 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge transfer complexes and radical cation salts of chiral methylated organosulfur donors†
Songjie
Yang
a,
Flavia
Pop
b,
Caroline
Melan
b,
Andrew C.
Brooks
ac,
Lee
Martin
a,
Peter
Horton
d,
Pascale
Auban-Senzier
e,
Geert L. J. A.
Rikken
f,
Narcis
Avarvari
*b and
John D.
Wallis
*a
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK. E-mail: john.wallis@ntu.ac.uk; Tel: +44 (0)115 848 8053
bLaboratoire MOLTECH-Anjou, Université d'Angers, CNRS, UMR 6200, UFR Sciences, Bât. K, 2 Bd. Lavoisier, 49045 Angers, France. E-mail: narcis.avarvari@univ-angers.fr; Fax: +33 02 41 73 54 05; Tel: +33 02 41 73 50 84
cDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
dNational Crystallography Service, School of Chemistry, University of Southampton, Highfield Campus, Southampton, SO17 1BJ, UK
eLaboratoire de Physique des Solides, UMR 8502, Université Paris-Sud, Bât. 510, 91405 Orsay, France
fLaboratoire National des Champs Magnétiques Intenses, UPR3228 CNRS/INSA/UJF/UPS, Toulouse & Grenoble, France
First published on 17th February 2014
Abstract
The single crystal X-ray structure of the all-axial conformer of the (R,R,R,R) enantiomer of the chiral donor tetramethyl-BEDT-TTF (TM-BEDT-TTF) was described and compared to the all-equatorial conformer. (S,S,S,S)-Tetramethyl-BEDT-TTF formed crystalline 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with TCNQ and TCNQ-F4, as well as a THF solvate of the TCNQ complex. Donors bis((2S,4S)-pentane-2,4-dithio)tetrathiafulvalene and (ethylenedithio)((2S,4S)-pentane-2,4-dithio)tetrathiafulvalene, which contain seven-membered rings bearing chirally oriented methyl groups, only formed complexes with TCNQ-F4. The TCNQ-F4 complexes contain planar organosulfur systems, in contrast to the TCNQ complexes in which there is minimal charge transfer. A variety of crystal packing modes were observed. Electrocrystallization experiments with both enantiomers and the racemic form of tetramethyl-BEDT-TTF afforded mixed valence radical cation salts with the AsF6 and SbF6 anions formulated as (TM-BEDT-TTF)2XF6 (X = As, Sb). Electrical conductivity was only found in one charge transfer complex, while the radical cation salts are all semiconducting.
:1 complexes with TCNQ and TCNQ-F4, as well as a THF solvate of the TCNQ complex. Donors bis((2S,4S)-pentane-2,4-dithio)tetrathiafulvalene and (ethylenedithio)((2S,4S)-pentane-2,4-dithio)tetrathiafulvalene, which contain seven-membered rings bearing chirally oriented methyl groups, only formed complexes with TCNQ-F4. The TCNQ-F4 complexes contain planar organosulfur systems, in contrast to the TCNQ complexes in which there is minimal charge transfer. A variety of crystal packing modes were observed. Electrocrystallization experiments with both enantiomers and the racemic form of tetramethyl-BEDT-TTF afforded mixed valence radical cation salts with the AsF6 and SbF6 anions formulated as (TM-BEDT-TTF)2XF6 (X = As, Sb). Electrical conductivity was only found in one charge transfer complex, while the radical cation salts are all semiconducting.
Introduction
One of the first discoveries of conductivity in organic systems was in the molecular complex of tetrathiafulvalene (TTF) 1 with TCNQ 2, which forms separate stacks with ca. 59% electron transfer from the TTF stack to the TCNQ stack.1 Conducting materials have been prepared by grinding the two components together,2 and more recently, conductivity has been observed at the interface between crystals of these two components.3 These two components have been used as models for the design of new electron donors and acceptors and have even been associated in the same molecule.4 The reduction potential of TCNQ can be modified by the introduction of substituents, e.g. fluorine, culminating in the much more oxidizing tetrafluoro-TCNQ 3 for which the reduction potential is ca. 0.53 V to 0.17 V for TCNQ relative to SCE.
BEDT-TTF 4, with an oxidation potential ca. 0.2 V higher than TTF, has been reported to form three 1:1 molecular complexes with TCNQ.5 A monoclinic phase, composed of stacks of alternating BEDT-TTF and TCNQ molecules, has a high resistivity of ca. 106 ohm cm but is semiconducting with the charge transfer estimated to be 20–30%.5c Two triclinic phases have segregated stacks, with the donor and acceptor planes roughly parallel in one case or perpendicular in the other.5b,d The former is metallic with a room temperature resistivity of 10−1 to 10−2 ohm cm and the charge transfer estimated to be ca. 74%. In contrast, the 1:1 complex of BEDT-TTF with TCNQ-F4 is a magnetic insulator with full charge transfer to the acceptors, which behaves as isolated spins and undergoes antiferromagnetic ordering at TN of 14 K.6 Although methyl substituted BEDT-TTF derivatives possessing stereogenic centers, such as the enantiopure tetramethyl-BEDT-TTF (S,S,S,S)-5, abbreviated henceforth as (S)-5, have been known for more than 25 years,7 to date there has been, surprisingly, no example of a charge transfer complex with TCNQ or TCNQ-F4. It was reported, however, that donor (S)-5 forms 2:1 and 3:2 radical cation salts, in which the donor is only partially oxidized and the dithiin rings adopt approximate envelope conformations with methyl groups organized equatorially,8 as well as a 1:1 complete family of salts with triiodide, comprising both (S) and (R) enantiomers and the racemic form, in which the methyl groups adopt also equatorial positions.9 Moreover, the same situation was observed in the solid state structure of neutral (S)-5 and (R)-5, although theoretical calculations suggest that the axial conformer is slightly more stable.9 The use of chiral TTF precursors, such as 5, in charge transfer complexes or radical cation salts in which a combination of chirality and conductivity might allow the observation of a synergistic effect referred to as the electrical magneto-chiral anisotropy (eMChA) effect10 when the transport is measured under an applied parallel magnetic field, presents a great opportunity to search for this synergy in bulk conductors. It is therefore of crucial importance to devote much effort towards the synthesis of chiral TTFs and derived materials, in which the chiral information is addressed in different ways, e.g. stereogenic centers, axial chirality, helical chirality, supramolecular chirality, or chirality on the anions.11 Moreover, the presence of stereogenic centers favours the occurrence of original crystal structures when compared to the achiral precursors and materials. The analogue of (S)-5 with external seven-membered rings, namely S,S,S,S-bis(pentane-2,4-dithio)TTF 6, and a hybrid donor containing one dimethylated seven-membered ring and one unsubstituted six membered ring 7 have been described more recently.12 Donor 7 forms a 1:1 salt with triiodide in which the seven-membered ring adopts a chair conformation with one axial methyl group and one equatorial methyl group12 as observed in the neutral donors 6 and 7. Other enantiopure families of donors include TTF-oxazolines,13 TTF-bis(oxazolines),14 diversely substituted BEDT-TTFs,15 TTF-sulfoxides,16 bis(pyrrolo)-TTFs,17 TTF-binaphthyls,18 tris(TTFs) showing supramolecular chirality,19 halogen containing derivatives,20 TTF-amides,21 TTF-helicenes,22 DM-EDT-TTF,23 and so on. However, complete series of conducting salts, comprising both enantiomers and the racemic form, are still very rare.9,13c,e,23
As a continuation of our effort in the field of chiral TTFs and derived molecular conductors, we report herein our investigations into TCNQ and TCNQ-F4 charge transfer complexes with the chiral donors 5, 6 and 7, together with two complete series of semiconducting salts based on the donor 5 and the anions AsF6− and SbF6−. Note that both enantiopure salts [(S)-5]2XF6 (X = As, Sb) have been mentioned in a previous report, but no structural analysis was provided.8 The conducting properties of the charge transfer complexes and radical cation salts are described. Moreover, the solid state structure of the axial conformer of (R)-5 is reported, together with a comparison to that of the equatorial conformer which was recently reported.9 The conformational issue related to the disposition of the methyl groups and understanding of the factors governing the occurrence of one form or the other are certainly of great importance since they have a massive impact on the packing of the donors, which ultimately influences the physical properties.
Results and discussion
The donors 5,7,9610 and 710 have been prepared according to the published procedures. The solid state structures of (S)- and (R)-5 as equatorial (eq) conformers were previously described by some of us as a triclinic P1 phase with two independent molecules in the unit cell.9 In the same report, DFT calculations have shown the slightly higher stability of the axial conformer, also supported by the experimental CD spectra in solution. In the present work we have succeeded in growing single crystals of the axial (ax) conformer of (R)-5 by slow diffusion of cyclohexane into a solution of compound in carbon disulfide. While crystals of 5-eq were orange needles,9 those of 5-ax are deep red prismatic blocks. As a matter of fact, a polycrystalline powder of 5 clearly shows the presence of two types of solid phases, an orange one and a pink-red one, of which the proportion in between depends on the solvent used in the mother liquor. For example, evaporation of toluene solutions yields a mixture of the orange and pink-red solids in roughly equal proportions, while the solid that resulted from carbon disulfide solutions consists almost exclusively of the pink-red phase. This clearly proves that both forms, i.e. equatorial and axial, coexist in solution and depending on the crystallization conditions, either of them crystallizes. The donor (R)-5-ax crystallizes in the monoclinic system, non-centrosymmetric space group P21, with one independent molecule in the asymmetric unit (Fig. 1). | ||
| Fig. 1 Molecular structures of (R)-5-eq (top) and (R)-5-ax (bottom). | ||
The six-membered rings adopt sofa type conformations, with opposite displacements of the sp3 carbon atoms of the rings (Table 1). Note the quasi-planarity of the dithiole rings, with dihedral angles of 2.4–4.5° along the internal S⋯S axes, in sharp contrast with the strong distortions observed in the structure of (R)-5-eq ranging between 15.8 and 27.2°.9
| Compound | Orientation of the methyl group | Displacements of CH atoms/Å |
|---|---|---|
| (R)-5-ax | Axial | +0.563, −0.331 |
| Axial | +0.285, −0.593 | |
| (R)-5-eq | Equatorial | +0.195, +0.909 |
| Equatorial | −0.697, +0.133 | |
| Equatorial | +0.083, −0.682 | |
| Equatorial | −1.028, −0.029 | |
| TMET·TCNQ | Axial | +0.517, −0.332 |
| Equatorial | +1.028, +0.364 | |
| Axial | +0.469, −0.415 | |
| Equatorial | +1.138, +0.523 | |
| TMET·TCNQ·0.5THF | Axial | +0.463, −0.407 |
| Equatorial | +1.028, +0.376 | |
| Axial | +0.503, −0.370 | |
| Equatorial | +1.096, +0.441 | |
| TMET·TCNQ-F4 | Equatorial | +0.681, −0.055 |
| Equatorial | +0.529, −0.341 | |
| Equatorial | +0.695, −0.172 | |
| Equatorial | +0.529, −0.340 |
Crystalline 1:1 charge transfer (CT) complexes with TCNQ-F4 were obtained from all three donors of all (S) configurations, i.e. (S)-5, 6 and 7, but only donor 5 gave crystalline complexes with TCNQ: a 1:1 complex and a 1:1 + 0.5THF complex. Crystal structures of all five complexes were measured at 120 K. In all of the complexes, the donors and acceptors pack together to form layers where the longest axes of the donor and the acceptor lie perpendicular to the layers; however, the ways in which the molecules are organized within the layer vary considerably.
Complexes of (S)-5
The reaction of donor (S)-5 with TCNQ in 1,1,2-trichloroethane or THF gave 1:1 complexes whose structures differ by the latter having a molecule of THF per every two donor and two acceptor molecules. The crystal structures are closely related; they are both triclinic systems with space group P1 with very similar unit cell dimensions apart from the solvate having a longer c axis. In both complexes, molecules of donor 5 and TCNQ are stacked alternately along the a axis, with stacks lying side by side in the −b and b directions to complete the layers in the ab plane. In the solvated complex, the THF molecules are sandwiched between the layers (Fig. 2–3). In the stacks of both complexes, the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the donor lies between the six-membered ring of a TCNQ above and an exocyclic CC bond of a TCNQ below.
C bond of the donor lies between the six-membered ring of a TCNQ above and an exocyclic CC bond of a TCNQ below.
 | ||
| Fig. 2 Structures of (S)-5·TCNQ (above) and its solvate with THF (below) showing stacking along the a axis, and the location of THF molecules in the solvate phase. | ||
 | ||
| Fig. 3 Packing for (S)-5·TCNQ·0.5THF showing the relative alignment of molecular components between stacks. | ||
The conformations of the donor molecules in the two crystal structures are very similar. The donors adopt gently bowed structures with flexing about the S⋯S vectors across each dithiole ring of 11.4–14.3°, a feature observed in the crystal structure of the neutral donor (R)-5-eq though with a wider range of angles (15.8–27.2°),9 while in (R)-5-ax both dithioles are practically planar (vide supra). The donor's methyl groups take equatorial orientations on one ethylene bridge, but axial orientations on the other ethylene bridge. A similar situation was encountered in 1:1 cycloadducts between 5 and tetrachlorocatecholate, for which both conformations were observed in the same molecule.24 The dithiin rings bearing axially oriented methyl groups adopt half chair conformations, while in contrast, all of the dithiin rings bearing equatorial methyl groups take a conformation with both sp3 ring carbon atoms displaced to the same side of the mean plane of the four other ring atoms, with one displacement being at least double the other (Table 1). Using the empirical correlation between molecular geometry and oxidation state for BEDT-TTF developed by Guionneau et al.,25 the averaged molecular geometry for the two crystallographically unique donors in each complex predicts net charges of 0.0 and +0.14 for the unsolvated and THF solvated complexes, respectively, suggesting little net charge transfer in the ground state.
In this respect, a useful comparative analysis can be done between (S)-5·TCNQ or (S)-5·TCNQ·0.5THF, both showing practically no charge transfer, and the monoclinic phase of BEDT-TTF·TCNQ which also has mixed donor–acceptor stacks.5c However, a charge transfer of approximately 20–30% is estimated in the latter. This is consistent with the observed planar geometry of the donor, while (S)-5 in both crystalline phases we describe herein is bent about the S⋯S axes. Since the oxidation potentials of BEDT-TTF and TM-BEDT-TTF are essentially the same, the difference in charge transfer is very likely due to the less favourable contacts or packing provoked by the additional four methyl groups.
The reaction of donor (S)-5 with the more oxidizing acceptor TCNQ-F43 produced a 1:1 complex which has quite different structural characteristics from the two TCNQ complexes. The crystal structure is triclinic with space group P1, with two donor molecules and two acceptor molecules arranged pseudo- and exactly centrosymmetrically, respectively, in the unit cell. The donor and acceptor molecules pack together with their best planes roughly parallel to form layers whose composition is shown in Fig. 4.
 | ||
| Fig. 4 Packing of (S)-5·TCNQ-F4 showing the zigzag stacking of donor cations and acceptor anions. | ||
Within the layer there are lines composed of donor and TCNQ-F4 molecules packed alternately side by side, and the relative orientation of successive lines in the layer leads to zigzag stacks of donors or acceptors running in the perpendicular direction through the layer. There is interpenetration between layers by the chiral hydrocarbon termini of the donors whose major axes are considerably longer than that of TCNQ-F4 (Fig. 5). All four methyl groups of the donors lie in pseudo-equatorial positions, and there is a range of conformations for the dithiin ring which all lie between an envelope and a half chair. Along the zigzag stacks of donors there is minimal face-to-face overlap with edge-to-edge S⋯S contacts in the range 3.43–3.79 Å. In contrast, for the acceptors, there are pairs which partially overlap face-to-face, so that a F and a N atom make short contacts to the central ring of the other (F⋯C, 3.38; N⋯C, 3.47 Å); however there is no such overlap between pairs for which the shortest contacts are between edges (F⋯CF, 3.32; F⋯CN, 3.41; and F⋯N, 3.37 Å). Bond length data for the two donors predict a charge of +0.97 in each case suggesting a complete charge transfer in this salt, and, in support of this, the organosulfur cores of the donor molecules are almost planar in contrast to those of the TCNQ complexes of 5.
 | ||
| Fig. 5 View down the stacking axis in (S)-5·TCNQ-F4. | ||
Complexes of donors 6 and 7 with TCNQ-F4
Donors 6 and 7 both form complexes with TCNQ-F4, though no crystalline complexes could be isolated by reaction with unsubstituted TCNQ. This may be related to the significant non-planarity of the neutral donors (Fig. 6), which are not compatible with the planar TCNQ, which would not be expected to oxidize donors 6 or 7. The first oxidation potentials of donors 6 and 7 (0.50–0.51 V vs. Ag/AgCl)10 are similar to that of TM-BEDT-TTF (+0.49 V vs. SCE).9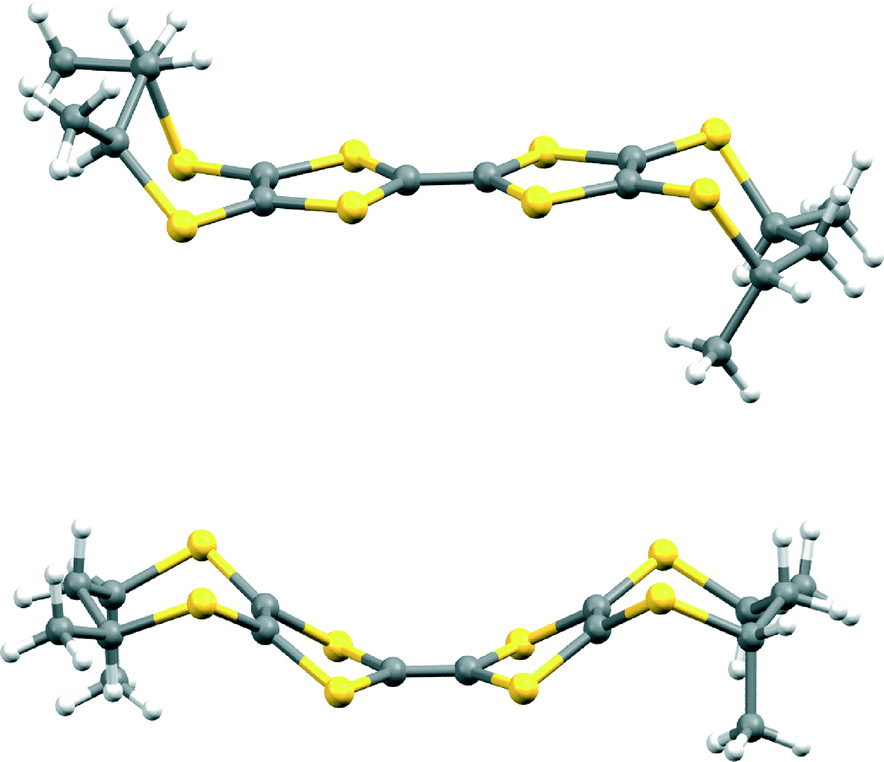 | ||
| Fig. 6 Structures of the donor cation in 6·TCNQ-F4 (above) and the neutral donor 6 (ref. 10) (below). | ||
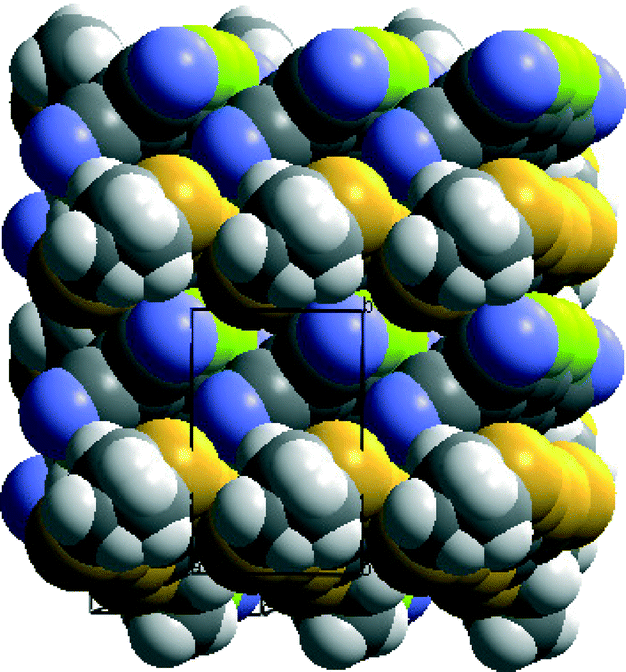
The crystal structure of the 1:1 molecular complex of the tetramethylated donor 6 with TCNQ-F4 is particularly interesting since it shows separate stacks for the donors and the acceptors (Fig. 7).
 | ||
| Fig. 7 Crystal packing of 6·TCNQ-F4 viewed with the b axis vertical and c axis horizontal. | ||
The crystal system is monoclinic and the space group is P21 with stacking organized along the b axis. In contrast to the neutral donor, the organosulfur portion of donor 6 is planar (Fig. 6), strongly suggesting a change in the oxidation state. One seven-membered ring adopts a well ordered chair conformation with one axial and one equatorial methyl group, and the two methine carbons and the methylene carbon displaced by similar amounts from the organosulfur plane (1.433–1.510 Å). There is disorder in the other seven-membered ring between two conformations in the ratio ca. 3:1. The major conformation is twisted and unsymmetrical with the three ring sp3 carbon atoms displaced to the same side of the organosulfur plane – with the methylene carbon displaced the most (2.190 Å) and the two methine carbon atoms displaced by quite different amounts (1.747 Å and 1.242 Å) (Fig. 8). The minor conformation is a chair, but displaced to the opposite side of the organosulfur plane with respect to the other end of the molecule.
 | ||
| Fig. 8 Molecular geometries of the cations 6 (above) and 7 (below) in their TCNQ-F4 complexes. | ||
According to the correlation between charge and bond lengths in the TTF portion of the donor, in this complex the donor carries a charge of +1.04, and so there has been full charge transfer. Thus, the crystal structure contains stacks of donor cations 6+ and acceptor anions 3−. Indeed the organosulfur region of the donor has undergone a major structural change upon oxidation from a bowed structure to an almost planar one (Fig. 6). The planes of the cations and anions lie at 23.2° to each other, and each stack of cations is surrounded on four sides by stacks of acceptor anions. Within the donor cation stacks the main axis of the organosulfur plane lies at 46° to the stacking axis with six S⋯S contacts in the range 3.61–3.86 Å. In the acceptor anion stacks, the main axes of the acceptors lie at 41° to the stacking axis, such that two exocyclic “double bonds” lie opposite to each other but oriented anti, so cyano groups of one molecule lie over the edge of the six-membered ring of the other and there are six C⋯C contacts in the range 3.33–3.44 Å.
In contrast, the crystal structure of the complex of the unsymmetrical dimethylated donor 7 adopts stacks of alternating donors and acceptors (Fig. 9).
 | ||
| Fig. 9 Alternate stacking of donor cations and acceptor anions along the b axis in 7·TCNQ-F4. | ||
However, each species is tilted so that the shorter axis of the molecular plane lies at ca. 50° to the stacking axis, which leads to a lateral overlap between donor molecules (and between acceptor molecules) in adjacent stacks (Fig. 10). The crystal system is monoclinic with space group P21, with stacking along the b axis, and the lateral overlap of donors or acceptors in the a direction forms layers perpendicular to the c axis. Between donors the shortest S⋯S contacts are 3.51 and 3.54 Å. The overlap between acceptors produces two fluorine/nitrile interactions (F⋯C: 3.22 & 3.35 Å; F⋯N: 3.19 & 3.41 Å) and two C–F bonds overlapping with C⋯F contacts of 3.34 and 3.37 Å. The seven-membered ring does not adopt the chair conformation but rather the two methine C atoms are displaced to opposite sides of the organosulfur plane (Fig. 8). The organosulfur portion of the donor is planar, in contrast to the neutral donor, and correlation of the molecular geometry of the TTF portion with the oxidation state suggests that it bears a charge of +0.92, and thus is in the monocation form.
 | ||
| Fig. 10 Side to side overlap between donors and between acceptors in 7·TCNQ-F4 viewed down the c axis. | ||
Radical cation salts (5)2XF6 (X = As, Sb)
Electrocrystallization of both enantiomers and the racemic mixture (obtained by mixing equimolar amounts of (S) and (R)) of TM-BEDT-TTF 5 in the presence of [NBu4]AsF6 and [NBu4]SbF6 as supporting electrolytes gave the corresponding 2:1 salts, (5)2AsF6 and (5)2SbF6. The (S) enantiomeric salts have been previously reported, although their structures were not detailed since they were isostructural to the [(R)-5]2PF6 salt described in more detail.8 [(R)-5]2AsF6 and [(R)-5]2SbF6 salts are isostructural and crystallize in the triclinic system, space group P1, with two independent donor units for one molecule of anion. The racemic salts [(rac)-5]2AsF6 and [(rac)-5]2SbF6 crystallize also in the triclinic system, centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with the anions situated on the inversion centre. The cell parameters are the same throughout the respective AsF6 and SbF6 series, except for the space groups, i.e. P1 for the enantiopure forms and P for the racemic ones. A slight increase of the cell volume (≈1.2%) is observed when passing from AsF6 to SbF6, in agreement with the larger size of the latter.
, with the anions situated on the inversion centre. The cell parameters are the same throughout the respective AsF6 and SbF6 series, except for the space groups, i.e. P1 for the enantiopure forms and P for the racemic ones. A slight increase of the cell volume (≈1.2%) is observed when passing from AsF6 to SbF6, in agreement with the larger size of the latter.
In all cases the methyl groups adopt pseudo-equatorial positions, probably as a means to maximize the intermolecular interactions. The dimethylethylene bridge is disordered at 70:30 in the structure of the racemic salt (Fig. 11), such that both enantiomers are present on the same crystallographic site, a feature already encountered with (rac)-5.9 The values of the central CC and internal C–S bonds are indicative of the mixed valence state of the donor molecules (Table 2).
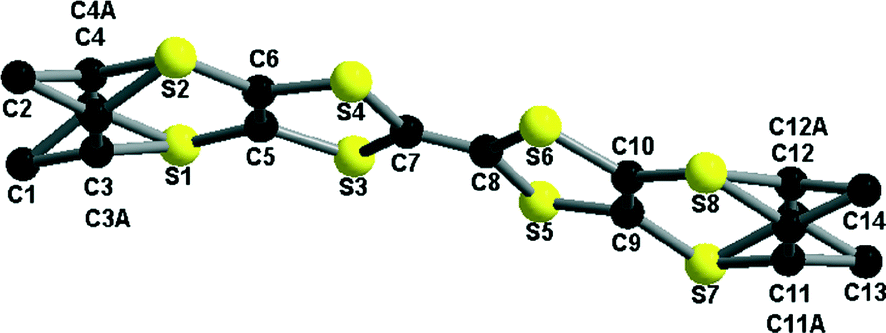 | ||
| Fig. 11 Donor molecule in the structure of [(rac)-5]2SbF6 together with the atom numbering scheme. The disorder model of the donor involves common positions for the methyl groups and disordered methine carbon atoms at s.o.f. 0.7:0.3. | ||
| Compound | [(S)-5]2AsF6 | [(R)-5]2AsF6 | [(rac)-5]2AsF6 | |||
|---|---|---|---|---|---|---|
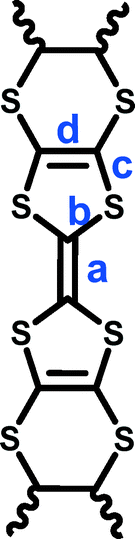 δ = (b + c) − (a + d) Q = 6.347 − 7.4638δ
δ = (b + c) − (a + d) Q = 6.347 − 7.4638δ |
a | 1.353(11) | 1.370(11) | 1.352(9) | 1.366(9) | 1.363(4) |
| b | 1.744(9) | 1.743(10) | 1.732(8) | 1.727(7) | 1.737(3) | |
| 1.747(9) | 1.735(9) | 1.753(6) | 1.740(6) | 1.740(3) | ||
| 1.744(9) | 1.736(9) | 1.743(6) | 1.741(7) | 1.741(3) | ||
| 1.746(9) | 1.740(10) | 1.754(8) | 1.749(8) | 1.741(3) | ||
| c | 1.765(8) | 1.744(9) | 1.748(6) | 1.742(7) | 1.743(3) | |
| 1.780(9) | 1.735(9) | 1.754(7) | 1.734(7) | 1.748(3) | ||
| 1.769(9) | 1.725(9) | 1.756(7) | 1.742(7) | 1.746(3) | ||
| 1.748(9) | 1.738(9) | 1.753(6) | 1.755(6) | 1.749(3) | ||
| d | 1.338(11) | 1.352(12) | 1.367(8) | 1.361(9) | 1.349(4) | |
| 1.338(12) | 1.353(12) | 1.347(9) | 1.332(8) | 1.347(4) | ||
| δ | 0.820 | 0.752 | 0.789 | 0.770 | 0.775 | |
| Q | 0.249 | 0.738 | 0.456 | 0.600 | 0.561 | |
| [(S)-5]2SbF6 | [(R)-5]2SbF6 | [(rac)-5]2SbF6 | ||||
| a | 1.378(10) | 1.364(10) | 1.355(7) | 1.370(7) | 1.360(4) | |
| b | 1.753(7) | 1.753(7) | 1.731(5) | 1.724(5) | 1.741(3) | |
| 1.726(8) | 1.726(8) | 1.738(4) | 1.736(5) | 1.741(2) | ||
| 1.733(9) | 1.733(9) | 1.742(5) | 1.737(5) | 1.741(2) | ||
| 1.729(7) | 1.729(7) | 1.755(5) | 1.753(5) | 1.741(3) | ||
| c | 1.738(7) | 1.738(7) | 1.736(5) | 1.741(4) | 1.743(2) | |
| 1.744(7) | 1.744(7) | 1.745(5) | 1.756(6) | 1.746(2) | ||
| 1.715(7) | 1.715(7) | 1.746(5) | 1.755(5) | 1.747(3) | ||
| 1.735(8) | 1.735(8) | 1.752(5) | 1.757(5) | 1.750(2) | ||
| d | 1.339(10) | 1.339(10) | 1.360(7) | 1.347(7) | 1.354(3) | |
| 1.383(11) | 1.383(11) | 1.342(7) | 1.338(7) | 1.350(3) | ||
| δ | 0.729 | 0.743 | 0.78 | 0.777 | 0.760 | |
| Q | 0.904 | 0.800 | 0.5230 | 0.546 | 0.559 | |
The donors form parallel columns reminiscent of a β phase, with short intra- and inter-stack S⋯S contacts (Fig. 12 and ESI†), with a classical organic–inorganic segregation (ESI†).26
 | ||
| Fig. 12 Packing diagram of [(S)-5]2SbF6 with emphasis on the S⋯S short contacts. SbF6 anions and H atoms were omitted. | ||
Bond length data for the donors, reported in Table 2, clearly indicate a mixed valence state for all of the salts, with a mean charge not far from +0.5. The higher discrepancy for the (S) salts arises from the somewhat lower quality of the crystal data set measurements.
Conducting properties
Measurements of resistivity for the charge transfer complexes show that all of them except 7·TCNQ-F4 have a room temperature resistance too high to be measured (>1.2 Mohm limit of the machine). For comparison, the monoclinic form of BEDT-TTF·TCNQ which has mixed stacks similar to the TMET·TCNQ complexes, but with 20–30% charge transfer, shows a resistivity ca. 106 ohm cm.5c7·TCNQ-F4 was found to be a semiconductor but with very low conductivity. Upon cooling from room temperature to just 290 K the resistance quickly increased but below 280 K the resistance was above the measurement limit. The activation energy is estimated to be 0.025 eV.In a previous report the [(S)-5]XF6 (X = P, As, Sb) series of salts was found to be semiconducting.8 With the complete series of AsF6 and SbF6 in our hands we proceeded to the measurement of their conducting properties. In spite of the structural disorder present in the racemic salts, which in general disfavors the transport properties,9 the enantiopure and racemic compounds within the same family, i.e. AsF6 or SbF6, show practically the same semiconducting behavior, with activation energies ranging between 920 and 1340 K, and similar room temperature conductivity values of around 0.5–1.0 S cm−1 (Fig. 13).
 | ||
| Fig. 13 Temperature dependence of the electrical resistivity ρ for single crystals of [5]2AsF6 (left) and [5]2SbF6 (right). | ||
Transport property measurements under an applied parallel magnetic field for both series of salts did not allow the detection of the electrical magneto-chiral anisotropy effect,11 probably because the chiral information is only poorly expressed at the crystal level.
Conclusions
Three chiral donors have been investigated in this work, namely the tetramethyl-BEDT-TTF 5 as enantiopure and racemic forms and the related seven-membered ring analogues 6 and 7 as the (S) enantiomers. The conformation of the enantiomerically pure donor 5 adopts all-axial or all-equatorial methyl groups in two crystalline polymorphs. Its salts with TCNQ and TCNQ-F4 show very little or complete charge transfer, respectively, and do not have any transport properties, in comparison to BEDT-TTF which forms three conducting salts with TCNQ. The radical cation salts of both enantiomers and the racemate of TM-BEDT-TTF with XF6− (X = As, Sb) all adopt very similar crystal structures, and, consistent with this, there was little difference between the conductivities of the racemate and enantiomers in the two families. The methyl groups adopted equatorial positions in all of these materials except in the TCNQ complexes where one pair was oriented axially. The donors 6 and 7 containing two or one seven-membered ring are significantly non-planar in their outer rings, which appears not compatible to the formation of TCNQ salts, but both form salts with TCNQ-F4, and that with 7 showed weak semiconducting properties. BEDT-TTF with only small hydrogen substituents is particularly well suited to forming tightly packed radical cation salts, but inclusion of other groups at both ends of the molecule makes such close packing more difficult. Nevertheless, interesting electroactive materials are likely to be produced if those groups bring attractive intermolecular interactions, more ordered crystal structures, and other physical properties resulting from the combination of chirality with conductivity and magnetism.Experimental
Donors 5,7,9610 and 710 have been prepared following the literature procedures.(R)-5-ax
Suitable single crystals for X-ray diffraction were obtained by slow diffusion of cyclohexane into a solution of compound in carbon disulfide. The crystals of the all-axial conformer are deep red prismatic blocks while those of the all-equatorial one are orange needles.Preparation of TCNQ complexes
(b) A hot solution of donor 5 (10 mg) in THF (2 ml) was added to a hot solution of TCNQ (5 mg) in THF (4 ml) and the mixture was heated at 65 °C for 1 h. Cooling gave crystals as long black plates.
Electrocrystallization of donor 5 with AsF6 and SbF6 anions
X-ray crystallography
Crystal structures for the charge transfer complexes with TCNQ and TCNQ-F4 were measured using MoKα radiation at 120 K on a Nonius Kappa CCD area-detector diffractometer located at the window of a Nonius FR591 rotating-anode X-ray generator, and solved and refined using the SHELX program suite.27 X-ray diffraction measurements for (R)-5-ax and the salts (5)2XF6 (X = As, Sb) were performed on a Bruker Kappa CCD diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å) at room temperature. The structures were solved by direct methods and refined by full-matrix least squares techniques based on F2. The non-H atoms were refined with anisotropic displacement parameters. Calculations were performed using the SHELX-97 crystallographic software package. CCDC reference numbers: CCDC 976560 ((S)-5·TCNQ), CCDC 976561 ((S)-5·TCNQ·0.5THF), CCDC 976562 ((S)-5·TCNQ-F4), CCDC 976563 (6·TCNQ-F4), CCDC 976564 (7·TCNQ-F4), CCDC 976361 ([(S)-5]2AsF6), CCDC 976362 ([(R)-5]2AsF6), CCDC 976363 ([(rac)-5]2AsF6), CCDC 976364 ([(S)-5]2SbF6), CCDC 976365 ([(R)-5]2SbF6), CCDC 976366 ([(rac)-5]2SbF6), CCDC 976367 ((R)-5-ax).286 unique reflections, 10819 with F > 4σ(F), Flack parameter: 0.08(6), R = 0.040, wR = 0.083.
840 unique reflections, 10784 with F > 4σ(F), Flack parameter: 0.14(6), R = 0.037, wR = 0.083.
, Dc = 1.741 g cm−3, μ(MoKα) = 1.703 mm−1, T = 293(2) K, 3702 unique reflections, 3025 with F > 4σ(F), R = 0.052, wR = 0.101.
, Dc = 1.792 g cm−3, μ(MoKα) = 1.525 mm−1, T = 293(2) K, 4363 unique reflections, 3839 with F > 4σ(F), R = 0.040, wR = 0.090.
Resistivity measurements
For the charge transfer salts, two-probe DC transport measurements were made on several crystals of each salt using a HUSO HECS 994C multi-channel conductometer. Gold wires (15 μm diameter) were attached to the crystal, and the attached wires were connected to an eight-pin integrated circuit plug with gold conductive cement. The machine had an upper limit for resistance measurement of 1.2 MOhm.For the radical cation salts [5]2XF6 (X = As, Sb), four-probe transport measurements were performed on crystals of each salt using an AC technique with an applied current Iac = 1 μA and low-frequency lock-in detection. Annular contacts were made by gold evaporation on which gold wires were attached with silver paste. Low temperature was achieved using a cryocooler equipment.
Acknowledgements
We thank the EPSRC for grant EP/C510488/1 and for a studentship, the EPSRC National Crystallography Service for datasets, and the EPSRC Mass Spectrometry Service for measurements. The work has benefited from support from the ESF COST action D35. This work was supported in France by the National Agency for Research (ANR, Project 09-BLAN-0045-01), the CNRS, the University of Angers and the Ministry of Education and Research (grant to C.M.).References
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 9, 948–949 CrossRef.
- F. Toda and H Miyamoto, Chem. Lett., 1995, 861 CrossRef CAS.
- (a) H. Alves, A. S. Molinari, H. Xie and A. F. Morpurgo, Nat. Mater., 2008, 7, 574–580 CrossRef CAS PubMed; (b) J. E. Kirtley and J. Mannhart, Nat. Mater., 2008, 7, 522–521 CrossRef PubMed; (c) S. Wen, W.-Q. Deng and K.-L. Han, Chem. Commun., 2010, 46, 5133–5135 RSC.
- F. Otón, V. Lloveras, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, Angew. Chem., Int. Ed., 2011, 50, 10902–10906 CrossRef PubMed.
- (a) G. Saito, H. Hayashi, T. Enoki and H. Inokuchi, Mol. Cryst. Liq. Cryst., 1985, 120, 341–344 CrossRef CAS; (b) T. Mori and H. Inokuchi, Solid State Commun., 1986, 59, 355–359 CrossRef CAS; (c) T. Mori and H. Inokuchi, Bull. Chem. Soc. Jpn., 1987, 60, 402–404 CrossRef CAS; (d) H. M. Yamamoto, M. Hagiwara and R. Kato, Synth. Met., 2003, 133–134, 449–451 CrossRef CAS.
- T. Hasegawa, K. Inukai, S. Kagoshima, T. Sugawara, T. Mochida, S. Sugiura and Y. Iwasa, Synth. Met., 1997, 86, 1801–1802 CrossRef CAS.
- (a) J. D. Dunitz, A. Karrer and J. D. Wallis, Helv. Chim. Acta, 1986, 69, 69–70 CrossRef; (b) B. Chen, F. Deilacher, M. Hoch, H. J. Keller, P. Wu, P. Armbruster, R. Geiger, S. Kahlich and D. Schweitzer, Synth. Met., 1991, 42, 2101–2105 CrossRef CAS; (c) S. Matsumiya, A. Izuoka, T. Sugawara, T. Taruishi and Y. Kawada, Bull. Chem. Soc. Jpn., 1993, 66, 513–522 CrossRef CAS.
- A. Karrer, J. D. Wallis, J. D. Dunitz, B. Hilti, C. W. Mayer, M. Bürkle and J. Pfeiffer, Helv. Chim. Acta, 1987, 70, 942–953 CrossRef CAS.
- F. Pop, S. Laroussi, T. Cauchy, C. J. Gómez-García, J. D. Wallis and N. Avarvari, Chirality, 2013, 25, 466–474 CrossRef CAS PubMed.
- (a) G. L. J. A. Rikken, J. Fölling and P. Wyder, Phys. Rev. Lett., 2001, 87, 236602 CrossRef CAS PubMed; (b) V. Krstic, S. Roth, M. Burghard, K. Kern and G. L. J. A. Rikken, J. Chem. Phys., 2002, 117, 11315–11319 CrossRef CAS.
- N. Avarvari and J. D. Wallis, J. Mater. Chem., 2009, 19, 4061–4076 RSC.
- S. Yang, A. C. Brooks, L. Martin, P. Day, M. Pilkington, W. Clegg, R. W. Harrington, L. Russo and J. D. Wallis, Tetrahedron, 2010, 66, 6977–6989 CrossRef CAS.
- (a) C. Réthoré, M. Fourmigué and N. Avarvari, Chem. Commun., 2004, 1384–1385 RSC; (b) C. Réthoré, M. Fourmigué and N. Avarvari, Tetrahedron, 2005, 61, 10935–10942 CrossRef; (c) C. Réthoré, N. Avarvari, E. Canadell, P. Auban-Senzier and M. Fourmigué, J. Am. Chem. Soc., 2005, 127, 5748–5749 CrossRef PubMed; (d) C. Réthoré, A. Madalan, M. Fourmigué, E. Canadell, E. B. Lopes, M. Almeida, R. Clérac and N. Avarvari, New J. Chem., 2007, 31, 1468–1483 RSC; (e) A. M. Madalan, C. Réthoré, M. Fourmigué, E. Canadell, E. B. Lopes, M. Almeida, P. Auban-Senzier and N. Avarvari, Chem.–Eur. J., 2010, 16, 528–537 CrossRef CAS PubMed.
- (a) F. Riobé and N. Avarvari, Chem. Commun., 2009, 3753–3755 RSC; (b) F. Riobé and N. Avarvari, Coord. Chem. Rev., 2010, 254, 1523–1533 CrossRef.
- (a) J. D. Wallis and J.-P. Griffiths, J. Mater. Chem., 2005, 15, 347–365 RSC; (b) J.-P. Griffiths, H. Nie, R. J. Brown, P. Day and J. D. Wallis, Org. Biomol. Chem., 2005, 3, 2155–2166 RSC.
- (a) M. Chas, M. Lemarié, M. Gulea and N. Avarvari, Chem. Commun., 2008, 220–222 RSC; (b) M. Chas, F. Riobé, R. Sancho, C. Minguíllon and N. Avarvari, Chirality, 2009, 21, 818–825 CrossRef CAS PubMed.
- S. Yang, A. C. Brooks, L. Martin, P. Day, H. Li, P. Horton, L. Male and J. D. Wallis, CrystEngComm, 2009, 11, 993–996 RSC.
- (a) R. Gómez, J. L. Segura and N. Martin, J. Org. Chem., 2000, 65, 7566–7574 CrossRef; (b) Y. Zhou, D. Zhang, L. Zhu, Z. Shuai and D. Zhu, J. Org. Chem., 2006, 71, 2123–2130 CrossRef CAS PubMed; (c) A. Saad, F. Barrière, E. Levillain, N. Vanthuyne, O. Jeannin and M. Fourmigué, Chem.–Eur. J., 2010, 16, 8020–8028 CrossRef CAS PubMed; (d) A. Saad, O. Jeannin and M. Fourmigué, Tetrahedron, 2011, 67, 3820–3829 CrossRef CAS; (e) A. Saad, O. Jeannin and M. Fourmigué, New J. Chem., 2011, 35, 1004–1010 RSC.
- (a) I. Danila, F. Riobé, J. Puigmartí-Luis, Á. Pérez del Pino, J. D. Wallis, D. B. Amabilino and N. Avarvari, J. Mater. Chem., 2009, 19, 4495–4504 RSC; (b) I. Danila, F. Riobé, F. Piron, J. Puigmartí-Luis, J. D. Wallis, M. Linares, H. Ågren, D. Beljonne, D. B. Amabilino and N. Avarvari, J. Am. Chem. Soc., 2011, 133, 8344–8353 CrossRef CAS PubMed; (c) I. Danila, F. Pop, C. Escudero, L. N. Feldborg, J. Puigmartí-Luis, F. Riobé, N. Avarvari and D. B. Amabilino, Chem. Commun., 2012, 48, 4552–4554 RSC.
- J. Lieffrig, R. Le Pennec, O. Jeannin, P. Auban-Senzier and M. Fourmigué, CrystEngComm, 2013, 15, 4408–4412 RSC.
- Y. Tatewaki, T. Hatanaka, R. Tsunashima, T. Nakamura, M. Kimura and H. Shirai, Chem.–Asian J., 2009, 4, 1474–1479 CrossRef CAS PubMed.
- T. Biet, A. Fihey, T. Cauchy, N. Vanthuyne, C. Roussel, J. Crassous and N. Avarvari, Chem.–Eur. J., 2013, 19, 13160–13167 CrossRef CAS PubMed.
- F. Pop, P. Auban-Senzier, A. Frąckowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS PubMed.
- F. Pop, J. Lacour and N. Avarvari, Rev. Roum. Chim., 2012, 57, 457–462 CAS.
- P. Guionneau, C. J. Kepert, G. Bravic, D. Chasseau, M. R. Truter, M. Kurmoo and P. Day, Synth. Met., 1997, 86, 1973–1974 CrossRef CAS.
- T. Mori, Bull. Chem. Soc. Jpn., 1998, 71, 2509–2526 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 976361–976367, 976560–976564. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42539h |
| This journal is © The Royal Society of Chemistry 2014 |