coordination geometry of the Cu1 center can be described as tetragonal pyramid geometry, in which four oxygen atoms (O2, O3, O4 and O11) comprise the equatorial plane, while one oxygen atom (O12A) occupies the apex position. The coordination geometry of the Cu2 can be regarded as five-coordinated, containing two oxygen atoms (O1 and O8) belonging to two different carboxylate groups of two separated ligands, two bridging oxygen atoms (O11 and O12) in the μ2 mode and one N atom (N8). Cu3 is five coordinated by three atoms (O6, O7 and O9) from two ligands, one μ-oxygen atom (O12) and one nitrogen atom (N9) from a different ligand to provide a tetragonal pyramid geometry (Fig. 2a). The bond lengths of Cu–O fall between 1.878(3) and 2.338(3) Å and the Cu–N bond is 2.361(4) Å, which is in good agreement with previous studies.23 The ligands possess two modes in 2 (Fig. 2b). In mode 1, the tetrazole group links two copper in the μ-2 mode, and two diverse carboxyl groups are in the chelate and bridging syn–syn mode. In mode 2, the carboxyl groups are in a coordinated fashion similar to mode 1. In 2, the three Cu(II) centers (Cu1, Cu2 and Cu3) are held together by two μ-oxygen atoms (O11 and O12), which bind the three metal centers (Fig. 2c). Cu1A shares the μ3 oxygen atom (O12) with Cu2 and Cu3 to form a hexanuclear structure (Fig. 2d), affording a stable planar building unit [Cu6O4(CO2)8], which further forms infinite 1D belts along the a-axis through the connection of the carboxyl groups in the L2− ligands. The 3-amino-tetrazole groups, the second functional moieties in the L2− ligands, are in a μ1,2 bridging fashion which connect the Cu6 cluster. Carboxyl groups connect the neighboring 1D belts distributing symmetrically on both sides of the belts (Fig. 2e). Finally, a 2D framework of 2 is formed through the connection of one belt with neighboring ones (Fig. 2f). Similar to the topological analysis of 1, compound 2 can be identified as a new (2,3,10)-connected (416·614·811·104)(43)2(4)2 net (Fig. 2g).
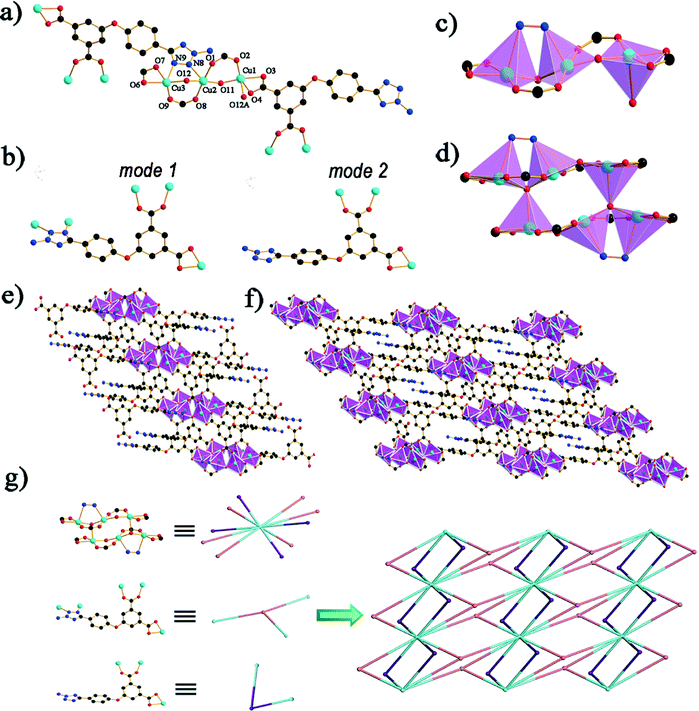 |
| | Fig. 2 (a) The coordination environment of Cu(II) and the bonding mode of H2atpia in the coordination polymer. All H atoms are omitted for clarity; (b) the two coordination modes in 2; (c) trinuclear copper (d) the hexanuclear-like copper SBU; (e) the 1-D belt in 2; (f) the 2D framework connected by neighboring belts; (g) the (2,3,10)-connected topology of 2. Black, C; red, O; blue, N; cyan, Cu. | |
The crystal structures of [Mn3(OH)2(atpia)3]·1.25H2O (3)
The single-crystal X-ray analysis reveals that compound 3 crystallizes in the monoclinic space group C2/c. In this structure, the asymmetric unit consists of three MnII atoms, three atpia2− ligands, two hydroxyl groups and 1.25 guest water molecules. Mn1 and Mn3 exhibit similar octahedral environment and each is coordinated by six carboxylate oxygen atoms from six atpia ligands. For Mn2, the distorted trigonal bipyramid coordination geometry is completed by four carboxylate oxygen atoms from four different atpia ligands and one oxygen atom from a hydroxyl group. Mn4 is penta-coordinated with a distorted trigonal bipyramid coordination geometry, and the metal center is surrounded by four carboxylate oxygens from three atpia ligands and one oxygen atom from a hydroxyl group. The Mn–O bond distances in the range of 2.088(3)–2.296(3) Å are in good accordance with the previous report. The linkages between Mn1 and Mn2, and Mn3 and Mn4 are achieved by two and a half carboxylate groups, while in the case of Mn2 and Mn4, the centers are connected by two carboxylate units. This sequence of Mn4A, Mn3, Mn4, Mn2, Mn1 and Mn2C is repeated to form an infinite Mn–O chain (Fig. 3a), which is further interlinked by atpia ligands to form a 2D architecture (Fig. 3b). Compared with the topology networks of 1 and 2, compound 3 exhibits an unprecedented (3,3,8,8)-connected new topology with a vertex symbol of (3·42)2(34·46·56·68·73·8) (Fig. 3c).
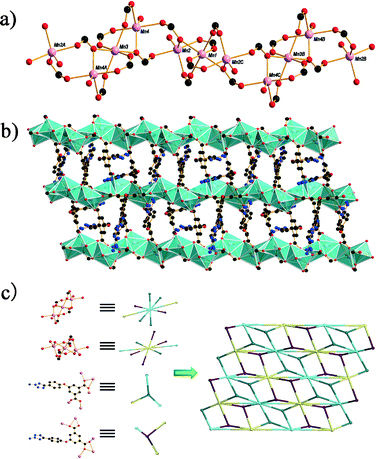 |
| | Fig. 3 (a) The one-dimensional Mn–O chain in 3; (b) the 2D framework of 3 generated from the linkage of the Mn–O chains via atpia2−; (c) the (3,3,8,8)-connected topology of 3. Black, C; red, O; blue, N; pink, Mn. | |
The influence of metal ions on the structures of the compounds
It is generally known that metal ions play a vital role in tuning the structural topology. Although different metal ions may have different charges, electron configurations, and ionic radii, hence exhibiting different coordination geometries, their coordination geometries are basically predictable. Compared with single metal ions, since many kinds of discrete metal clusters, especially the polygonal and polyhedral ones, are structurally well-defined, they can be more efficiently used as nodes or secondary building units (SBUs).24 Based on the similar synthetic system, compounds 1–3 can be obtained with the only difference of changing the metal ions (Cd2+, Cu2+ and Mn2+). As the center ions have the same charge, the ionic radii may be the chief factor that affects the coordination number. The ionic radii of Cd2+, Cu2+ and Mn2+ are 95 pm, 73 pm and 67 pm, respectively. With the corresponding gravity, the larger the radius of the central ion is, the more ligands it can hold around. In other words, it may have a higher coordination number to generate a framework with high dimensions. Combined with the concrete characteristics of the different metal ions, compounds 1–3 form diverse SBUs respectively. The hexanuclear-like copper and trinuclear manganese building units are used as nodes when linking with bridging ligands to construct the 2D layers of compounds 2 and 3. The layers are dense structures and thus, interpenetration is avoided. The tetranuclear-like cadmium building unit is employed to build a 2D layer that has ligating sites at both sides which are further linked by the ligands into 3D architectures (Table 2).
Table 2 The dimensionality and coordination number of different structures with respect to the metal centers
| Name |
Central ion |
Ionic radii (pm) |
Coordination number |
Dimensionality |
| 1 |
Cd2+ |
95 |
6,7 |
3D |
| 2 |
Cu2+ |
73 |
5 |
2D |
| 3 |
Mn2+ |
67 |
5,6 |
2D |
Thermal stability analysis and powder X-ray diffraction (PXRD) analysis
The thermal stability of 1–3 was determined by thermogravimetric analysis (TGA) (Fig. S3†). The thermal analysis of 1 shows that compound 1 is stable up to 302 °C. The thermal analysis of 2 shows that the weight is almost unchanged in the temperature range of 30–290 °C. Above 290 °C, the residue begins to decompose. This means that compound 2 is stable up to the high temperature of 290 °C. As for 3, a weight loss of 1.64% is observed in the range of 20–140 °C, corresponding to the removal of 1.25 water molecules (calcd: 1.83%), and it did not decompose until 273 °C. The purity of compounds 1–3 is confirmed by powder X-ray diffraction (PXRD) analyses, in which each experimental PXRD pattern is well consistent with the one obtained from the stimulated PXRD based on the single-crystal samples at room temperature (Fig. S4–S6†).
Luminescent properties
The luminescent properties of compounds with d10 metal centers have attracted intense interest because of their potential applications in chemical sensors, photochemistry and electroluminescent display.25–28 The solid state luminescent properties of the free H2atpia ligand and compound 1 were investigated at room temperature under the same experimental conditions and their emission spectra and excitation spectra are given (Fig. 4). An emission band is observed at 469 nm upon excitation at 356 nm for H2atpia, which can be ascribed to the ligand centered electronic transitions, that is, the π* → n or π* → π transition in nature according to the reported literature.29,30 Compound 1 shows an emission peak at 464 nm (λex = 365 nm), which is close to that of the free H2atpia ligand. Thus the emission peak of compound 1 can probably be attributed to the intraligand fluorescent emission.29b,31 In comparison with the free ligand, a blue-shift of about 5 nm is found for 1. In addition, the fluorescent intensity of compound 1 dramatically increases compared with the H2atpia ligand. This may be attributed to the ligand coordination to the metal center, which enhances the rigidity of the ligand and thus reduces the loss of energy through a radiationless pathway.32
 |
| | Fig. 4 The fluorescence spectra of compound 1 and the ligand in the solid state at room temperature. | |
Magnetic properties
The solid state, variable-temperature dc magnetic susceptibility measurements for compounds 2 and 3 have been carried out in an applied magnetic field of 1000 Oe in the temperature range 2–300 K using a quantum designed SQUID magnetometer. The magnetic properties of 2 in the form of cMT versus T and cM−1versus T per Cu6 unit are presented in Fig. 5. The cMT value per Cu6 unit at 300 K is 2.24 emu K mol−1, which is in good agreement with that expected for six uncoupled Cu2+ ions. The cMT value keeps almost constant until about 50 K, and then decreases sharply upon further cooling. The decrease of cMT in the low temperature range indicates the weak antiferromagnetic interaction between the Cu2+ centers within the Cu6 unit. Moreover, the cM−1versus T data over the whole temperature range obey the Curie–Weiss law very well, and give the best-fitting parameters C = 2.35 emu K mol−1 and q = −1.30 K. The negative q value also indicates the existence of weak antiferromagnetic interactions.
 |
| | Fig. 5 The magnetic susceptibility of 2 plotted as cMT vs. T and cM−1vs. T curves. The red line indicates the fitting of the cM−1vs. T data using the Curie–Weiss equation in the range 2–300 K. | |
The magnetic properties of 3 in the form of cMT versus T and cM−1versus T per Mn2+ ion are presented in Fig. 6. The cMT value per Mn2+ center was found to be 4.31 emu K mol−1 which corresponds exactly to the spin only value of 4.34 emu K mol−1 expected for a magnetically isolated Mn2+ ion. Upon cooling, the cMT value shows a continuous steady decrease, falling to 1.93 emu K mol−1 at 2 K. The decrease of cMT could be ascribed to the antiferromagnetic coupling between the Mn2+ ions. The magnetic data above 25 K could be well fitted by the Curie–Weiss law with C = 4.40 emu K mol−1 and q = −14.37 K. The negative q value also confirms the antiferromagnetic coupling within the Mn2+ chain.
 |
| | Fig. 6 The magnetic susceptibility of 3 plotted as cMT vs. T and cM−1vs. T curves. The red line indicates the fitting of the cM−1vs. T data using the Curie–Weiss equation in the range 2–300 K. | |
Conclusion
In summary, by the usage of the H2atpia ligand with different transition metal salts, three novel metal–organic frameworks, namely 1–3, have been constructed under hydrothermal conditions. They show diverse structures and dimensionalities from a 2D network to a 3D coordination architecture and all display new topologies. The crystal structure of 1 possesses a 3D framework, which is connected by H2atpia and a Cd4 cluster to generate a (2,3,10)-connected net. Compound 2 is a 2D framework with a (2,3,10)-connected net based on hexanuclear metal centers as the SBU. Compound 3 represents a 2D architecture generated from the linkage of the Mn–O chains via the H2atpia ligands with a (3,3,8,8)-connected net. The photoluminescence studies reveal that compound 1 displays an intense fluorescent emission band in the solid state at room temperature and thus may be a good candidate for a potential photoactive material. The variable-temperature magnetic susceptibility measurements indicate the existence of an antiferromagnetic interaction in 2 and 3. Work along this line using other transition metals and semi-rigid ligands with both carboxylic acid and tetrazole groups is in progress in our lab.
Acknowledgements
This work was supported by the Foundation of the National Natural Science Foundation of China (no. 21371069), the Specialized Research Fund for the Doctoral Program of Higher Education (no. 20110061110015) and the National High Technology Research and Develop Program (863 program) of China (no. 2013AA031702).
Notes and references
-
(a) D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975 CrossRef CAS;
(b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed;
(c) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136 CrossRef CAS PubMed;
(d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
-
(a) N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS PubMed;
(b) B. T. N. Pham, L. M. Lund and D. Song, Inorg. Chem., 2008, 47, 6329 CrossRef CAS PubMed;
(c) S. Su, W. Chen, C. Qin, S. Song, Z. Guo, G. Li, X. Song, M. Zhu, S. Wang, Z. Hao and H. Zhang, Cryst. Growth Des., 2012, 12, 1808 CrossRef CAS;
(d) V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378 RSC;
(e) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef CAS PubMed.
-
(a) J. P. Zhang, Y. B. Zhang, J. B. Lin and X. M. Chen, Chem. Rev., 2012, 112, 1001 CrossRef CAS PubMed;
(b) Z. M. Hao and X. M. Zhang, Cryst. Growth Des., 2008, 8, 3586 CrossRef.
-
(a) J. Chen, Y. L. Feng, Z. G. Jiang and J. W. Cheng, CrystEngComm, 2011, 13, 6071 RSC;
(b) S. Wang, X. H. Ding, J. L. Zuo, X. Z. You and W. Huang, Coord. Chem. Rev., 2011, 255, 1713 CrossRef CAS PubMed;
(c) G. Q. Kong and C. D. Wu, Cryst. Growth Des., 2010, 10, 4590 CrossRef CAS;
(d) F. Yu, S. J. Zhang, X. Zhao, D. R. Yuan, C. M. Wang and T. R. Shrout, Cryst. Growth Des., 2010, 10, 1871 CrossRef CAS.
-
(a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed;
(b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed;
(c) S. Natarajan and P. Mahata, Chem. Soc. Rev., 2009, 38, 2304 RSC.
-
(a) L. J. Murray, A. M. Dinc and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC;
(b) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC;
(c) G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380 RSC.
-
(a) A. U. Czaja, N. Trukhan and U. Meuller, Chem. Soc. Rev., 2009, 38, 1284 RSC;
(b) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC;
(c) D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502 CrossRef CAS PubMed.
-
(a) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC;
(b) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
-
(a) M. P. Suh, Y. E. Cheon and E. Y. Lee, Chem.–Eur. J., 2007, 13, 4208 CrossRef CAS PubMed;
(b) J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2008, 130, 6010 CrossRef CAS PubMed.
-
(a) O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016 CrossRef CAS PubMed;
(b) Y. Yan, S. Yang, A. J. Blake, W. Lewis, E. Poirier, S. A. Barnett, N. R. Champness and M. Schröder, Chem. Commun., 2011, 47, 9995 RSC;
(c) D. Yuan, D. Zhao, D. Sun and H. C. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5357 CrossRef CAS PubMed.
-
(a) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC;
(b) M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed;
(c) A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2009, 43, 58 CrossRef PubMed;
(d) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
-
(a) H. Zhao, Z. R. Qu, H. Y. Ye and R. G. Xiong, Chem. Soc. Rev., 2008, 37, 84 RSC;
(b) Q. Ye, Y. M. Song, G. X. Wang, K. Chen, D. W. Fu, P. W. H. Chan, J. S. Zhu, S. D. Huang and R. G. Xiong, J. Am. Chem. Soc., 2006, 128, 6554 CrossRef CAS PubMed;
(c) M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef PubMed;
(d) W. Ouellette, A. V. Prosvirin, K. Whitenack, K. R. Dunbar and J. Zubieta, Angew. Chem., Int. Ed., 2009, 48, 2140 CrossRef CAS PubMed;
(e) X. M. Zhang, J. Lv, F. Ji, H. S. Wu, H. Jiao and P. V. R. Schleyer, J. Am. Chem. Soc., 2011, 133, 4788 CrossRef CAS PubMed.
-
(a) D. C. Zhong, M. Meng, J. Zhu, G. Y. Yang and T. B. Lu, Chem. Commun., 2010, 46, 4354 RSC;
(b) M. Dincă, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904 CrossRef PubMed;
(c) Z. Zhang, S. Xiang, Q. Zheng, X. Rao, J. U. Mondal, H. D. Arman, G. D. Qian and B. L. Chen, Cryst. Growth Des., 2010, 10, 2372 CrossRef CAS;
(d) J. Tao, Z. J. Ma, R. B. Huang and L. S. Zheng, Inorg. Chem., 2004, 43, 6133 CrossRef CAS PubMed;
(e) W. C. Song, Q. Pan, P. C. Song, Q. Zhao, Y. F. Zeng, T. L. Hu and X. H. Bu, Chem. Commun., 2010, 46, 4890 RSC;
(f) T. T. Luo, H. L. Tsai, S. L. Yang, Y. H. Liu, R. D. Yadav, C. C. Su, C. H. Ueng, L. G. Lin and K. L. Lu, Angew. Chem., Int. Ed., 2005, 44, 6063 CrossRef CAS PubMed.
-
(a) Y. Li, G. Xu, W. Q. Zou, M. S. Wang, F. K. Zheng, M. F. Wu, H. Y. Zeng, G. C. Guo and J. S. Huang, Inorg. Chem., 2008, 47, 7945 CrossRef CAS PubMed;
(b) W. C. Song, J. R. Li, P. C. Song, Y. Tao, Q. Yu, X. L. Tong and X. H. Bu, Inorg. Chem., 2009, 48, 3793 Search PubMed;
(c) L. Hou, L. N. Jia, W. J. Shi, Y. Y. Wang, B. Liu and Q. Z. Shi, Dalton Trans., 2013, 42, 3653 RSC;
(d) J. Y. Sun, L. Wang, D. J. Zhang, D. Li, Y. Cao, L. Y. Zhang, S. L. Zeng, G. S. Pang, Y. Fan, J. N. Xu and T. Y. Song, CrystEngComm, 2013, 15, 3402 RSC;
(e) J. Xiang, Y. Luo, L. L. Zhao, C. H. Wang and J. S. Wu, Inorg. Chem. Commun., 2013, 31, 23 CrossRef CAS PubMed.
-
(a) X. F. Wang, Y. B. Zhang and W. Xue, Cryst. Growth Des., 2012, 12, 1626 CrossRef CAS;
(b) H. X. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846 CrossRef CAS PubMed.
- S. M. Hawxwell, G. M. Espallargas, D. Bradshaw, M. J. Rosseinsky, T. J. Prior, A. J. Florence, J. van de Streek and L. Brammer, Chem. Commun., 2007, 1532 RSC.
- J. P. Collman, J. I. Brauman, J. P. Fitzgerald, P. D. Hampton, Y. Naruta, J. W. Sparapany and J. A. Ibers, J. Am. Chem. Soc., 1988, 110, 3477 CrossRef CAS.
- P. Lama, A. Aijaz, E. C. Sañudo and P. K. Bharadwaj, Cryst. Growth Des., 2010, 10, 283 CAS.
-
G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
-
(a) W. Clegg, J. T. Cressey, A. McCamley and B. P. Straughan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 234 CrossRef;
(b) R. H. Wang, M. C. Hong, J. H. Luo, R. Cao, Q. Shi and J. B. Weng, Eur. J. Inorg. Chem., 2002, 2904 CrossRef CAS.
- R. Wang, D. Yuan, F. Jiang, L. Han, Y. Gong and M. Hong, Cryst. Growth Des., 2006, 6, 1351 CAS.
-
V. A. Blatov, Multipurpose crystallochemical analysis with the program package TOPOS, IUCr Comp. Comm. Newsl., 2006, vol. 7, p. 4 Search PubMed.
-
(a) J. Carranza, C. Brennan, J. Sletten, J. M. Clemente-Juan, F. Lloret and M. Julve, Inorg. Chem., 2003, 42, 8716 CrossRef CAS PubMed;
(b) S. Koner, S. Saha, T. Mallah and K. Okamoto, Inorg. Chem., 2004, 43, 840 CrossRef CAS PubMed.
- D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhaktaa and R. J. T. Houka, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- M. A. Braverman and R. L. LaDuca, Cryst. Growth Des., 2007, 7, 2343 CAS.
- X. Q. Yao, M. D. Zhang, J. S. Hu, Y. Z. Li, Z. J. Guo and H. G. Zheng, Cryst. Growth Des., 2011, 11, 3039 CAS.
- A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334 CrossRef CAS PubMed.
-
(a) Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed;
(b)
A. W. Adamson and P. D. Fleischauer, Concepts of Inorganic Photo chemistry, John Wiley, New York, 1975 Search PubMed.
-
(a) W. Zhao, H. F. Zhu, T. A. Okamura and W. Y. Sun, Supramol. Chem., 2003, 15, 345 CrossRef CAS;
(b) Y. J. Mu, G. Han, S. Y. Ji, H. W. Hou and Y. T. Fan, CrystEngComm, 2011, 13, 5943 RSC;
(c) P. P. Cui, J. L. Wu, X. L. Zhao, D. Sun, L. L. Zhang, J. Guo and D. F. Sun, Cryst. Growth Des., 2011, 11, 5182 CrossRef CAS.
-
(a) X. L. Zheng, Y. Liu, M. Pan, X. Q. Lv, J. Y. Zhang, C. Y. Zhao, Y. X. Tong and C. Y. Su, Angew. Chem., Int. Ed., 2007, 46, 7399 CrossRef CAS PubMed;
(b) Y. Q. Xiao, Y. J. Cui, Q. Zheng, S. C. Xiang, G. D. Qian and B. L. Chen, Chem. Commun., 2010, 46, 5503 RSC;
(c) D. Y. Ma, W. X. Wang, Y. W. Li, J. Li, C. Daiguebonne, G. Calvez and O. Guillou, CrystEngComm, 2010, 12, 4372 RSC;
(d) W. Q. Kan, B. Liu, J. Yang, Y. Y. Liu and J. F. Ma, Cryst. Growth Des., 2012, 12, 2288 CrossRef CAS.
- J. Tao, M. L. Tong, J. X. Shi, X. M. Chen and W. N. Seik, Chem. Commun., 2000, 2043 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: the supplementary crystallographic data for 1–3. The PXRD patterns, a table of selected bond distances and angles for 1–3, the FTIR spectra figures for the H2atpia ligand and the TGA plot of compounds 1–3. CCDC numbers 972184–972186. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ce00105b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 





