 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic frameworks as heterogeneous photocatalysts: advantages and challenges
M. A.
Nasalevich
,
M.
van der Veen
,
F.
Kapteijn
and
J.
Gascon
*
Catalysis Engineering, Department of Chemical Engineering, Delft University of Technology, Julianalaan 136, Delft, 2628 BL, The Netherlands. E-mail: J.Gascon@tudelft.nl; Fax: +31 1527 85006; Tel: +31 1527 84851
First published on 1st April 2014
Abstract
The use of metal organic frameworks (MOFs) as heterogeneous photocatalysts is critically reviewed. First we revisit the general assumption of MOFs behaving truly as semiconductors, demonstrating that such semiconducting behaviour only occurs in a very limited subset of materials. Further, the main approaches for efficient light harvesting and active site engineering in MOF-based photocatalysts are discussed. Finally, the main advantages of MOFs as photocatalysts and the challenges that need to be addressed in order to improve catalytic performance are evaluated.
Introduction
Since Fujishima and Honda reported in 1972 the first example of photocatalysis,1 the scientific community has not stopped dreaming about developing efficient synthetic catalysts that would ultimately mimic photosynthesis. The use of visible light as an energy source for chemical transformations is indeed one of the biggest challenges we face nowadays. With water purification being the most successful application of photocatalysis2 and some emerging applications like photocatalytic coatings and air purifiers, a great deal of effort has been put forward in order to develop photocatalysts able to perform more selective catalytic transformations, from water splitting for energy applications3 to synthesis of important chemical building blocks and fuels from CO24 or even asymmetric photocatalysis.5To date, TiO2 has been the most successful photocatalyst due to its relatively high efficiency, low cost and availability. Recent research on new photocatalytic materials, either semiconductor or not, has delivered a number of possible substitutes, especially in the case of applications making use of the solar spectrum. Semiconductors based on cations with the d0 configuration such as Ta5+ or Nb5+, as well as oxides or nitrides of d10 elements such as Bi3+, In3+ or Ga3+, are among the most interesting novel photocatalysts. In addition, some classical semiconductors like ZnO or CdS, initially disregarded as a consequence of their poor stability under irradiation, have been reconsidered.6
Metal Organic Frameworks (MOFs) are crystalline compounds consisting of infinite lattices built up of the inorganic secondary building unit (SBU, metal ions or clusters) and organic linkers, connected by coordination bonds of moderate strength.7 Distinct from traditional inorganic materials, MOFs can be synthesized from well-defined molecular building blocks thanks to both the reliability of molecular synthesis and the hierarchical organization via crystal engineering. MOFs can therefore be understood as molecules arranged in a crystalline lattice.8 High adsorption capacities and easy tunability have spurred applications in gas storage, separation and molecular sensing.9–13 Bio-compatible scaffolds hold promise for medical applications.14,15 The easy compatibilization of MOFs with either organic or inorganic materials may result in composites with applications varying from (opto)electronic devices to food packaging materials and membrane separation.16–19 Last but not least, their tunable adsorption properties, high dispersion of components and pore size and topology, along with their intrinsic hybrid nature, all point at applications in heterogeneous catalysis.20–22
The a priori clear similarities between MOFs and bulk transition metal oxides triggered a decade ago the first examples of the application of MOFs in photocatalysis. The best known example is MOF-5, where the Zn4O clusters forming the structure have been seen as semiconductor clusters spaced by organic moieties that act as photon antennas.23–27 However, as the field has matured, it is now clear that the chemistry and electronic properties of these hybrid materials may be radically different from those of classical semiconductors. Consequently, strategies different from those applied in classical semiconductors to improve photocatalytic performance should be developed for MOFs.
This highlight article seeks to complement the recent reviews on MOF catalysis and photocatalysis.20,22,28–33 In contrast to a classical review, we first critically reconsider the concept of “MOFs as semiconductors”, we then highlight the most important advantages of MOFs as light harvesters and discuss different approaches for the design of more efficient photocatalysts. Finally, we share our personal opinion on future directions for the development of efficient MOF photocatalysts.
MOFs: semiconductors or insulators?
MOFs have been labelled as semiconductors based on their optical transitions and electrochemical and photochemical activity.33–35 Yet such activity does not necessarily imply semiconductivity. Inorganic semiconductors are characterized by a delocalized valence and a conduction band through which the charge carriers are mobile. Organic semiconductors are typically characterized by delocalized orbitals via extended conjugated π-bonds, allowing for charge carrier mobility. Also in metal–organic frameworks a certain degree of delocalization is necessary to show a semiconductive behaviour.36 Of course, only the measurement of the current through the material or the charge carrier mobility directly determines whether a material is (semi)conductive.For a small subset of metal–organic frameworks, the conductivity has been reported. The reported frameworks typically have small but discernible conductivities of 10−9–10−3 S cm−1.37 These frameworks are based on Cu(I) or Ag(I) ions37 or dithiolene based frameworks.38 Another rare example of a 3-D porous metal–organic framework that shows conductivity is based on triazole ligands. Out of an isoreticular series of different divalent metal ions, only the conductivity of the framework with Fe(II) ions has been reported.39 At this point, it should be mentioned that electroconductivity is related to the ability of a given material to conduct electric current. When it comes to photo-catalysis and light harvesting, photo-conductivity, the mobility of electrons and holes generated upon electromagnetic radiation, is much more important. For a few MOFs the photo-induced time-resolved microwave conductivity (TRMC) is reported (see Fig. 1). This technique probes the local charge mobility and is an essential tool when studying materials for photocatalysis. In such measurements the product of the charge carrier density and the charge carrier mobility is given.40 The signal is obtained with nanosecond time resolution after light absorption. The reported mobility values are in the range between 1 × 10−5 and 4 × 10−5 cm2 V−1 s−1. This is quite low compared to conjugated polymers where values of 10−3 cm2 V−1 s−1 and higher are common.40,41 These values have been reported for a MOF with stacked thiafulvalene ligands,42 a MOF that contains infinite Mn–S chains that should facilitate charge carrier mobility,43 and MIL-125, a Ti(IV)44 containing structure that has also been studied for its photocatalytic behaviour.45 For thiafulvalene and Mn–S chain structures, the amount of photogenerated mobile charges was determined. The quantum yields are in the order of 10−4–10−3. This meant thus a high intrinsic charge mobility of 0.2 cm2 V−1 s−1 for the thiafulvalene framework and 0.02 cm2 V−1 s−1 for the Mn–S chain framework. For the latter structure this corresponds to charge delocalization over 8–12 Mn–S units. Yet only a very small fraction (10−4–10−3) of the absorbed photons leads to charges that are mobile. Taking MIL-125(Ti) as the most representative example of photo-active MOFs, it shows a poor photoconductance (mobility ~10−5 cm2 V−1 s−1 upon 340 nm illumination, see Fig. 1).44 This conductance is significantly suppressed upon lowering the temperature, in clear contrast to pure TiO2 with mobilities in the range of 1 cm2 V−1 s−1 nearly independent of temperature.46,47 This difference suggests thermally activated hopping as the main mechanism for the charge transport due to the isolation of the Ti clusters by the organic linkers in the MOF.48 Indeed, such clusters in MOFs are too far apart to fulfil the Mott transition conditions, being approximately 4 Bohr radii.49,50 Moreover, in most MOFs, the distance between linkers is too large as to allow efficient π–π stacking51 and there is hardly any orbital overlap, keeping the electrons preferentially in a localized state. This fact demonstrates, as recently rationalized by Lin and co-workers, that MOFs have to be understood as molecules arranged in a crystalline lattice.8 In case of photocatalysis, materials like MIL-125(Ti) should therefore be seen as an array of self-assembled molecular catalysts rather than as classical semiconductors. Therefore, optical absorption spectra should be considered as sets of individual discrete absorption bands, and the HOMO–LUMO gap terminology should be used in order to describe the discrete character of the light-induced transitions in these coordination compounds. For MOF-5, the most prominent MOF to which semiconductive behaviour has been ascribed due to the electro- and photochemical behaviour, Walsh et al. calculated the electronic band structure. No band dispersion was observed, which is consistent with localized carriers and low levels of conductivity.36 These results are in line with experimental observations by our research group comparing photo-catalytic performance of iso-reticular MOFs and their corresponding monodentate analogues.27
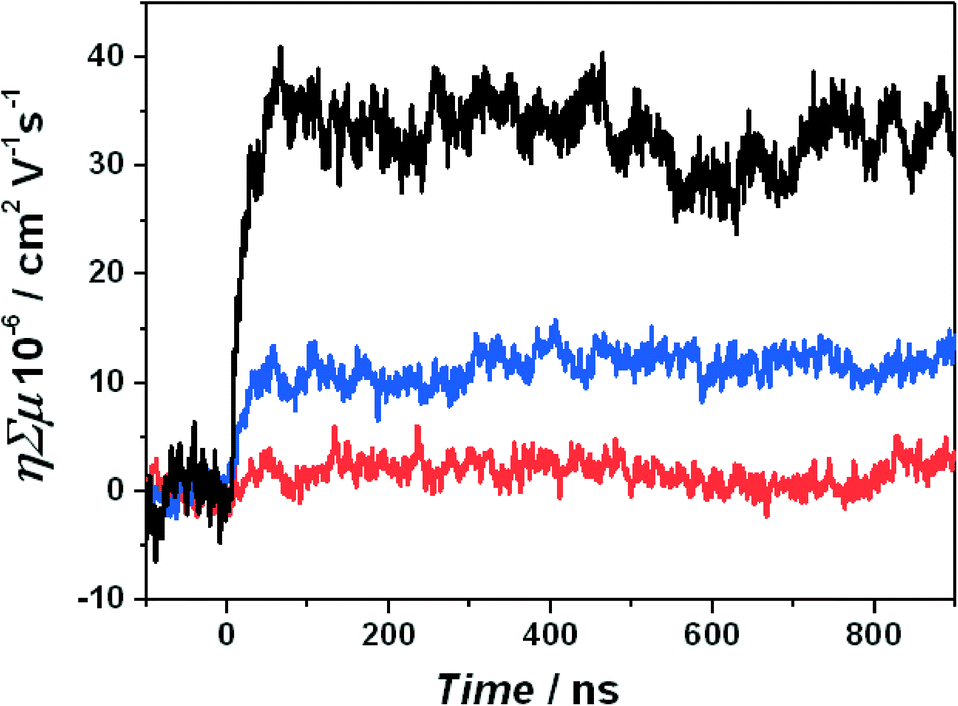 | ||
| Fig. 1 Time-resolved microwave conductivity traces for MIL-125(Ti). The experiments were carried out at an excitation wavelength of 340 nm and at different temperatures: −120 °C (red), −70 °C (blue) and −30 °C (black). Adapted from Nasalevich et al. (2013).44 | ||
Semiconductivity in metal–organic frameworks seems thus to occur only in a limited subset and is so far of relatively low magnitude. This notwithstanding, it has indeed been shown in the literature that upon absorption of light, electrons and holes can be generated in MOFs with reductive and oxidative power, respectively. Yet, for most MOFs these charges are not mobile. This has implications for photocatalysis. Photocatalysis consists of a reduction and an oxidation half reaction. When the photo-generated holes and electrons are not mobile, this implies that the oxidation and reduction sites need to be present in close vicinity to the location where the photo-excited charges are generated. In contrast to bulk solids, the crystalline nanoporous structure of metal–organic frameworks allows such a multi-modal construction. The spatial proximity of the photo-generated charge carriers though might favour charge recombination competing with the desired redox reactions. On the other hand, the porosity of MOFs facilitates diffusion of reactants and products throughout their crystals, which can compensate for that.
Tuning light harvesting properties
In contrast to classical semiconductors such as TiO2, CdS, and ZnO, MOFs exhibit excellent optical tunability. First of all, the organic linkers can be selected prior to the MOF synthesis, delivering a desired absorption wavelength. Generally, the optical transitions of interest are ligand-to-metal charge transfer (LMCT) in character. The energy required to induce these transitions depends on the level of conjugation of the aromatic system of the ligand and on the metal to which the ligand is coordinated. Predicted first by Civalleri52 this effect found its first experimental proof in a series of isoreticular MOFs adopting a MOF-5 topology with different organic bidentate ligands.26 The general strategy consists of using functionalized linkers like aminoterephthalic acid (ATA). For example, the amino-substituent, once introduced to the ligand, provides the lone pair of nitrogen for the interaction with the π*-orbitals of the benzene ring, donating electron density to the antibonding orbitals. This results in a new, higher HOMO level that brings absorption to the visible region.53 This concept was first realized by Garcia and co-workers for the case of Zr-based UiO-type materials.54 The use of ATA allowed sensitizing the originally deep-UV absorbing MOF (when synthesized with unsubstituted terephthalic acid) to the visible region. This red-shift in absorption resulted in an enhanced photocatalytic activity. The addition of a second amino-group in the linker was calculated to follow a similar trend: the absorption edge was found at 1.3 eV for the diaminated MIL-125(Ti) against 2.4 and 3.6 eV for the mono-aminated and amino-free framework, respectively.55However, introducing the desired functionalization in a framework of choice is not always synthetically feasible.7 For the MIL-125(Ti) topology this was achieved only when 10% of diaminoterephthalic acid was mixed with 90% of ATA; the attempts to obtain pure (NH2)2-MIL-125(Ti) were unsuccessful. Therefore, the use of post-synthetic modifications (PSM)56–60 can certainly help introduce functionalities not achievable by direct synthesis. An example of such a PSM was recently reported by our group.44 In this instance amino groups of the ATA linkers were converted to dye-like molecular fragments after MOF synthesis. This transformation delivered a material exhibiting a significant red-shift in light absorption with respect to the parent NH2-MIL-125(Ti) (Fig. 2). In fact, the solid is able to absorb 100% more photons emitted by the Xe-lamp used in the study. The improved light absorption resulted in a higher activity of the framework in benzyl alcohol oxidation.44
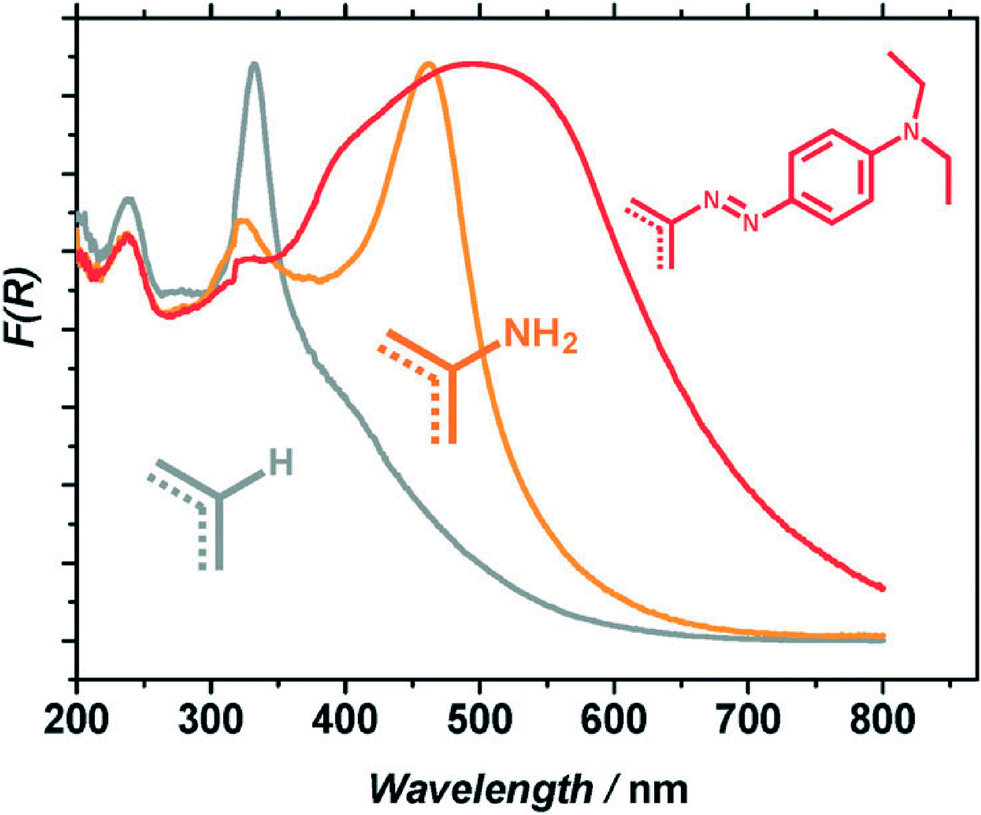 | ||
| Fig. 2 Normalized diffuse reflectance spectra of: MIL-125(Ti) (grey), NH2-MIL-125(Ti) (orange) and MR-MIL-125(Ti) (red).44 Reproduced with permission from the Royal Society of Chemistry. | ||
Another interesting approach to alter optical properties of MOFs is to change the metal in the nodes of a given framework (i.e. MIL-101(Cr) vs. MIL-101(Fe)).61 Obviously, the main optical transitions ascribed to LMCT are affected by orbitals of the metal centres. Materials containing Fe-, Cd-, Co-, and Ni-clusters were reported to have visible light photocatalytic activity associated with the corresponding transitions originating from the metals.62,63 Obviously, rationalizing the behaviour of such materials is only possible when the topology of the frameworks is similar. For example, the UiO-66(Zr) and MIL-125(Ti) series possess very similar topologies and d0 metal-based clusters. Independent studies revealed that the corresponding transitions in NH2-MIL-125(Ti) (ref. 64 and 65) are red shifted with respect to the ones in NH2-UiO-66(Zr) (ref. 54 and 66) by circa 40 nm. Noteworthy, the LMCT nature of the transitions introduced by the amino groups was confirmed by detecting the EPR-signatures of both Ti3+ and Zr3+.
In summary, optical absorption of MOFs can be easily tuned either by choosing an appropriate linker or post-synthetically. Alternatively, engineering the optical response by fine-tuning the cluster-forming metal or even by using mixed metal clusters might be in the future a powerful tool, but at this moment rational design is still out of the question. Such manipulations with absorptive properties lead to improvements in the photocatalytic activity of the frameworks. However, the activity of so far reported MOF photocatalysts is very modest. Given the fact that tuning optical absorption does not seem to be a problem, it is clear that active site engineering, explained below, is the path to follow in order to improve catalytic activity.
Active site engineering
The different approaches to induce photocatalytic activity in a MOF scaffold are summarized in Fig. 3. The MOF organic linkers can be considered as light-harvesting units transferring the energy of excited states to inorganic MexOy clusters consisting of only a few metal atoms. At this level of quantization the cluster size is too small to impose distinct periodic features from those found in bulk semiconductors. Such approach usually results in the generation of free charges upon illumination at the appropriate wavelength and in moderate to low photocatalytic performance. A more elaborate approach to employ MOFs for photocatalysis is to use them as carriers for photocatalytically active species. This approach can be used for the encapsulation of a variety of active sites: from semiconductor nanoparticles67 to molecular catalysts based on transition metal complexes,68 they all have been successfully encapsulated in MOFs.69 In this case, the MOF can act either as a mere container or participate in the charge transfer process (see Fig. 4 where ligand to cluster charge transfer (LCCT) is indicated).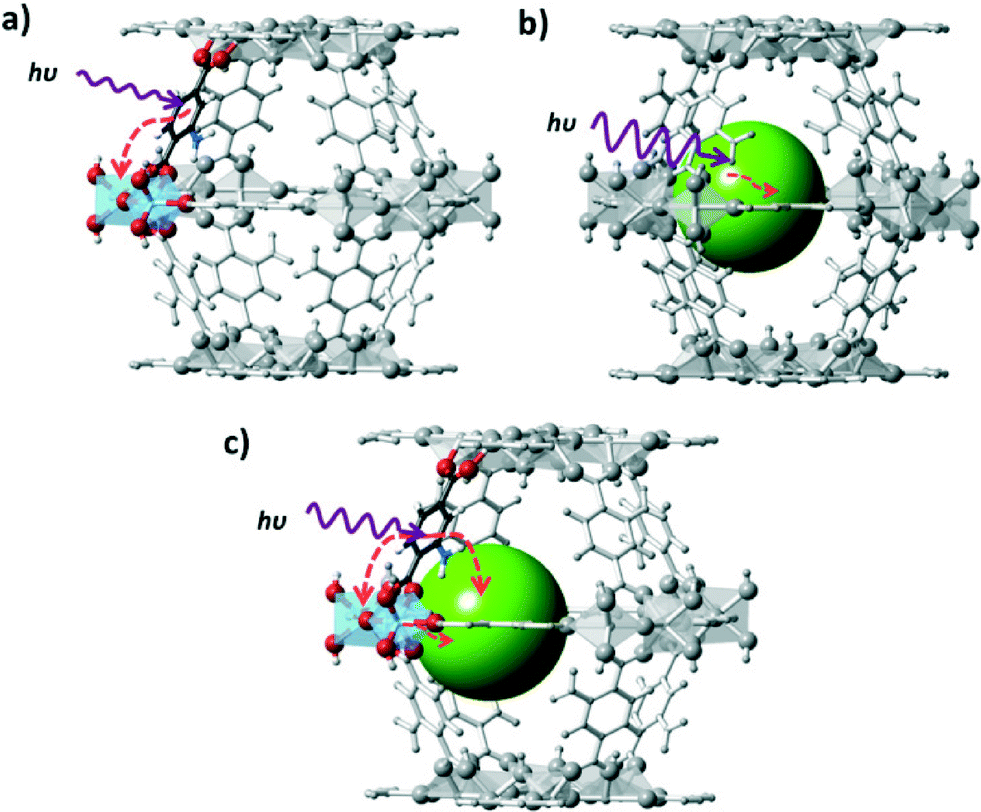 | ||
| Fig. 3 Different approaches to induce photocatalytic activity in a MOF scaffold: a) the organic linker is used as an antenna for light sensitizing and charge transfer to the inorganic cluster occurs; b) the MOF is used as a container for the encapsulation of a photocatalyst that directly absorbs light and c) charge transfer occurs between the MOF scaffold and the encapsulated catalyst. | ||
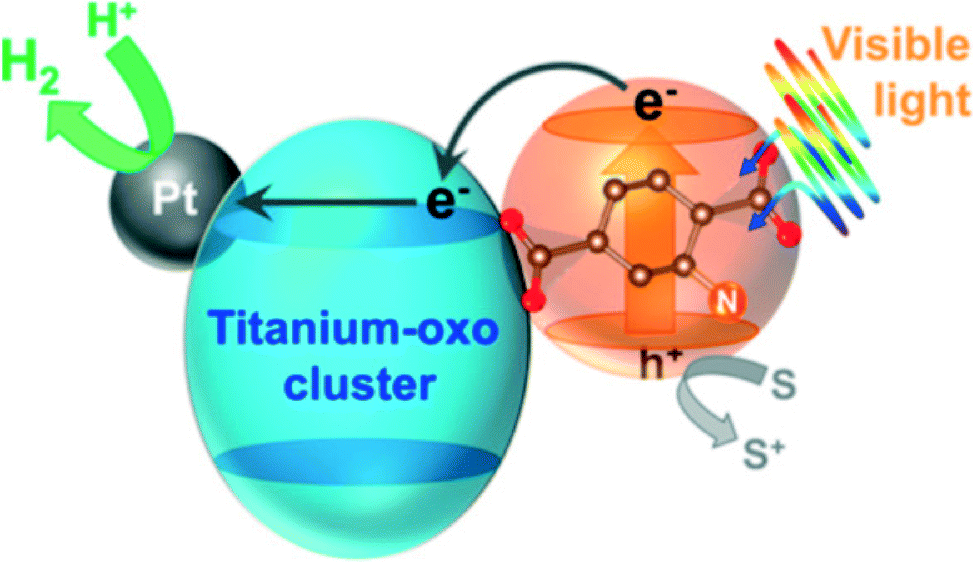 | ||
| Fig. 4 A schematic illustration of photocatalytic hydrogen production reaction over Pt-supported Ti-MOF-NH2 on the basis of the LCCT mechanism.60 Reproduced with permission from the American Chemical Society. | ||
One of the main advantages of using metal–organic frameworks for supporting active species is that these moieties can be either covalently bonded to the framework or encapsulated in its cavities. This strategy was proven to prevent leaching of homogeneous catalysts, often consisting of precious metals and being soluble under given reaction conditions. In 2011 Lin and co-workers reported a series of UiO-67(Zr) materials doped with Ir-, Ru- and Re-complexes that were subsequently applied for water oxidation, aza-Henry transformations and CO2 reduction, respectively.70 The solids exhibit outstanding photocatalytic performance, comparable to that of the corresponding homogeneous analogues. In addition, the catalysts were confirmed to be recyclable, proving their heterogeneous nature. Another interesting catalyst was introduced in 2012; in this case, a framework with the UiO-66 topology was synthesized following a mixed-linker strategy. Biphenyl-4,4′-dicarboxylic acid being the primary linker was combined with [Ir(ppy)2(bpy)]Cl-derived dicarboxylic acid (see Fig. 5).71
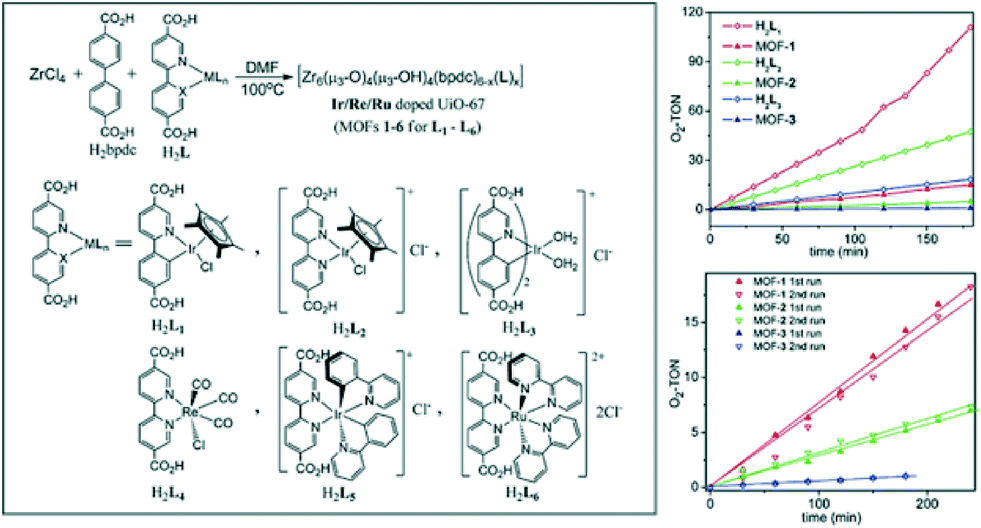 | ||
| Fig. 5 Doping of UiO-67(Zr) frameworks with molecular catalysts: schematic view (left), plots of O2 evolving turnover number (O2-TON) vs. time for doped MOFs and the corresponding homogeneous analogues (top right), and recyclability studies (bottom right).71 Reproduced with permission from the American Chemical Society. | ||
This yielded a crystalline porous solid with an iridium complex content of 2 wt.%. In addition to the molecular catalyst, platinum nanoparticles (Pt NPs) were deposited within the cavities of the MOF by photodeposition (PD). This bi-functional catalyst, in which the charges generated by the Ir-complex are injected into Pt NPs, showed a remarkable activity in H2 evolution from H2O (3400 TONs), exceeding that of the homogeneous system. The enhanced activity was attributed to the more efficient electron transfer favoured in the confined space of the MOF cavities. A similar way to introduce desirable active sites was implemented in the case of porphyrin-based MOFs. In these catalysts, the frameworks are built of porphyrin-like linkers responsible for photocatalysis. Rosseinsky and co-workers reported a MIL-60(Al) type framework containing meso-tetra(4-carboxyl-phenyl) porphyrin that was active in H2 evolution in combination with Pt.72 The solid was synthesized in a base-free form, conserving the possibility of tuning the active sites by changing the metal coordinated to the porphyrin rings. Recently, an Al-based framework constructed by Cu–porphyrin building units and Zn-based Sn(IV)–porphyrin MOFs were proven to catalyse the reduction of carbon dioxide to methanol73 with very moderate production rates and the oxidation of phenols and sulphides,74 respectively.
The use of MOFs for the encapsulation of polyoxometalates (POMs) has been explored for several years for different catalytic applications.75–77 A Ln3+-based MOF containing [BW12O40]5− anions was recently applied for photocatalytic oxidation of thiophene with molecular oxygen.78 The UV light-driven transformation was speculated to be assisted by the charge separation within the Keggin anions. Another example of a photocatalytically active POM-based metal–organic framework contains [Mo6O18(O3AsPh)2]4− polyoxoanions and Cu(I)–organic moieties. The solid behaves as a photocatalyst in methylene blue degradation.79
In all of the examples above, the role of the MOF is limited to that of a container or ‘nanoreactor’ for species that are active in various photocatalytic transformations. This approach allows the controlled anchoring or heterogenization of active sites. To the best of our knowledge, it has yielded the best activities so far. However, it should be emphasized that following this approach, the outstanding ability of MOFs to separate charges upon light illumination is largely unutilised. One of the few materials reported to date which takes advantage of the MOF component was reported by Matsuoka and colleagues.65,80 This catalyst is composed of Pt NPs photo-deposited on NH2-MIL-125(Ti). Platinum surfaces are known to be among the best platforms for H–H bond formation and in this particular instance Pt-nanoparticles introduce active sites required for efficient hydrogen evolution in combination with the light-absorbing unit, NH2-MIL-125(Ti).
Mechanistic studies suggest that absorption of visible light by the organic linkers is followed by LMCT, yielding Ti3+ species. These surplus electrons are likely to be injected into Pt NPs that act as electron-‘reservoirs’ (see Fig. 4). The reaction takes place at the surface of these nanoparticles. A similar effect was observed for Pt@NH2–UiO-66(Zr).66
Ray and co-workers presented another composite where the MOF can be seen as multifunctional. This photocatalyst is based on TiO2 nanotubes (TiO2 NTs) that are photo-sensitized with ZIF-8 crystals and Pt NPs are added as electron-trapping species, also within the MOF porosity.81 In this case the MOF acts as a photo-sensitizer and TiO2 as the photocatalyst. CdS nanoparticles embedded in MIL-101(Cr) were speculated to be sensitized by the d–d transitions of the chromium centres of the MOF.67 However, this mechanism was not supported experimentally.
Conclusions and future perspectives
Based on their optical transition and electrochemical and photochemical activity, MOFs have been long considered as semiconductors. However, such semiconducting behaviour only occurs in a very limited subset of materials and is so far of relatively low magnitude. In spite of such low conductivities, upon absorption of light, electrons and holes are generated in many MOF structures that result in reductive and oxidative power, respectively. In view of these evidences, MOFs should be considered as an array of self-assembled molecular catalysts rather than as classical semiconductors, and the HOMO–LUMO gap terminology should be used in order to describe the discrete character of the light-induced transitions in these coordination compounds.The molecular nature of MOFs offers unprecedented advantages for the design of efficient photocatalysts due to their highly porous nature. While the dye sensitizing approach has been commonly applied to traditional semiconductors, its main limitation lies in the fact that the available external surface for reaction is very limited. In the case of MOFs, efficient sensitizing can be combined with an extremely high concentration of active sites in their pore space. It is also worth highlighting that in spite of the porous crystalline nature of MOFs, light penetration and scattering do not seem to be a problem; as the wavelength of UV and visible light is much larger than the pore diameter of most MOFs (~0.3–3 nm), the light is not scattered by the pores. Instead the MOF structure is ‘seen’ by the light as a homogeneous structure. Absorbance occurs via the metal–organic complexes of the MOF, leading to a ligand-to-metal charge separated complex.
Important progress has been made to extend the absorption of light by MOFs from the UV to the optical spectrum. However, little attention has been paid to the absolute extinction coefficient of the ligand-to-metal charge complexes in metal–organic frameworks. Typical dyes in dye-sensitized solar cells and artificial photosynthesis have extinction coefficients of 104–105 M−1 cm−1.82 Moreover, in natural photosynthesis, several dyes cooperate in antennae structures to pool the light energy.83 When several antennae complexes transfer the photo-excited charges to a single reaction centre, high charge fluxes occur. It can be argued that such high energy fluxes are necessary to drive the multi-electron transfer processes involved in generating solar fuels in a way that is competitive with charge recombination.83,84 Yet, currently the extinction coefficients per chromophoric unit of the metal–organic framework are typically not reported. Determining the extinction coefficients and improving them via synthesizing new MOF structures or post-functionalization could be a fruitful strategy to achieve higher efficiencies in solar fuel generation.
In this highlight, we have summarized the main strategies for the design of photo-active MOFs and MOF containing composites (see Fig. 3). When catalysis takes place at the inorganic cluster, the so far reported activities are low. On the opposite, when the MOF scaffold is used as a container or as a light harvester, superior catalytic performance, already comparable to the state of the art homogeneous systems, and higher selectivities than in other solid photocatalysts have been reported. Now it is time for the suitable design of active sites and matching of functionalities. This should result in an efficient use of the number of photo-generated charges. Matching chemical reaction velocity with a lifetime of generated charges to minimize recombination and maximize the amount of energy utilized for the photo-chemical reaction will be the key. In this sense, combination of advanced characterization techniques and proper photo-catalytic testing will be a must for the successful development of efficient MOF based photocatalysts. We believe that homogeneous photo- and electro-catalysts will be the source of inspiration in constructing new photoactive scaffolds and composites. Moreover, the synthetic tools developed during the last decades by the MOF community will certainly help introduce such functional sites.
Regarding testing and reporting of kinetic data on photocatalytic performance, a lot of work is needed, not only for MOFs but for photocatalytic solids in general. Whereas in well stirred photoreactors concentrations are independent of location and light distribution is still inhomogeneous. This makes it impossible to use the common stirred tank (CSTR) approximation whose reactor conditions are equal to outflux conditions. In most reactors, gradients in photon absorption exist from high rates at the lamp side to low rates far from the lamp. Guidelines to properly measure and interpret kinetic data from well-mixed photoreactors have been recently published and should be considered by the photocatalysis community when reporting kinetic performance.85 Adopting such guidelines will certainly help compare photocatalytic performance reported in different publications, something not possible in most cases due either to the lack of reported information or to the different conditions applied.
Last but not least, we are confident that the next decade will witness an explosion in the number of optical applications of MOFs. Such applications will not be restricted to photo-catalysis; but also in optical based molecular sensing,86 light harvesting87 and even lighting, MOFs may find applications as excellent carrier materials.
Acknowledgements
The Dutch National Research School Combination Catalysis Controlled by Chemical Design (NRSC-Catalysis) is gratefully acknowledged for funding this research.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112 CAS.
- A. Nijland and S. R. Harutyunyan, Catal. Sci. Technol., 2013, 3, 1180 CAS.
- M. D. Hernández-Alonso, F. Fresno, S. Suárez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231 Search PubMed.
- M. G. Goesten, F. Kapteijn and J. Gascon, CrystEngComm, 2013, 15, 9249 RSC.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- G. Ferey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. De Weireld, A. Vimont, M. Daturi and J.-S. Chang, Chem. Soc. Rev., 2011, 550–562 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastián, C. Casado and J. Coronas, Chem. Mater., 2012, 24, 2829–2844 CrossRef CAS.
- B. Zornoza, C. Tellez, J. Coronas, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2013, 166, 67–78 CrossRef CAS.
- T. Rodenas, M. van Dalen, E. García-Pérez, P. Serra-Crespo, B. Zornoza, F. Kapteijn and J. Gascon, Adv. Funct. Mater., 2014, 24, 249–256 CrossRef CAS.
- T. Rodenas, M. van Dalen, P. Serra-Crespo, F. Kapteijn and J. Gascon, Microporous Mesoporous Mater., 2013 DOI:10.1016/j.micromeso.2013.08.049.
- A. Corma, H. Garcia and F. X. L. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- S. Bordiga, C. Lamberti, G. Ricchiardi, L. Regli, F. Bonino, A. Damin, K. P. Lillerud, M. Bjorgen and A. Zecchina, Chem. Commun., 2004, 2300–2301 RSC.
- M. Alvaro, E. Carbonell, B. Ferrer, F. Xamena and H. Garcia, Chem. – Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- F. Xamena, A. Corma and H. Garcia, J. Phys. Chem. C, 2007, 111, 80–85 Search PubMed.
- J. Gascon, M. D. Hernandez-Alonso, A. R. Almeida, G. P. M. van Klink, F. Kapteijn and G. Mul, ChemSusChem, 2008, 1, 981–983 CrossRef CAS PubMed.
- H. Khajavi, J. Gascon, J. M. Schins, L. D. A. Siebbeles and F. Kapteijn, J. Phys. Chem. C, 2011, 115, 12487–12493 CAS.
- F. Llabres i Xamena and J. Gascon, Metal Organic Frameworks as Heterogeneous Catalysts, The Royal Society of Chemistry, 2013 Search PubMed.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro, A. Corma and H. Garcia, Dalton Trans., 2011, 40, 6344–6360 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- J.-L. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630–2640 CrossRef CAS.
- C. G. Silva, A. Corma and H. Garcia, J. Mater. Chem., 2010, 20, 3141–3156 RSC.
- F. X. Llabres i Xamena, A. Abad, A. Corma and H. Garcia, J. Catal., 2007, 250, 294–298 CrossRef CAS.
- H. A. Lopez, A. Dhakshinamoorthy, B. Ferrer, P. Atienzar, M. Alvaro and H. Garcia, J. Phys. Chem. C, 2011, 115, 22200–22206 CAS.
- C. H. Hendon, D. Tiana and A. Walsh, Phys. Chem. Chem. Phys., 2012, 14, 13120–13132 RSC.
- G. Givaja, P. Amo-Ochoa, C. J. Gomez-Garcia and F. Zamora, Chem. Soc. Rev., 2012, 41, 115–147 RSC.
- Y. Kobayashi, B. Jacobs, M. D. Allendorf and J. R. Long, Chem. Mater., 2010, 22, 4120–4122 CrossRef CAS.
- F. Gandara, F. J. Uribe-Romo, D. K. Britt, H. Furukawa, L. Lei, R. Cheng, X. F. Duan, M. O'Keeffe and O. M. Yaghi, Chem. – Eur. J., 2012, 18, 10595–10601 CrossRef CAS PubMed.
- T. J. Savenije, A. J. Ferguson, N. Kopidakis and G. Rumbles, J. Phys. Chem. C, 2013, 117, 24085–24103 CAS.
- A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193–1202 CrossRef CAS PubMed.
- T. C. Narayan, T. Miyakai, S. Seki and M. Dinca, J. Am. Chem. Soc., 2012, 134, 12932–12935 CrossRef CAS PubMed.
- L. Sun, T. Miyakai, S. Seki and M. Dinca, J. Am. Chem. Soc., 2013, 135, 8185–8188 CrossRef CAS PubMed.
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 90, 10575–10577 RSC.
- Y. H. Fu, D. R. Sun, Y. J. Chen, R. K. Huang, Z. X. Ding, X. Z. Fu and Z. H. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- M. C. Fravventura, D. Deligiannis, J. M. Schins, L. D. A. Siebbeles and T. J. Savenije, J. Phys. Chem. C, 2013, 117, 8032–8040 CAS.
- T. J. Savenije, A. Huijser, M. J. W. Vermeulen and R. Katoh, Chem. Phys. Lett., 2008, 461, 93–96 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- M. Schiavello, Heterogeneous photocatalysis, Wiley, Chichester, U.K., 1997 Search PubMed.
- N. F. Mott, Can. J. Phys., 1956, 34, 1356–1368 CrossRef CAS.
- V. Barone, M. Casarin, D. Forrer, M. Pavone, M. Sambi and A. Vittadini, J. Comput. Chem., 2009, 30, 934–939 CrossRef CAS PubMed.
- B. Civalleri, F. Napoli, Y. Noel, C. Roetti and R. Dovesi, CrystEngComm, 2006, 8, 364–371 RSC.
- P. M. Wojciechowski, W. Zierkiewicz, D. Michalska and P. Hobza, J. Chem. Phys., 2003, 118, 10900–10911 CrossRef CAS.
- C. G. Silva, I. Luz, F. X. Llabrés I Xamena, A. Corma and H. García, Chem. – Eur. J., 2010, 16, 11133–11138 CrossRef CAS PubMed.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'Arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed.
- D. Venkataraman, G. B. Gardner, S. Lee and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 11600–11601 CrossRef CAS.
- C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS PubMed.
- Z. Q. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- H. L. Jiang, D. Feng, T. F. Liu, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 14690–14693 CrossRef CAS PubMed.
- P. Á. Szilágyi, P. Serra-Crespo, I. Dugulan, J. Gascon, H. Geerlings and B. Dam, CrystEngComm, 2013, 15, 10175–10178 RSC.
- K. G. M. Laurier, F. Vermoortele, R. Ameloot, D. E. De Vos, J. Hofkens and M. B. J. Roeffaers, J. Am. Chem. Soc., 2013, 135, 14488–14491 CrossRef CAS PubMed.
- L. Wen, J. Zhao, K. Lv, Y. Wu, K. Deng, X. Leng and D. Li, Cryst. Growth Des., 2012, 12, 1603–1612 CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CAS.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19(42), 14279–14285 CrossRef CAS PubMed.
- J. He, Z. Yan, J. Wang, J. Xie, L. Jiang, Y. Shi, F. Yuan, F. Yu and Y. Sun, Chem. Commun., 2013, 49, 6761–6763 RSC.
- J. L. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630–2640 CrossRef CAS.
- J. Juan-Alcaniz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102–10118 RSC.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- C. Wang, K. E. Dekrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- Y. Liu, Y. Yang, Q. Sun, Z. Wang, B. Huang, Y. Dai, X. Qin and X. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 7654–7658 CAS.
- M. H. Xie, X. L. Yang, C. Zou and C. D. Wu, Inorg. Chem., 2011, 50, 5318–5320 CrossRef CAS PubMed.
- J. Juan-Alcañiz, E. V. Ramos-Fernandez, U. Lafont, J. Gascon and F. Kapteijn, J. Catal., 2010, 269, 229–241 CrossRef.
- J. Juan-Alcaniz, M. Goesten, A. Martinez-Joaristi, E. Stavitski, A. V. Petukhov, J. Gascon and F. Kapteijn, Chem. Commun., 2011, 47, 8578–8580 RSC.
- E. V. Ramos-Fernandez, C. Pieters, B. van der Linden, J. Juan-Alcaniz, P. Serra-Crespo, M. W. G. M. Verhoeven, H. Niemantsverdriet, J. Gascon and F. Kapteijn, J. Catal., 2012, 289, 42–52 CrossRef CAS.
- W.-N. Li, F. Lin, X.-X. Li, L.-C. Zhang, W.-S. You and Z.-X. Jiang, J. Coord. Chem., 2013, 66, 2829–2842 CrossRef CAS.
- B. Liu, J. Yang, G.-C. Yang and J.-F. Ma, Inorg. Chem., 2012, 52, 84–94 CrossRef PubMed.
- T. Toyao, M. Saito, Y. Horiuchi, K. Mochizuki, M. Iwata, H. Higashimura and M. Matsuoka, Catal. Sci. Technol., 2013, 3, 2092–2097 CAS.
- T. T. Isimjan, H. Kazemian, S. Rohani and A. K. Ray, J. Mater. Chem., 2010, 20, 10241–10245 RSC.
- P. D. Frischmann, K. Mahata and F. Wurthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- D. L. Andrews and D. S. Bradshaw, J. Chem. Phys., 2004, 121, 2445–2454 CrossRef CAS PubMed.
- M. Motegh, J. Cen, P. W. Appel, J. R. van Ommen and M. T. Kreutzer, Chem. Eng. J., 2012, 207–208, 607–615 CrossRef CAS.
- J. Ferrando-Soria, H. Khajavi, P. Serra-Crespo, J. Gascon, F. Kapteijn, M. Julve, F. Lloret, J. Pasan, C. Ruiz-Perez, Y. Journaux and E. Pardo, Adv. Mater., 2012, 24, 5625–5629 CrossRef CAS PubMed.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |