 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of the drug methimazole and its iodine adduct with elemental zinc†
Francesco
Isaia
*a,
M. Carla
Aragoni
a,
Massimiliano
Arca
a,
Claudia
Caltagirone
a,
Alessandra
Garau
a,
Peter G.
Jones
b,
Vito
Lippolis
a and
Riccardo
Montis
a
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Cittadella Universitaria, 09042 Monserrato (CA), Italy. E-mail: isaia@unica.it; Fax: +39 070 6754456; Tel: +39 070 6754496
bInstitut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany
First published on 17th February 2014
Abstract
The reactivity of zinc complexes with N,S-donor molecules may be of relevance to the study of Zn-metalloproteins and -metalloenzymes. In this context, the zinc complex [Zn(MeImSH)2I2] was synthesised by the reaction of zinc powder with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 iodine adduct of the drug methimazole [(MeImSH)·I2]. The molecular structure of the complex, elucidated by X-ray diffraction analysis, showed a tetrahedral zinc(II) centre coordinated by two neutral methimazole units (through the sulfur atoms) and two iodides. From the reaction of MeImSH and Zn powder, the complex [Zn(MeImSH)(MeImS)2] (MeImS = deprotonated form of methimazole) was separated and characterised. An analysis of the crystal packing of the neutral complexes [Zn(MeImSH)2X2] (X = I, Br and Cl) and the ionic complex [Zn(MeImSH)3I]I showed that in all of the complexes the sulfur atom, in addition to binding to the metal centre, contributes to the formation of 1-D chains built via C(4)–H⋯S and N–H⋯X interactions in the neutral complexes, and via C(4)–H⋯S and N–CH3⋯S interactions in the ionic complex [Zn(MeImSH)3I]I. The deprotonation/protonation of the coordinated methimazole units can modulate the coordination environment at the Zn core. From the reaction of complex [Zn(MeImSH)3I]I with a strong non-coordinating organic base, we have shown that, as a consequence of the NH deprotonation of methimazole S-coordinated to zinc(II), the ligand coordination mode changes from S-monodentate to N,S-bridging. Correspondingly, in the complex [Zn(MeImSH)(MeImS)2], the MeImS that displays the N,S-bridging mode at zinc can be N-protonated and thereby changes to the S-monodentate coordination.
:1 iodine adduct of the drug methimazole [(MeImSH)·I2]. The molecular structure of the complex, elucidated by X-ray diffraction analysis, showed a tetrahedral zinc(II) centre coordinated by two neutral methimazole units (through the sulfur atoms) and two iodides. From the reaction of MeImSH and Zn powder, the complex [Zn(MeImSH)(MeImS)2] (MeImS = deprotonated form of methimazole) was separated and characterised. An analysis of the crystal packing of the neutral complexes [Zn(MeImSH)2X2] (X = I, Br and Cl) and the ionic complex [Zn(MeImSH)3I]I showed that in all of the complexes the sulfur atom, in addition to binding to the metal centre, contributes to the formation of 1-D chains built via C(4)–H⋯S and N–H⋯X interactions in the neutral complexes, and via C(4)–H⋯S and N–CH3⋯S interactions in the ionic complex [Zn(MeImSH)3I]I. The deprotonation/protonation of the coordinated methimazole units can modulate the coordination environment at the Zn core. From the reaction of complex [Zn(MeImSH)3I]I with a strong non-coordinating organic base, we have shown that, as a consequence of the NH deprotonation of methimazole S-coordinated to zinc(II), the ligand coordination mode changes from S-monodentate to N,S-bridging. Correspondingly, in the complex [Zn(MeImSH)(MeImS)2], the MeImS that displays the N,S-bridging mode at zinc can be N-protonated and thereby changes to the S-monodentate coordination.
Introduction
Iodine adducts of S-donor containing ligands (DS·I2) have recently been employed as oxidizing agents towards elemental metals (M0),1a whereby the one-step reaction (1) proceeds in non-aqueous solvents under mild reaction conditions. This reaction is currently being exploited for the recovery of precious and/or toxic metals from industrial waste.2 Another interesting aspect of this oxidation concerns the possibility of synthesizing transition- and main-group metal complexes with DS ligands, as these complexes are often not obtainable by conventional synthetic procedures. A great number of I2 adducts of thioamide-containing molecules have been synthesized to date.1b Though all of them can potentially be considered good candidates as oxidising reagents, only a few of them were successful in the oxidative dissolution of metals as summarised in Table 1. | (1) |
| Adducta | Metal | Solvent | Main product/s | Ref. |
|---|---|---|---|---|
| a MeImSH = methimazole, 1-methyl-3H-imidazole-2-thione, PTU = propylthiouracil, 6-propyl-2-sulfanylpyrimidin-4-one, mbtt = 3-methyl-benzothiazole-2-thione, mbit = 1-methyl-1H-benzimidazole-2(3H)-thione, bmitm = 1,1′-bis(3-methyl-4-imidazoline-2-thione)methane, Me2dazdt = N,N′-dimethylperhydro-1,4-diazepine-2,3-dithione. b mtbiH = 2-methylthiobenzimidazolium cation. | ||||
| MeImSH·I2 | Hg | CH2Cl2 | [Hg2I4(MeImSH)2] | 2a |
| PTU·I2 | Hg | CH2Cl2 | [Hg(PTU)2I2·HgI2] | 2a |
| mbtt·I2 | Au | Et2O | [Au(mbtt)2]I3 | 2b |
| mbit·I2 | Au | Et2O | (mtbiH)2[AuI4]I3b | 2b |
| bmitm·2I2 | Sn | Et2O | [Sn(bmitm)2I2](I3)2∙2/3I2 | 2c |
| Me2dazdt·2I2 | Au | THF | [Au(Me2dazdt)I2]I3 | 2d |
| Me2dazdt·2I2 | Hg | THF | [Hg(Me2dazdt)I2] | 2e |
| Me2dazdt·2I2 | Pd | THF | [Pd(Me2dazdt)2](I3)2 | 2f |
The dual role played by the S-donors (DS) in the reaction (1) has recently been recognised.2a,b In addition to polarising the bound I2 molecule, DS acts as a good coordinating agent towards the oxidised metal ion. In previous studies, we have identified some factors that influence the course of the reaction (1). First, the DS·I2 adduct should be inherently stable to avoid the oxidation of the donor and the formation of (poly)iodides, or iodine(I) derivatives (Scheme 1), since under these circumstances, the oxidation/complexation of metals is not observed. Moreover, if the adduct DS·I2 tends to dissociate, either the equilibrium species DS or I2 can react with the oxidized/complexed metal, with the result that the outcome of reaction (1) becomes very unpredictable. The second important factor concerns the nature of the metal. Even if the oxidation of metals whose ions are “hard” often occurs under the experimental conditions outlined in reaction (1), the corresponding complexes with the ligand DS are difficult to isolate since the “soft” S-donor atom is intrinsically a poor coordinating agent towards these ions, and the formation of non-crystallizable oils is commonly observed. Thirdly, both the reaction conditions (solvent and temperature) and the DS·I2 to M0 molar ratio influence the outcome of the reaction and need to be optimised in order to favour the formation of solid compounds.
 | ||
| Scheme 1 Top: in non-polar solvents, compounds containing S-donor Lewis base (DS) generally form only 1:1 charge–transfer complexes with I2. The formation constant of the 1:1 complex (Kf) depends on the equilibrium molar concentrations according to the equation Kf = [DS·I2]/[DS][I2].3 Bottom: the interaction between DS and I2 to give 1:1 adducts containing an almost linear S–I–I fragment can be seen as a charge–transfer process n → σ* (i.e. MO combination of a lone pair from the donor with the empty σ* orbital of I2). Depending on the charge density transferred by the DS donor molecule to I2 and the nature of the solvent, this can result in lengthening of the I–I bond and finally the breaking of this bond and formation of new species.1b | ||
Among the thioamide derivatives, the antithyroid drug methimazole4b (1-methyl-3H-imidazole-2-thione) (MeImSH), as shown in Fig. 1, is of interest because it forms the very stable adduct (MeImSH)·I2 both in solution and in the solid state.4b,c The large formation constant (Kf of 92433 M−1 in CH2Cl2 at 25 °C)4b ensures that the (MeImSH)·I2 adduct is the only “active” oxidant species in reaction (1). This adduct proved to be capable of dissolving and complexing liquid mercury in CH2Cl2, leading to good yield of the neutral complex [Hg2I4(MeImSH)2].2a In light of these good results, we have turned our attention to the lightest element of group 12. Zinc is one of the essential chemical elements for living organisms, and the presence of this element in a very low concentration5a is fundamental in catalytic, structural, and regulatory living processes.5b Since the discovery in 1939 that the enzyme carbonic anhydrase contains stoichiometric amounts of zinc,5c more than 3000 zinc proteins have been identified.5d,e With so many proteins requiring zinc, the concentration range in which Zn(II) ions are maintained at specific cellular concentrations to exert cellular regulation, transport, and homeostasis is the result of many processes, most of which have yet to be clarified.5f For these reasons, the exposure to chemicals, or the therapeutic consumption of metal chelating agents that interfere with zinc buffering can either result in zinc deficiency, and/or in the potential reduction of drugs efficacy.6 Methimazole, which is widely used for the long-term treatment of hyperthyroidism and Graves' disease, can potentially interact/interfere with the zinc-buffering system and Zn-metalloenzyme activities.4b,6a Experimental evidence on this point has been reported by Ferrer et al.6b who determined for methimazole a moderate inhibitor effect on alkaline phosphatase activity (IC50 = 70 μM).
 | ||
| Fig. 1 Chemical structures of methimazole (MeImSH) and its anion (MeImS). | ||
Investigation of the coordination chemistry of zinc with methimazole can therefore provide useful indications of the coordination number, the geometry at the metal centre, and the elements involved, since methimazole can behave either as a neutral or an anionic ligand7 (Fig. 1) and can coordinate through the sulfur and/or the nitrogen atoms. Moreover, the characterization of zinc complexes with N,S-donor molecules provides information for modelling the binding between the zinc metal ion and N- and S- donor sites and thus predicting the structure of Zn-metalloproteins and -metalloenzymes. We report here the reactivity of adduct (MeImSH)·I2 in dichloromethane with zinc powder and the characterisation, also by means of X-ray diffraction, of the isolated neutral zinc(II) complex [Zn(MeImSH)2I2]. The S-coordination of MeImSH to the ZnX2 moiety and also the crystal packing in complexes [Zn(MeImSH)2X2] [X = I, Br, and Cl] and [Zn(MeImSH)3I]I have been compared to identify the nature of the intermolecular interactions. The reactivity of MeImSH towards zinc powder and the characterisation of the resulting complex [Zn(MeImSH)(MeImS)2] are also reported. Moreover, the coordinating mode of methimazole in zinc complexes with respect to its neutral (thione) or anionic (thionate) form is discussed.
Results and discussion
Reactivity of the adduct (MeImSH)·I2 with Zn powder
The reaction between the adduct (MeImSH)·I2 and elemental zinc powder (CH2Cl2, 25 °C) in 1:1 and 2:1 molar ratios was carried out for two days. During this time the zinc dissolves completely and the dark red colour of the mixture due to the CT-complex (MeImSH)·I2 turned to pale red; no separation of solid products was observed. Evaporation of the solution in air resulted in dark red sticky oily products that we failed to crystallize. To overcome this point, the same reaction was also carried out, but increasing the amount of MeImSH to obtain a molar ratio MeImSH:(MeImSH)·I2:Zn of 1:1:1; under these reaction conditions the solution gradually turned from dark red to pale yellow within 48 h, thereafter affording the pale yellow crystalline zinc(II) complex of stoichiometry [Zn(MeImSH)2I2] by slow evaporation in air. It is noteworthy that Fujisawa et al.8 were not able to synthesise this complex by reacting ZnI2 with MeImSH in MeOH, the unexpected ionic complex of stoichiometry [Zn(MeImSH)3I]I was instead obtained. We also tested whether the complex [Zn(MeImSH)2I2] could be obtained from the reaction of ZnI2 with MeImSH (1:2 molar ratio) in CH2Cl2, but we again obtained the complex [Zn(MeImSH)3I]I.
The experimental data show that the adduct (MeImSH)·I2 in CH2Cl2 can successfully oxidize zinc powder to Zn(II) by a two-electron transfer process according to the overall reaction (2).
| (MeImSH)·I2 + MeImSH + Zn → Zn(MeImSH)2I2 | (2) |
This reaction represents a forward step with respect to reaction (1), as it has allowed us to improve the oxidative-dissolution process avoiding the formation of oily products arising from an incorrect molar ratio of the reagents. In previous studies, we recognised that the oxidising and complexing properties of DS·I2 adducts towards zero-valent metals could be interpreted considering the charge separation along the S–Ib–It moiety induced by the S → I2 interaction to form the reactive polarized [DS–I]δ+⋯Iδ− system. In the case of the adduct (MeImSH)·I2, the calculated NBO charge distribution2a showed a sulfur atom which is slightly positive (0.078 e) and a permanent charge separation between the iodine atoms of 0.348 e (NBO charges: −0.367 and −0.19 e for It and Ib, respectively), with a consequent increase of the reactivity with respect to that of molecular iodine.9 As similarly proposed for the oxidation of liquid mercury to yield the neutral mercury(II) complex [Hg2(MeImSH)2I4],2a it is reasonable to hypothesise that the oxidation/complexation of zinc proceeds via a mechanism of oxidative addition10 with the (MeImSH-I)δ+ moiety acting as an electrophilic agent. Based on the stoichiometry of the isolated complex [Zn(MeImSH)2I2], a second unit of MeImSH binding to the zinc(II) center provides a stable tetrahedral coordination geometry.
Crystal structure of the complex [Zn(MeImSH)2I2]
The complex [Zn(MeImSH)2I2] crystallises in a monoclinic crystal system (space group P21/n). A summary of the crystal data is reported in Table 2.| Compound | [Zn(MeImSH)2I2] |
|---|---|
| Formula | C8H12I2N4S2Zn |
| M. W. | 547.51 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 10.7697(11) |
| b/Å | 10.1580(8) |
| c/Å | 29.054(3) |
| α/° | 90.00 |
| β/° | 91.111(4) |
| γ/° | 90.00 |
| V/Å3 | 3177.8(5) |
| T/K | 133(2) |
| Crystal shape | Tablet |
| Colour | Pale yellow |
| Z | 8 |
| All reflns | 64301 |
| Un. reflns | 9699 |
| R int | 0.0275 |
| R 1,obs [I > 2σ(I)] | 0.0181 |
| R 1,all | 0.0234 |
| wR2 (obs) | 0.0395 |
| wR2 (all) | 0.0412 |
The asymmetric unit of [Zn(MeImSH)2I2] (Fig. 2) consists of two independent molecules (1 and 2), each featuring a Zn2+ cation adopting a slightly distorted tetrahedral geometry with two MeImSH units, coordinating via sulfur atoms S(1) and S(2) or S(3) and S(4), respectively, and by two I− anions, coordinating via iodides I(1) and I(2) or I(3) and I(4), respectively. Selected bond distances and angles for units 1 and 2 are presented in Table 3. The molecules differ slightly in ring orientations; a least-squares fit of all non-H atoms gave an r.m.s. deviation of 0.18 Å. For the sake of completeness, we have also included the data concerning the structures previously published [Zn(MeImSH)2Br2],8 [Zn(MeImSH)2Cl2],8 and [Zn(MeImSH)3I]I,8 CSD-REFCOD: GARPIZ, GARPEV, and GARPOF, respectively.
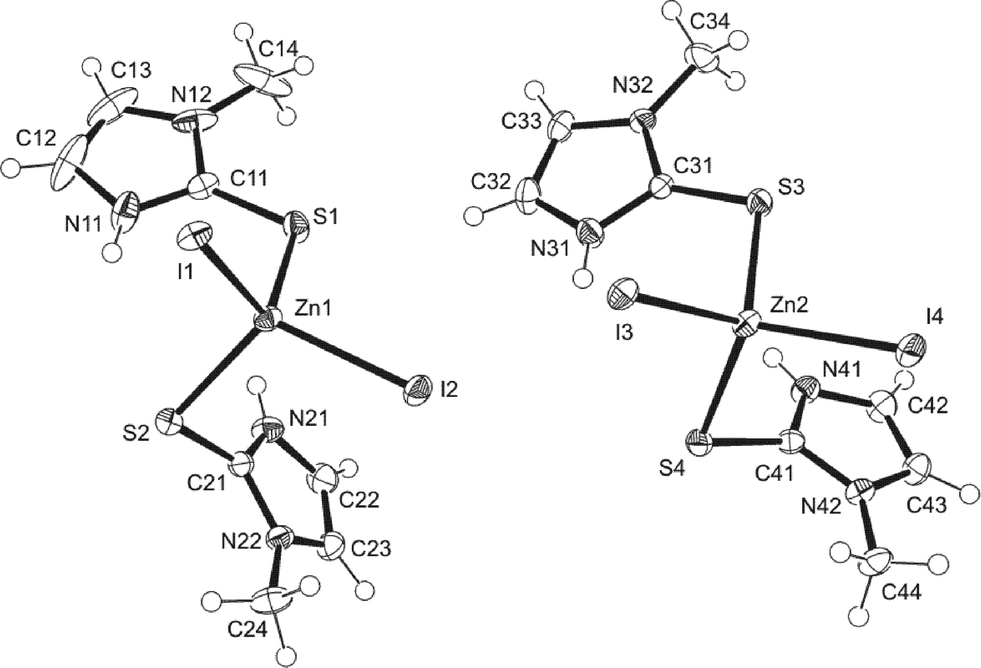 | ||
| Fig. 2 The two independent molecules 1 (left) and 2 (right) that constitute the asymmetric unit of complex [Zn(MeImSH)2I2]. | ||
| 1 | 2 | [Zn(MeImSH)2Br2] | [Zn(MeImSH)2Cl2] | [Zn(MeImSH)3I]I | |
|---|---|---|---|---|---|
| Zn–S1 | 2.3392(5) | 2.3562(5) | 2.338(2) | 2.336(2) | 2.376(3) |
| Zn–S2 | 2.3703(5) | 2.3669(5) | 2.342(2) | 2.345(1) | 2.375(3) |
| Zn–X1 | 2.6011(3) | 2.5807(3) | 2.390(1) | 2.252(1) | 2.574(2) |
| Zn–X2 | 2.5864(3) | 2.5824(3) | 2.390(1) | 2.259(1) | — |
| C1–S1 | 1.706(2) | 1.707(2) | 1.702(9) | 1.709(4) | 1.71(1) |
| C1–S2 | 1.721(2) | 1.722(2) | 1.720(7) | 1.714(5) | 1.71(1) |
| X1–Zn–X2 | 117.89(1) | 116.58(1) | 109.57(4) | 110.07(5) | — |
| X1–Zn–S1 | 101.48(1) | 103.87(1) | 118.67(6) | 102.54(5) | 112.98(8) |
| X2–Zn–S1 | 111.57(2) | 110.17(2) | 106.93(6) | 115.39(5) | — |
| X1–Zn–S2 | 109.45(1) | 110.26(1) | 101.28(6) | 118.19(5) | 112.74(9) |
| X2–Zn–S2 | 107.51(2) | 107.04(2) | 115.77(6) | 107.00(5) | — |
| S1–Zn–S2 | 108.60(2) | 108.74(2) | 104.91(7) | 103.83(5) | 105.6(1) |
In particular, we are interested in the comparison of molecules 1–2 with the two isostructural complexes [Zn(MeImSH)2Br2] and [Zn(MeImSH)2Cl2], which only differ in the coordinating halides. However, since it is relevant for the scope of this paper, we also comment on the complex [Zn(MeImSH)3I]I, which features a different structure with three S-monodentate MeImSH units and one iodide tetrahedrally coordinated to the zinc(II), and a second iodide ion acting as the counter-ion. The comparison of the geometrical parameters shows small differences for the [Zn(MeImSH)2X2] complexes (X = I, Br, and Cl). In particular, units 1 and 2 show differences mainly involving the Zn–X distances. These increase along the series Cl–Br–I. The bond angles are similar and all of the structures adopt a slightly distorted tetrahedral geometry. The C–S bond distances are very similar for all of the structures, with values lying in the range 1.70–1.73 Å. However, the analysis of the conformations reveals differences concerning the orientations adopted by the two MeImSH moieties. These are very similar for the Cl and Br complexes, in which the conformation is locked via N–H⋯X intra-molecular interactions (N–H⋯X distances are 2.47 Å for [Zn(MeImSH)2Cl2] and 2.60 Å for [Zn(MeImSH)2Br2]), but differ for the two symmetrically independent units of 1 and 2, in which no intramolecular interactions are observed. This is represented in Fig. 3 in which the units 1 and 2, and the tetrahedral unit of [Zn(MeImSH)2Cl2] (reported as representative of the isostructural set) are shown.
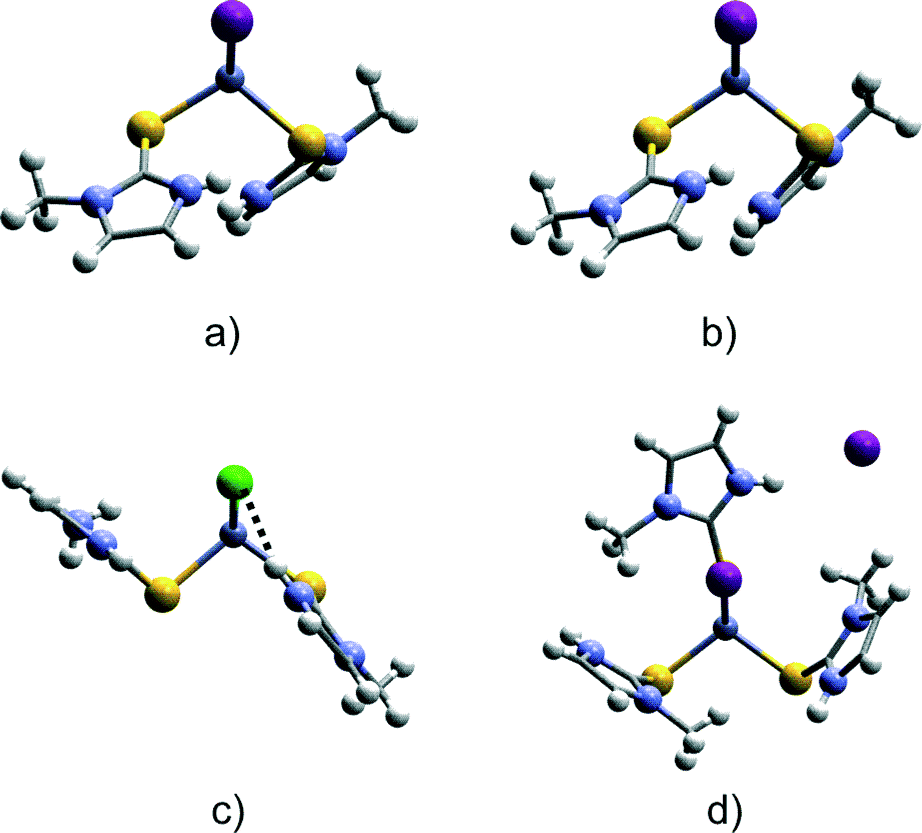 | ||
| Fig. 3 Conformational comparison of a) independent molecule 1, b) independent molecule 2, c) complex [Zn(MeImSH)2Cl2], and d) complex [Zn(MeImSH)3I]I. The molecules are viewed along the direction parallel to the plane X1–Zn–X2 for a), b), and c) and viewed along the direction parallel to the plane X1–Zn–S3 for d). This results in the superimposition of the two halogens for a), b), and c) and of I and S atoms in d). | ||
[Zn(MeImSH)2Cl2] and [Zn(MeImSH)2Br2] are compared. In Fig. 3, the tetrahedral unit of [Zn(MeImSH)3I]I is also reported for completeness.
Packing analysis
The units 1 and 2 are alternately connected to each other via weak C–H⋯S interactions (C–H⋯S distances are 2.86 and 2.73 Å) assisted by N–H⋯I contacts [N–H⋯I distances are 3.20(3) and 3.21(2) Å] to form 1-D chains (Fig. 4a). A further set of C–H⋯S interactions (depicted as red dashed lines in Fig. 4b; C–H⋯S distances are 3.14 and 3.01 Å) connects adjacent 1-D chains to form a more complex arrangement, labelled as A. | ||
| Fig. 4 Representation of the main molecular arrangements identified for the structure of the complex [Zn(MeImSH)2I2]. a) Single 1-D chain, built by intermolecular interactions involving the two independent molecules 1 and 2; b) molecular arrangement A. The molecular arrangements are oriented to better show the intermolecular interactions. Bond distances are in Å. | ||
The resulting crystal packing could be easily described as an assembly of 1-D chains of type A along the remaining two dimensions. In particular, adjacent 1-D molecular arrangements A are assembled along the [001] direction via C43–H⋯S interactions (C43–H⋯S distance is 3.09 Å) assisted by a set of weaker C(Me)–H⋯I interactions (C–H⋯I distances are 3.30, 3.32, and 3.29 Å) and related by n-glide planes parallel to the (101) plane which generates a change in their direction of propagation from [110] to [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] (respectively, orange and blue in Fig. 5a and b). The chains propagate along the [010] direction under the effect of 21 screw axes, resulting in a herringbone motif (Fig. 5c).
0] (respectively, orange and blue in Fig. 5a and b). The chains propagate along the [010] direction under the effect of 21 screw axes, resulting in a herringbone motif (Fig. 5c).
 | ||
| Fig. 5 Representation of the crystal packing of the complex [Zn(MeImSH)2I2]. a) Crystal packing viewed along the [010] direction (21 screw axes are indicated in black). Intermolecular interactions connecting adjacent chains are indicated by black dashed lines. Red colour is used for interactions within the molecular arrangements A; b) development of the 1-D chains A along the [001] direction under the effect of n-glide symmetry; c) crystal packing viewed along the [001] direction. The molecules are colour coded depending on the direction of propagation of the 1D chains: orange as the [110] direction, blue as the [10] direction. The differences in the orientation of the chains are also indicated by using different labels (A and A*) and coloured arrows. | ||
In order to keep the comparative approach undertaken at the molecular level in the previous section, we have also extended the crystal packing analysis to the three crystal structures [Zn(MeImSH)2Cl2], [Zn(MeImSH)2Br2], and [Zn(MeImSH)3I]I. The comparison of this set of closely related compounds is intended to assess the importance of various factors, such as intermolecular interaction, shape and conformation, in the resulting crystal packing. In particular, we adopt a retro-synthetic approach, aiming to identify recurring structural motifs (e.g. specific intermolecular interactions and/or specific molecular arrangements) that might have a role in determining crystal packing similarities and/or differences.11 Selected intermolecular and intramolecular distances are summarized in Table 4.
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) | |
|---|---|---|---|---|---|
| [Zn(MeImSH)2I2] | |||||
| — | N(41)–H(04)⋯I(1) | 0.81(2) | 2.83(2) | 3.583(2) | 157(2) |
| Fig. 4a, 6 | N(31)–H(03)⋯I(2) | 0.81(2) | 3.21(2) | 3.827(2) | 135(2) |
| — | N(21)–H(02)⋯I(3) | 0.81(2) | 2.93(2) | 3.673(2) | 153(3) |
| Fig. 4a, 6 | N(11)–H(01)⋯I(4) | 0.80(2) | 3.20(3) | 3.779(2) | 132(3) |
| — | N(11)–H(01)⋯S(2) | 0.80(2) | 2.93(3) | 3.604(2) | 143(3) |
| — | N(31)–H(03)⋯S(4) | 0.81(2) | 3.16(2) | 3.781(2) | 135(2) |
| Fig. 4a, 6, 5a | C(12)–H(12)⋯S(3) | 0.95 | 2.73 | 3.676(3) | 172 |
| Fig. 4a, 6, 5a | C(32)–H(32)⋯S(1) | 0.95 | 2.86 | 3.767(2) | 159 |
| Fig. 5a | C(43)–H(43)⋯S(4) | 0.95 | 3.09 | 3.76 | 129 |
| Fig. 5a | C(24)–H(24B)⋯I(1) | 0.98 | 3.29 | 4.25 | 165 |
| Fig. 5a | C(44)–H(44B)⋯I(2) | 0.98 | 3.32 | 3.81 | 112 |
| Fig. 5a | C(44)–H(44C)⋯I(3) | 0.98 | 3.30 | 4.26 | 167 |
| [Zn(MeImSH)2Br2] | |||||
| Fig. 6b | N(11)–H(11A)⋯Br(1) | 0.95 | 2.69 | 3.616(7) | 164 |
| Fig. 3c (showed for Cl) | N(21)–H(21A)⋯Br(2) | 0.95 | 2.60 | 3.485(6) | 155 |
| — | C(23)–H(23A)⋯Br(2) | 0.95 | 2.84 | 3.559(8) | 133 |
| — | C(24)–H(24C)⋯S(2) | 0.95 | 2.68 | 3.169(9) | 113 |
| Fig. 6b | C(12)–H(12A)⋯S(2) | 0.95 | 3.12 | 3.71 | 122 |
| [Zn(MeImSH)2Cl2] | |||||
| Fig. 6c | N(21)–H(21A)⋯Cl(1) | 0.95 | 2.55 | 3.484(5) | 167 |
| — | C(13)–H(13A)⋯Cl(2) | 0.95 | 2.73 | 3.498(6) | 139 |
| Fig. 3c | N(11)–H(11A)⋯Cl(2) | 0.95 | 2.47 | 3.346(5) | 154 |
| Fig. 6c | C(22)–H(22A)⋯S(1) | 0.95 | 3.14 | 3.72 | 121 |
| [Zn(MeImSH)3I]I | |||||
| — | N(11)–H(11A)⋯I(2) | 0.95 | 2.75 | 3.631(8) | 155 |
| — | N(21)–H(21A)⋯I(2) | 0.97 | 2.76 | 3.65(4) | 154 |
| — | N(31)–H(31A)⋯I(2) | 0.95 | 2.65 | 3.55(3) | 159 |
| — | C(32)–H(32A)⋯S(2) | 0.95 | 2.86 | 3.78(4) | 175 |
| Fig. 6d | C(12)–H(12A)⋯S(1) | 0.95 | 2.82 | 3.74(1) | 163 |
| Fig. 6d | C(22)–H(22A)⋯S(3) | 0.95 | 2.83 | 3.71(1) | 155 |
| Fig. 6d | C(34)–H(34B)⋯S(3) | 0.95 | 3.20 | 3.87 | 130 |
| Fig. 6d | C(14)–H(14C)⋯S(2) | 0.95 | 3.17 | 3.85 | 130 |
With the exception of the isostructural neutral complexes, a preliminary comparison of the unit cell parameters shows major differences. This is not surprising that the different conformations (Fig. 3) and, most importantly, the different number of independent molecules in the asymmetric unit (Z′) are taken into account. However, the analysis reveals some analogies, mainly concerning the common tendency to form specific intermolecular interactions. Similar to the observations for the complex [Zn(MeImSH)2I2] (Fig. 4a), for all of the structures, it is possible to identify 1-D chains built via weak interactions of C(4)–H⋯S (Fig. 6a–d) involving C–H adjacent to the MeImSH N–H group (C–H⋯S distances lie in the range 2.8–3.2 Å). Furthermore, for the Br and Cl complexes (Fig. 6b and c), these are assisted by N–H⋯halogen contacts (N–H⋯Cl and N–H⋯Br distances are 2.55 Å and 2.69 Å), in a similar manner as observed for complex [Zn(MeImSH)2I2]. In the case of the ionic complex [Zn(MeImSH)3I]I, the N–H⋯halogen interaction is replaced by a set of C(Me)–H⋯S interactions (C(Me)–H⋯S distances are 3.17 and 3.20 Å). The result is a set of 1D chains developing along the [010] direction for the isostructural set [Zn(MeImSH)2Cl2] and [Zn(MeImSH)2Br2] and along the [110] direction for [Zn(MeImSH)3I]I.
 | ||
| Fig. 6 Representation of the main molecular arrangements identified for the structure of the complexes: a) [Zn(MeImSH)2I2], C–H⋯S and N–H⋯I; b) [Zn(MeImSH)2Br2], C–H⋯S and N–H⋯Br; c) [Zn(MeImSH)2Cl2], C–H⋯S and N–H⋯Cl; d) [Zn(MeImSH)3I]+, C(Me)–H⋯S and C(4)–H⋯S. The interactions are depicted in blue, bond distances are in Å. | ||
These chains only differ from a geometrical point of view, arising from the different shape of the tetrahedral units. In fact, the different orientations of the MeImSH moieties in the three types of structures (Fig. 3) expose the NH and CH hydrogen bond donors along different directions generating differences in the resulting patterns. This is represented in Fig. 6, in which C–H⋯S and N–H⋯halogen distances are also included.
Again, as already seen for the complex [Zn(MeImSH)2I2] (Fig. 5), the crystal packing of the structures [Zn(MeImSH)2Cl2], [Zn(MeImSH)2Br2], and [Zn(MeImSH)3I]I can be rationalised starting from these 1-D chains. In the isostructural complexes [Zn(MeImSH)2Cl2] and [Zn(MeImSH)2Br2], the 1-D chains are assembled via 21 screw axes (Fig. 7) along the [100] direction via weak C(Me)–H⋯S (C–H⋯S distances are 2.94 Å for the Cl derivative and 3.20 Å for the Br derivative), and C(4)–H⋯Hal interactions (intermolecular distances 2.73 Å for the Cl derivative and 2.94 Å for the Br derivative). The chains develop along the [001] direction by simple translation, with no intermolecular interactions occurring between adjacent chains. A similar behaviour is observed in [Zn(MeImSH)3I]I, where the different instances of the 1-D chains develop along the [001] direction via 21 screw axes. Also in this case (Fig. 8), the adjacent chains are connected to each other via C(Me)–H⋯S interactions (C–H⋯S intermolecular distance is 3.06 Å).
 | ||
| Fig. 7 Representation of the crystal packing of the isostructural set [Zn(MeImSH)2Cl2] and [Zn(MeImSH)2Br2] showed for the Cl derivative as representative. a) 1-D chain B viewed along the 001 direction (left) and 1-D chain B viewed along the [010] direction (right); b) development of the crystal packing along the [100] direction. The 1-D chains B are assembled under the effect of 21 screw axis (red arrows) and viewed along the [010] direction; c) 3-D packing viewed along the [010] direction. Instances of the 1-D chains B are colour coded according to the different orientations of propagation. The differences in the orientation of the chains are also indicated by using different labels (B and B*). | ||
 | ||
| Fig. 8 Representation of the crystal packing of the complex [Zn(MeImSH)3I]I. a) 1-D chain C viewed along the [010] direction (left) and 1-D chain C viewed along the [0] direction (right); b) development of the crystal packing along the [001] direction and the [10] direction. The 1-D chains C are assembled via the 21 screw axis (red arrows) operating along the [001] direction and by simple translation along the [10] direction. The portion of crystal packing is viewed along the [0] direction; c) 3-D packing viewed along the [0] direction. Instances of the 1-D chains C are colour coded according to the different orientations of propagation. The differences in the orientation of the chains are also indicated by using different labels (C and C*). | ||
As described above, the structural behaviour of closely related compounds has certainly highlighted major differences in their structures, but has also identified some similarities. In particular, the common tendency to interact via C–H⋯S and N–H⋯X (X = Cl, Br and I) interactions results in slightly different molecular arrangements. This fact indicates the important role of specific directional intermolecular interactions in directing packing modes.
The structural analysis clearly shows some features that deserve to be commented. First of all, as previously mentioned, the [Zn(MeImSH)2I2] complex crystallises in the monoclinic crystal system (space group P21/c, Z′ = 2) while the isostructures [Zn(MeImSH)2Cl2] and [Zn(MeImSH)2Br2] are orthorhombic (space group P212121, Z′ = 1). Such differences are not uncommon and could arise from the different conditions of crystallization.8 It is known that the nature of the solvent can play an important role during the nucleation processes and, consequently, in the resulting crystal structure. Depending on the reaction conditions, the same compound can crystallise with different crystal packing modes (as polymorphs or different solvates). In this specific case, the most relevant departure consists of a different number of independent molecules in the asymmetric unit (Z′ = 2 for [Zn(MeImSH)2I2] and Z′ = 1 for the isostructural set [Zn(MeImSH)2Cl2] and [Zn(MeImSH)2Br2]). Crystal structures with Z′ > 1 are quite common; among others, Anderson et al.12 point out that, although many factors may contribute to the crystallization of molecule with Z′ > 1, there is no general rule to explain this phenomenon. In this case, the absence of any specific intramolecular interaction in the [Zn(MeImSH)2I2] complex, which generally might favour some specific conformations,13 could determine a certain degree of conformational flexibility in solution. Desiraju14 defined a crystal with Z′ > 1 as a “kinetic form which has been trapped before the molecules have adjusted themselves in their final orientations”. Certainly, the condition of crystallization can have an important role in this context.
Reactivity of MeImSH with Zn powder
The reaction in CH2Cl2 between MeImSH and Zn powder (4:1 molar ratio) leads to the complete dissolution/oxidation of Zn in about 7 days. In the course of the reaction, a whitish solid powder precipitated. The electrospray ionization mass spectrum (ESI-MS), recorded in the positive ion mode, showed for this compound the highest peak at m/z = 405 (100%) with an isotopic pattern consistent with the zinc-containing species Zn(MeImSH)(MeImS)2 + H+ (see the ESI†). Unfortunately, because of its low solubility in common organic solvents and in water, it was not possible to carry out any study in solution nor to isolate suitable crystals for X-ray determination; in these circumstances MAS 13C-NMR spectroscopy proved to be a useful tool. In Table 5, the 13C NMR data of MeImSH, complexes [Zn(MeImSH)2I2] and [Zn(MeImSH)(MeImS)2] are listed. As a consequence of S-coordination to the zinc(II) centre, effects on chemical shifts were found for all carbon nuclei. In complex [Zn(MeImSH)2I2], the thioamido carbon proves to be most sensitive to complexation as confirmed by the markedly upfield shift (≈10 ppm); this is associated with the increased double bond character at the C(S)–N linkage. Conversely, carbons C(4), and C(5) (see Fig. 1 for atom numbering) are slightly deshielded compared to free methimazole. The MAS spectrum of complex [Zn(MeImSH)(MeImS)2] shows interesting features since both the resonances related to methimazole in its neutral and anionic forms are observed. The resonance of thioamido carbon in MeImS is further shifted upfield with respect to that of the MeImSH moiety, since NH deprotonation causes an increase in the π-electron density delocalisation involving the carbon and the nitrogen atoms of the thioamide group. The experimental data suggest that oxidation/complexation of elemental zinc by MeImSH proceeds in accordance with the overall process:| 3 MeImSH + Zn → Zn(MeImSH)(MeImS)2 + H2 | (3) |
| Compound | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S S |
C5 | C4 | N–CH3 | Solvent |
|---|---|---|---|---|---|
| MeImSH | 163.3 | 120.0 | 114.2 | 34.0 | CDCl3–CH3CN (4:1 (v:v)) |
| [Zn(MeImSH)2I2] | 152.5 | 120.6 | 115.4 | 34.1 | CDCl3–CH3CN (4:1 (v:v)) |
| [Zn(MeImSH)(MeImS)2] | 150.8 | 122.6 | 117.9 | 32.5 | Solid state |
| 146.7 | 120.3 | 113.6 | 30.5 |
The reaction (3) is in accordance with the results reported by Sousa et al.15 about the electrochemical oxidation of a zinc anode in a solution of MeImSH in acetonitrile. Moreover, Sousa reported on the crystal structure of complex [Zn4O(MeImS)6] that features MeImS acting as a (N,S)-bridging ligand that binds pairs of non-interacting zinc atoms.15
Reactivity of complex [Zn(MeImSH)(MeImS)2] with HI
Complex [Zn(MeImSH)(MeImS)2] shows a low solubility in the most common organic solvents and in water; however, it is readily solubilized in hydroiodic acid (HI) solutions. The reaction of [Zn(MeImSH)(MeImS)2], suspended in a mixture of CH3CN–CH2Cl2 with a ratio of 1:1 (v/v), and HI (57 wt.% in water) (molar ratio of the Zn complex to HI is 1:2) readily causes the dissolution of the suspended powder with formation of a clear solution. By slow evaporation of the solvents, a pale yellow powder was obtained; the compound was identified by elemental analysis and IR spectroscopy as the complex [Zn(MeImSH)3I]I isolated by Fujisawa8 (Scheme 2). We also verified that the protonation process was reversible; indeed, the addition to [Zn(MeImSH)3I]I of three equivalents of the strong organic base 1,8-bis(dimethylamino)naphthalene (DMAN) in a mixture of CH3CN–CH2Cl2 with a ratio of 1:1 (v/v), resulted in the formation of a whitish powder identified as the expected complex [Zn(MeImSH)(MeImS)2] (Scheme 2). The deprotonation/protonation of the coordinated methimazole units can modulate the coordination environment at the Zn-core; as a consequence of methimazole NH-deprotonation, the binding mode changes from S-monodentate to an N,S-bridging mode.
 | ||
| Scheme 2 Deprotonation/protonation assisted conversion of Zn-coordinated methimazole from thionato to thione forms. | ||
Conclusions
The oxidative dissolution of zinc powder by either the I2 adduct of antithyroid drug methimazole or methimazole alone is easily accomplished in CH2Cl2, leading to novel stable complexes of zinc(II), [Zn(MeImSH)2I2] and [Zn(MeImSH)(MeImS)2], respectively. The X-ray crystal structure of the former complex shows two independent zinc(II) ions, each coordinated by two neutral ligands (via the S-atoms) and two iodides in a distorted tetrahedral coordination geometry. The crystal packing highlights the formation of 1-D chains built via weak interactions of C(4)–H⋯S and N–H⋯halogen. These interactions also lead to the formation of 1-D chains in complexes [Zn(MeImSH)2X2] (X = Br, and Cl), whereas in the ionic complex [Zn(MeImSH)3I]I, the 1-D chain formation is based on weak interactions of C(4)–H⋯S and N–CH3⋯S. The results reported in this study confirm the propensity of the drug methimazole to form both a strong Zn–S bond, and to activate intermolecular interactions in relation to the other ligands completing the zinc(II) coordination sphere. The average Zn–S bond distance [2.3492(5) Å] in complexes [Zn(MeImSH)2X2] (X = I, Br, and Cl) is reasonably similar to the average Zn–cysteine bond lengths [2.32 ± 0.16 Å] found in zinc proteins determined by NMR,5f this fact supports the hypothesis that methimazole could block one or more cysteine/histidine binding sites and thereby affect the multifunctional role of the zinc in the proteins.The characterization of complex [Zn(MeImSH)(MeImS)2] shows neutral (thione) and anionic (thionate) methimazole units coordinating to the metal centre. Based on the reactivities of complexes [Zn(MeImSH)(MeImS)2] and [Zn(MeImSH)3I]I, we have shown that the NH deprotonation of methimazole S-coordinated to the Zn(II) ion modifies the coordinating characteristics of this ligand from S-monodentate to N,S-bridging. In the latter case, the anionic methimazole could bind two Zn(II) centres in close proximity, as reported in the complex [Zn4O(MeImS)6] where d(Zn–Zn) ≈ 3.2 Å.15 Within the scope of the study of the interaction of molecules of biological/pharmacological interest with zinc, the results underline that methimazole may in some ways be a dangerous metal-coordinating compound, connected with the formation of a stable zinc(II)–thioamide moiety and the formation of hydrogen bonds via NH, N–CH3, and C(4)H groups. Moreover, the Zn-coordinated methimazole can markedly modify the coordination environment when changing from its thione to thionate form, and vice versa.
Experimental
Materials and instrumentation
Reagents were used as purchased from Aldrich. Elemental analyses were carried out using a Fisons Instruments 1108 CHNS elemental analyser. FT-Infrared spectra of powdered samples were measured using a Thermo-Nicolet 5700 spectrometer from 4000–400 cm−1 in the form of pressed KBr pellets. 13C NMR spectra were recorded using a Varian 400 MHz spectrometer, chemical shifts are reported in ppm (δ) downfield from TMS using the same solvent as the internal reference; 13C NMR MAS spectra were calibrated such that the observed up field peak in the spectrum of adamantane is set to δ = 31.47. All of the low resolution mass spectra were recorded by electrospray ionisation using a Fisons VG platform instrument or a Waters ZMD quadrupole mass spectrometer in HPLC grade acetonitrile.Synthesis of complex [Zn(MeImSH)2I2]
A mixture of MeImSH·I2 (0.7874 g, 2.139 mmol), MeImSH (0.2442 g, 2.139 mmol), and zinc dust (<10 μm) (0.1398 g, 2.139 mmol) in CH2Cl2 (50 mL) was stirred at 25 °C for 2 days. The resultant clear, pale yellow solution was slowly concentrated to dryness, and the white solid powder was washed with a 1:1 (v:v) CH2Cl2–n-hexane mixture (2 × 5 mL) and then dried in vacuo. Yield: 0.6440 g, 55.0% referring to zinc. Pale yellow single crystals for X-ray analysis were obtained by dissolving the title compound in a 2:1 (v:v) mixture of CH2Cl2–n-hexane and cooling the solution at 5 °C for a few days. C8H12I2N4S2Zn (547.519): calcd. C 17.54, H 2.21, N 10.23, S 11.68; found: C 17.4, H 2.2, N 10.1, S 11.7. IR (KBr, ν cm−1): 3287br, 3163m, 3133m, 1682w, 1572s, 1468s, 1450s, 1404m, 1350w, 1280m, 1155m, 1086m, 1015w, 920w, 839w, 733s, 685m, 667s, 627s, 595m, 510m, 418w, 406w.
Synthesis of complex [Zn(MeImSH)(MeImS)2]
A mixture of MeImSH (1.403 g, 12.294 mmol) and zinc dust (<10 μm) (0.2610 g, 4.098 mmol) in CH2Cl2 (40 mL) was sonicated for 5 min, then stirred at 20 °C for seven days. The whitish solid that precipitated in the course of the reaction was collected by suction filtration, washed with water and dried in vacuo. Yield 1.0740 g, 64.8% referring to zinc. C12H16N6S3Zn (405.89): calcd. C 35.51, H 3.98, N 20.72, S 23.66; found: C 35.8, H 4.0, N 20.8, S 23.7. IR (KBr, ν cm−1): 3105m, 2940w, 1457vs, 1369vs, 1314vs, 1285vs, 1144vs, 14001m, 1368s, 1314s, 1245s, 1149s, 1084m, 1014w, 953w, 740s, 691s, 674s, 528w, 514w.
X-ray structure determination of complex [Zn(MeImSH)2I2]
A crystal ca. 0.25 × 0.25 × 0.13 mm was mounted in inert oil on a glass fibre and transferred to the cold gas stream of a Bruker SMART 1000 CCD diffractometer. Data were collected to 2θmax = 61° using monochromated Mo Kα radiation (λ = 0.71073 Å). An absorption based on multi-scans was performed. The structure was refined anisotropically on F2 using the program SHELXL-97.16 NH hydrogens were refined freely but with N–H distance restraints (SADI); methyls as idealized rigid groups were allowed to rotate but not tip; and other H atoms were refined using a riding model starting from calculated positions. Complete crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 976347.Acknowledgements
We would like to thank Regione Autonoma della Sardegna for financial support.Notes and references
- (a) V. Lippolis and F. Isaia, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, RSC Publishing, Cambridge, 2013, ch. 8.2, pp. 448–472 Search PubMed; (b) W. T. Pennington, T. W. Hanks and H. D. Arman, in Halogen Bonding with Dihalogens and Interhalogens: Fundamentals and Applications, ed. P. Metrangolo and G. Resnati, Spinger-Verlag, Berlin, 2008, pp. 65–104, and references therein Search PubMed.
- (a) F. Isaia, M. C. Aragoni, M. Arca, C. Caltagirone, C. Castellano, F. Demartin, A. Garau, V. Lippolis and A. Pintus, Dalton Trans., 2011, 40, 4505–4513 RSC; (b) F. Isaia, M. C. Aragoni, M. Arca, C. Caltagirone, F. Demartin, A. Garau and V. Lippolis, Dalton Trans., 2013, 42(2), 492–498 RSC; (c) F. Bigoli, P. Deplano, F. A. Devillanova, V. Lippolis, M. L. Mercuri, M. A. Pellinghelli and E. F. Trogu, Inorg. Chim. Acta, 1998, 267, 115–121 CrossRef CAS; (d) F. Bigoli, P. Deplano, M. L. Mercuri, M. A. Pellinghelli, G. Pintus, A. Serpe and E. F. Trogu, Chem. Commun., 1998, 2351–2352 RSC; (e) F. Bigoli, M. C. Cabras, P. Deplano, M. L. Mercuri, L. Marchiò, A. Serpe and E. F. Trogu, Eur. J. Inorg. Chem., 2004, 5, 960–963 CrossRef; (f) A. Serpe, F. Bigoli, M. C. Cabras, P. Fornasiero, M. Graziani, M. L. Mercuri, T. Montini, L. Pilia, E. F. Trogu and P. Deplano, Chem. Commun., 2005, 1040–1042 RSC.
-
K
f values can spread over a very wide range of values depending on the nature of DS and solvent, i.e. 1:1 adduct methimazole–I2, Kf = 92433 L mol−1, CH2Cl2, 25 °C, 1:1 adduct dimethyl sulfide–I2, Kf = 74.0 L mol−1, CCl4, 25 °C. M. C. Aragoni, M. Arca, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis and G. Verani, Coord. Chem. Rev., 1999, 184, 271–290 CrossRef CAS ; and references therein.
- (a) The drug methimazole, preventing iodine and thyroperoxidase enzyme from their normal interactions with thyroglobulin to form thyroid hormones thyroxine (T4) and triiodothyronine (T3), decreases thyroid hormone production. D. S. Cooper, N. Engl. J. Med., 2005, 352, 905–917 CrossRef CAS PubMed; (b) C. Laurence, M. J. El Ghomari, J.-Y. Le Questel and R. Mokhlisse, J. Chem. Soc., Perkin Trans. 2, 1998, 1545–1551 RSC , and references therein; (c) H. N. Po and M. C. Keindl, J. Am. Chem. Soc., 1988, 110, 2586–2591 CrossRef; (d) F. Isaia, M. C. Aragoni, M. Arca, F. Demartin, F. A. Devillanova, G. Floris, A. Garau, M. B. Hursthouse, V. Lippolis, R. Medda, F. Oppo, M. Pira and G. Verani, J. Med. Chem., 2008, 51, 4050–4053 CrossRef CAS PubMed.
- (a) Toxicological profile for Zinc, Agency for Toxic Substances and Disease Registry (ATSDR), U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA, 2005 Search PubMed; (b) D. S. Auld, BioMetals, 2001, 14, 271–273 CrossRef CAS PubMed; (c) S. Hughes and S. Saran, J. Am. Coll. Nutr., 2006, 25, 285–291 CrossRef CAS PubMed; (d) D. Keilin and T. Mann, Nature, 1939, 144, 442–443 CrossRef CAS; (e) W. Maret, Adv. Nutr., 2013, 4, 82–91 CrossRef CAS PubMed; (f) M. Laitaoja, J. Valjakka and J. Jänis, Inorg. Chem., 2013, 52, 10983–10991 CrossRef CAS PubMed.
- (a) A. A. Alturfan, E. Zengin, N. Dariyerli, E. E. Alturfan, M. K. Gumustas, E. Aytac, M. Aslan, N. Balkis, A. Aksu, G. Yigit, E. Uslu and E. Kokoglu, Folia Biol., 2007, 53, 183–188 CAS; (b) N. M. Urquiza, S. G. Manca, M. A. Moyano, R. A. Dellmans, L. Lezama, T. Rojo, L. G. Naso, P. A. M. Williams and E. G. Ferrer, BioMetals, 2010, 23, 255–264 CrossRef CAS PubMed.
- (a) E. S. Raper, Coord. Chem. Rev., 1997, 165, 475–567 CrossRef CAS; (b) A. Rodríguez, J. A. García-Vázquez, A. Sousa-Pedrares, J. Romero and A. Sousa, Inorg. Chem. Commun., 2003, 6, 619–622 CrossRef; (c) A. Rodríguez, A. Sousa-Pedrares, J. A. García-Vázquez, J. Romero and A. Sousa, Eur. J. Inorg. Chem., 2005, 2242–2254 CrossRef; (d) A. Rodríguez, A. Sousa-Pedrares, J. A. García-Vázquez, J. Romero and A. Sousa, Eur. J. Inorg. Chem., 2007, 1444–1456 CrossRef.
- Y. Matsunaga, K. Fujisawa, N. Amir, Y. Miyashita and K.-I. Okamoto, J. Coord. Chem., 2005, 58, 1047–1061 CrossRef CAS.
- No reaction is observed between zinc powder and iodine in CH2Cl2 at 25 °C.
- (a) R. H. Crabtree, in Oxidative Addition and Reductive Elimination The Organometallic Chemistry of the Transition Metals, Wiley-Interscience, Hoboken, USA, 4th edn, 2005, pp. 159–182 Search PubMed; (b) J. Halpern, Acc. Chem. Res., 1970, 3, 386–392 CrossRef CAS.
- (a) R. Montis and M. B. Hursthouse, CrystEngComm, 2012, 14(21), 7466–7478 RSC; (b) M. B. Hursthouse, R. Montis, L. Niitsoo, J. Sarson, T. L. Threlfall, A. M. Asiri, S. A. Khan, A. Y. Obaid and L. M. Al-Harbi, CrystEngComm, 2014, 16, 2205–2219 RSC.
- K. M. Anderson, S. E. Tallentire, M. R. Probert, A. S. E. Goeta, B. G. Mendis and J. W. Steed, Cryst. Growth Des., 2011, 11(3), 820–826 CAS.
- M. Olivari, C. Caltagirone, A. Garau, F. Isaia, M. E. Light, V. Lippolis, R. Montis and M. A. Scorciapino, New J. Chem., 2013, 37(3), 663–669 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46(44), 8342–8356 CrossRef CAS PubMed.
- R. Castro, J. A. Garcia-Vasquez, J. Romero, A. Sousa, Y. D. Chang and J. Zubieta, Inorg. Chim. Acta, 1995, 237, 143–146 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electrospray Ionization Mass Spectroscopy (ESI-MS) data. CCDC 976347. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42601g |
| This journal is © The Royal Society of Chemistry 2014 |