 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and solid-state structures of a macrocyclic receptor based on the 2,6-bis(2-anilinoethynyl)pyridine scaffold†
Jeffrey M.
Engle
,
Pushpinder S.
Singh
,
Chris L.
Vonnegut
,
Lev N.
Zakharov
,
Darren W.
Johnson
* and
Michael M.
Haley
*
Department of Chemistry & Biochemistry and Materials Science Institute, University of Oregon, Eugene, Oregon 97403-1253, USA. E-mail: haley@uoregon.edu; dwj@uoregon.edu; Fax: +1 541 346 0487; Tel: +1 541 346 0456, +1 541 346 1695
First published on 6th January 2014
Abstract
A fluorescent macrocyclic anion receptor based on the 2,6-bis(2-anilinoethynyl)pyridine scaffold has been synthesized to investigate the mechanism of fluorescence quenching in this class of compounds. X-ray crystallography reveals that the binding pocket of the receptor is a natural host to both H2O and HCl, accommodating either molecule in nearly identical environments. Our studies show that protonation, not collisional quenching, is responsible for the observed fluorescence quenching response.
The seminal crown ether studies of Pedersen in the mid-1960s heralded the importance of the field of artificial macrocyclic ion receptors.1,2 Much of the work in this area has been inspired by nature where many ionophores, such as valinomycin (Fig. 1, left), are preorganized with a specific size and shape to selectively bind ions.3 Since these pioneering discoveries, the field of macrocyclic receptors has expanded rapidly as advanced synthetic techniques, ranging from ring closing metathesis4 to molecular templating,5–7 have permitted access to a wide range of macrocyclic topologies in high yields.
 | ||
| Fig. 1 Structures of valinomycin, a potassium ionophore (left) and a doubly protonated azacryptand binding an anion (right). | ||
Most of the early studies on macrocyclic ionophores focused on cations since they have relatively low energies of hydration, compared to anions, are less polarizable, and have predictable binding geometries. The first examples of macrocyclic receptors for anions (ionophores that are much less explored) appeared during the study of crown ether derivatives. Replacing one or more of the oxygen atoms with a more basic nitrogen afforded aza-crown macrocycles that exhibited a high affinity for anions upon protonation.8 This idea was expanded further by the study of azacryptands, where macrobicyclic, three-dimensional aza-crown ethers were designed to encapsulate specific anions (Fig. 1, right).9,10 While these compounds were of great interest from a fundamental supramolecular standpoint, the early molecules lacked distinct spectral features for their different binding conformations. To have utility as an anion sensor, a receptor should ideally have easy to read spectral changes upon a binding event. Specifically, anion sensors that exhibit a fluorescence response have received considerable attention for their potential use in cellular imaging, pollution/hazard detection, and waste remediation.11–16
Recently our team has focused on the synthesis and binding properties of a family of inherently fluorescent receptors based on the 2,6-bis(anilinoethynyl)pyridine scaffold.17–22 Interestingly, phenylurea derivatives substituted with electron-withdrawing groups (e.g., 1a, Fig. 2) displayed an “off–on” response in the presence of chloride, which typically quenches fluorescence.19,23 We hypothesized that in the absence of solvophobic aggregation, free rotation about the sp–sp2 single bonds resulted in non-radiative decay of the molecule in its unbound state; however, anion binding could restrict rotation and afford increased conjugation via a nearly planar U-shaped conformation (Fig. 2).
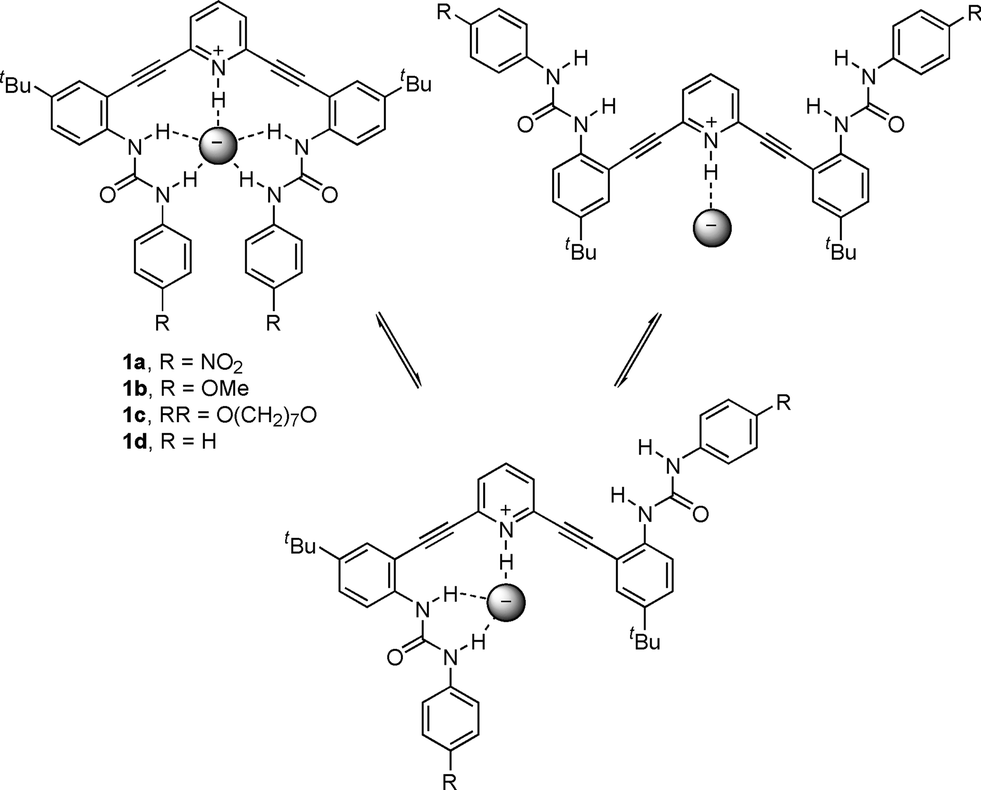 | ||
| Fig. 2 Proposed binding conformations in 2,6-bis(2-anilinoethynyl)-pyridine receptors. | ||
Conversely, anion binding by receptors with electron-donating-substituted phenylureas (e.g., 1b) quenched fluorescence. This is likely associated with the less acidic nature of the urea hydrogens, but the exact mechanism of quenching was unclear. We speculated that a macrocyclic alkoxyphenylurea receptor such as 1c would enforce the U-shaped conformation and thus limit the role of non-radiative decay in the observed fluorescence quenching. We report herein the synthesis of macrocycle 1c, its excitation and emission spectra, and its solid state structure binding either H2O or HCl.
The preparation of receptor 1c is shown in Scheme 1. Williamson ether synthesis between 1,7-dibromoheptane and p-nitrophenol under basic conditions afforded bis-nitrophenyl compound 2, which was reduced via catalytic hydrogenation to give bis-aniline 3. Interestingly, when 3 was treated with triphosgene, yields of the desired bis-phenylisocyanate 4 were poor and unreliable. Counterintuitively, higher yields of 4 were obtained by first protonating 3 with anhydrous HCl followed by the addition of phosgene (as a solution in toluene), presumably due to slower reaction kinetics and reduced oligomerization. Unstable 4 was concentrated under reduced pressure to remove excess phosgene and HCl and subsequently reacted with previously reported 2,6-bis(2-anilinoethynyl)pyridine core 517,22 to afford the desired macrocycle 1c in low yield.
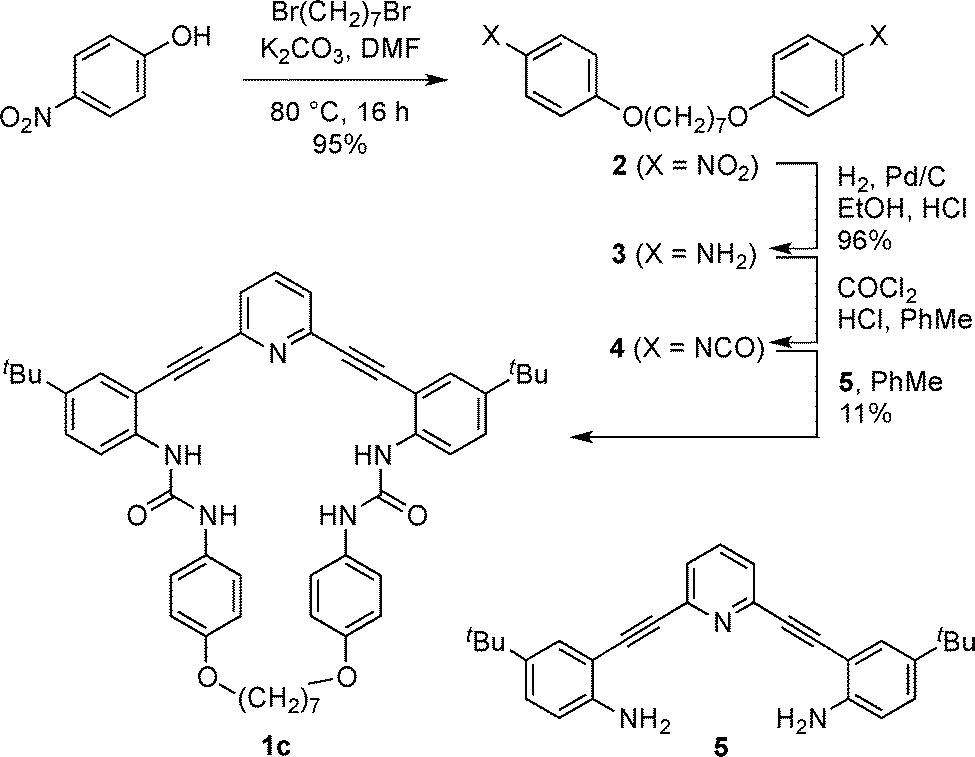 | ||
| Scheme 1 Synthesis of receptor 1c. | ||
Colourless single crystals of 1c were grown via slow evaporation from a CH2Cl2/hexanes solution. The resulting X-ray structure (Fig. 3, top) shows the desired macrocycle with an unanticipated water molecule bound within the cavity.24 The urea hydrogens in each host form a tetradentate hydrogen bond network to the oxygen of the encapsulated water molecule; the pyridine nitrogen and urea carbonyls of neighbouring molecules accept the remaining hydrogen bond from the bound water molecule, resulting in an H-bonded 1-D chain within the crystal structure (Fig. 3, bottom). One of the H atoms from the water molecule and one of the H atoms from a urea group in 1c·H2O make intermolecular O–H⋯O and N–H⋯O H-bonds with O⋯O and N⋯O distances and angles at H atoms of 2.943(3) Å, 2.924(3) Å and 168(5)°, 169(2)°, respectively.
 | ||
| Fig. 3 Front view (top) and representative crystal packing (bottom) of 1c·H2O; non-coordinating H atoms and hexane solvent molecule omitted for clarity; ellipsoids drawn at the 50% probability level. | ||
Gratifyingly, orange crystals of the HCl salt of 1c were also obtained by bubbling HCl gas through a CHCl3 solution of the macrocycle, which was then subjected to slow vapour diffusion of pentane at −25 °C.25 The crystal structure (Fig. 4, top) shows the chloride anion stabilised by five H-bonds, similar to what was observed in the structures of parent phenylurea 1d17 and a benzene-core analogue of 1b;26 however, the urea N–H⋯Cl bond distances (Table 1) in 1c·HCl are all shorter (Δd = 0.023–0.196 Å) than the analogous distances found in 1d·HCl, clearly illustrating the tighter binding of chloride when encapsulated by the macrocycle. This result shows that the binding pocket can accommodate a molecule of either HCl or H2O in the solid state along with minor changes in conformation, analogous to what was observed with our initial class of sulphonamide receptors.20 Nonetheless, without the additional hydrogen bond donor found in the binding pocket of 1c·H2O, the crystal packing of 1c·HCl is dramatically different: dimeric units form with intermolecular interactions dominated by van der Waals' forces (Fig. 4, bottom). The crystal structure of 1c·HCl also includes a CHCl3 solvent molecule that fills the empty space between these dimers.
 | ||
| Fig. 4 Front view (top) and representative crystal packing (bottom) of 1c·HCl; non-coordinating H atoms and solvent molecules omitted for clarity; ellipsoids drawn at the 50% probability level. | ||
| Bond | 1c·HCl | 1d·HClb | Benzene-core analogue of 1b·HClc |
|---|---|---|---|
| a Distances in Å between heavy atoms; atom numbering shown in Fig. 4. b Ref. 17. c Ref. 26. d C1–H1⋯Cl for benzene-core analogue of 1b. | |||
| N1–H1⋯Cl | 3.025(3) | 3.033(3) | 3.579(3)d |
| N2–H2⋯Cl | 3.500(3) | 3.640(3) | 3.523(3) |
| N3–H3⋯Cl | 3.194(3) | 3.227(3) | 3.212(3) |
| N4–H4⋯Cl | 3.194(3) | 3.279(3) | 3.214(3) |
| N5–H5⋯Cl | 3.536(3) | 3.600(3) | 3.732(3) |
The neutral state of macrocycle 1c is fluorescent with an emission band at 400 nm; however, when HCl is added the fluorescence is red shifted and significantly quenched (Fig. 5). Interestingly, if 1c is protonated using an acid containing a non-coordinating counteranion (e.g., HBF4, which typically has a lower quenching proficiency than other anions),27 the fluorescence is quenched to the same degree as with HCl. In line with the observed change in emission spectra, the excitation spectra of both 1c·HCl and 1c·HBF4 are blue shifted from the neutral species 1c by the same degree. These findings suggest that protonation plays the key role in the observed quenching behaviour instead of collisional quenching, as is typically encountered in the presence of halides. Furthermore, bound anions can affect the pKa of adjacent functional groups, indicating that bis(arylethynyl)pyridines tailored to bind a specific anion could selectively trigger a protonation-based quenching response. This could allow for selective sensors that overcome typical Stern–Volmer quenching trends (I− > CNS− > Br− > Cl− > C2O42− > CH3CO2− > SO42− > NO3− > F−). What is still unclear is why electron-withdrawn phenyl ureas display an “off–on” response in the presence of chloride.19,22 One would presume that quenching to some degree would be observed and investigation into this behaviour is ongoing.
 | ||
| Fig. 5 Fluorescence excitation and emission spectra of receptor 1c in CHCl3 solution and the response upon exposure to selected acids. Neutral 1c was excited at 347 nm and the protonated receptors at 300 nm. | ||
In summary, we have disclosed the synthesis of macrocyclic receptor 1c for comparison with its acyclic counterparts. X-ray structural analysis shows that chloride binding in the cavity of 1c affords shorter hydrogen bonds and thus tighter binding within the pocket compared to its acyclic counterpart 1d. The binding pocket of receptor 1c is a natural host to both H2O and HCl, accommodating either one in similar environments. Emission studies indicate that the quenching behaviour observed with electron donating substituents in this class of bis(arylethynyl)pyridine receptors is due to protonation and not collisional quenching. This could allow for selective quenching-based sensors that overcome typical Stern–Volmer quenching trends.
Acknowledgements
This research was supported by the NIH (GM087398), which also funded early stage intellectual property that was licensed by SupraSensor Technologies, a company co-founded by the PIs. J.M.E. and P.S.S. acknowledge the NSF for a GK-12 fellowship (DGE-0742540) and a summer undergraduate STEP fellowship (DUE-0622620), respectively. University of Oregon NMR facilities are supported by a grant from the NSF (CHE-0923589). This publication was made possible, in part, by the Mass Spectrometry Facilities and Services Core of the Environmental Health Sciences Center, Oregon State University, grant no. P30 ES00210, National Institute of Environmental Health Sciences, National Institutes of Health.Notes and references
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- M. C. Rose and R. W. Henkens, Biochim. Biophys. Acta, Gen. Subj., 1974, 372, 426–435 CrossRef CAS.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed.
- S. Anderson and H. L. Anderson, in Templated Organic Synthesis, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 2007, pp. 1–38 Search PubMed.
- T. J. Hubin and D. H. Busch, Coord. Chem. Rev., 2000, 200–202, 5–52 CrossRef CAS.
- Templating, Self-Assembly and Self-Organization, ed. J.-P. Sauvage and M. W. Hosseini, vol. 9, Pergamon, 1st edn, Oxford, 1999 Search PubMed.
- C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431–2432 CrossRef CAS.
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS PubMed.
- S. Kubik, C. Reyheller and S. Stüwe, J. Inclusion Phenom. Macrocyclic Chem., 2005, 52, 137–187 CrossRef CAS.
- S. Mizukami, T. Nagano, Y. Urano, A. Odani and K. Kikuchi, J. Am. Chem. Soc., 2002, 124, 3920–3925 CrossRef CAS PubMed.
- A. Parola, J. Lima, C. Lodeiro and F. Pina, in Fluorescence of Supermolecules, Polymers, and Nanosystems, ed. M. N. Berberan-Santos, Springer, Berlin, 2008, vol. 4, pp. 117–149 Search PubMed.
- T. Gunnlaugsson, M. Glynn, P. Kruger and F. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS.
- C. N. Carroll, J. J. Naleway, M. M. Haley and D. W. Johnson, Chem. Soc. Rev., 2010, 39, 3875–3888 RSC.
- L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993–1017 RSC.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- C. N. Carroll, O. B. Berryman, C. A. Johnson II, L. N. Zakharov, M. M. Haley and D. W. Johnson, Chem. Commun., 2009, 2520–2522 RSC.
- C. A. Johnson, O. B. Berryman, A. C. Sather, L. N. Zakharov, M. M. Haley and D. W. Johnson, Cryst. Growth Des., 2009, 9, 4247–4249 CAS.
- C. N. Carroll, B. A. Coombs, S. P. McClintock, C. A. Johnson II, O. B. Berryman, D. W. Johnson and M. M. Haley, Chem. Commun., 2011, 47, 5539–5541 RSC.
- O. B. Berryman, C. A. Johnson II, L. N. Zakharov, M. M. Haley and D. W. Johnson, Angew. Chem., Int. Ed., 2008, 47, 117–120 CrossRef CAS PubMed.
- J. M. Engle, P. S. Lakshminarayanan, C. N. Carroll, L. N. Zakharov, M. M. Haley and D. W. Johnson, Cryst. Growth Des., 2011, 11, 5144–5152 CAS.
- J. M. Engle, C. N. Carroll, D. W. Johnson and M. M. Haley, Chem. Sci., 2012, 3, 1105–1110 RSC.
- In Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer, New York, 2006, pp. 277–330 Search PubMed.
- Crystallographic data for 1c·H2O (CCDC 968268): C53H62N5O5 [(C50H53N5O4)(H2O)0.5(C6H14)], M = 849.08, 0.19 × 0.17 × 0.07 mm, T = 193(3) K, orthorhombic, space group Pbca, a = 24.0752(18) Å, b = 9.9609(7) Å, c = 45.509(3) Å, V = 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 913.6(14) Å3, Z = 8, Dc = 1.034 Mg m−3, μ = 0.067 mm−1, F(000) = 3640, 2θmax = 50.00°, 59833 reflections, 9605 independent reflections [Rint = 0.0709], R1 = 0.0636, wR2 = 0.1759 and GOF = 1.010 for 9605 reflections (699 parameters) with I > 2σ(I), R1 = 0.1058, wR2 = 0.2000 and GOF = 1.014 for all reflections, max/min residual electron density +0.309/−0.219 e Å3.
913.6(14) Å3, Z = 8, Dc = 1.034 Mg m−3, μ = 0.067 mm−1, F(000) = 3640, 2θmax = 50.00°, 59833 reflections, 9605 independent reflections [Rint = 0.0709], R1 = 0.0636, wR2 = 0.1759 and GOF = 1.010 for 9605 reflections (699 parameters) with I > 2σ(I), R1 = 0.1058, wR2 = 0.2000 and GOF = 1.014 for all reflections, max/min residual electron density +0.309/−0.219 e Å3. - Crystallographic data for 1c·HCl (CCDC 968267): C56H67Cl4N5O4 [(C50H53N5O4)(HCl)(HCCl3)(C5H12)], M = 1015.95, 0.19 × 0.06 × 0.02 mm, T = 100(2) K, monoclinic, space group P21/n, a = 14.6699(8) Å, b = 13.9019(8) Å, c = 27.0087(15) Å, β = 102.838(3)°, V = 5370.4(5) Å3, Z = 4, Dc = 1.257 Mg m−3, μ = 2.392 mm−1, F(000) = 2152, 2θmax = 135.66°, 37694 reflections, 9580 independent reflections [Rint = 0.0531], R1 = 0.0891, wR2 = 0.2627 and GOF = 1.059 for 9580 reflections (577 parameters) with I > 2σ(I), R1 = 0.1023, wR2 = 0.2771 and GOF = 1.059 for all reflections, max/min residual electron density +1.824/−1.053 e Å3.
- B. W. Tresca, L. N. Zakharov, C. N. Carroll, D. W. Johnson and M. M. Haley, Chem. Commun., 2013, 49, 7240–7242 RSC.
- J. T. Stock, Bull. Hist. Chem., 2002, 27, 57–61 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data and X-ray analysis of receptor. CCDC 968267 & 968268. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/ccce42307g |
| This journal is © The Royal Society of Chemistry 2014 |