DOI:
10.1039/C3CE42646G
(Paper)
CrystEngComm, 2014,
16, 3540-3547
Morphology tuning in the formation of vaterite crystal thin films with thermoresponsive poly(N-isopropylacrylamide) brush matrices†
Received
29th December 2013
, Accepted 26th February 2014
First published on 26th February 2014
Abstract
The use of a thermoresponsive polymer brush matrix resulted in the formation and morphology tuning of CaCO3 crystal thin films. Poly(N-isopropylacrylamide) (PNIPAm) brushes with thickness of approximately 500 nm were synthesized and employed as crystallization templates for CaCO3. The PNIPAm brush matrix induced the formation of vaterite thin films in the presence of poly(acrylic acid). Moreover, crystallization of CaCO3 at temperatures above and below the lower critical solution temperature of the PNIPAm brush matrices led to the preparation of vaterite films with distinctly different crystallographic orientations. The approach of using a stimuli-responsive polymer brush matrix may provide a novel way for the development of hybrid coating materials.
Introduction
Biominerals such as the nacre of shells, bones and teeth are organic/inorganic composites synthesized naturally by living organisms.1–3 These biominerals exhibit elaborate structures that function as structural supports, protection and mineral storage. The biomineralization process has been inspiring materials scientists for the last two decades to develop functional composites under mild conditions.4–19 Cooperation of insoluble macromolecular templates and soluble acidic biomacromolecules is crucial in biomineralization.20–22 For synthetic materials, CaCO3 crystal thin films have also been successfully synthesized via the bio-inspired approach.4,7,23–25
A variety of polymer templates have been used as templates for biomimetic synthesis of inorganic crystals.26,27 Our previous studies have shown that insoluble polymer matrices, such as poly(vinyl alcohol) (PVA), chitin and chitosan, promoted the formation of thin films of calcium carbonate crystals.7,28,29
Among the coating methods of polymer templates which are subsequently used as crystallization matrices for inorganic crystals,7 polymer brushes are attractive as new templates because of the density of their functional groups on the surface. Polymer brushes are thin polymer coatings consisting of polymer chains with the termini covalently fixed to a substrate, and such polymer layers possess excellent long-time stability in various solvents at a wide range of temperatures.30–32 Recent advancements on surface-initiated controlled radical polymerization techniques, such as atom transfer radical polymerization (ATRP), enabled the precise control of the thickness, composition and architecture of the polymer brushes.33–35 Surface-initiated ATRP allows the modification of various surfaces, such as silicon, metal oxides36 and polymer surfaces,37 by covalently attached polymer brushes. Thus, a study of the crystallization behavior of inorganic minerals on polymer brush matrices may provide useful information toward the design of organic/inorganic functional hybrid coating materials. However, only very limited examples of inorganic crystallization on polymer brush matrices have been reported.23,38
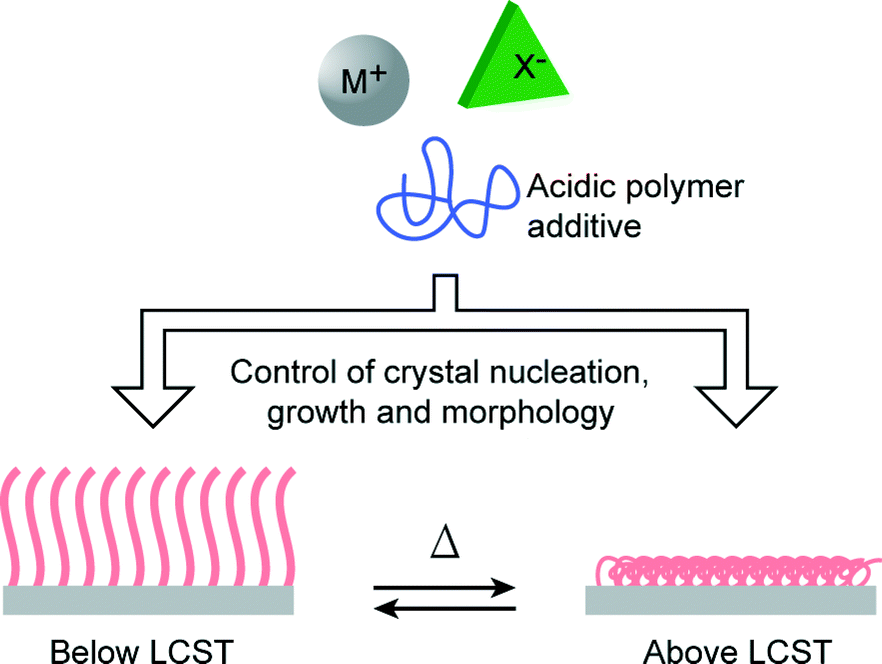
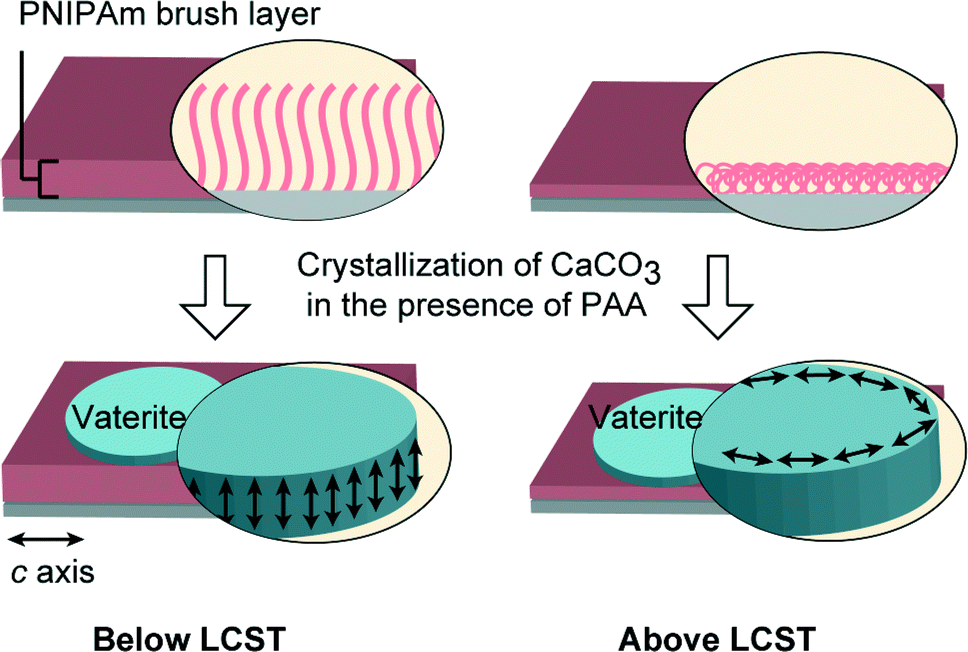
In biomineralization, morphological and orientational selection is of significant importance to living organisms since it is closely related to the functional properties of the resultant biominerals.1–3,39,40 Recently, stimuli-responsive polymers have attracted a great deal of attention.41–43 In artificial biomimetic systems, the use of stimuli-responsive polymer matrices is a promising method for the realization of the morphological and orientational control of mineral crystals. We have previously reported the use of a photoresponsive polymer matrix for the successful control of the morphologies of CaCO3 crystal thin films.25 On the other hand, PNIPAm is a thermoresponsive polymer that exhibits lower critical solution temperature (LCST) in water at around 32 °C.44 PNIPAm brushes exhibit an aggregated conformational structure due to the intramolecular hydrogen bonding between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H at temperatures above its LCST, while at temperatures below the LCST, a stretched conformational structure is adopted because of hydrogen bonding interactions with water molecules.45,46 PNIPAm coatings have been widely used as bioactive matrices for cell cultivation, showing reversible cell attachment and detachment properties at temperatures above and below the LCST.47–49 When PNIPAm brushes were used as crystallization matrices, our previous study showed that the use of PNIPAm brush matrices induced the formation of unidirectionally oriented fibrous calcium carbonate at a temperature above the LCST.23 Generally, many polymer template surfaces could promote the formation of inorganic nuclei by decreasing the activation energy of nucleation,50,51 and the conformational change of polymer chains in the template is likely to influence the crystallization behavior of minerals. Thus, the conformational rearrangements of the PNIPAm brush chains across the LCST are expected to induce the formation of crystals with different morphologies (Fig. 1). Herein, we report on the study of the crystallization of CaCO3 on thermoresponsive PNIPAm brush matrices at temperatures above and below the LCST.
O and N–H at temperatures above its LCST, while at temperatures below the LCST, a stretched conformational structure is adopted because of hydrogen bonding interactions with water molecules.45,46 PNIPAm coatings have been widely used as bioactive matrices for cell cultivation, showing reversible cell attachment and detachment properties at temperatures above and below the LCST.47–49 When PNIPAm brushes were used as crystallization matrices, our previous study showed that the use of PNIPAm brush matrices induced the formation of unidirectionally oriented fibrous calcium carbonate at a temperature above the LCST.23 Generally, many polymer template surfaces could promote the formation of inorganic nuclei by decreasing the activation energy of nucleation,50,51 and the conformational change of polymer chains in the template is likely to influence the crystallization behavior of minerals. Thus, the conformational rearrangements of the PNIPAm brush chains across the LCST are expected to induce the formation of crystals with different morphologies (Fig. 1). Herein, we report on the study of the crystallization of CaCO3 on thermoresponsive PNIPAm brush matrices at temperatures above and below the LCST.
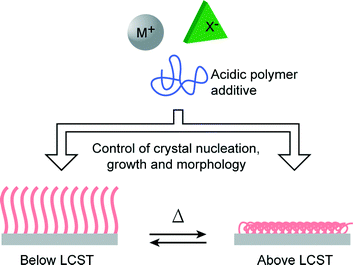 |
| | Fig. 1 Schematic illustration of the approach of using thermoresponsive PNIPAm brush matrices for controlling crystal nucleation, growth and morphology. | |
Experimental
Materials
N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) and poly(acrylic acid) (PAA, Mw = 2.0 × 103) were purchased from Aldrich. Triethylamine (99%), (3-aminopropyl)triethoxysilane (99%), 2-bromo-2-methylpropionyl bromide (98%), and N-isopropylacrylamide (NIPAm, 97%) were purchased from Tokyo Kasei. Copper(I) bromide (CuBr, 99.7%), methanol (MeOH, 99.5%), calcium chloride, ammonium carbonate and ethanol (anhydrous, 99.5%) were obtained from Wako. NIPAm was purified by vacuum sublimation. PMDETA was vacuum-distilled prior to use. All other chemicals were used as received without further purification.
Preparation of ATRP-initiator deposited silicon and glass substrates
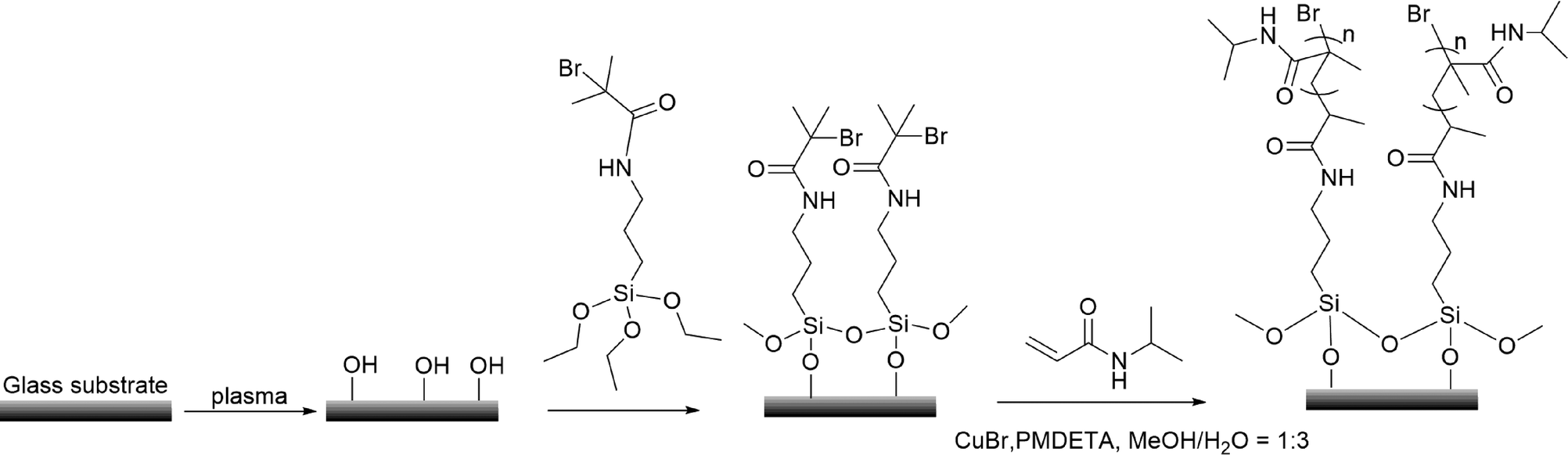
The synthesis of PNIPAm brushes followed the procedures given in Scheme 1. Firstly, 2-bromo-2-methyl-N-(3-triethoxysilyl-propyl)-propionamide (ATRP-initiator) was synthesized by the condensation of (3-aminopropyl)trimethoxysilane and 2-bromo-2-methylpropionyl bromide. The silicon and glass substrates were placed in an oxygen plasma treatment chamber (Cute-1MP, Femto Science) filled with pure oxygen to clean the surface for 5 min (100 W). Subsequently, the slides were immersed in 30 mM solution of the ATRP-initiator in anhydrous ethanol for 24 h at 25 °C. The resultant slides were washed with ethanol, sonicated in ethanol for 1 min, and dried with a stream of nitrogen.
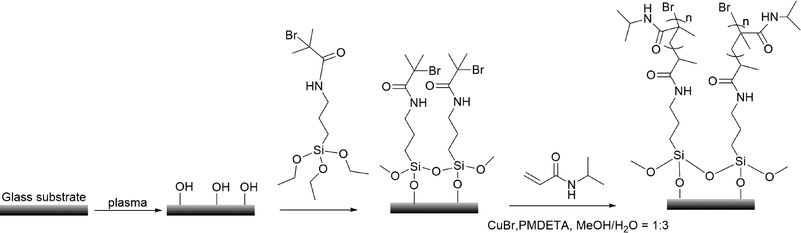 |
| | Scheme 1 Procedures for the synthesis of PNIPAm brushes via surface-initiated ATRP. | |
Synthesis of PNIPAm brushes from ATRP-initiator modified substrates
In a typical ratio of [NIPAm]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CuBr]:[PMDETA] = 200:1:5, 2 g of NIPAm, 6 ml of deionized water and 2 ml of methanol were added into a Schlenk flask, and the solution was degassed by three freeze–pump–thaw cycles. CuBr (12 mg) and PMDETA (60 mg) were added into the deoxygenated solution. Subsequently, the solution was transferred into a nitrogen purged reaction vessel containing the ATRP-initiator modified substrates. The reaction was allowed to proceed for 1 h. After polymerization, substrates grafted with PNIPAm brushes were taken from the polymerization solution, rinsed with large quantities of methanol and deionized water, and dried with a stream of nitrogen.
:[CuBr]:[PMDETA] = 200:1:5, 2 g of NIPAm, 6 ml of deionized water and 2 ml of methanol were added into a Schlenk flask, and the solution was degassed by three freeze–pump–thaw cycles. CuBr (12 mg) and PMDETA (60 mg) were added into the deoxygenated solution. Subsequently, the solution was transferred into a nitrogen purged reaction vessel containing the ATRP-initiator modified substrates. The reaction was allowed to proceed for 1 h. After polymerization, substrates grafted with PNIPAm brushes were taken from the polymerization solution, rinsed with large quantities of methanol and deionized water, and dried with a stream of nitrogen.
Crystallization of calcium carbonate on substrates grafted with PNIPAm brushes
Purified water obtained from an Auto pure WT100 purification system (Yamato, relative resistivity: maximum 1.8 × 107 Ω cm) was employed for CaCO3 crystallization. Calcium chloride aqueous solution ([Ca2+] = 10 mM) containing PAA (2.5 × 10−3 wt%) was used to carry out CaCO3 crystallization. The solution was transferred into vessels containing the matrices grafted with PNIPAm brushes. The vessels were then placed in a closed desiccator together with ammonium carbonate. An incubator (Fukushima) was used to maintain a constant crystallization temperature. CaCO3 crystallization at 20 °C (below the LSCT) and 40 °C (above the LSCT) was carried out in this study.
Characterization
Fourier transform infrared (FTIR) spectra were recorded using a Jasco FT/IR-6100 spectrometer (Jasco, Tokyo). The brush thickness was determined using a Jasco M-500 ellipsometer (Jasco, Tokyo) equipped with a He–Ne laser (λ = 632.8 nm). The thickness was further examined by tapping-mode atomic force microscopy (AFM) measurements (Bruker, Santa Barbara). Scanning electron microscopy (SEM) images were obtained using a Hitachi S-4700 field-emission SEM (Hitachi, Tokyo) operated at 3–5 kV. Samples were coated with a layer of platinum using a Hitachi E-1030 ion sputter (Hitachi, Tokyo). Polarizing optical microscope images were taken using an Olympus BX51 polarizing optical microscope (Olympus, Tokyo). Raman spectra were obtained using a Jasco Model NR-1800 spectrometer (Jasco, Tokyo) with an excitation source of Nd:YAG laser (532 nm). X-ray diffraction (XRD) measurements were performed using a Rigaku SmartLab Intelligent X-ray Diffraction System (Rigaku, Tokyo) with filtered Cu Kα radiation (λ = 1.5406 Å, operating at 40 kV and 40 mA). Transmission electron microscopy (TEM) images were taken using a JEOL JEM-2010HC operated at 200 kV.
Results and discussion
Characterization of PNIPAm brush matrices synthesized via surface-initiated ATRP
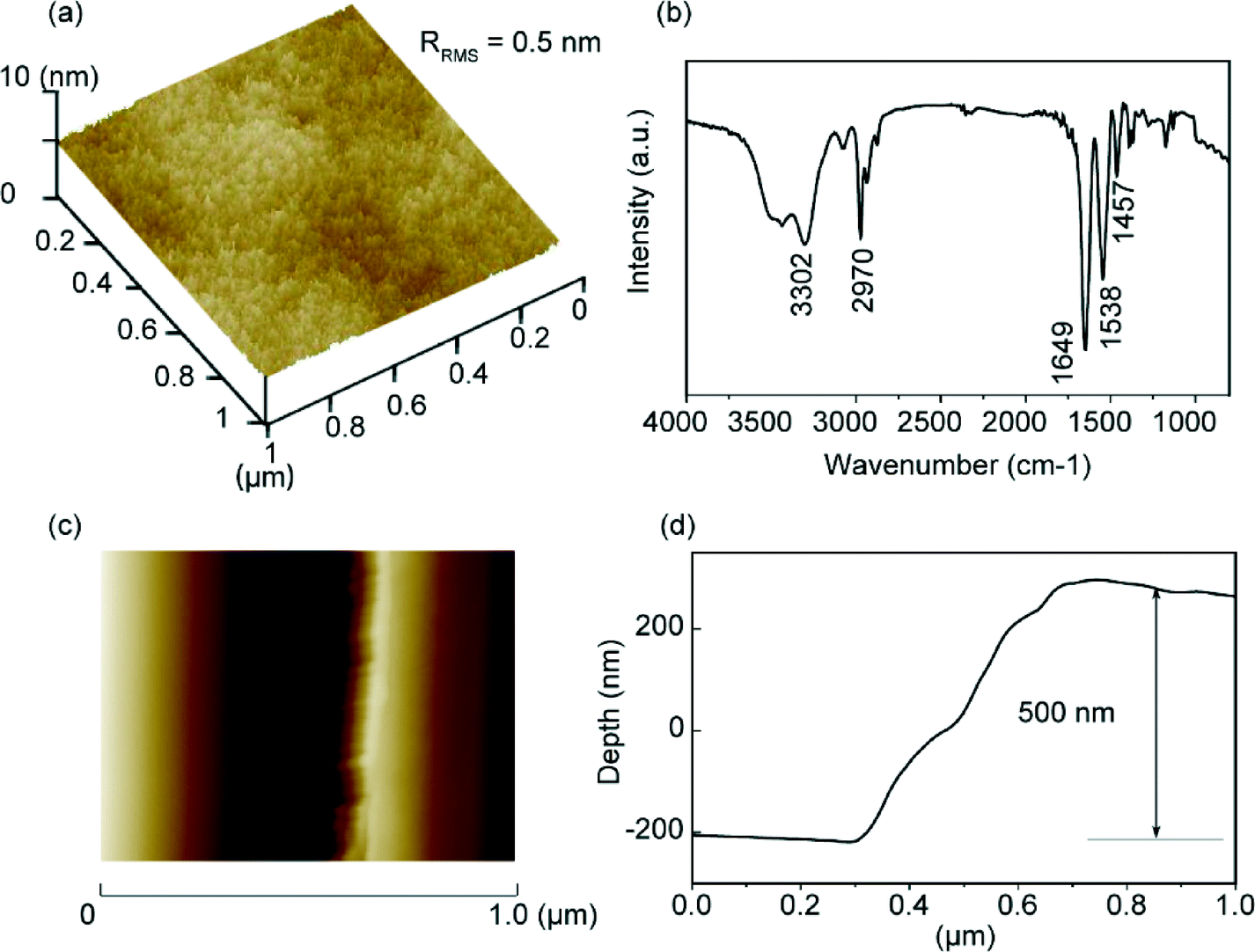
The surface morphology of the PNIPAm brush substrates synthesized via surface-initiated ATRP was measured by AFM. Fig. 2a shows the three-dimensional AFM image of the PNIPAm grafted substrate surface. The synthesized PNIPAm brushes have a root-mean-square roughness (RRMS) value of about 0.5 nm over a 1 μm × 1 μm area. No aggregation of the chains was observed, which shows that the brush surface is uniform and homogeneous. The formation of PNIPAm brushes was also confirmed by IR spectroscopy (Fig. 2b). The absorption peak at 3302 cm−1 corresponds to the stretching of the amide N–H group. The anti-symmetric stretching vibration of the CH3 group gives rise to the peak at 2970 cm−1. The strong absorption peak at 1649 cm−1 can be attributed to the secondary amide CO stretch. The two peaks at 1386 cm−1 and 1365 cm−1 correspond to the vibration of the two methyl groups in –C(CH3)2.52 Brush thickness was determined by AFM measurements performed across the scratched and unscratched boundaries of the brush substrate (Fig. 2c), and the corresponding depth graph shows that the brush has a thickness of approximately 500 nm in air (Fig. 2d).
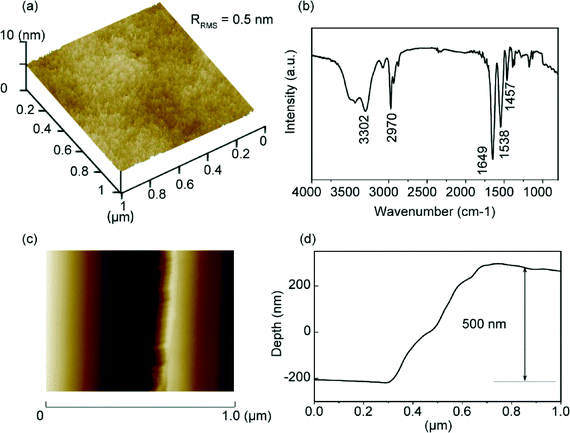 |
| | Fig. 2 Characterization of PNIPAm-grafted substrates: (a) three-dimensional AFM image of the PNIPAm-grafted silica surface, (b) IR spectrum of the PNIPAm brush, (c) AFM image across the scratched and unscratched boundaries, and (d) corresponding depth figure across the boundaries. | |
Crystallization of CaCO3 on PNIPAm brush matrices below the LCST
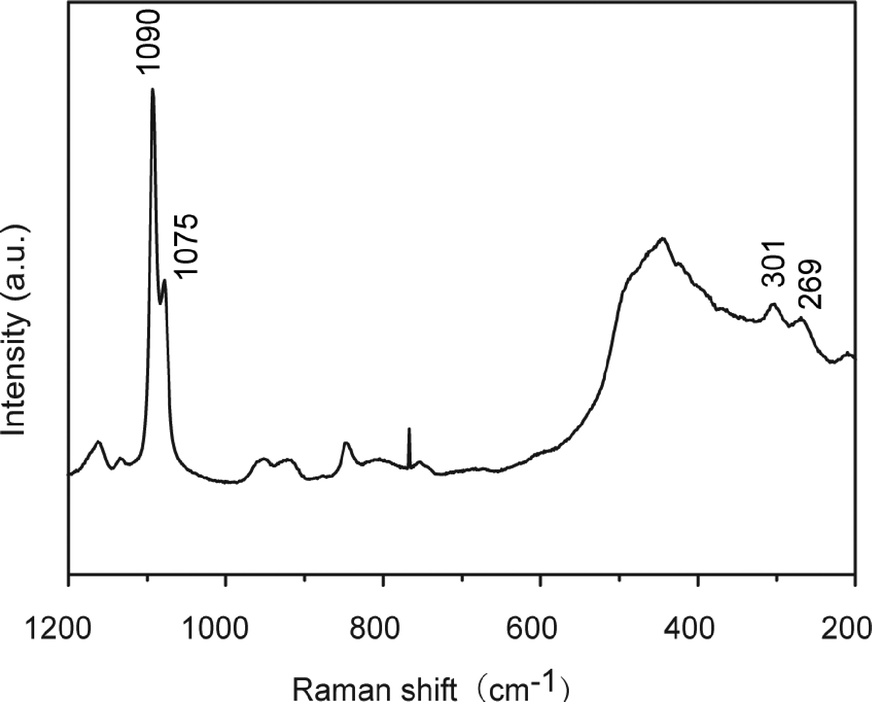
Thin films of calcium carbonate with crystal domains approximately 200 μm in diameter were formed on PNIPAm brush matrices in the presence of PAA (2.5 × 10−3 wt%) when crystallization was carried out at 20 °C (Fig. 3a). The CaCO3 thin films have a flat surface, although cracks along the crystal growth directions of the thin films were observed under SEM observation (Fig. 3b). The cross-sectional SEM image (Fig. 3c) reveals that the resultant thin films with thickness of about 1 μm is composed of particles approximately 50 nm in size. The Raman spectrum (Fig. 4) shows that the thin films formed at 20 °C are mainly vaterite because the corresponding Raman bands of CO32− are observed at 1090 cm−1 (ν1, symmetric stretching mode), 1075 cm−1 (ν1), and 301 cm−1 (lattice mode).53
 |
| | Fig. 3 CaCO3 crystals developed on PNIPAm brush matrices in the presence of PAA (2.5 × 10−3 wt%) after crystallization for 10 h at 20 °C: (a) polarizing optical microscope image, (b) SEM image (top view), and (c) cross-sectional SEM image of the thin film. | |
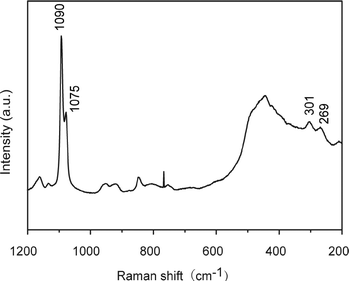 |
| | Fig. 4 Raman spectrum of CaCO3 crystal thin films developed on PNIPAm brush matrices in the presence of PAA (2.5 × 10−3 wt%) after crystallization for 10 h at 20 °C. | |
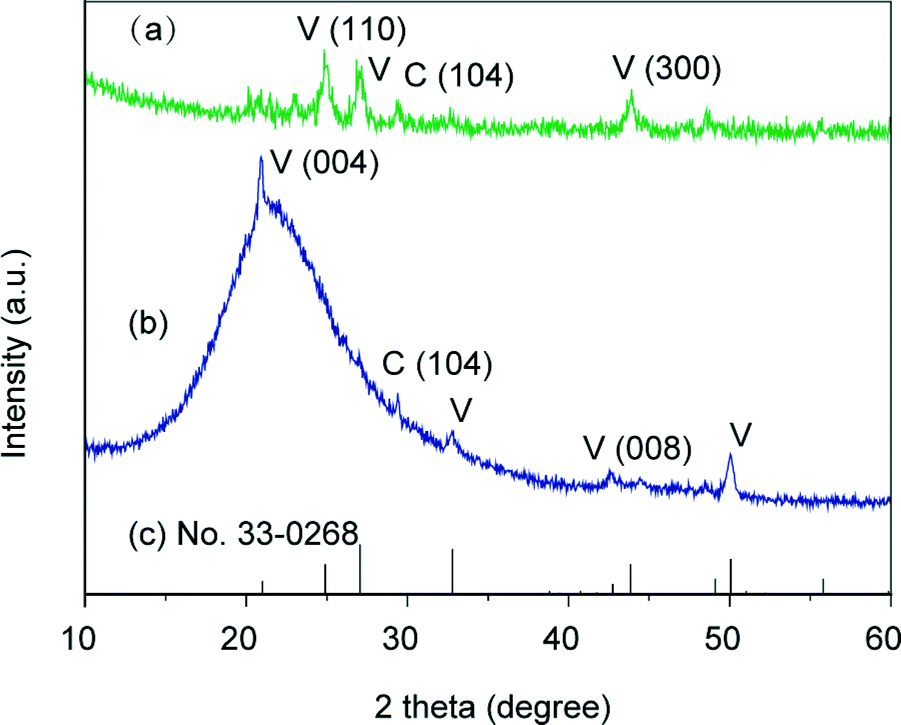
In-plane and out-of-plane XRD measurements were conducted to study the polymorph and the preferential orientation of the CaCO3 crystal thin films formed at 20 °C (Fig. 5). The diffraction patterns in the XRD spectra show that the samples are mainly vaterite in morphology, which is consistent with the results obtained by Raman spectroscopy. For the thin films formed at 20 °C, diffraction peaks of the (110) and (300) planes, which are parallel to the c axis of the crystals, are only observed in the in-plane XRD pattern (Fig. 5a). In contrast, compared to the standard powder diffraction pattern (pdf no. 33-0268, Fig. 5c), the intensified peak assignable to the (004) plane, which is perpendicular to the c axis, is observed in the out-of-plane XRD pattern (Fig. 5b). These results suggest that the c axis in the crystal thin films formed at 20 °C is preferentially perpendicular to the substrate. The perpendicular orientation of the c axis was also confirmed by selected area electron diffraction (SAED) (Fig. 6), and such a preferential orientation led to dark patterns with very weak birefringence under polarizing optical microscope observation (Fig. 3a).54 The bright-field TEM image (Fig. 6a) and the corresponding SAED pattern (Fig. 6b) also indicate that the thin films favoured the [1 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] growth direction.
0] growth direction.
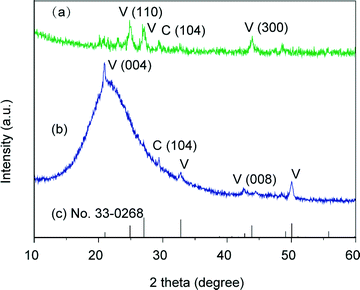 |
| | Fig. 5 XRD patterns of the CaCO3 crystal thin films formed on PNIPAm brush matrices at 20 °C: (a) in-plane XRD pattern, (b) out-of-plane XRD pattern, and (c) vaterite standard powder XRD pattern from JCPDS (no. 33-0268). V and C stand for vaterite and calcite, respectively. | |
 |
| | Fig. 6 (a) Bright-field TEM image and (b) corresponding SAED pattern of an isolated vaterite film grown on PNIPAm brush matrices in the presence of PAA at 20 °C. The SAED pattern shows that the thin films have a zone axis of [001]. | |
Crystallization of CaCO3 on PNIPAm brush matrices above the LCST
Crystallization of calcium carbonate at 40 °C was also carried out to study the effect of the entangled state of the brushes on the crystallization behavior. Fig. 7a shows the polarizing optical microscope image of CaCO3 crystal thin films after 6 h of crystallization at 40 °C, which is a temperature above the LSCT of PNIPAm brush matrices. Concentric spherulitic crystal thin films that exhibit mosaic patterns due to the regular orientation of the c axis were observed. The crystal domains are about several hundreds of micrometers in diameter. Fig. 7b shows the SEM image of a homogeneous film surface. The thin films with thickness of about 1.5 μm are composed of polycrystalline particles approximately several tens of nanometers in size (Fig. 7c).
 |
| | Fig. 7 CaCO3 crystal thin films formed on PNIPAm brush matrices in the presence of PAA (2.5 × 10−3 wt%) after crystallization at 40 °C for 6 h: (a) polarizing optical microscope image, (b) SEM image (top view) and (c) cross-sectional SEM image. | |
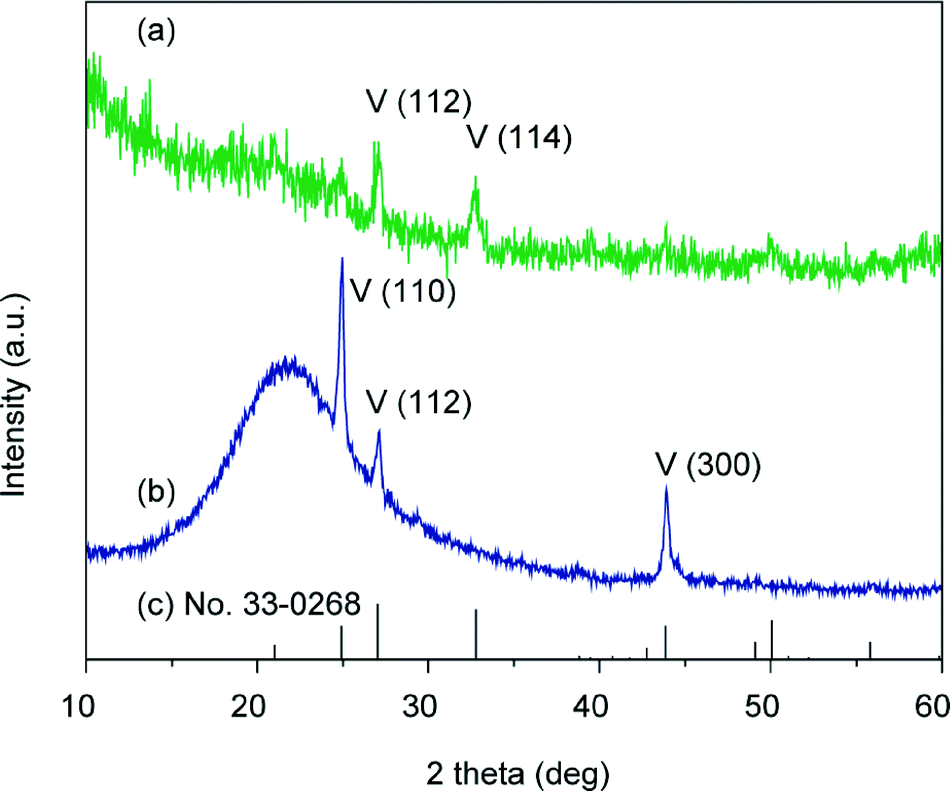
Both the Raman spectrum (Fig. S1 in the ESI†) and the XRD patterns of the thin films formed at 40 °C show that the crystals are of vaterite polymorph (Fig. 8). In comparison with the standard powder diffraction pattern (pdf no. 33-0268, Fig. 8c), intensified peaks of the (110) and (300) planes, which are parallel to the c axis, are observed in the out-of-plane XRD pattern (Fig. 8b), while the intensities of these two peaks are evidently weak in the in-plane XRD pattern (Fig. 8a). These results suggest that the CaCO3 crystal thin films formed at 40 °C preferentially oriented with the c axis in the crystal thin films parallel to the substrate.
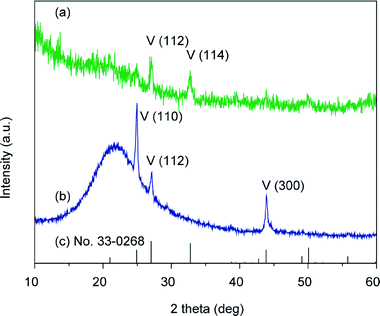 |
| | Fig. 8 XRD patterns of the CaCO3 crystal thin films formed on PNIPAm brush matrices at 40 °C: (a) in-plane XRD pattern, (b) out-of-plane XRD pattern, and (c) vaterite standard powder XRD pattern from JCPDS (no. 33-0268); V stands for vaterite. | |
The orientation of the vaterite thin films formed at 40 °C was also examined by electron diffraction analysis (Fig. 9). The directions of the crystal growth and the c axis of the crystal thin films are shown in Fig. 9a. The SAED pattern (Fig. 9b) shows that the vaterite thin films formed at 40 °C have a zone axis of [110] and the c axis is aligned parallel to the surface plane of these thin films and perpendicularly oriented to the [1 0] growth direction.
 |
| | Fig. 9 (a) Bright-field TEM image and (b) corresponding SAED pattern of an isolated vaterite film grown on PNIPAm brush matrices in the presence of PAA at 40 °C. | |
Effects of stimuli-responsive PNIPAm brush matrices on CaCO3 crystallization
It is noteworthy that vaterite, thermodynamically the least stable polymorph of anhydride CaCO3,55,56 was formed using PNIPAm brushes as crystallization matrices both above and below the LCST in the presence of PAA. For crystallization of minerals, the existence of polymer matrices may significantly influence the morphologies and polymorphs of crystals. For instance, our previous studies showed that PVA matrices preferentially induced the formation of aragonite thin films in the presence of PAA.29,57,58 Calcite crystal thin films were obtained using chitosan,59 chitin28,60 and cellulose28 matrices in the presence of PAA additives. In addition, Volkmer et al. reported that the use of poly(methacrylic acid) brush matrices induced the formation of amorphous calcium carbonate.38
We have found that the use of PNIPAm brush matrices led to the formation of vaterite thin films with distinct differences in the orientational behavior above and below the LCST (Fig. 10). For nonthermal switching systems, biomimetic vaterite thin films with preferential orientation have been reported previously by our group.54,61 For instance, vaterite thin films with the c axis oriented perpendicular and parallel to the substrate in an alternating manner were formed by the cooperative effect of the chitosan matrix and the poly(aspartate) additive.54 We have also recently reported that the synergistic effect of carboxylated polyrotaxane additives and poly(vinyl alcohol) matrices led to the formation of vaterite thin films with the c axis parallel to the substrate.61 In comparison with those systems, the use of stimuli-responsive PNIPAm brush matrices enabled the possibility of tuning the crystal orientation by external stimuli.
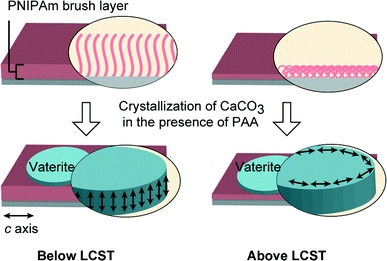 |
| | Fig. 10 Schematic illustration of vaterite thin film formation on PNIPAm brush matrices at temperatures below (20 °C) and above (40 °C) the LCST in the presence of PAA additives. Left: vaterite thin films with the c axis perpendicular to the substrate; right: vaterite thin films with the c axis parallel to the substrate. | |
Although the vaterite thin films formed on PNIPAm brush matrices at temperatures above and below the LCST both favoured the [1 0] growth direction, they have distinct preferential orientations. For thin films developed at a temperature below the LCST, the (001) plane is parallel to the film surface (Fig. 6). However, for vaterite thin films developed at a temperature above the LCST, the (110) plane is parallel to the thin film surface (Fig. 9). It is assumed that the orientational difference is attributable to the conformational change of PNIPAm chains. In addition, acidic additives such as PAA, which interact with cationic ions, are essential for the biomimetic development of hybrid thin films. The interaction between PAA and PNIPAm has also been reported.62,63 Recent studies have also shown that acidic polymer additives induce the formation of the amorphous calcium carbonate (ACC) precursor or the polymer-induced liquid precursor (PILP).22 The induced precursors are ionic and have fluidic character, which allow them to penetrate into PNIPAm brush matrices, resulting in the formation of vaterite thin films.
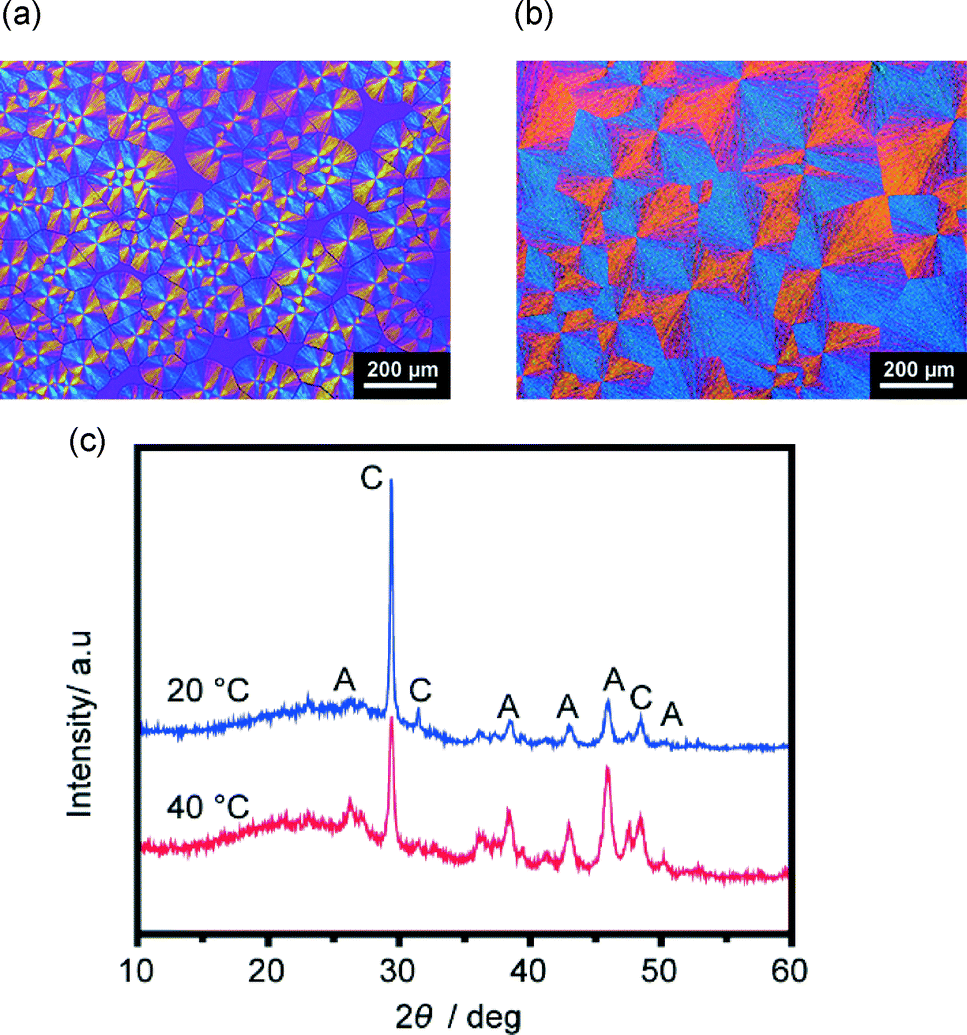
Spin-coated PNIPAm matrices cross-linked by glycerol were also employed for the crystallization of CaCO3 to further examine the effect of brush structures on CaCO3 crystallization (Fig. 11). For crystal thin films formed on the spin-coated PNIPAm matrices at temperatures above and below the LCST, only aragonite and calcite polymorphs are observed (Fig. 11c). To prepare a spin-coated PNIPAm matrix, a cross-linking agent is necessary to anchor the water-soluble PNIPAm. In contrast, thin films formed on PNIPAm brush matrices at temperatures above and below the LCST have vaterite polymorphs and show obvious differences in the crystallographic orientation. For the spin-coated gel and the corresponding polymer brush matrices, the difference in the conformational structures of the chains of the polymer matrices is assumed to be influential to their swelling behavior64 and affinity with ions in aqueous solutions, thus leading to different crystallization behaviors of CaCO3.
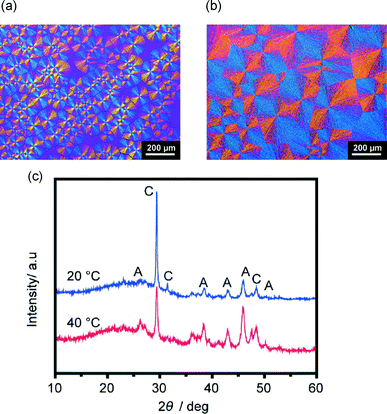 |
| | Fig. 11 Polarizing optical microscope images of CaCO3 thin films formed on spin-coated PNIPAm substrates: (a) crystallization at 20 °C, (b) crystallization at 40 °C. (c) XRD patterns of corresponding thin films. A and C in XRD patterns standard for aragonite and calcite, respectively. | |
Conclusion
We have found that thermoresponsive PNIPAm brushes with thickness of approximately 500 nm act as effective polymer matrices for tuning the orientation and morphology of CaCO3 crystal thin films. CaCO3 thin films with the c axis preferentially perpendicular to the substrates were obtained below the LCST, while crystallization above the LCST induced the formation of vaterite thin films with the c axis parallel to the substrates. Oriented vaterite tablets have been found in biominerals,56 and this study may deepen our understanding of biomineralization and could lead to a new way for developing hybrid materials.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (no. 22107003) on the Innovative Areas: “Fusion Materials: Creative Development of Materials and Exploration of Their Function through Molecular Control” (area no. 2206) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Notes and references
- L. Addadi and S. Weiner, Angew. Chem., Int. Ed. Engl., 1992, 31, 153 CrossRef PubMed.
-
E. Baeuerlein, P. Behrens and M. Epple, Handbook of biomineralization, Wiley-VCH, Weinheim, 2007 Search PubMed.
-
S. Mann, Biomineralization: principles and concepts in bioinorganic materials chemistry, Oxford University Press, New York, 2001 Search PubMed.
- T. Kato, Adv. Mater., 2000, 12, 1543 CrossRef CAS.
- S. H. Yu, H. Cölfen, K. Tauer and M. Antonietti, Nat. Mater., 2005, 4, 51 CrossRef CAS PubMed.
- F. Nudelman and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2012, 51, 6582 CrossRef CAS PubMed.
- T. Kato, T. Sakamoto and T. Nishimura, MRS Bull., 2010, 35, 127 CrossRef CAS.
- C. Ohtsuki, M. Kamitakahara and T. Miyazaki, J. Tissue Eng. Regener. Med., 2007, 1, 33 CrossRef CAS PubMed.
- H. B. Yao, H. Y. Fang, X. H. Wang and S. H. Yu, Chem. Soc. Rev., 2011, 40, 3764 RSC.
- A. Sugawara-Narutaki, Polym. J., 2013, 45, 269 CrossRef CAS.
- W. L. Noorduin, A. Grinthal, L. Mahadevan and J. Aizenberg, Science, 2013, 340, 832 CrossRef CAS PubMed.
- Y. Oaki, R. Adachi and H. Imai, Polym. J., 2012, 44, 612 CrossRef CAS.
- Y. Y. Kim, K. Ganesan, P. C. Yang, A. N. Kulak, S. Borukhin, S. Pechook, L. Ribeiro, R. Kroger, S. J. Eichhorn, S. P. Armes, B. Pokroy and F. C. Meldrum, Nat. Mater., 2011, 10, 890 CrossRef CAS PubMed.
- Y. Y. Kim, L. Ribeiro, F. Maillot, O. Ward, S. J. Eichhorn and F. C. Meldrum, Adv. Mater., 2010, 22, 2082 CrossRef CAS PubMed.
- Y. Oaki, S. Kajiyama, T. Nishimura, H. Imai and T. Kato, Adv. Mater., 2008, 20, 3633 CrossRef CAS PubMed.
- Y. Oaki, M. Kijima and H. Imai, J. Am. Chem. Soc., 2011, 133, 8594 CrossRef CAS PubMed.
- A. Finnemore, P. Cunha, T. Shean, S. Vignolini, S. Guldin, M. Oyen and U. Steiner, Nat. Commun., 2012, 3, 966 CrossRef PubMed.
- D. K. Keum, K. M. Kim, K. Naka and Y. Chujo, J. Mater. Chem., 2002, 12, 2449 RSC.
- Y. Tanaka and K. Naka, Polym. J., 2012, 44, 586 CrossRef CAS.
- A. Berman, L. Addadi and S. Weiner, Nature, 1988, 331, 546 CrossRef CAS.
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, Science, 2009, 325, 1388 CrossRef CAS PubMed.
- L. B. Gower, Chem. Rev., 2008, 108, 4551 CrossRef CAS PubMed.
- S. Kumar, T. Ito, Y. Yanagihara, Y. Oaki, T. Nishimura and T. Kato, CrystEngComm, 2010, 12, 2021 RSC.
- A. Sugawara, T. Ishii and T. Kato, Angew. Chem., Int. Ed., 2003, 42, 5299 CrossRef CAS PubMed.
- T. Sakamoto, Y. Nishimura, T. Nishimura and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 5856 CrossRef CAS PubMed.
- N. A. J. M. Sommerdijk and G. de With, Chem. Rev., 2008, 108, 4499 CrossRef CAS PubMed.
- T. Kato, A. Sugawara and N. Hosoda, Adv. Mater., 2002, 14, 869 CrossRef CAS.
- N. Hosoda and T. Kato, Chem. Mater., 2001, 13, 688 CrossRef CAS.
- T. Sakamoto, A. Oichi, Y. Oaki, T. Nishimura, A. Sugawara and T. Kato, Cryst. Growth Des., 2009, 9, 622 CAS.
-
R. C. Advincula, Polymer brushes: synthesis, characterization, applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schuwer, C. Sugnaux, S. Tugulu and H. A. Klok, Chem. Rev., 2009, 109, 5437 CrossRef CAS PubMed.
- A. Nomura, K. Okayasu, K. Ohno, T. Fukuda and Y. Tsujii, Macromolecules, 2011, 44, 5013 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed.
- J. Pyun and K. Matyjaszewski, Chem. Mater., 2001, 13, 3436 CrossRef CAS.
- M. Kobayashi and A. Takahara, Chem. Rec., 2010, 10, 208 CrossRef CAS PubMed.
- C. R. Vestal and Z. J. Zhang, J. Am. Chem. Soc., 2002, 124, 14312 CrossRef CAS PubMed.
- A. Carlmark and E. Malmstrom, J. Am. Chem. Soc., 2002, 124, 900 CrossRef CAS PubMed.
- S. Tugulu, M. Harms, M. Fricke, D. Volkmer and H. A. Klok, Angew. Chem., Int. Ed., 2006, 45, 7458 CrossRef CAS PubMed.
- A. M. Belcher, X. H. Wu, R. J. Christensen, P. K. Hansma, G. D. Stucky and D. E. Morse, Nature, 1996, 381, 56 CrossRef CAS.
- M. A. Meyers, J. McKittrick and P. Y. Chen, Science, 2013, 339, 773 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- T. P. Russell, Science, 2002, 297, 964 CrossRef CAS PubMed.
- T. Chen, R. Ferris, J. M. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- T. Sun, G. Wang, L. Feng, B. Liu, Y. Ma, L. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 43, 357 CrossRef CAS PubMed.
- M. Kaholek, W. K. Lee, S. J. Ahn, H. W. Ma, K. C. Caster, B. LaMattina and S. Zauscher, Chem. Mater., 2004, 16, 3688 CrossRef CAS.
- C. Y. Xue, B. C. Choi, S. Choi, P. V. Braun and D. E. Leckband, Adv. Funct. Mater., 2012, 22, 2394 CrossRef CAS PubMed.
- Y. Hou, A. R. Matthews, A. M. Smitherman, A. S. Bulick, M. S. Hahn, H. Hou, A. Han and M. A. Grunlan, Biomaterials, 2008, 29, 3175 CrossRef CAS PubMed.
- Y. Kumashiro, M. Yamato and T. Okano, Ann. Biomed. Eng., 2010, 38, 1977 CrossRef PubMed.
-
P. M. Dove, J. De Yoreo and S. Weiner, Biomineralization, Mineralogical Society of America, Washington, DC, 2003 Search PubMed.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef PubMed.
- H. Suzuki, H. M. Nurul, T. Seki, T. Kawamoto, H. Haga, K. Kawabata and Y. Takeoka, Macromolecules, 2010, 43, 9945 CrossRef CAS.
- U. Wehrmeister, A. L. Soldati, D. E. Jacob, T. Hager and W. Hofmeister, J. Raman Spectrosc., 2010, 41, 193 CAS.
- A. Sugawara, A. Oichi, H. Suzuki, Y. Shigesato, T. Kogure and T. Kato, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5153 CrossRef CAS PubMed.
- L. Kabalah-Amitai, B. Mayzel, Y. Kauffmann, A. N. Fitch, L. Bloch, P. U. P. A. Gilbert and B. Pokroy, Science, 2013, 340, 454 CrossRef CAS PubMed.
- L. Qiao, Q. L. Feng and Z. Li, Cryst. Growth Des., 2007, 7, 275 CAS.
- T. Sakamoto, A. Oichi, T. Nishimura, A. Sugawara and T. Kato, Polym. J., 2009, 41, 522 CrossRef CAS.
- N. Hosoda, A. Sugawara and T. Kato, Macromolecules, 2003, 36, 6449 CrossRef CAS.
- T. Kato, T. Suzuki, T. Amamiya, T. Irie and N. Komiyama, Supramol. Sci., 1998, 5, 411 CrossRef CAS.
- T. Kato, T. Suzuki and T. Irie, Chem. Lett., 2000, 186 CrossRef CAS.
- F. Zhu, T. Nishimura, H. Eimura and T. Kato, CrystEngComm, 2014, 16, 1496 RSC.
- M. Koussathana, P. Lianos and G. Staikos, Macromolecules, 1997, 30, 7798 CrossRef CAS.
- G. Staikos, G. Bokias and K. Karayanni, Polym. Int., 1996, 41, 345 CrossRef CAS.
- A. Karim, J. F. Douglas, F. Horkay, L. J. Fetters and S. K. Satija, Phys. B, 1996, 221, 331 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ce42646g |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence