 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Salt formation affects the conformational and assembly properties of p-carboxylatocalix[4]arenes†
Stuart
Kennedy
a,
Christine M.
Beavers
b,
Simon J.
Teat
b and
Scott J.
Dalgarno
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh, EH14 4AS, Scotland, UK. E-mail: S.J.Dalgarno@hw.ac.uk; Fax: +44 (0)131 451 3180; Tel: +44 (0)131 451 8025
bLawrence Berkeley National Laboratory, 1 Cyclotron Road, MS 6R2100, Berkeley, California 94720, USA
First published on 12th March 2014
Abstract
The conformational properties and self-assembly behaviour of the p-carboxylatocalix[4]arenes, when in the presence of a pyridine template, are well understood. Salt formation with these molecules, a process driven by the introduction of an amino group to the 2-position of the pyridine template, has dramatic consequences on both building block conformation and the resulting self-assembly.
The p-carboxylatocalix[n]arenes (pCO2[n]s, where n represents the number of aromatic rings in the calixarene) have received relatively little attention as molecular building blocks in supramolecular chemistry. This is surprising when one considers a) their structural relation to the p-sulfonato analogues which have been studied extensively,1 b) the fact that the benzoate moiety has featured heavily in MOF formation2 and c) that well-established synthons may be used to drive self-assembly with carboxylic acids.3 Despite this these molecules show great promise for exploitation in areas such as targeted self-assembly4 (through careful choice of synthon employed) and the formation of novel metal–organic systems.5 The latter can be discrete5a,c,d,f,h or polymeric5b in nature, and we have recently shown that these can be designed so as to specifically contain inherently cavity-containing pCO2[4]s.5h
The basic calix[4]arene (C[4]) framework adopts a cone conformation due to the presence of concerted H-bonding interactions at the polyphenolic lower-rim. This can be readily modulated through C[4] lower-rim alkylation to afford, amongst others, partially pinched-cone (via di-O-alkylation) and fully pinched-cone (via tetra-O-alkylation) conformers as shown in Fig. 1A; tetra-O-alkylation with methyl or ethyl chains allows for conformational inversion through the C[4] annulus, and chains greater or equal to propyl in length are known to lock the molecule in the fully pinched-cone conformation.6 Once the desired conformer (or structurally mobile C[4] for OMe and OEt derivatives) has been synthesised, the introduction of upper-rim carboxylic acid functionality is relatively straightforward. We have investigated the assembly of a range of pCO2[4]s containing two4b,d,f,g or four4c,e lower-rim alkyl chains in the presence of pyridine (Py), picoline and ethylpyridine templates.
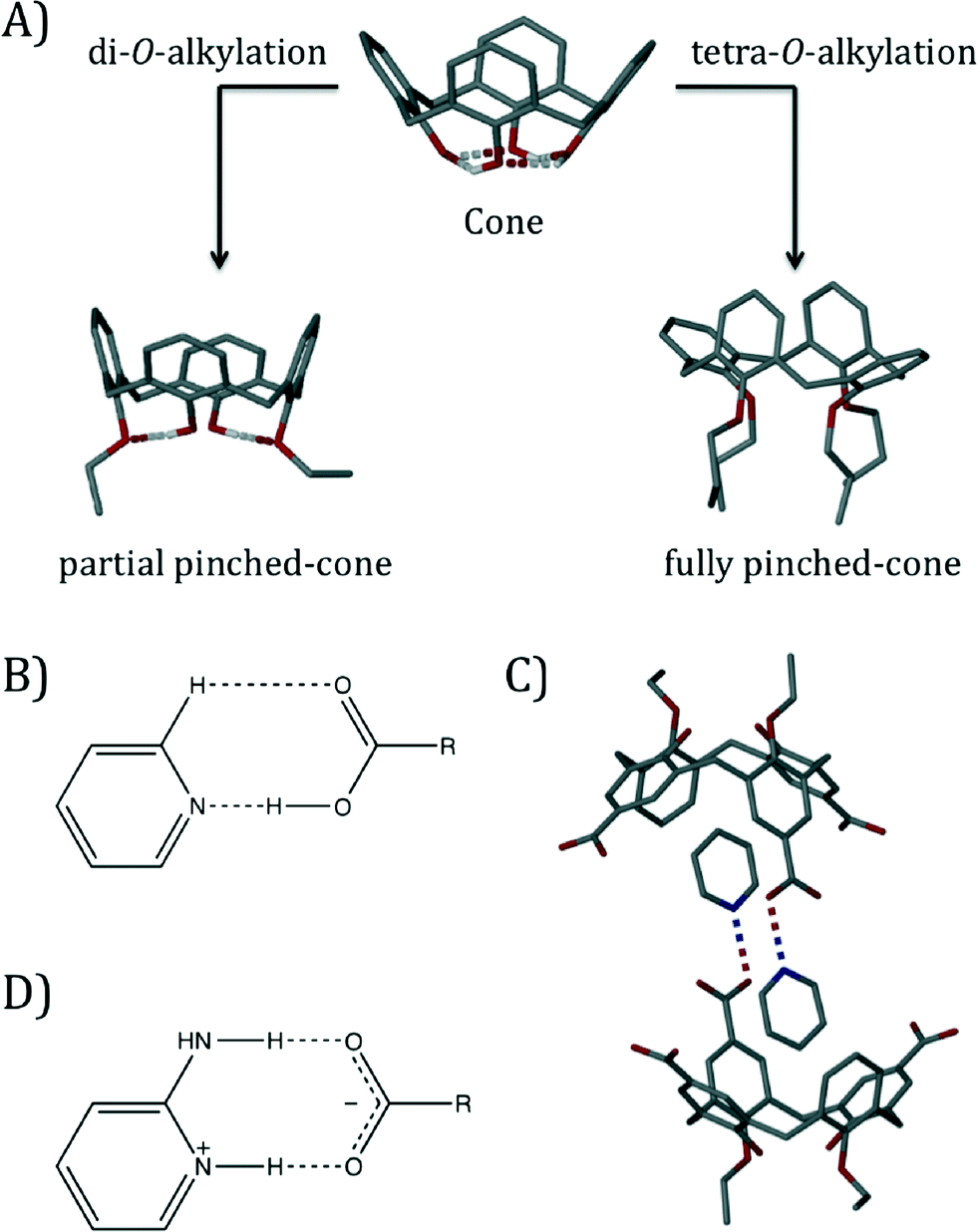 | ||
| Fig. 1 A) Control over C[4] conformation by di- and tetra-O-alkylation at the lower-rim. B) Schematic of the Py⋯CO2H heterosynthon. C) Head-to-head H-bonded capsule formed by crystallisation of a di-O-alkylated pCO2[4] in the presence of a Py template.4f D) Schematic of the 2-aminopyridinium-carboxylate heterosynthon. | ||
In the aforementioned series of studies we found that a) the components generally formed the expected Py⋯CO2H heterosynthon shown in Fig. 1B, b) if a cavity is present (e.g. in a lower-rim di-O-alkyl pCO2[4]) it will be occupied by a Py or Py derivative guest template,4b,d,f,g c) host–guest pCO2[4]/Py (or Py derivative) arrangements dimerise to form capsules4a,b,d,f,g (Fig. 1C) and d) Py or Py derivative templates form the aforementioned heterosynthon but do not force access to the potential cavity of a lower-rim tetra-O-alkyl pCO2[4] (fully pinched-cone conformation, Fig. S1†).4c,e Here we report interesting effects on the conformational properties of a series of tetra-O-alkyl pCO2[4]s, as well as their assembly preferences, once salt formation has been performed via introduction of an amino group to the Py template (using 2-aminopyridine, 2-AP).7 This well-known heterosynthon was targeted for structural comparison with related Py⋯CO2H driven assembly of pCO2[4]s (compare Fig. 1B and D), and it was anticipated that deprotonation of the upper-rim would cause deviation from the fully pinched-cone conformation due to repulsion of CO2− groups. We screened salt formation in methanol between 2-aminopyridine (2-AP) and the series of p-carboxylato-O-alkylcalix[4]arenes shown in Fig. 2;‡ single crystals of salts formed for 1, 3 and 4, and the interesting structural features of these assemblies are described in order below.
 | ||
| Fig. 2 Tetra-O-alkyl pCO2[4]s used in salt formation with 2-aminopyridine. | ||
Single crystals of the salt (5) formed between 1 and 2-AP were found to be in a monoclinic cell and structure solution was performed in the space group P21/n.† The asymmetric unit in 5 consists of one molecule of 1 (with three of four upper-rim CO2H groups deprotonated), four 2-AP molecules (three of which are protonated, H2-AP) and two and a half methanol molecules (the half being disordered by symmetry) of crystallisation (Fig. 3). The most noticeable feature upon inspection is that 1 unexpectedly adopts a partial pinched-cone conformation; as stated above, lower-rim OMe groups allow for conformational mobility, and crystallisation of 1 from 4-picoline affords a structure in which the calixarene adopts a partial-cone conformation with inversion of one aromatic ring.4e,8 One of the aromatic rings of 1 in 5 is slightly splayed in comparison to the others within the framework. One H2-AP cation occupies the resulting host cavity, forming various intermolecular interactions as shown in Fig. 3; there are CH⋯π and π-stacking interactions between guest and host, occurring with respective H[42]⋯aromatic centroid and aromatic centroid⋯aromatic centroid distances of 2.6 Å and 3.975 Å. There is also an HNH⋯OCO H-bonding interaction between the cationic guest and an upper-rim CO2− group of 1 with an H[43A]⋯O[6] distance of 2.2 Å. Symmetry expansion around the guest in 5 results in formation of a H-bonded head-to-head dimer/capsule as shown in Fig. 4. This occurs through formation of the targeted heterosynthon shown in Fig. 1D,7 with an NH⋯OCO H-bonding interaction between H[38] and symmetry equivalent (s.e.) O[7] (H[38]⋯O[7]s.e. distance of 1.8 Å), as well as an HNH⋯OCO H-bonding interaction between H[43B] and O[8]s.e. (H[43B]⋯O[8]s.e. distance of 2.0 Å). The two remaining 2-AP cations in 5 also interact with upper-rim CO2− groups of 1 through formation of the same heterosynthon (Fig. 3); this occurs with two NH⋯OCO (respective H[45]⋯O[6] and H[52]⋯O[12] distances of 1.7 and 1.9 Å) and two HNH⋯OCO H-bonding interactions (respective H[50B]⋯O[5] and H[57B]⋯O[11] distances of 2.0 and 1.9 Å). The neutral 2-AP in the asymmetric unit interacts with two upper-rim CO2− groups of 1via two HNH⋯OCO interactions with H[64A]⋯O[6] and H[64B]⋯O[7] distances of 2.1 and 2.1 Å. The three methanol molecules of crystallisation form a range of OH⋯OCO and OH⋯O H-bonding interactions; these occur between neighbouring MeOH of crystallisation, or CO2H/CO2− groups of 1 (Fig. 3).
 | ||
| Fig. 3 Asymmetric unit found in the crystal structure of the salt (5) formed between 1 and 2-AP. H-bonding and CH⋯π interactions are shown as split-colour and red dashed lines respectively. Hydrogen atoms (apart from those involved in H-bonding and CH⋯π interactions) are omitted for clarity. | ||
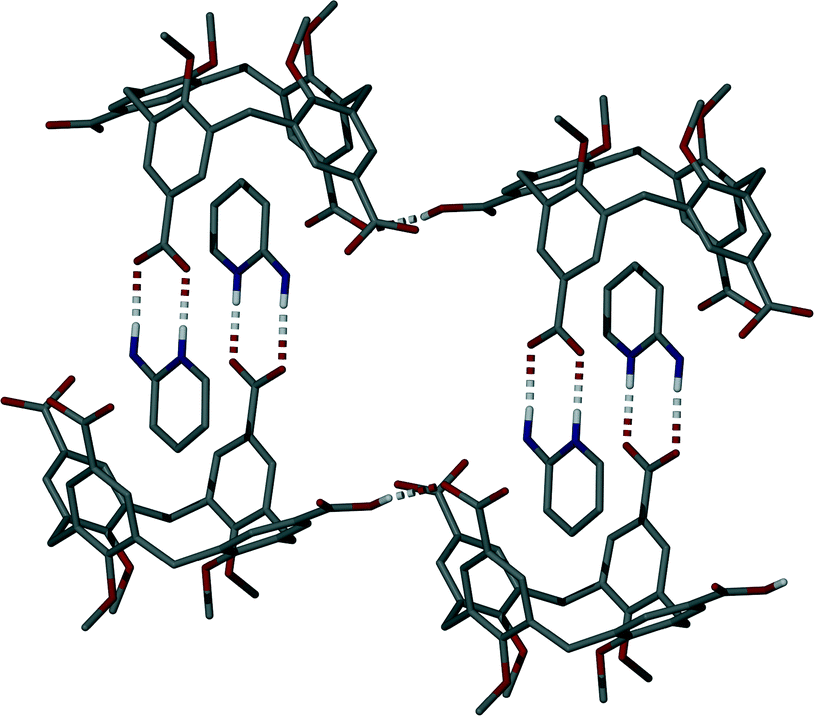 | ||
| Fig. 4 H-bonding interactions (shown as split-colour dashed lines) both within and between neighbouring H-bonded head-to-head capsules in 5. Hydrogen atoms (apart from those involved in H-bonding interactions) are omitted for clarity. | ||
Further symmetry expansion of the asymmetric unit around the splayed/protonated upper-rim CO2H group in 1 reveals an OCOH⋯OCO H-bonding interaction to an upper-rim CO2− group of an s.e. molecule (Fig. 4); this unique OCOH⋯OCO interaction occurs with an H[9]⋯O[12] distance of 1.8 Å. Examination of the extended structure reveals that these capsules, linked by H-bonding interactions, pack in an anti-parallel bi-layer motif (Fig. S2†) akin to several previously reported for the Py derivative solvates of pCO2[4]s.4e The final noteworthy feature of 5 is that the closest distance between C atoms of the upper-rim CO2− groups in 1 was found to be ~7.4 Å. This is significantly greater than values typically observed for the distance between upper-rim CO2H groups in tetra-O-alkyl pCO2[4]s where the lower-rim alkyl chain is greater or equal to propyl in length; the respective distances observed for 3 and 4 are ~4.1 and ~3.7 Å upon crystallisation from Py or the picolines.4e Unfortunately we have been unable to crystallise 1 in a fully pinched-cone conformation to date so it is therefore not possible to make direct structural comparison between neutral and anionic forms, or draw conclusions relating to upper-rim charge repulsion and or associated cationic guest inclusion in this particular case.
Single crystals of the salt (6) formed between 3 and 2-AP were found to be in a triclinic cell and structure solution was performed in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .† The asymmetric unit in 6 is large and comprises two molecules of 3 (with all upper-rim CO2H groups deprotonated), eight molecules of 2-AP (all of which are protonated and one of which is disordered over two positions), two molecules of methanol, six and a half water molecules and a disordered methanol/water of crystallisation (0.33 and 0.67 occupancies respectively). Simple inspection of 6 reveals that each calixarene cavity is occupied by a H2-AP cation, but to differing extents (Fig. 5). In the first of these there are two CH⋯π interactions observed between both the meta hydrogens of the H2-AP and aromatic rings on the anionic host (Fig. 5A); these occur with H[92]⋯aromatic centroid and H[94]⋯aromatic centroid distances of 3.1 and 2.8 Å respectively. As is the case in 5 there is concomitant HNH⋯OCO H-bonding between the cationic guest and an upper-rim CO2− group of the host; this occurs with an H[95B]⋯O[7] distance of 2.2 Å. Any CH⋯π interactions in the second host–guest arrangement can be considered to be very long (Fig. 5B),9 and as a result there is no evidence of analogous HNH⋯OCO H-bonding with the calixarene upper-rim. Rather the H2-AP cation forms an HNH⋯O H-bonding interaction with a water of crystallisation with an NH⋯O distance of 2.1 Å.
.† The asymmetric unit in 6 is large and comprises two molecules of 3 (with all upper-rim CO2H groups deprotonated), eight molecules of 2-AP (all of which are protonated and one of which is disordered over two positions), two molecules of methanol, six and a half water molecules and a disordered methanol/water of crystallisation (0.33 and 0.67 occupancies respectively). Simple inspection of 6 reveals that each calixarene cavity is occupied by a H2-AP cation, but to differing extents (Fig. 5). In the first of these there are two CH⋯π interactions observed between both the meta hydrogens of the H2-AP and aromatic rings on the anionic host (Fig. 5A); these occur with H[92]⋯aromatic centroid and H[94]⋯aromatic centroid distances of 3.1 and 2.8 Å respectively. As is the case in 5 there is concomitant HNH⋯OCO H-bonding between the cationic guest and an upper-rim CO2− group of the host; this occurs with an H[95B]⋯O[7] distance of 2.2 Å. Any CH⋯π interactions in the second host–guest arrangement can be considered to be very long (Fig. 5B),9 and as a result there is no evidence of analogous HNH⋯OCO H-bonding with the calixarene upper-rim. Rather the H2-AP cation forms an HNH⋯O H-bonding interaction with a water of crystallisation with an NH⋯O distance of 2.1 Å.
 | ||
| Fig. 5 Sections of the asymmetric unit in the crystal structure of the salt (6) formed between 3 and 2-AP. Occupation of the calixarene cavities by H2-AP cations is observed to different extents as shown in A and B; this is indicated by the presence of host–guest CH⋯π interactions in the former but not the latter. H-bonding and CH⋯π interactions are shown as split-colour and red dashed lines respectively. Hydrogen atoms (apart from those involved in H-bonding and CH⋯π interactions) have been omitted for clarity. Selected atoms have been labelled according to discussion. | ||
Symmetry expansion of the asymmetric unit around the upper-rim of both calixarenes reveals that, irrespective of the degree of cavity occupation by the H2-AP guests described above, formation of H-bonded head-to-head dimers akin to those found in 5 occurs via formation of the targeted heterosynthon.7 In both cases this is facilitated by analogous HNH⋯OCO and NH⋯OCO H-bonding interactions as shown in Fig. 6 (capsule shown for expansion of the arrangement shown in Fig. 5B). There are two NH⋯OCO hydrogen-bonding interactions between both H-bonded head-to-head dimers with respective H[90]⋯O[6]s.e. and H[97]⋯O[22]s.e. distances of 1.8 and 1.9 Å. Additionally there are two HNH⋯OCO H-bonding interactions with respective H[95A]⋯O[5] and H[10A]⋯O[20] distances of 2.0 and 1.9 Å.
 | ||
| Fig. 6 One H-bonded head-to-head dimer found in the crystal structure of 6. H-bonding interactions are shown as split-colour dashed lines. Hydrogen atoms (apart from those involved in the hydrogen-bonding interactions) are omitted for clarity. | ||
Five of the remaining H2-AP cations in the asymmetric unit (including one of which is disordered over two positions) interact with upper-rim CO2− groups via one HNH⋯OCO and one NH⋯OCO H-bonding interaction in each case. The NH⋯OCO H-bonding interactions have distances ranging from 1.7–1.9 Å, while the HNH⋯OCO interactions range from 1.9–2.0 Å (Fig. 5). The final H2-AP cation in the asymmetric unit interacts with the remaining upper-rim CO2− group via only one NH⋯OCO H-bonding interaction with an H[118]⋯O[10] distance of 1.7 Å (Fig. 5A). The additional solvent of crystallisation (MeOH/H2O) forms a wide range of H-bonding interactions, but inspection shows that none appear to dictate extended assembly in an influential manner (as is the case for the cationic H2-AP template). Examination of the extended structure of 6 reveals that the components assemble as capsules within a wave-like bi-layer motif as shown in Fig. S3.†
As stated above, the average closest distance between C atoms of upper-rim CO2H groups in 3 (when crystallised in neutral form from Py or the picolines) was found to be ~4.1 Å.4e Examination of the analogous distances between C atoms of the upper-rim CO2− groups in 6 reveals that these are significantly greater with respective C[41]⋯C[43] and C[85]⋯C[87] distances of ~6.7 and ~7.1 Å (average distance of ~6.93 Å, Fig. 7).
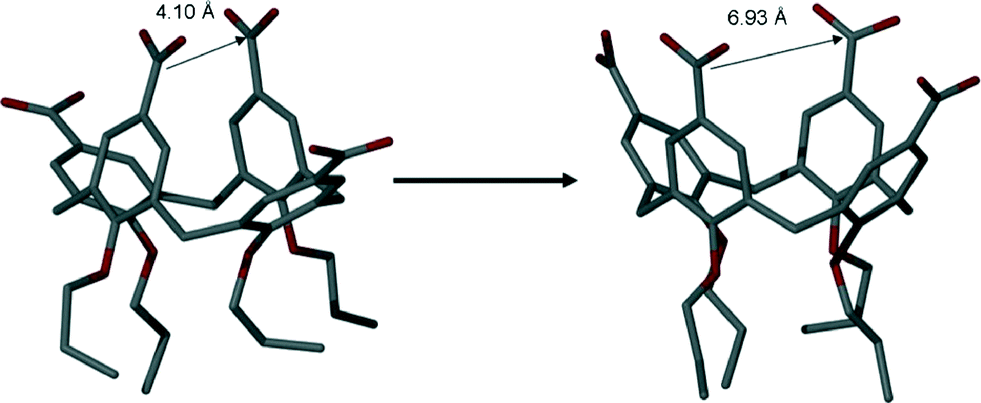 | ||
| Fig. 7 Increase in the average distance between proximal upper-rim carbon atoms in neutral and tetra-anionic 3 (in 6). Hydrogen atoms omitted for clarity. | ||
Single crystals of the salt (7) formed between 4 and 2-AP were found to be in a monoclinic cell and structure solution was performed in the space group C2/c.† The asymmetric unit in 7 comprises one half of a molecule of 4 (with upper-rim CO2H groups deprotonated) and two 2-AP molecules (both of which are protonated, Fig. 8).
 | ||
| Fig. 8 Symmetry expansion of the asymmetric unit in the crystal structure of the salt (7) formed between 4 and 2-AP. H-bonding interactions are shown as split-colour and red dashed lines respectively. Hydrogen atoms (apart from those involved in H-bonding) have been omitted for clarity. Selected atoms have been labelled according to discussion. | ||
The most noticeable feature upon inspection is that the H2-AP cations do not occupy the cavity presented by the calixarene, nor do they force access within this particular structure. One H2-AP cation in 7 forms the targeted heterosynthon7 with the splayed symmetry unique upper-rim calixarene CO2− group via one NH⋯OCO and one HNH⋯OCO H-bonding interaction; this occurs with respective H[25]⋯O[3] and H[31A]⋯O[4] distances of 1.9 Å and 1.7 Å. The remaining H2-AP cation is disordered over two positions and was modelled at 50% occupancy in each. In one position this interacts with the pinched upper-rim calixarene CO2− group via one NH⋯OCO and one HNH⋯OCO hydrogen-bonding interaction; this occurs with respective H[113]⋯O[5] and H[10C]⋯O[6] distances of 1.9 Å and 2.5 Å. Symmetry expansion of the asymmetric unit around the other position reveals two HNH⋯OCO hydrogen-bonding interactions between the NH2 group on the H2-AP cation and both the splayed and pinched upper-rim CO2− groups on the half-calixarene; this occurs with H[10A]⋯O[6] and H[10B]⋯O[3] distances of 2.8 and 2.3 Å. Examination of the extended structure reveals that the components pack in an anti-parallel bi-layer array as shown in Fig. S4.†
The average closest distance between C atoms of upper-rim CO2H groups in 4 (when crystallised in neutral form from Py or the picolines) was found to be ~3.8 Å. Symmetry expansion of the asymmetric unit around the half calixarene in 7 reveals that the proximal upper-rim s.e. carbon atoms are separated by a distance of ~4.8 Å, representing a significant increase in the size of the cavity present (Fig. 9). Given that cavity occupation by a H2-AP cation is not observed, we propose that this may arise from a combination of upper-rim CO2− charge repulsion and intermolecular interactions between neighbouring cations; this is difficult to state for the latter as there is disorder present in this region of the structure.
 | ||
| Fig. 9 Increase in the average distance between proximal upper-rim carbon atoms in neutral and tetra-anionic 4 (in 7). Hydrogen atoms omitted for clarity. | ||
Conclusions
The p-carboxylatocalix[4]arenes have been shown to be versatile supramolecular building blocks in the presence of pyridine (or pyridine derivative) templates through exploitation of a) the Py⋯CO2H heterosynthon and b) the availability of a cavity for template/guest occupation.4a,b,d,f,g Here we have extended this template chemistry to include salt and related heterosynthon formation through the use of 2-aminopyridine. The more basic template fully deprotonated the calixarenes studied in two out of three cases, with the other forming a tri- rather than tetra-anionic building block, a feature possibly relating to poor solubility based on the short lower-rim alkyl group present. Given the observation of cavity accessibility in two cases, or the increase in cleft size relative to the neutral forms of 3 and 4 (structural comparison possible), it is reasonable to assume that the presence of the H2-AP cations markedly influences both conformational and assembly properties of the lower-rim tetra-O-alkyl-p-carboxylatocalixarenes in general. Work continues in further tailoring the assembly behaviour of these useful hosts through targeted heterosynthon formation with a view to controlling the formation of nanoscale spherical and tubular architectures.Acknowledgements
We thank the EPSRC for financial support of this work. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.Notes and references
- For relatively recent reviews on p-sulfonatocalix[4]arene see: J. L. Atwood, L. J. Barbour, M. J. Hardie and C. L. Raston, Coord. Chem. Rev., 2001, 222, 3 CrossRef CAS; S. J. Dalgarno, J. L. Atwood and C. L. Raston, Chem. Commun., 2006, 4567 RSC.
- For examples see: S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC; H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS PubMed; D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC; X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schroder, J. Am. Chem. Soc., 2009, 131, 2159 CrossRef PubMed; Z. Zhang, L. Zhang, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 928 CrossRef PubMed; W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443 CrossRef PubMed; A. Schoedel, A. J. Cairns, Y. Belmabkhout, L. Wojtas, M. Mohamed, Z. Zhang, D. M. Proserpio, M. Eddaoudi and M. J. Zaworotko, Angew. Chem., Int. Ed., 2013, 52, 2902 CrossRef PubMed.
- For example see: J. W. Steed and J. L. Atwood, in Supramolecular Chemistry, John Wiley and Sons, Sussex, UK, 2nd edn, 2009, ch. 8 Search PubMed.
- (a) S. J. Dalgarno, J. E. Warren, J. Antesberger, T. E. Glass and J. L. Atwood, New J. Chem., 2007, 31, 1891 CAS; (b) S. Kennedy and S. J. Dalgarno, Chem. Commun., 2009, 5275 RSC; (c) S. Kennedy, S. J. Teat and S. J. Dalgarno, Dalton Trans., 2010, 39, 384 RSC; (d) S. Kennedy, C. M. Beavers, S. J. Teat and S. J. Dalgarno, New J. Chem., 2011, 35, 28 RSC; (e) S. Kennedy, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2012, 12, 679 CrossRef CAS; (f) S. Kennedy, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2012, 12, 688 CrossRef CAS; (g) S. Kennedy, P. P. Cholewa, R. D. McIntosh and S. J. Dalgarno, CrystEngComm, 2013, 15, 1520 RSC.
- (a) F. A. Cotton, P. Lei, C. Lin, C. A. Murillo, X. Wang, S. Y. Yu and Z. X. Zhang, J. Am. Chem. Soc., 2004, 126, 1518 CrossRef CAS PubMed; (b) S. J. Dalgarno, K. M. Claudio-Bosque, J. E. Warren, T. E. Glass and J. L. Atwood, Chem. Commun., 2008, 1410 RSC; (c) S. Kennedy, G. Karotsis, C. M. Beavers, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Angew. Chem., Int. Ed., 2010, 49, 4205 CrossRef CAS PubMed; (d) S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 785 CrossRef PubMed; (e) S. P. Bew, A. D. Burrows, T. Düren, M. F. Mahon, P. Z. Moghadam, V. M. Sebesteyen and S. Thurston, Chem. Commun., 2012, 48, 4824 RSC; (f) P. P. Cholewa, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Chem. Commun., 2013, 49, 3203 RSC; (g) P. P. Cholewa, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2013, 13, 2703 CrossRef CAS; (h) P. P. Cholewa, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2013, 13, 5165 CrossRef CAS.
- C. D. Gutsche, in Calixarenes, Royal Society of Chemistry, Cambridge, UK, 1989, ch. 4 Search PubMed.
- J. A. Bis and M. J. Zaworotko, Cryst. Growth Des., 2005, 5, 1169 Search PubMed; C. B. Akeröy, A. Rajbanshi, Z. J. Li and J. Desper, CrystEngComm, 2012, 12, 4231 RSC.
- Compound 1 is poorly soluble in most solvents and we propose that the degree of protonation may relate to this.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic Information Files (CIFs) for 5–7 and additional figures for structural description and powder X-ray diffraction. CCDC 971748–971750. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42523a |
‡ Compounds 1–4 were prepared according to literature procedure.4 Synthesis of 5: compound 1 (71 mg, 0.11 mmol) was suspended in methanol (3 ml) and excess 2-aminopyridne (309 mg, 3.3 mmol) was added. The mixture was then heated gently until the suspension became a clear solution. Slow evaporation of the solution over several hours afforded colourless single crystals suitable for diffraction studies in near-quantitative yield. Removal from the mother liquor, followed by drying and powder diffraction studies, showed that the crystals were sensitive to this procedure via significant differences in calculated vs. observed powder patterns. As this was the case three unit cell checks were run on single crystals taken from the sample vial in order to confirm bulk purity. These consistently corresponded to the single crystal parameters listed below for 5. Synthesis of 6: an analogous procedure to that followed for 5 was undertaken, using an equimolar amount of 3 as a starting material and an analogous excess ratio of 2-AP. Slow evaporation of the solution over several hours again afforded single crystals suitable for diffraction studies in near-quantitative yield. Synthesis of 7: an analogous procedure to that followed for 5 was undertaken, using an equimolar amount of 4 as a starting material and an analogous excess ratio of 2-AP. Slow evaporation of the solution over several hours again afforded single crystals suitable for diffraction studies in near-quantitative yield. Removal from the mother liquor, followed by drying and powder diffraction studies, showed that the crystals were stable towards solvent loss via agreement between calculated and observed powder patterns (Fig. S5†). General crystallographic details: data for 5 and 7 were collected on a Bruker Apex II CCD Diffractometer operating at 100(2) K with synchrotron radiation (λ = 0.77490 Å). Data for 6 were collected on a Bruker Apex II CCD Diffractometer operating at 100(2) K with Mo-Kα radiation (λ = 0.71073 Å). Crystal data for 5 (CCDC 971748): C59H67N8O14.5, M = 1120.21, colourless block, 0.11 × 0.06 × 0.04 mm3, monoclinic, space group P21/n, a = 12.2773(9), b = 28.901(2), c = 15.6350(12) Å, β = 93.101(2), V = 5539.5(7) Å3, Z = 4, 2θmax = 50.4°, 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 609 reflections collected, 7704 unique (Rint = 0.0692). Final GooF = 1.011, R1 = 0.0505, wR2 = 0.1342, R indices based on 5616 reflections with I > 2σ(I) (refinement on F2). Crystal data for 6 (CCDC 971749): C130.65H167.30N16O34, M = 2505.90, colourless block, 0.40 × 0.40 × 0.30 mm3, triclinic, space group P, a = 16.5671(14), b = 19.7029(17), c = 20.3946(18) Å, α = 87.763(4), β = 84.240(4), γ = 77.864(3)°, V = 6474.5(10) Å3, Z = 2, 2θmax = 44.3°, 66296 reflections collected, 15780 unique (Rint = 0.0360). Final GooF = 1.893, R1 = 0.0809, wR2 = 0.2388, R indices based on 12285 reflections with I > 2σ(I) (refinement on F2). Crystal data for 7 (CCDC 971750): C68H80N8O12, M = 1201.40, colourless block, 0.12 × 0.06 × 0.01 mm3, monoclinic, space group C2/c, a = 13.1086(4), b = 38.2571(17), c = 12.9509(6) Å, β = 99.220(2), V = 6410.9(5) Å3, Z = 4, 2θmax = 55.3°, 34712 reflections collected, 7377 unique (Rint = 0.1098). Final GooF = 1.389, R1 = 0.1289, wR2 = 0.2471, R indices based on 3229 reflections with I > 2σ(I) (refinement on F2). 609 reflections collected, 7704 unique (Rint = 0.0692). Final GooF = 1.011, R1 = 0.0505, wR2 = 0.1342, R indices based on 5616 reflections with I > 2σ(I) (refinement on F2). Crystal data for 6 (CCDC 971749): C130.65H167.30N16O34, M = 2505.90, colourless block, 0.40 × 0.40 × 0.30 mm3, triclinic, space group P, a = 16.5671(14), b = 19.7029(17), c = 20.3946(18) Å, α = 87.763(4), β = 84.240(4), γ = 77.864(3)°, V = 6474.5(10) Å3, Z = 2, 2θmax = 44.3°, 66296 reflections collected, 15780 unique (Rint = 0.0360). Final GooF = 1.893, R1 = 0.0809, wR2 = 0.2388, R indices based on 12285 reflections with I > 2σ(I) (refinement on F2). Crystal data for 7 (CCDC 971750): C68H80N8O12, M = 1201.40, colourless block, 0.12 × 0.06 × 0.01 mm3, monoclinic, space group C2/c, a = 13.1086(4), b = 38.2571(17), c = 12.9509(6) Å, β = 99.220(2), V = 6410.9(5) Å3, Z = 4, 2θmax = 55.3°, 34712 reflections collected, 7377 unique (Rint = 0.1098). Final GooF = 1.389, R1 = 0.1289, wR2 = 0.2471, R indices based on 3229 reflections with I > 2σ(I) (refinement on F2). |
| This journal is © The Royal Society of Chemistry 2014 |